

Abstract
Trichorhinophalangeal syndrome type I (TRPSI) is a rare disorder that causes distinctive ectodermal, facial, and skeletal features affecting the hair (tricho-), nose (rhino-), and fingers and toes (phalangeal) and is inherited in an autosomal dominant pattern. TRPSI is caused by loss of function variants in TRPS1, involved in the regulation of chondrocyte and perichondrium development. Pathogenic variants in TRPS1 include missense mutations and deletions with variable breakpoints, with only a single instance of an intragenic duplication reported to date. Here we report an affected individual presenting with a classic TRPSI phenotype who is heterozygous for a de novo intragenic ∼36.3-kbp duplication affecting exons 2–4 of TRPS1. Molecular analysis revealed the duplication to be in direct tandem orientation affecting the splicing of TRPS1. The aberrant transcripts are predicted to produce a truncated TRPS1 missing the nuclear localization signal and the GATA and IKAROS-like zinc-finger domains resulting in functional TRPS1 haploinsufficiency. Our study identifies a novel intragenic tandem duplication of TRPS1 and highlights the importance of molecular characterization of intragenic duplications.
Keywords: bulbous nose, cone-shaped epiphyses of phalanges 2 to 5, depigmentation/hyperpigmentation of skin, long philtrum, prominent ear helix, short stature, thick eyebrow
INTRODUCTION
Trichorhinophalangeal syndrome (TRPS) is a rare disorder, inherited in an autosomal dominant pattern, which is characterized by facial (bulbous nose, long philtrum, thin upper lip, and prominent ears), ectodermal (sparse, thin, and depigmented hair, thick eyebrows with lateral thinning, and dystrophic nails), and skeletal abnormalities (short stature, short hands and feet, hip dysplasia, and cone-shaped epiphyses at the phalanges) (Maas et al. 1993). Two distinct clinical types have been described for TRPS, with type I (TRPSI; OMIM#190350) caused by pathogenic variants in TRPS1, and type II (TRPSII, also known as Langer–Giedion syndrome; OMIM#150230). TRPSI is caused by loss of function mutations in TRPS1 and clinical features present with significant phenotypic variability among affected individuals and family members (Maas et al. 2015), including a form characterized by severe brachydactyly, previously proposed as a separate entity (TRPSIII; OMIM#190351) (Niikawa and Kamei 1986; Lüdecke et al. 2001). TRPSII stems from contiguous gene deletions encompassing TRPS1, RAD21, and EXT1 (Maas et al. 1993). In addition to clinical features described in TRPSI, individuals with TRPSII present with exostoses and an increased risk of intellectual disability (Langer et al. 1984).
TRPS1 (zinc-finger transcription factor TRPS1; OMIM#604386) is a seven-exon transcriptional repressor involved in the regulation of chondrocyte and perichondrium development (Napierala et al. 2008). The most common pathogenic variants found in TRPS1 are nonsense and frameshift variants scattered along exons 4–7, which effectively truncate/alter the protein sequence (Maas et al. 2015). Intragenic and full gene deletions have also been reported with variable breakpoints, together with missense variants primarily clustering in exon 6 (Maas et al. 2015). In contrast to the more frequent deletions and single-nucleotide variants (SNVs), duplication events involving TRPS1 are rare and mostly involve single or few (<10) nucleotides (Maas et al. 2015). Only a large pathogenic duplication of at least 600 bp (c.2097−?_c.2700+?dup) in TRPS1 has been reported in an individual presenting with TRPSI, but no further characterization of this variant at the molecular or protein level has been reported (Maas et al. 2015). At the nucleotide level, duplication breakpoints could disrupt TRPS1 sequence or generate gene fusions, and when intragenic, they could give rise to new transcripts through splicing alterations or truncate proteins by introducing stop codons (Newman et al. 2015), all of which could contribute to TRPS pathogenesis.
