

Abstract
Diagnostic exome sequencing yields a single genetic diagnosis in ∼30% of cases, and according to recent studies the prevalence of identifying two genetic conditions in a single individual range between 4.6% and 7%. We present a patient diagnosed with three different rare conditions, each explained by a pathogenic variant in a different gene. A 17-yr-old female was evaluated for a history of motor and speech delay, scoliosis, distinctive craniofacial features, and dry skin in the Department of Clinical Genomics at Mayo Clinic. Her distinctive features included prominent forehead, epicanthus, depressed nasal bridge, narrow mouth, prognathism, malar flattening, and oligodontia. Family history was notable for dry skin in her mother and missing teeth in the paternal grandmother. Previous diagnostic testing was unrevealing including biochemical testing, echocardiogram, abdominal ultrasound, and electroencephalogram. Previous genetic testing included a microarray-based comparative genomic hybridization that was reported normal. Three pathogenic loss-of-function heterozygous variants were identified by exome trio sequencing, each linked to different genetic conditions: SIN3A (Witteveen–Kolk syndrome), FLG (dermatitis), and EDAR (ectodermal dysplasia). Together, these three genetic alterations could explain the patient's overall phenotype. This patient demonstrates the importance of performing a thorough curation of exome data when presented with a complex phenotype. Although phenotypic variability can explain some of these situations, the hypothesis of multiple diseases coexisting in a single patient should never be disregarded completely.
Keywords: abnormality of the eyebrow, abnormality of the eyelashes, attention deficit hyperactivity disorder, autism, central hypotonia, cervical ribs, depressed nasal bridge, dry skin, high forehead, hypodontia, joint laxity, narrow mouth, perioral eczema, prominent epicanthal folds, short stature, thickened ears, thoracic scoliosis
CASE PRESENTATION
A 17-yr-old Caucasian female presented to the Department of Clinical Genomics at Mayo Clinic for an evaluation of a history of motor and speech delay, scoliosis, and recurrent rash. The patient's neonatal and perinatal history was unremarkable with the exception of mild bilateral hip clicks noted at birth. History of gross motor and speech delay was noted with the patient walking and pronouncing her first words at 18 mo. A reevaluation of the patient's speech comprehension at Mayo Clinic at age 14 showed weaknesses in attention processes. The patient also met the criteria for attention deficit hyperactivity disorder (ADHD)-inattentive subtype based on the Conners Parent Rating Scale-Revised (T = 82), the Conners ADHD Index (T = 74), and the BASC Parent Report (T = 74). As such, it was thought that the patient's speech and learning difficulties were due to inattentiveness and ADHD rather than cognitive impairment. Also reported was decreased muscle tone, daytime enuresis, and rashes that were refractory to treatment. The patient displayed distinctive craniofacial features (Fig. 1A) not noted in the parents or siblings (Table 1; Fig. 1D). Skeletal findings were significant for short stature, bilateral cervical ribs at C7 resulting in 13 pairs of matched ribs, and progressive scoliosis (Fig. 1B). At age 12, the patient underwent a posterior spinal fusion with instrumentation and bone grafting to correct a left thoracolumbar curve of 70°. She had several dental anomalies with narrow palate, frequent cavities, class III malocclusion, missing permanent teeth (Nos. 7 and 10), and impacted molars (Nos. 17 and 32) (Fig. 1C).
Figure 1.
