

Abstract
The PROM1 (prominin 1) gene encodes an 865-amino acid glycoprotein that is expressed in retinoblastoma cell lines and in the adult retina. The protein is localized to photoreceptor outer segment disc membranes, where it plays a structural role, and in the retinal pigment epithelium (RPE), where it acts as a cytosolic protein that mediates autophagy. Mutations in PROM1 are typically associated with cone-rod dystrophy 12 (OMIM#3612657), autosomal dominant retinal macular dystrophy 2 (OMIM#608051), autosomal recessive retinitis pigmentosa 41 (OMIM#612095), and Stargardt disease 4 (OMIM#603786). Here we describe the first case of PROM1-associated Leber congenital amaurosis (LCA) in a 12-yr-old Asian male, caused by two not previously described deleterious frameshift variants in the compound heterozygous state. Clinical features include the presence of bull's eye maculopathy, pendular horizontal nystagmus, and photodysphoria consistent with the clinical diagnosis of LCA. The patient was evaluated using ophthalmic imaging, electroretinography, and whole-exome sequencing. Electroretinography revealed extinguished retinal activity.
Keywords: congenital horizontal nystagmus, congenital visual impairment, severe visual impairment
INTRODUCTION
Leber congenital amaurosis (LCA; OMIM#204000) is one of the most common causes of childhood blindness, accounting for 5% of all inherited retinal dystrophies and occurring in 2–3 out of 100,000 newborns (Koenekoop et al. 2004). It is characterized by the onset of severe visual impairment at birth or during infancy and is often accompanied by nystagmus, sluggish pupils, and severely abnormal or nondetectable electroretinogram (ERG) (Koenekoop et al. 2004; Thompson et al. 2017). LCA is phenotypically and genetically heterogeneous, with as many as 24 known genes involved in its pathogenesis (Thompson et al. 2017). Understanding the underlying genetic basis of LCA is therefore crucial to determining an appropriate and effective treatment course, including potential gene-specific, pharmacological, or pharmacogenetic therapies and interventions (Kumaran et al. 2017).
Mutations in PROM1 (NM_006017.2; OMIM#604365) have been documented in the literature as manifesting in myriad complications involving the visual system and specifically result in inherited retinal dystrophies such as autosomal dominant and autosomal recessive retinitis pigmentosa (RP) and cone-rod dystrophy (Michaelides et al. 2010). In this study, we identify two novel frameshift variants in PROM1 and expand the phenotypes of PROM1-associated disease to include the clinical manifestation of LCA.
RESULTS
A 12-yr-old male of Asian descent was referred to the Edward S. Harkness Eye Institute at Columbia University Irving Medical Center because of suspected presence of rod-cone dystrophy. The patient presented with slight photodysphoria and nystagmus beginning at the age of 2 yr and also experienced poor vision at the age of 3 yr and began wearing glasses at this time. He demonstrated a history of continually decreased visual acuity. His medical history was unremarkable with the exception of visual abnormalities. There was no known family history of rod-cone dystrophy or other eye diseases. The patient had not been taking any ocular or systemic medications. Upon examination, the patient's best-corrected visual acuity was 20/80 in the right eye (OD) and 20/70 in the left eye (OS). Pendular horizontal nystagmus was observed. When examined, anterior segments appeared to be quiet, and the lenses appeared clear. Intraocular tension was 15 mmHg (OD) and 14 mmHg (OS). The patient's dilated fundus examination demonstrated that the optic nerves had good rims. An extensive mottled appearance outside the maculae and a bull's eye pattern of continuous atrophy in the maculae were observed (Fig. 1).
Figure 1.

Retinal phenotype of a patient with Leber congenital amaurosis (LCA) due to compound heterozygous mutations in the PROM1 gene. (A) Wide-angle fundus photograph of the right and left eyes showing thinning of the vessels. No pigmentary clumps were seen. (B) Short-wavelength autofluorescence images showing absent signal on the periphery and a bull's eye appearance at the fovea. (C) SD-OCT unveils a blurred ellipsoid zone line at the fovea that is absent on the periphery in both eyes. The internal layers are well delineated.