In this report, we present an 11-yr-old male with a history of short stature, mild facial dysmorphism, and ectodermal and skeletal abnormalities consistent with a diagnosis of TRPSI. Chromosomal microarray analysis identified a de novo intragenic ∼36.3-kbp duplication affecting exons 2–4 of TRPS1 (NM_014112). Molecular characterization of the duplication by mate-pair sequencing (MPseq) revealed it to be in direct tandem orientation, and RNA sequencing (RNA-seq) discovered abnormal transcripts including the duplicated exons leading to out-of-frame products with predicted protein truncations. The present study is the first full characterization of an intragenic TRPS1 duplication leading to TRPSI and a valuable example for the study of intragenic tandem duplication effects in genomic transcriptional outputs.
RESULTS
Case Presentation
An 11-yr-old male (II-3, Fig. 1A) was referred for genetic evaluation because of a history of nonspecific skin hyperpigmentation involving left shoulder and axilla, short stature (139.5 cm, 5.1%, Z = −1.63 based on CDC length-for-age data), and scoliosis (Fig. 1B,C). He was mildly dysmorphic with a bulbous nose, prominent ears, a thin upper lip, and a long philtrum; his hair was noted to be fine and sparse, and his eyebrows were thick with lateral rarefaction. X-ray imaging revealed the presence of cone-shaped epiphyses at the phalanges (Fig. 1D,E). Similar skin lesions were present in his otherwise healthy mother. None of these features were present in his fraternal twin brother (II-4) or other full and half siblings (Fig. 1A).
Figure 1.
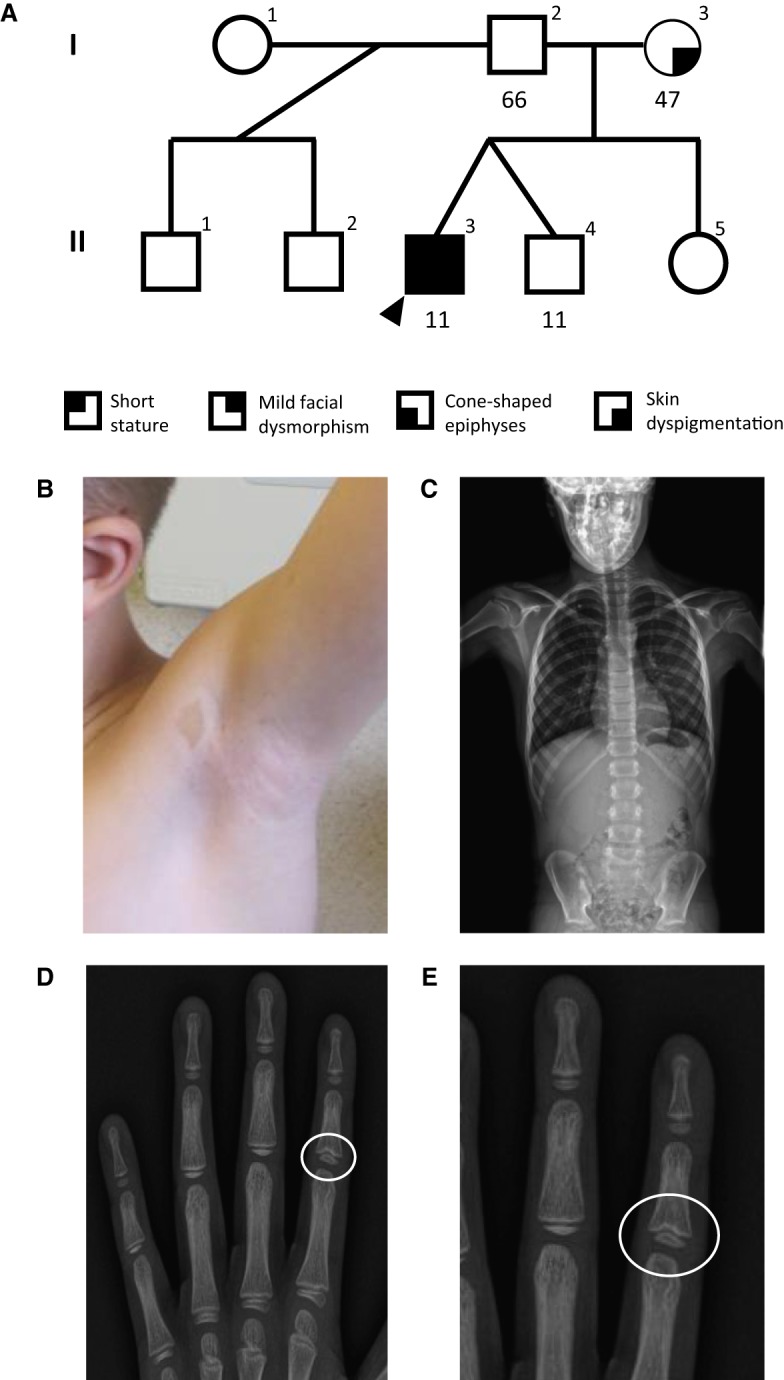
(A) Family pedigree for the analyzed proband. Proband is indicated with a black arrowhead. Numbers underneath indicate years of age. The clinical features of proband include (B) skin hyperpigmentation, (C) scoliosis, and (D,E) cone-shaped epiphyses at the phalanges as indicated with the white circle (E is the zoom-in view of D).