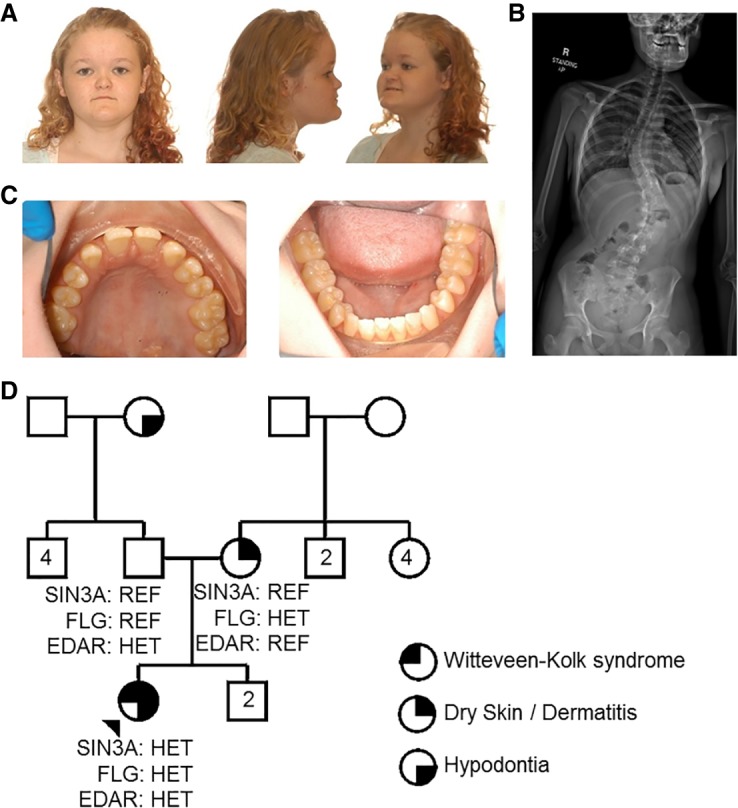
(A) Dysmorphic features and (B) scoliosis presented by the patient compatible with Witteveen–Kolk syndrome caused by pathogenic variants in SIN3A. (C) Patient's dentition abnormalities and hypodontia compatible with ectodermal dysplasia caused by pathogenic variants in EDAR. (D) Pedigree summarizing major genetic and phenotypic findings in the proband (arrowhead) and her family. Only the proband and her parents were genetically tested. (HET) Heterozygous, (REF) homozygous reference.
Table 1.
Clinical findings
| Features | Proband | Mother | Father | Comments |
|---|---|---|---|---|
| Witteveen–Kolk syndrome clinical featuresa | ||||
| Short stature | Yes | No | No | |
| Low birth weight | No | No | No | |
| Obesity | No | No | No | |
| Intrauterine growth retardation | No | No | No | |
| Poor postnatal growth | Yes | No | No | |
| Prominent forehead | Yes | No | No | |
| Microcephaly | No | No | No | |
| Long philtrum | No | No | No | |
| Smooth philtrum | No | No | No | |
| Microretrognathia | No | No | No | |
| Facial asymmetry | No | No | No | |
| Long face | No | No | No | |
| Large ears | Yes | No | No | |
| Cupped ears | No | No | No | |
| Hypertelorism | No | No | No | |
| Downslanting palpebral fissures | No | No | No | |
| Strabismus | No | No | No | |
| Microphthalmia, mild | No | No | No | |
| Epicanthus | Yes | No | No | |
| Hypopigmentation of the iris | No | No | No | |
| Prominent nasal bridge | No | No | No | |
| Wide nasal bridge | No | No | No | |
| Depressed nasal bridge | Yes | No | No | |
| Enlarged naris | No | No | No | |
| Short nose | No | No | No | |
| Narrow mouth | Yes | No | No | |
| Thick lower lip | No | No | No | |
| High palate | No | No | No | |
| Open mouth | No | No | No | |
| Diaphragmatic hernia | No | No | No | |
| Hypospadias | N/A | N/A | No | |
| Microphallus | N/A | N/A | No | |
| Cryptorchidism | No | No | No | |
| Joint laxity | Yes | No | No | |
| Scoliosis | Yes | No | No | |
| Brachydactyly | No | No | No | |
| Partial absence of thumbs | No | No | No | |
| Proximally placement of thumbs | No | No | No | |
| Clinodactyly | No | No | No | |
| Long, slender fingers | No | No | No | |
| Abnormal insertion of the toes | No | No | No | |
| Sparse scalp hair | No | No | No | |
| High anterior hairline | No | No | No | |
| Broad eyebrows | Yes | No | No | |
| Loose connective tissue | No | No | No | |
| Delayed development | Yes | No | No | |
| Expressive speech deficiency | Yes | No | No | |
| Intellectual disability, mild | No | No | No | |
| Mental retardation | No | No | No | |
| Hypotonia | Yes | No | No | |
| Seizures (rare) | No | No | No | |
| Cortical abnormalities | N/T | N/T | N/T | |
| Enlarged ventricles | N/T | N/T | N/T | |
| Thin corpus callosum | N/T | N/T | N/T | |
| Attention deficit | Yes | No | No | |
| Hyperactivity | No | No | No | |
| Autistic features | Yes | No | No | |
