

Abstract
Effective vaccines inducing lifelong protection against many important infections such as respiratory syncytial virus (RSV), human immunodeficiency virus (HIV), influenza and Epstein-Barr virus (EBV) are not yet available despite decades of research. As an alternative to a protective vaccine we developed a genetic engineering strategy in which CRISPR/Cas9 was utilized to replace endogenously-encoded antibodies with antibodies targeting RSV, HIV, influenza or EBV in primary human B cells. The engineered antibodies were expressed efficiently in primary B cells under the control of endogenous regulatory elements, which maintained normal antibody expression and secretion. Using engineered mouse B cells, we demonstrated that a single transfer of B cells engineered to express an antibody against RSV resulted in potent and durable protection against RSV infection in RAG1-deficient mice. This approach offers the opportunity to achieve sterilizing immunity against pathogens for which traditional vaccination has failed to induce or maintain protective antibody responses.
One Sentence Summary:
B cells reprogrammed to express pathogen-specific antibodies using CRISPR/Cas9 protect against infection.
Introduction
Protective vaccines have reduced morbidity and mortality from several infectious diseases in large part by activating the humoral immune response and subsequent production of high affinity pathogen-specific antibodies produced by B cells. Unfortunately, vaccines for many common diseases are not yet available despite considerable research efforts. One example is RSV, a common pathogen that infects the upper and lower respiratory tracts. RSV is a serious threat to infants, the elderly, those with cardiopulmonary disease, and those undergoing hematopoietic stem cell transplant, where it is a significant cause of morbidity and mortality (1–3). The first RSV vaccine trial occurred in 1966 but proved harmful when subsequent natural infection caused severe lower respiratory disease and two deaths (4). The second RSV vaccine trial occurred in 2016 and also failed to protect against infection (5). While several other candidate RSV vaccines are being developed, it is unclear if any will elicit a protective response.
Despite the failure of vaccine trials, evidence exists for antibody-mediated protection against RSV. Several monoclonal antibodies including the RSV-specific monoclonal antibody palivizumab have been shown to protect against RSV infection in vitro or in vivo (6, 7). Similarly, monoclonal antibodies protective against HIV, influenza, EBV, human metapneumovirus virus, Dengue, Zika, Ebola and many other pathogens are also being developed (8). However, the infusion of monoclonal antibodies like palivizumab is limited to high risk populations because monthly reinfusion is required to maintain protection. While new approaches to increase the antibody half-life after injection have been developed (9), even the most promising of these strategies would require lifelong reinfusion to maintain protection.
To overcome the need for reinfusion, alternative strategies to generate long-term immunity have been explored. One approach involves viral transduction of muscle cells with an adenoviral vector encoding a protective antibody (10, 11). Another approach is transduction of hematopoietic stem cells with a lentivirus-encoded secreted antibody, which are differentiated into antibody-secreting plasma cells in vitro prior to infusion, or allowed to differentiate in vivo after infusion (12, 13). A shared limitation of both the adenoviral/muscle and lentiviral/stem cell approaches is that the level of antibody produced is fixed and unresponsive to infection. In contrast, protective vaccines elicit both long-lived memory B cells and antibody-secreting plasma cells. Memory B cells express a membrane bound form of antibody that allows these cells to rapidly respond and differentiate into additional antibody-secreting cells upon infection.
In an effort to mimic the protective B cell response, we developed a genetic engineering strategy that allowed for the expression of protective antibodies against RSV, HIV, influenza or EBV in mouse or human B cells under endogenous regulatory elements. This was challenging because fully functional B cells require alternative splicing and polyadenylation to produce membrane bound as well as secreted antibodies, a process which is difficult to recapitulate in a viral transgene (14, 15). Adding an additional level of difficulty, antibodies are produced as the product of two genes, heavy chain gene (IgH) and either the kappa (Igκ) or lambda (Igλ) light chain gene. Targeting the heavy chain locus is complicated by the large size and extreme genetic heterogeneity of this area in antibody-expressing B cells. Each developing B cell undergoes recombination of V, D, and J segments over more than a megabase of DNA within the heavy chain locus, and this results in variable regions that are essentially unique to each cell (16). This sequence variability makes directly targeting antibody coding regions challenging. One group recently bypassed this limitation by replacing the entire heavy chain locus with the heavy chain VDJ of their choosing (17). This approach is promising but limited to antibodies that bind antigens without light chain involvement (17). Another recent study inserted the full light chain into the light chain V region loci and a secreted version of the heavy chain into the heavy chain V region loci (18). This work is limited in that only secreted antibody was expressed, and it was unclear from this work if expression of the endogenous antibody was eliminated (18). To build upon this previous work, we developed a single cut approach where the full light chain linked to the heavy chain VDJ was inserted into an intronic region of the heavy chain locus. Using this approach, we find that both murine and human B cells can be efficiently engineered to express antibodies targeting pathogens. Further, a single transfer of murine B cells engineered to express an RSV-specific antibody can protect RAG1−/− mice from infection for several months.