Genetic Testing
A next-generation sequencing (NGS) RASopathy panel targeting 18 genes (NF1, SPRED1, LZTR1, PTPN11, PPP1CB, BRAF, CBL, HRAS, KRAS, NRAS, MAP2K1, MAP2K2, RAF1, RIT1, RASA2, SHOC2, SOS1, and SOS2) and deletion/duplication analysis of LZTR1, NF1, and SPRED1 (University of Alabama at Birmingham) was performed because of the proband's short stature and familial skin hyperpigmentation, the result of which was negative. Subsequent chromosome microarray (CMA) testing of the proband and his parents revealed the presence of a heterozygous 36.3-kbp duplication at 8q23.3 in the proband (arr[hg19] 8q23.3(116603667_116640019) × 3 dn), including exons 2–4 from TRPS1 (NM_014112) (Fig. 2A; Supplemental Fig. 1A,B). Parental CMA studies demonstrated that this duplication was not inherited from either parent, and it was therefore classified as a de novo event. Given the overlap between the proband's phenotype and TRPSI, it was hypothesized that the duplication could result in the functional disruption of the duplicated allele. To distinguish between a tandem and an insertional duplication, we analyzed the proband's blood using MPseq, a specialized NGS technique for the detection of structural rearrangements and copy-number abnormalities (Drucker et al. 2014; Johnson et al. 2018; Smadbeck et al. 2018). MPseq established the duplication to be in direct tandem orientation within TRPS1 (Supplemental Fig. 1C,D), with estimated breakpoints at Chr 8:115,591,898–115,628,547 (hg38). The identified MPseq duplication breakpoints were confirmed with Sanger sequencing and mapped to Chr 8:115,591,769–115,628,731 (hg38) (Table 1). Analysis of the Sanger sequence revealed microhomology between the rearranged segments and at the breakpoint junction, with a nearby sequence corresponding to transposon repetitive elements (SINE and hAT-Charlie) (Supplemental Data).
Figure 2.

Abnormal transcripts associated with intragenic duplication in TRPS1. (A) Schematic of TRPS1 (NM_014112) with tandem duplication of exons 2–4 as found in proband. The observed splicing patterns are shown by lines connecting the exons, and abnormal splicing is shown in bold black lines. Asterisks indicate premature termination codons caused by abnormal splicing in duplicated region. (B) Schematic of TRPS1 with reported domains for the reference protein and for the predicted truncated proteins from each abnormal splicing event (product a and b from A). Integrative Genomics Viewer (IGV) screenshots of the RNA-seq paired reads supporting abnormal splicing aligned to the proband-specific synthetic reference sequence of (C) product a: exons 4–2–3, and (D) product b: exons 4–3 without intronic sequence included.
Table 1.