| Aggression | No | No | No | |
| Growth hormone deficiency (2 patients) | No | No | No | |
| Hypogonadotropic hypogonadism (1 patient) | No | No | No | |
| Ectodermal dysplasia 10A clinical featuresb | ||||
| Oligodontia | Yes | No | No | Paternal grandmother reported to present hypodontia as well. |
| Microdontia | No | No | No | |
| Misshapen teeth | No | No | No | |
| Hypohidrosis | No | No | No | |
| Smooth, thin, dry skin | No | No | No | |
| Onychodysplasia (40%) | No | No | No | |
| Hypotrichosis | No | No | No | |
| Fine, slow-growing hair | No | No | No | |
| Sparse eyebrows and eyelashes | Yes | No | No | |
| Intolerance to heat and fever | No | No | No | |
| Other clinical features | ||||
| Perioral dermatitis | Yes | Yes | No | This is consistent with reported milder manifestations reported in heterozygous carriers of FLG variants. |
| Dry skin | Yes | Yes | No | |
| Bilateral cervical ribs at C7 resulting in 13 pairs of matched ribs | Yes | No | No | This is probably related to the patient's Witteveen–Kolk syndrome. |
Phenotypes detected in the proband and her parents as reported in previous diagnosed patients.
(N/T) Not tested, (N/A) not applicable.
aThe list of clinical features is based on the OMIM clinical synopsis (#613406, Witteveen–Kolk syndrome).
bThe list of clinical features is based on the OMIM clinical synopsis (#129490, Ectodermal dysplasia 10A).
The family history was remarkable (Fig. 1D) with dry skin noted in the proband's mother and absent upper lateral incisor on one side reported in the paternal grandmother. Neither the proband nor the mother was noted to have asthma, though the proband was noted to have more frequent and prolonged respiratory illnesses in comparison with her siblings. Other laboratory testing performed on the patient, including biochemical testing, echocardiogram, abdominal ultrasound, and electroencephalogram, were unremarkable. Previous genetic testing included a microarray-based comparative genomic hybridization that was reported normal.
TECHNICAL ANALYSIS
Exome sequencing was performed by GeneDx using CLIA standards (XomeDx; https://www.genedx.com/test-catalog/available-tests/xomedx-whole-exome-sequencing-trio/). A proprietary capture system was used and targets were sequenced with paired-end reads on an Illumina platform and aligned to GRCH37/UCSC hg19. Sequence variants and deletions or duplications involving three or more coding exons were called with XomeAnalyzer, an analysis software developed by GeneDX (Retterer et al. 2015). Variants were confirmed by capillary sequencing and reported according to the Human Genome Variation Society (HGVS) guidelines. Although genetic testing of more family members would have been very helpful to assess more confidently the pathogenicity of the variants reported, this was unfortunately not possible since they were not available for testing. The mean depth of coverage was 84×, and the quality threshold was 98.3%.