Full-field electroretinography (ffERG) demonstrated that scotopic rod-specific responses, maximum ERG, photopic 30-Hz flicker ERG, and transient photopic ERG were extinguished (Fig. 2).
Figure 2.

Electroretinogram testing. Scotopic and photopic responses were extinguished in both eyes.
The patient's presentation of early-onset nystagmus, poor vision, and extinguished ERG were consistent with a form of LCA. The patient was diagnosed with LCA at the age of 12 yr old with ERG, imaging, genetic testing, and ophthalmic examination conducted at this same time. Whole-exome sequencing results from the patient and his parents revealed that the patient was compound heterozygous for two deleterious variants, c.139del:p.His47Ilefs*12 and c.1877_1878del:p.Ile626Argfs*6, in the PROM1 gene. The c.139del variant was inherited maternally, and the c.1877_1878del variant was inherited paternally (Table 1; Fig. 3).
Table 1.
Summary of the variants reported in this study
| Gene | Chromosome | HGVS DNA reference | HGVS protein reference | Variant type | Predicted effect (substitution, deletion, etc.) | dbSNP/dbVar ID | Genotype (heterozygous/homozygous) | Sample | Percentage of reads aligned | Average read coverage | Percentage of SLC52A 2 sites (1354 bases) with ≥10-fold coverage |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PROM1 | Chr 4: 16077391delG (GRCh37/hg19) | c.139del | p.His47Ilefs*l2 | Frameshift | Frameshift deletion | rs747512450 | Heterozygous | Proband | 99.003 | 181.53 | 98.936 |
| PROM1 | Chr 4: 15993904delTA (GRCh37/hg19) | c.1877_1878del | p.Ile626Argfs*6 | Frameshift | Frameshift deletion | rs1300041533 | Heterozygous |
Figure 3.
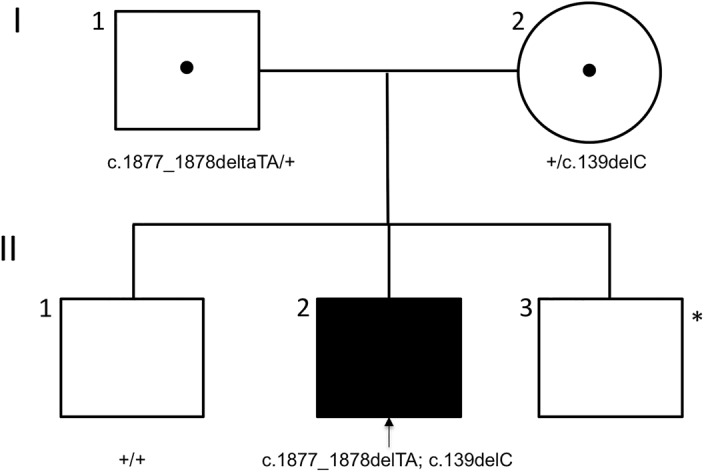
The affected patient's inheritance of two heterozygous frameshift variants in the PROM1 gene. Family pedigree with arrow indicating the proband. Black symbol represents clinically affected subject. Black dot in center of symbols represents carriers. An asterisk indicates that this subject was not genetically tested.
DISCUSSION
In this report, we present a unique case of an individual with two not previously described deleterious variants in the PROM1 gene that cause LCA. The patient's early childhood onset of severe visual impairment, along with his clinical presentation of nystagmus and extinguished ffERG, were consistent with the symptoms of LCA. Differential diagnoses of severe early childhood–onset retinal dystrophy (SECORD) and early-onset RP were eliminated based on the patient's extinguished ffERG results, as ffERG is typically detectable (although diminished) in patients with SECORD or early-onset RP (Kumaran et al. 2017).