Variant table
| Gene | Chromosome | HGVS variant | Variant type | Predicted effect (ACMG) | MPseq coverage | Genotype (heterozygous/homozygous) | Parent of origin | Comments |
|---|---|---|---|---|---|---|---|---|
| TRPS1 | 8 | GRCh38 g.115591769_ 115628731 | Duplication | Likely pathogenic | 24× | Heterozygous | De novo | Variant was confirmed by Sanger |
Functional Studies
To assess the functional impact of the intragenic duplication on TRPS1 transcription, we performed NGS whole-transcriptome sequencing from blood RNA. The schematic of the duplicated exons is shown in Figure 2A and protein domains in Figure 2B. We observed in proband's sample and unrelated samples (data not shown) skipping of exon 2, which occurs in a known transcript (NM_001282902) that is lowly expressed in the GTEx data (www.gtexportal.org). We created a proband-specific coding reference including exons 4–2–3 consecutively together and another reference for exons 4–3. By aligning the RNA-seq reads to the synthetic reference sequences (Fig. 2C,D, respectively), the reads supporting these aberrant junctions caused by the duplication event in proband can be visualized. A total of 52 read pairs support splicing from exon 4 to either exon 2 or 3, with 13 of those having one read in the pair spanning the exon 4–2 junction and 15 having a one read in the pair spanning the exon 4–3 junction. The consequence to the transcript in either event is a premature termination codon (Fig. 2A). Exon 2 has 121 bp of 5′ UTR and when splicing from exon 4, translates to 16 amino acids followed by a premature termination codon. When splicing from exon 4 to 3, the transcript is out of frame and translates to 14 amino acids and a premature termination codon in the duplicated exon 3. At the exon level, reads per kilobase of transcript per million mapped reads (RPKM) revealed a higher number of reads mapping to exons 2–4 compared to a cohort of 127 differently affected individuals or unaffected relatives of subjects with suspected monogenetic disease (Fig. 3). An expression analysis using OUTRIDER (Brechtmann et al. 2018) showed only a modest increase in expression for TRPS1 (0.38 log2 fold change), most likely owing to the presence of the third copy of exons 2–4 (∼50% of TRPS1 nucleotides) being transcribed and leading to additional reads contributing to overall gene expression. However, the nonduplicated exons in TRPS1 support normal expression levels and suggest the abnormal transcript with the duplicated exons may be escaping nonsense-mediated decay (NMD) that would be predicted from the premature termination codons. Given the proband's phenotype and the derived functional evidence of the intragenic duplication effect on the TRPS1 transcript, the proband was diagnosed with TRPSI.
Figure 3.

Gene expression analysis. (A) TRPS1 exonic expression levels in blood RNA of the proband compared to a cohort of 127 individuals differently affected with, or unaffected individuals related to individuals with, suspected rare monogenetic disease. (B) Gene expression changes called by OUTRIDER in this individual compared to the cohort represented as −log10(unadjusted P-value) versus Z-score.
DISCUSSION
We described a novel 36.3-kbp intragenic duplication in TRPS1 in an individual presenting with the classic ectodermal, facial, and skeletal abnormalities of TRPSI. CMA and MPseq analyses showed the duplication to be de novo and in direct tandem orientation within TRPS1 in the affected subject, effectively duplicating exons 2–4. Although more than 130 pathogenic variants have been reported in TRPS1, to our knowledge, this is the first characterization of a large intragenic duplication in TRPS1 associated with TRPSI.
The clinical features of TRPSI are caused by haploinsufficiency of TRPS1, a transcriptional repressor involved in the regulation of chondrocyte and perichondrium development. The identified intragenic duplication of exons 2–4 was shown to produce abnormal transcripts with splicing patterns including exons 4–2 and 4–3. Both of these abnormal transcripts are predicted to (1) undergo NMD or (2) produce a likely nonfunctional protein missing the GATA zinc finger, the IKAROS-like zinc finger, and the nuclear localization signal (NLS) (Fig. 2B). In a NMD scenario, a lower-than-average number of mapped reads was expected for the transcripts. RPKM values of the duplicated exons were higher compared to normal male controls, whereas nonduplicated exons had RPKM ranges similar to controls. Such observations suggest that the aberrant intragenic duplication transcripts may not undergo NMD; further inspection of RNA-seq data failed to reveal any informative sequence variants for detecting allelic expression bias that would result from one transcript undergoing NMD. Either the allele with the duplicated exons is undergoing NMD, but overall gene transcription is up-regulated to compensate, or the abnormal transcripts escape NMD, are expressed at normal levels, and are translated into truncated nonfunctional protein lacking the functional GATA and IKAROS-like zinc fingers necessary for its correct genomic binding and repression activities. Such truncations can effectively cause functional haploinsufficiency of TRPS1 in the reported individual and thus be a cause of TRPSI. Future proteomic experiments could evaluate the presence of truncated versions lacking the aforementioned domains.