VARIANT INTERPRETATION
Three pathogenic variants in the genes SIN3A, FLG, and EDAR (Table 2) were reported and further investigated at the Center for Individualized Medicine (Mayo Clinic). A de novo (inferred by the absence of the variant in the proband's confirmed parents) pathogenic heterozygous [NM_001145358.1] c.2809_2810delAA (p.Lys937Glufs*2) deletion was detected in the SIN3A gene. The gene was predicted to be highly intolerant to loss of function variation based on gnomAD gene constraints (pLI = 1) (Lek et al. 2016). This variant is located in exon 16 of 22, was predicted to cause a frameshift resulting in nonsense-mediated decay of the transcript according to NMDEscPredictor (Coban-Akdemir et al. 2018), and has not been associated with disease prior to this study. It was absent in the healthy population, although one individual with a similar truncating variant (p.Lys937ArgfsTer19) has been reported in gnomAD (Lek et al. 2016). Therefore, it is possible that disorders associated with this gene display variable expressivity. Witteveen–Kolk syndrome (MIM:613406, Table 1) is a recently described disorder caused by pathogenic variants in SIN3A. Patients diagnosed with Witteveen–Kolk syndrome present with attention deficit, delayed development, and dysmorphic facial features (Witteveen et al. 2016) that resemble those observed in this patient (Table 1). Other overlapping features noted include joint laxity, scoliosis, and autistic features. However, oligodontia and dermatological issues have not been previously reported for this disorder. Based on the predicted damaging effect of the variant, the strong phenotypic overlap with previously reported patients, and ACMG classification as pathogenic (Richards et al. 2015), we were confident in diagnosing this patient with Witteveen–Kolk syndrome.
Table 2.
Genomic findings: pathogenic variants detected in our patient through exome sequencing
| Gene | Genomic location | HGBVS cDNA | HGVS protein | Zygosity | Parent of origin | ACMG variant classification |
|---|---|---|---|---|---|---|
| SIN3A | Chr 15:75684624_75684625del (GRCh37) Chr 15:75392283_75392284del (GRCh38) |
NM_001145358.1: c.2809_2810delAA | p.Lys937Glufs*2 | Heterozygous | De novo | Pathogenic |
| FLG | Chr 1:152285861 (GRCh37) Chr 1:152313385 (GRCh38) |
NM_002016.1: c.1501C>T | p.Arg501* | Heterozygous | Maternal | Pathogenic |
| EDAR | Chr 2:109524376 (GRCh37) Chr 2:108907920 (GRCh38) |
NM_022336.3: c.903C>A | p.Cys301* | Heterozygous | Paternal | Pathogenic |
Exome sequencing also identified a pathogenic maternally inherited heterozygous nonsense variant [NM_002016.1] c.1501C>T (p.Arg501*) in the FLG gene, which was reported to cause ichthyosis vulgaris (MIM:146700; Table 1) in a semidominant manner (Smith et al. 2006). The gene was predicted to be tolerant to loss of function based on gene constraints reported by gnomAD (pLI = 0) (Lek et al. 2016) and the transcript containing the variant was not predicted to undergo nonsense-mediated decay according to NMDEscPredictor (Coban-Akdemir et al. 2018). Although 19 homozygous alleles of this variant have been detected in healthy populations (Lek et al. 2016) (with a minor allele frequency of 0.93%), this variant has been reported in ClinVar as pathogenic (Variation ID: 16319) by various submitters, in HGMD as a disease-causing mutation (CM061002) and numerous publications have previously found a strong genotype–phenotype association in several cohorts of patients that support this variant's pathogenicity (Marenholz et al. 2006; Palmer et al. 2006; Smith et al. 2006; Weidinger et al. 2006). Incomplete penetrance and variable expressivity associated with this gene may explain the presence of apparently unaffected homozygotes in the population. FLG heterozygous carriers have a susceptibility to dermatitis, whereas individuals with biallelic mutations are at risk for more severe features (Smith et al. 2006; McGrath and Uitto 2008; Brown et al. 2009). In view of the above information and using the ACMG criteria (Richards et al. 2015), we classified the variant as pathogenic, and thus we concluded that the patient and her mother are additional reported individuals with a mild phenotype presentation that could likely explain their dry skin and perioral dermatitis.