Whole-exome sequencing analysis of the affected individual identified two compound heterozygous frameshift variants, c.139del:p.His47Ilefs*12 in exon 1 and c.1877_1878del:p.Ile626Argfs*6 in exon 16, in PROM1 (NM_006017.2; OMIM#604365). Among the 27 total exons in PROM1, the c.139del variant was found to replace the His47 residue with Ile in the extracellular amino-terminal E1 domain in exon 1, whereas the c.1877_1878del variant replaced the Ile626 residue with Arg in the extracellular large glycosylated E2 domain in exon 16 (Fig. 4). Both changes result in the premature termination of translation and are predicted to undergo nonsense-mediated decay, leading to the loss of PROM1 protein.
Figure 4.

Deleterious frameshift variants in the PROM1 protein. Structural representation of the extracellular amino-terminal E1 domain in exon 1 and the Ile626 residue with Arg in the extracellular large glycosylated E2 domain with two residues affected by frameshift variants in our patient in red.
Previous studies have demonstrated that PROM1 may play an important role in photoreceptor morphology and autophagy in retinal pigment epithelium (RPE) cells. For example, PROM1 mutations in CHO cells have been suggested to impair synthesis of evaginations at the outer segments of rod photoreceptors or impair conversion of these evaginations to disks (Maw et al. 2000). In the RPE, PROM1 may be involved in mediating autophagy by impeding mTOR signaling and acting with p62 and HDAC6 in a macromolecular scaffold implicated in autophagosome maturation and trafficking (Bhattacharya et al. 2017). Thus, loss or absence of PROM1 caused by truncating mutations may disrupt these processes and lead to aberrant signaling that results in retinal disease. However, the mechanism by which PROM1 causes LCA specifically, rather than RP or other retinal dystrophies, has not yet been investigated and is currently unknown.
To our knowledge, this is the first report to associate mutations in the PROM1 gene with the LCA phenotype. Autosomal recessive inheritance of PROM1 variants is associated with RP, cone-rod dystrophy and RP with macular degeneration and autosomal dominant inheritance of PROM1 variants is associated with cone-rod dystrophy, Stargardt-like macular dystrophy, and bull's eye macular dystrophy (Zhang et al. 2007; Yang et al. 2008; Eidinger et al. 2015; Wawrock et al. 2018; Liang et al. 2019). For example, Permanyer et al. (2010) reports two patients with biallelic truncating mutations in PROM1 that manifest in severe RP. These patients differ from our patient because of the absence of nystagmus in one patient, and both cases report RP symptoms beginning in adulthood, whereas our patient presented with pendular horizontal nystagmus and vision loss starting at the age of 2–3 yr, as is consistent with the diagnosis of LCA (Permanyer et al. 2010). Other reports have also found that missense PROM1 mutations are linked to autosomal dominant Stargardt-like or bull's eye macular dystrophy, whereas nonsense and frameshift mutations result in RP, severe cone-rod dystrophy with macular degeneration, and night blindness (Permanyer et al. 2010). Our study highlights two novel PROM1 variants that expand the genetic basis of LCA and the possible clinical presentations that can be caused by PROM1 mutations. These findings may influence future gene-based therapies for LCA as well as pave the way for mechanistic studies that elucidate the pathogenesis of PROM1-mediated LCA.
METHODS
Genetic Analyses
Whole-exome sequencing was performed on peripheral blood obtained from the affected individual. Agilent SureSelectXT Human All Exon V5 + UTRs capture and Illumina HiSeq2500 sequencing technology were used for the detection of pathogenic mutations, which were analyzed with the NextGENe software from SoftGenetics and the proprietary analytical pipeline of the Laboratory of Personalized Genomic Medicine (PGM), Department of Pathology and Cell Biology, Columbia University.
This test was developed and its performance characteristics determined by the Molecular Pathology Laboratory of Columbia University. The test methodology has been validated in-house, and the methodology and validation data have been reviewed by the Clinical Laboratory Evaluation Program of the New York State Department of Health. The laboratory also participates in interlaboratory testing under the auspices of the College of American Pathologists, in keeping with the Clinical Laboratory Improvement Amendments of 1988 (CLIA 88). Therefore, this test is used for clinical purposes. It should not be regarded as investigational or for research on this basis alone.