Altogether, intragenic duplications within TRPS1 can have major clinical significance as illustrated in the currently studied individual. Identification and molecular characterization of other intragenic TRPS1 duplications will be necessary to assess genotype–phenotype correlations and to discover specific sites within the gene that are prone to further or recurrent chromosome rearrangements, as seen from our discovery of microhomology at the reported duplication junction. Microhomology-mediated mechanisms such as fork stalling and template switching (FoSTeS) (Lee et al. 2007) and microhomology-mediated break-induced replication (MMBIR) (Hastings et al. 2009) have been extensively associated with genomic germline duplications and deletions and other complex rearrangements. Both FoSTeS and MMBIR are plausible mechanisms to explain the origin of the intragenic TRPS1 duplication reported herein, given the detected junction microhomology and the presence of transposon sequences (SINE and hAT-Charlie) in the vicinity of the breakpoints, which can prime replication strand invasions upon double-strand breaks.
As we hope to have illustrated with this study, comprehensive molecular characterization of intragenic duplications is essential for the investigation of their pathogenicity and estimating potential recurrence risks. As NGS becomes a routine clinical test in clinical diagnosis, we surmise that additional intragenic duplications involving TRPS1 and many more genes will be uncovered, and their impact on protein structure assessed and correlated to diverse forms of human disease.
MATERIALS AND METHODS
Approval for this study was received by the Mayo Clinic Institutional Review Board, application number 12-009346; participants were consented for release of data included in this study. Chromosome microarray was performed on peripheral blood using the CytoScan HD Suite (Thermo Fisher Scientific), which evaluates allelic and copy-number information, according to the manufacturer's protocol. CMA data was analyzed using ChAS software version 3.1. MPseq was performed on DNA extracted from peripheral blood using AutoPure LS (QIAGEN). MPseq libraries were prepared using Nextera Mate Pair Library Preparation Kit (Illumina) and subsequently purified and processed for short-read library preparation using TruSeq DNA Library Prep kit (Illumina). Purified libraries were sequenced in an Illumina HiSeq 2500 using RapidRun mode to obtain 101-bp reads. MPseq data was processed with BIMAv3 and analyzed with SVAtools version 0.24.9. Polymerase chain reaction (PCR) experiments were performed to amplify breakpoint junctions identified by MPseq.
RNA was isolated using the miRNeasy Mini Kit (QIAGEN) following the standard protocol from blood drawn in a PAXgene Blood RNA Tube (QIAGEN). RIN and DV200 values were determined for starting RNA concentrations using the Agilent Bioanalyzer or TapeStation. RNA libraries were prepared according to the manufacturer's instructions for the TruSeq RNA Access Library Prep Kit (Illumina). Coding regions of the transcriptome were captured by pooling four of the cDNA libraries at 200 ng each following the manufacturer's instructions for the TruSeq RNA Access Library Prep Kit (Illumina). The concentration and size distribution of the final libraries were determined on an Agilent Bioanalyzer DNA 1000 chip (Agilent Technologies). A final quantification, using Qubit fluorometry (Invitrogen), was performed to confirm sample concentration.
Libraries were sequenced at ∼65 million fragment reads per sample (4 samples/lane) following Illumina's standard protocol using the Illumina cBot and HiSeq 3000/4000 PE Cluster Kit. The flow cells were sequenced as 100 × 2 paired-end reads on an Illumina HiSeq 4000 using HiSeq 3000/4000 sequencing kit and HCS v3.3.20 collection software. Base calling was performed using Illumina's RTA version 2.5.2.