Results
Targeting strategy and emAb cassette design
To circumvent the complexity of the antibody heavy chain gene, we focused upon a small 2600 nucleotide region of DNA present in all B cells between the last J gene segment and the region involved in class switching. This region was further limited due to the presence of a critical intronic Eμ enhancer, one of several strong enhancer elements that cooperate to drive high level expression of recombined VDJ genes despite the weak promoters of V gene segments (19, 20). Activity of these enhancers is regulated in part by the proximity of promoters relative to the Eμ enhancer, and insertion of a transgene between the recombined VDJ segments and the Eμ enhancer can completely block transcription of the upstream VDJ segment (21). We therefore inserted a synthetic VDJ under the control of a heavy chain promoter upstream of the Eμ enhancer would allow for physiological expression of the inserted engineered monoclonal antibody, which we termed an “emAb.”
To enable one-hit insertion, we designed an emAb cassette that contained a heavy chain promoter followed by a complete light chain linked to a recombined heavy chain VDJ containing a splice junction to allow for splicing to downstream endogenous heavy chain constant regions (Fig. 1A). Utilizing the endogenous heavy chain constant region reduced the insert size and allowed emAbs to be expressed in membrane-bound and secreted forms of all isotype classes under the control of endogenous regulatory elements. When expressed, the emAb light chain is physically linked to the heavy chain with a 57 amino acid glycine-serine linker (Fig. 1B), which has been used previously in single chain Fab fragments (22). The linker also contained three tandem Streptag-II motifs to facilitate the detection and enrichment of engineered cells (23). Physically linking the heavy and light chains also minimized the possibility of mispairing between an inserted emAb heavy chain and the endogenous light chain.
Figure 1. CRISPR/Cas9 mediated replacement of an endogenous antibody with an engineered monoclonal antibody (emAb).

(A) Diagram showing insertion of an emAb cassette containing a heavy chain promoter, full light chain, linker, and partial heavy chain into the intronic region between the terminal J segment and the Eμ enhancer, upstream of the endogenous heavy chain constant region exons. (B) Schematic showing normal transmembrane and secreted antibody versus transmembrane and secreted emAb with the endogenous heavy chain constant regions shown in grey.
CRISPR/Cas9-mediated emAb expression in RAMOS B cells
Insertion and expression of an emAb cassette was first tested in the Burkitt-lymphoma derived RAMOS B cell line that natively expresses membrane-bound and secreted antibodies. Analyzing the region between the terminal J segment and Eμ using the CrispRGold algorithm (24), several potential Cas9 guide RNA (gRNA) binding sites were identified. We focused upon gRNA huIgH296, which targeted a region 296 nucleotides downstream of IgHJ6 where single nucleotide polymorphisms (SNPs) with a frequency above 1% have not been reported (25). Electroporation of RAMOS cells with huIgH296 gRNA precomplexed with Cas9 protein resulted in efficient DNA cutting, with insertions or deletions (indels) at this site being detected in ~72% of genomic heavy chain DNA (Fig. 2A). After electroporation, we incubated cells with AAV encoding an engineered RSV-emAb cassette derived from palivizumab flanked by 450 nucleotide homology arms on either side of the huIgH296 target site. Since the AAV does not include the heavy chain constant regions which are essential for antibody expression, RAMOS cells would only gain the ability to bind RSV F antigen if the RSV-emAb cassette was successfully inserted into the heavy chain locus. Flow cytometry was used to assess RSV-emAb expression on the cell surface by measuring binding to fluorescent RSV F antigen and streptactin, a modified streptavidin with high affinity for the Streptag-II motifs in the linker (26). Using this approach, ~30% of RSV-emAb-engineered RAMOS cells bound RSV F antigen and streptactin compared to less than 0.3% of control RAMOS cells (Fig. 2B). To determine whether the endogenous heavy chain was silenced in emAb-expressing B cells, we examined the surface expression of the endogenous lambda light chain, which would only be present if it were able to pair with the endogenous heavy chain. For this we focused upon RAMOS cells expressing high levels of surface BCR by gating on B cells expressing high levels of CD79b (Fig. 2C). As expected, surface expression of endogenous lambda light chain expression was eliminated in RSV-emAb-engineered RAMOS cells that bound RSV F antigen (Fig. 2C) even though the expression of lambda gene was not inactivated by our strategy. These results indicate that emAb engineering replaces the endogenous antibody expressed by RAMOS B cells.