Electroretinography
ffERG (Diagnosys, LLC) was performed with Dawson, Trick, and Litzkow (DTL) fiber electrodes and Ganzfield stimulation according to international standards. Additionally, recordings were obtained for our patient for both eyes in accordance with the International Society for Clinical Electrophysiology of Vision (ISCEV) guidelines in both the scotopic and photopic states (McCulloch et al. 2015). The amplitudes and implicit times recorded for each eye of the patient were compared to age-matched control patients with normal values.
Imaging
All imaging procedures were performed according to the methodology described by Sujirakul et al. (2015). The patient's eyes were dilated and then ophthalmic assessment including funduscopic examination, digital fundus photography, and both fundus short-wavelength autofluorescence (SW-AF) and spectral domain optical coherence tomography (SD-OCT) imaging were performed. SW-AF (488-nm wavelength stimulus, barrier filtered transmitted light from 500 to 680 nm, 55° × 55° field) and SD-OCT images were acquired, utilizing a confocal scanning laser ophthalmoscope (cSLO; Spectralis HRA + OCT, Heidelberg Engineering). The patient's eyes were dilated by administering 1% tropicamide and 2.5% phenylephrine. SD-OCT images were obtained as horizontal 9 × 9-mm scans (870-nm light source and 7-µM axial resolution) positioned through the macula and obtained in high-resolution mode. The scans were recorded automatically by the use of recorded IR-R (820-nm light source) fundus images.
ADDITIONAL INFORMATION
Data Deposition and Access
The association between these two variants and LCA described in this study has been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). The NM_006017.2: p.(His47Ilefs*12) frameshift variant can be accessed through dbSNP rs148234606 and ClinVar SCV000996524. The NM_:p.(p.Ile626Argfs*6) frameshift variant can be accessed through dbSNP rs375088539 and ClinVar SCV000996523.
Ethics Statement
The family consented to this genetic study and gave permission for the publication of genetic and clinical results. A retrospective analysis of one patient diagnosed with LCA who presented to the Department of Ophthalmology at Edward S. Harkness Eye Institute, Columbia University was performed. The study followed the tenets of the Declaration of Helsinki and was approved by the Institutional Review Board at Columbia University Medical Center (protocol #AAAR0284). As per a retrospective study with no more than a minimal risk for the subject, this study did not require written consent by IRB policies.
Acknowledgments
Funding for this research was supported by the Global Ophthalmology Awards Program (GOAP), a Bayer-sponsored initiative committed to supporting ophthalmic research across the world. The Jonas Children's Vision Care and the Bernard & Shirlee Brown Glaucoma Laboratory are supported by the National Institutes of Health (P30EY019007, R01EY018213, R01EY024698, R01EY026682, R21AG050437), the National Cancer Institute Core (5P30CA013696), the Foundation Fighting Blindness (TA-NMT-0116-0692-COLU), the Research to Prevent Blindness (RPB) Physician-Scientist Award, and unrestricted funds from RPB, New York, New York. S.H.T. is a member of the RD-CURE Consortium and is supported by Kobi and Nancy Karp, the Crowley Family Fund, the Rosenbaum Family Foundation, the Tistou and Charlotte Kerstan Foundation, the Schneeweiss Stem Cell Fund, New York State (C029572), and the Gebroe Family Foundation.
Competing Interest Statement
The authors have declared no competing interest.