RNA-sequencing analysis was performed using MAP-RSeq (Kalari et al. 2014). Reads were aligned to the human genome (hg19) and transcriptome using TopHat2 (Kim et al. 2013) running Bowtie (v1) (Langmead 2010). Gene- and exon-level read counts were generated using HiSeq (Anders et al. 2015) and BEDTools (Quinlan 2014), respectively.
Custom reference creation: Two custom human reference sequences were constructed to represent the aberrant transcripts arising from the tandem duplication of TRPS1 exons 2–4. Constituent exon sequences were extracted from the UCSC hg19 FASTA sequence utilizing genomic coordinates for RefSeq transcript NM_014112.4. FASTA sequence for the exons were concatenated in the order Exon 4 → Exon 2 → Exon 3 and Exon 4 → Exon 3 in the 5′ to 3′ direction, mimicking the expected aberrant juxtaposition of the exons in the final transcripts. All other TRPS1 exons were excluded from the custom reference to ensure specificity of alignments supporting the aberrantly formed splice junctions. Alignment: Cutadapt (Martin 2011) was used to remove poly(A) tails from raw RNA-seq reads. Alignment to the custom reference sequence was performed using BWA MEM (Li 2013) with default parameters. SAMTools (Li et al. 2009) was used to remove reads with multiple mappings or improper read-pairing or to insert sizes varying from the expected range. Read pairs containing one mate crossing an aberrantly formed splice junction by fewer than 10 bases were filtered to prevent nonspecific alignments that might align with equal homology with the normal transcript. Integrative Genomics Viewer (IGV) (Thorvaldsdóttir et al. 2013) was used to generate snapshots of the final alignments. A custom annotation track was created to enable identification of exon order in the custom reference.
Outlier expression analyses: Expression counts and RPKMs were computed as above for a cohort of 128 samples from the Mayo Clinic Service Line 2 for rare and undiagnosed diseases. Genes with low expression in blood were filtered out. Specifically, genes whose 0.95 quantile FPKM was <1 or whose counts were 0 in more than one-quarter of the samples were excluded from the analysis. Outlier expression analysis was then performed using OUTRIDER (Brechtmann et al. 2018) with the Benjamini–Hochberg correction for multiple comparisons, which provided confounder-corrected z-scores, log2 fold changes, P-values, and adjusted P-values.
ADDITIONAL INFORMATION
Data Deposition and Access
The duplication position within TRPS1 was submitted to the Leiden Open Variation Database (Fokkema et al. 2011) under accession number 0000597835. Patient consent does not allow for deposition of raw data.
Ethics Statement
The studied family was counseled and consented under IRB 12-009346 from the Mayo Clinic Department of Laboratory Medicine and Pathology and Center for Individualized Medicine.
Acknowledgments
We thank the reported family for their enthusiasm and participation in this research report. We also thank the staff at Mayo Clinic Genomics Laboratory for their excellence in the clinical processing and testing of the samples from this family.
Author Contributions
C.J.Z.-M. and M.A.C. performed experiments and interpreted data; M.A.C., S.B., G.J., and G.O. analyzed RNA-seq data; S.T.P. and D.B.-V. provided clinical interpretations and consultation; L.A.B. interpreted CMA data; all authors read and approved the manuscript.
Funding
Funding was provided by the Mayo Clinic Department of Laboratory Medicine and Pathology and Center for Individualized Medicine.
Competing Interest Statement
The authors have declared no competing interest.