Figure 2. CRISPR/Cas9 mediated replacement of an endogenous antibody with an emAb targeting RSV in RAMOS B cells.

(A) The frequency of insertions or deletions (indels) in genomic heavy chain sequences in independent experiments (n=3) analyzed by decomposition two-four days following electroporation of huIgH296 gRNA/Cas9. (B) Representative flow cytometric analysis of RSV F antigen and Streptactin binding by control and engineered RAMOS cells two days following electroporation with huIgH296 gRNA/Cas9 followed by incubation with an RSV-emAb encoding AAV. Cell binding before (middle panel) and after FACS-purification and expansion (right panel) is displayed. The numbers on the plots represent the mean ± SD % of RAMOS cells binding both RSV F antigen and Streptactin from three independent experiments, and two separate assessments of sorted RSV-emAb RAMOS cells. (C) Representative flow cytometric analysis of two similar experiments examining the loss of Igλ expression by CD79b+ RSV F+ RAMOS B cells compared to CD79b+ RSV F− and CD79b− cells in the same culture. (D) Representative flow cytometric analysis and (E) peak fold increase in Fluo-4 fluorescence in RSV-emAb+ RAMOS cells and control RAMOS cells following stimulation with 1 μg/mL αIg F(ab’)2 or 1 μg/mL tetramerized RSV F antigen. Data points are combined from three independent experiments and the p value was determined using an unpaired two-tailed t-test with Welch’s correction.
In order to assess the functionality of the RSV-emAb, RAMOS cells binding RSV F antigen and streptactin were FACS-purified to create an RSV-emAb cell line. To confirm functional interaction between the RSV-emAb and the BCR signaling complex, control and RSV-emAb RAMOS cells were stimulated with tetramerized RSV F antigen or with polyclonal αIg F(ab’)2. Only the RSV-emAb cell line fluxed calcium in response to RSV F antigen, whereas the RSV-emAb and control RAMOS cell lines had similar responses to αIg (Fig. 2D, E). These results indicate that emAb engineering reprograms B cells with a functional monoclonal antibody.
CRISPR/Cas9-mediated emAb expression in primary human B cells
We next engineered human primary B cells using a multistep process of expansion and differentiation (Fig. 3A). Human CD19+ B cells were MACS-purified from PBMCs and stimulated with a cocktail of cytokines, a multimerized CD40 ligand and CpG for 48 hours prior to electroporation with huIgH296 gRNA/Cas9. Sequencing of genomic DNA revealed the presence of indels in ~67% of heavy chain DNA sequences from six independent donors (Fig. 3B). To test the performance of the emAb backbone across multiple independently derived antibodies, we designed three additional emAb cassettes encoding for the HIV-1 broadly neutralizing antibody VRC01, the influenza broadly neutralizing antibody MEDI8852, and the EBV neutralizing antibody AMM01 (27–29). All emAb cassettes were efficiently introduced into primary human B cells and resulted in the generation of populations which bound RSV F antigen, influenza hemagglutinin (Flu HA) stem, EBV gH/gL, or HIV-1 Envelope (Env) (Fig. 3C). Tests of RSV-emAb engineering in eight independent donors resulted in RSV antigen being bound by 16–44% of engineered B cells, compared to under 1% of mock engineered control B cells. The emAb expression efficiency ranged from 5–59% for the three additional antiviral emAbs (Fig. 3D). All four emAb B cell populations also secreted engineered antibodies when induced to expand and differentiate into CD38+ CD27+ antibody-secreting cells through additional culture (Fig. 3E–G). These data demonstrate the flexible nature of the emAb platform for engineering primary B cells to produce and secrete protective monoclonal antibodies.
Figure 3. CRISPR/Cas9 mediated replacement of endogenous antibodies with engineered emAbs targeting RSV, HIV-1, Influenza or EBV in primary human B cells.