A heterozygous pathogenic nonsense [NM_022336.3] c.903C>A (p.Cys301*) paternally inherited variant in EDAR was also reported. The EDAR gene was predicted to be tolerant to loss of function based on gene constraint (pLI = 0) (Lek et al. 2016) reported in gnomAD, and mutations in this gene have been associated with ectodermal dysplasia 10A and 10B (MIM:604095). This variant was present at a very low frequency in the healthy population (Lek et al. 2016) (minor allele frequency 0.0015% with no homozygotes listed) and reported as pathogenic in ClinVar (Variant ID: 449014) in association with hypohidrotic ectodermal dysplasia. Zeng et al. (2017) reported a patient with a heterozygous truncating variant in EDAR and a phenotype of oligodontia that resembles this patient. Although the variant was inherited from an asymptomatic father, high phenotypic variability, as well as reduced penetrance, has been described for this gene (Plaisancié et al. 2013). Since this is a null variant, present in low frequency in gnomAD, and reputable sources have reported the variant as pathogenic, we also classified the variant as pathogenic according to ACMG guidelines (Richards et al. 2015). We concluded that this variant likely explained the oligodontia observed in our patient and possibly in her paternal grandmother (Table 1; Fig. 1D).
SUMMARY
Diagnostic exome sequencing testing is one of the most comprehensive genetic tests available, resulting in diagnoses of 30%–50% of patients tested (Lazaridis et al. 2014). Additionally, 4.6%–7% of patients are diagnosed with two independent monogenic conditions (Yang et al. 2013; Yang et al. 2014; Posey et al. 2016). Here, we demonstrate an affected individual in which supernumerary phenotypes were associated with three separate genetic disorders explaining the constellation of symptoms presented by the patient. This report highlights the importance of detailed phenotyping and family history and considering the possibility of superimposed phenotypes when analyzing results from phenotypically complex individuals.
ADDITIONAL INFORMATION
Database Deposition and Access
All three variants reported in this patient were submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) with accession numbers SCV000619334.2 (SIN3A variant), SCV000321672.6 (FLG variant), and SCV000616705.1 (EDAR variant).
Ethics Statement
This study was approved by the Mayo Clinic Institutional Review Boards (IRBs). Informed consent for participation in this study was provided by the proband in person using an IRB approved form.
Acknowledgments
The authors thank the patient and her family for allowing us to perform and publish this research. We also thank Dr. Zaccariello for providing numerical scores from neurological assessments performed in the patient.
Author Contributions
R.H.G. visited the patient and provided clinical expertise including final diagnoses while J.L.K. offered genetic counseling. C.K. performed an in-depth genetic analysis of the patient's clinical report. E.W.K. directed this research. A.F. and L.S.-R. generated the manuscript, and all authors reviewed it prior to submission.
Funding
This research was funded internally by the Center for Individualized Medicine at Mayo Clinic.
Competing Interest Statement
The authors have declared no competing interest.