Referees
Scott E Hickey
Anonymous
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brechtmann F, Mertes C, Matusevičiūtė A, Yépez VA, Avsec Z, Herzog M, Bader DM, Prokisch H, Gagneur J. 2018. OUTRIDER: a statistical method for detecting aberrantly expressed genes in RNA sequencing data. Am J Hum Genet 103: 907–917. 10.1016/j.ajhg.2018.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drucker TM, Johnson SH, Murphy SJ, Cradic KW, Therneau TM, Vasmatzis G. 2014. BIMA V3: an aligner customized for mate pair library sequencing. Bioinformatics 30: 1627–1629. 10.1093/bioinformatics/btu078 [DOI] [PubMed] [Google Scholar]
- Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. 2011. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat 32: 557–563. 10.1002/humu.21438 [DOI] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR. 2009. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 5: e1000327 10.1371/journal.pgen.1000327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SH, Smadbeck JB, Smoley SA, Gaitatzes A, Murphy SJ, Harris FR, Drucker TM, Zenka RM, Pitel BA, Rowsey RA, et al. 2018. SVAtools for junction detection of genome-wide chromosomal rearrangements by mate-pair sequencing (MPseq). Cancer Genet 221: 1–18. 10.1016/j.cancergen.2017.11.009 [DOI] [PubMed] [Google Scholar]
- Kalari KR, Nair AA, Bhavsar JD, O'Brien DR, Davila JI, Bockol MA, Nie J, Tang X, Baheti S, Doughty JB, et al. 2014. MAP-RSeq: Mayo analysis pipeline for RNA sequencing. BMC Bioinformatics 27: 224 10.1186/1471-2105-15-224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer LO Jr, Krassikoff N, Laxova R, Scheer-Williams M, Lutter LD, Gorlin RJ, Jennings CG, Day DW. 1984. The tricho-rhino-phalangeal syndrome with exostoses (or Langer–Giedion syndrome): four additional patients without mental retardation and review of the literature. Am J Med Genet 19: 81–112. 10.1002/ajmg.1320190110 [DOI] [PubMed] [Google Scholar]
- Langmead B. 2010. Aligning short sequencing reads with bowtie. Curr Protoc Bioinformatics 32: 11.17.11–11.17.14. 10.1002/0471250953.bi1107s32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR. 2007. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131: 1235–1247. 10.1016/j.cell.2007.11.037 [DOI] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv e-prints. [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078-2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüdecke HJ, Schaper J, Meinecke P, Momeni P, Gross S, von Holtum D, Hirche H, Abramowicz MJ, Albrecht B, Apacik C, et al. 2001. Genotypic and phenotypic spectrum in tricho-rhino-phalangeal syndrome types I and III. Am J Hum Genet 68: 81–91. 10.1086/316926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas S, Shaw A, Bikker H, Hennekam, RCM. 1993. Trichorhinophalangeal syndrome. In GeneReviews® (ed. Adam MP, Ardinger HH, Pagon RA, et al. ). University of Washington, Seattle. [PubMed] [Google Scholar]
- Maas SM, Shaw AC, Bikker H, Lüdecke HJ, van der Tuin K, Badura-Stronka M, Belligni E, Biamino E, Bonati MT, Carvalho DR, et al. 2015. Phenotype and genotype in 103 patients with tricho-rhino-phalangeal syndrome. Eur J Med Genet 58: 279–292. 10.1016/j.ejmg.2015.03.002 [DOI] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17: 3 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- Napierala D, Sam K, Morello R, Zheng Q, Munivez E, Shivdasani RA, Lee B. 2008. Uncoupling of chondrocyte differentiation and perichondrial mineralization underlies the skeletal dysplasia in tricho-rhino-phalangeal syndrome. Hum Mol Genet 17: 2244–2254. 10.1093/hmg/ddn125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman S, Hermetz KE, Weckselblatt B, Rudd MK. 2015. Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints. Am J Hum Genet 96: 208–220. 10.1016/j.ajhg.2014.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niikawa N, Kamei T. 1986. The Sugio–Kajii syndrome, proposed tricho-rhino-phalangeal syndrome type III. Am J Med Genet 24: 759–760. 10.1002/ajmg.1320240420 [DOI] [PubMed] [Google Scholar]
- Quinlan AR. 2014. BEDTools: the Swiss-army tool for genome feature analysis. Curr Protoc Bioinformatics 47: 1–34. 10.1002/0471250953.bi1112s47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smadbeck JB, Johnson SH, Smoley SA, Gaitatzes A, Drucker TM, Zenka RM, Kosari F, Murphy SJ, Hoppman N, Aypar U, et al. 2018. Copy number variant analysis using genome-wide mate-pair sequencing. Genes Chromosomes Cancer 57: 459–470. 10.1002/gcc.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The duplication position within TRPS1 was submitted to the Leiden Open Variation Database (Fokkema et al. 2011) under accession number 0000597835. Patient consent does not allow for deposition of raw data.