REFERENCES
- Bhattacharya S, Jinggang Y, Winborn CS, Zhang Q, Yue J, Chaum E. 2017. Prominin-1 is a novel regulator of autophagy in the human retinal pigment epithelium. Invest Ophthalmol Vis Sci 58: 2366–2387. 10.1167/iovs.16-21162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eidinger O, Leibu R, Newman H, Rizel L, Perlman I, Ben-Yosef T. 2015. An intronic deletion in the PROM1 gene leads to autosomal recessive cone-rod dystrophy. Mol Vis 8: 1295–1306. [PMC free article] [PubMed] [Google Scholar]
- Koenekoop RK. 2004. An overview of Leber congenital amaurosis: a model to understand human retinal development. Surv Ophthalmol 49: 379–398. 10.1016/j.survophthal.2004.04.003 [DOI] [PubMed] [Google Scholar]
- Kumaran N, Moore AT, Welebe RG, Michaelides M. 2017. Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Br J Ophthalmol 101: 1147–1154. 10.1136/bjophthalmol-2016-309975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, She X, Chen J, Zhai Y, Liu Y, Zheng K, Gong Y, Zhu H, Luo X, Sun X. 2019. Identification of novel PROM1 mutations responsible for autosomal recessive maculopathy with rod-cone dystrophy. Graefes Arch Clin Exp Ophthalmol 257: 619–628. 10.1007/s00417-018-04206-w [DOI] [PubMed] [Google Scholar]
- Maw MA, Corbeil D, Koch J, Hellwig A, Wilson-Wheeler JC, Bridges RJ, Kumaramanickavel G, John S, Nancarrow D, Roper K, et al. 2000. A frameshift mutation in prominin (mouse)-like 1 causes human retinal degeneration. Hum Mol Genet 9: 27–34. 10.1093/hmg/9.1.27 [DOI] [PubMed] [Google Scholar]
- McCulloch DL, Marmor MF, Brigell MG, Hamilton R, Holder GE, Tzekov R, Bach M. 2015. ISCEV standard for full-field clinical electroretinography (2015 update). Doc Opthalmol 130: 1–12. 10.1007/s10633-014-9473-7 [DOI] [PubMed] [Google Scholar]
- Michaelides M, Gaillard MC, Escher P, Tiab L, Bedell M, Borruat FX, Barthelmes D, Carmona R, Zhang K, White E, et al. 2010. The PROM1 mutation p.R373C causes an autosomal dominant bull's eye maculopathy associated with rod, rod-cone, and macular dystrophy. Invest Ophthalmol Vis Sci 51: 4771–4780. 10.1167/iovs.09-4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Permanyer J, Navarro R, Friedman J, Pomares E, Castro-Navarro J, Marfany G, Swaroop A, Gonzàlez-Duarte R. 2010. Autosomal recessive retinitis pigmentosa with early macular affectation caused by premature truncation in PROM1. Invest Ophthalmol Vis Sci 51: 2656–2663. 10.1167/iovs.09-4857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sujirakul T, Davis R, Erol D, et al. Bilateral concordance of the fundus hyperautofluorescent ring in typical retinitis pigmentosa patients. 2015. Ophthalmic Genet 56: 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JA, De Roach JN, McLaren TL, Montgomery HE, Hoffmann LH, Campbell IR, Chen FK, Mackey DA, Lamey TM. 2017. The genetic profile of Leber congenital amaurosis in an Australian cohort. Mol Genet Genomic Med 5: 652–667. 10.1002/mgg3.321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawrocka A, Skorczyk-Werner A, Wicher K, Niedziela Z, Ploski R, Rydzanicz M, Sykulski M, Kociecki J, Weisschuh N, Kohl S, et al. 2018. Novel variants identified with next-generation sequencing in Polish patients with cone-rod dystrophy. Mol Vis 24: 326–339. [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Chen Y, Lillo C, Chien J, Yu Z, Michaelides M, Klein M, Howes KA, Li Y, Kaminoh Y, et al. 2008. Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J Clin Investig 118: 2908–2916. 10.1172/JCI35876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Zulfiqar F, Xiao X, Riazuddin SA, Ahmad Z, Caruso R, MacDonald I, Sieving P, Riazuddin S, Hejtmancik JF. 2007. Severe retinitis pigmentosa mapped to 4p15 and associated with a novel mutation in the PROM1 gene. Hum Genet 122: 293–299. 10.1007/s00439-007-0395-2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The association between these two variants and LCA described in this study has been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). The NM_006017.2: p.(His47Ilefs*12) frameshift variant can be accessed through dbSNP rs148234606 and ClinVar SCV000996524. The NM_:p.(p.Ile626Argfs*6) frameshift variant can be accessed through dbSNP rs375088539 and ClinVar SCV000996523.