REFERENCES
- Brown SJ, Relton CL, Liao H, Zhao Y, Sandilands A, McLean WHI, Cordell HJ, Reynolds NJ. 2009. Filaggrin haploinsufficiency is highly penetrant and is associated with increased severity of eczema: further delineation of the skin phenotype in a prospective epidemiological study of 792 school children. Br J Dermatol 161: 884–889. 10.1111/j.1365-2133.2009.09339.x [DOI] [Google Scholar]
- Coban-Akdemir Z, White JJ, Song X, Jhangiani SN, Fatih JM, Gambin T, Bayram Y, Chinn IK, Karaca E, Punetha J, et al. 2018. Identifying genes whose mutant transcripts cause dominant disease traits by potential gain-of-function alleles. Am J Hum Genet 103: 171–187. 10.1016/j.ajhg.2018.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis KN, McAllister TM, Babovic-Vuksanovic D, Beck SA, Borad MJ, Bryce AH, Chanan-Khan AA, Ferber MJ, Fonseca R, Johnson KJ, et al. 2014. Implementing individualized medicine into the medical practice. Am J Med Genet C Semin Med Genet 166C: 15–23. 10.1002/ajmg.c.31387 [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marenholz I, Nickel R, Rüschendorf F, Schulz F, Esparza-Gordillo J, Kerscher T, Grüber C, Lau S, Worm M, Keil T, et al. 2006. Filaggrin loss-of-function mutations predispose to phenotypes involved in the atopic march. J Allergy Clin Immunol 118: 866–871. 10.1016/j.jaci.2006.07.026 [DOI] [PubMed] [Google Scholar]
- McGrath JA, Uitto J. 2008. The filaggrin story: novel insights into skin-barrier function and disease. Trends Mol Med 14: 20–27. 10.1016/j.molmed.2007.10.006 [DOI] [PubMed] [Google Scholar]
- Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, Goudie DR, Sandilands A, Campbell LE, Smith FJ, et al. 2006. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet 38: 441–446. 10.1038/ng1767 [DOI] [PubMed] [Google Scholar]
- Plaisancié J, Bailleul-Forestier I, Gaston V, Vaysse F, Lacombe D, Holder-Espinasse M, Abramowicz M, Coubes C, Plessis G, Faivre L, et al. 2013. Mutations in WNT10A are frequently involved in oligodontia associated with minor signs of ectodermal dysplasia. Am J Med Genet A 161A: 671–678. 10.1002/ajmg.a.35747 [DOI] [PubMed] [Google Scholar]
- Posey JE, Rosenfeld JA, James RA, Bainbridge M, Niu Z, Wang X, Dhar S, Wiszniewski W, Akdemir ZH, Gambin T, et al. 2016. Molecular diagnostic experience of whole-exome sequencing in adult patients. Genet Med 18: 678–685. 10.1038/gim.2015.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retterer K, Scuffins J, Schmidt D, Lewis R, Pineda-Alvarez D, Stafford A, Schmidt L, Warren S, Gibellini F, Kondakova A, et al. 2015. Assessing copy number from exome sequencing and exome array CGH based on CNV spectrum in a large clinical cohort. Genet Med 17: 623–629. 10.1038/gim.2014.160 [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–423. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FJD, Irvine AD, Terron-Kwiatkowski A, Sandilands A, Campbell LE, Zhao Y, Liao H, Evans AT, Goudie DR, Lewis-Jones S, et al. 2006. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet 38: 337–342. 10.1038/ng1743 [DOI] [PubMed] [Google Scholar]
- Weidinger S, Illig T, Baurecht H, Irvine AD, Rodriguez E, Diaz-Lacava A, Klopp N, Wagenpfeil S, Zhao Y, Liao H, et al. 2006. Loss-of-function variations within the filaggrin gene predispose for atopic dermatitis with allergic sensitizations. J Allergy Clin Immunol 118: 214–219. 10.1016/j.jaci.2006.05.004 [DOI] [PubMed] [Google Scholar]
- Witteveen JS, Willemsen MH, Dombroski TCD, van Bakel NHM, Nillesen WM, van Hulten JA, Jansen EJR, Verkaik D, Veenstra-Knol HE, van Ravenswaaij-Arts CMA, et al. 2016. Haploinsufficiency of MeCP2-interacting transcriptional co-repressor SIN3A causes mild intellectual disability by affecting the development of cortical integrity. Nat Genet 48: 877–887. 10.1038/ng.3619 [DOI] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al. 2013. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 369: 1502–1511. 10.1056/NEJMoa1306555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, et al. 2014. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312: 1870–1879. 10.1001/jama.2014.14601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng B, Zhao Q, Li S, Lu H, Lu J, Ma L, Zhao W, Yu D. 2017. Novel EDA or EDAR mutations identified in patients with X-linked hypohidrotic ectodermal dysplasia or non-syndromic tooth agenesis. Genes (Basel) 8: 259 10.3390/genes8100259 [DOI] [PMC free article] [PubMed] [Google Scholar]