(A) Schematic representation of the human B cell engineering protocol. (B) Frequency of indels detected in genomic heavy chain sequences from B cells two days after electroporation with gRNA huIgH296/Cas9 (n=8 individuals). (C) Representative flow cytometric analysis and (D) quantitation of antigen binding to human B cells from different individuals (n=3–7) engineered to express emAbs based upon the HIV-1 broadly neutralizing antibody VRC01 (HIV-emAb), the influenza broadly neutralizing antibody MEDI8852 (Flu-emAb), the EBV neutralizing antibody AMM01 (EBV-emAb), or RSV-emAb. Cells were analyzed at day four shown in panel A and compared to control B cells that were mock electroporated and cultured similarly. (E) ELISA-mediated quantitation of antigen-specific antibodies in day 10 supernatants from 2–3 independent B cell cultures per specificity. (F) Combined data from two experiments displaying the number of emAb B cells in cultures at day 7, 11 and 14 displayed as a fold expansion over the number of cells sorted at day 4 (n=2–6). (G) Representative flow cytometric analysis of CD38, CD27 expression by B cells at the start of culture, day 4 and day 10. The mean ± SD% in each quadrant was generated from three individuals. Representative of three similar experiments.
Engineered B cells can co-express emAbs off of both heavy chain loci
Work with transgenic mice demonstrated that productive VDJ sequences on both heavy chain loci results in simultaneous transcription and translation of both heavy chains (30). These results indicate that if the emAb cassette was only inserted into the unproductive heavy chain, cells simultaneously expressing their endogenous antibodies and emAbs could be produced (Fig. 4A). This would be problematic if the endogenous antibody caused autoimmune tissue destruction due to binding self-antigens. This is a concern given that up to 20% of the naive B cell repertoire has been shown to express antibodies that can bind self-antigens (31). To determine whether B cells co-expressing both the emAb and endogenous antibodies were produced, we FACS-purified CD19+ B cells that expressed antibodies utilizing lambda light chains (Fig. 4B) prior to engineering with Flu-emAb, which utilized a kappa light chain. Since many cells in the culture downregulated surface BCR expression as a result of the culture conditions, we gated on cells expressing high levels of CD79b to focus on cells retaining high surface BCR expression (Fig. 4B). Within the Flu-emAb engineered CD79b+ B cells, most cells that gained the ability to bind influenza HA lost surface expression of the lambda light chain (Fig. 4B). The loss of lambda light chain expression indicated that the inserted emAb cassette blocked expression of the endogenous antibody in these cells. However, nearly half of Flu HA-binding B cells retained lambda light chain expression on the cell surface (Fig. 4B). These results suggested that in many of the cells, emAb insertion occurred on the non-productive heavy chain locus, resulting in co-expression of the emAb and the endogenous antibody. This was not detected in RAMOS experiments since RAMOS cells have a c-Myc translocation in one IgH loci between the Eμ and constant regions (32, 33).
Figure 4. Simultaneous emAb expression from both heavy chain loci.

(A) Representation of a productively recombined heavy chain and a non-productive heavy chain, both of which contain the huIgH296 gRNA target site. (B) Representative flow cytometric analysis of the loss of Igλ expression by FACS-purified Igλ+ primary cells engineered to express Flu-emAb, which utilizes Igκ. The numbers on the Igλ plots represent the mean ± SD% from two independent experiments. (C) Representative flow cytometric analysis of RSV F antigen and Flu HA binding by control B cells, B cells engineered separately with Flu-emAb and RSV-emAb and co-cultured for 24 hours prior to analysis, or B cells simultaneously double-engineered with Flu-emAb and RSV-emAb. Data are representative of two independent experiments.
The expression of emAb from both the productive and non-productive heavy chain offered the possibility of producing dual-emAb B cells by insertion of a different cassette into each locus. To test this possibility, we assessed B cells engineered simultaneously with AAVs encoding RSV-emAb and Flu-emAb cassettes. Simultaneous engineering of cells with RSV-emAb and Flu-emAb resulted in ~6% of cells binding both RSV F and Flu HA, a population which is not detected in control B cells, or cells that were engineered with the individual AAVs followed by 24 hours of co-culture prior to analysis (Fig. 4C). Taken together, these results demonstrated that emAbs can be simultaneously expressed by both heavy chain loci.
Murine emAb B cells protect against RSV infection
Having demonstrated the ability to engineer primary B cells, we next assessed the protective capability of these cells in a murine model of infection. Murine emAb-expressing B cells were produced using a process of priming, electroporation, and emAb cassette delivery similar to that used in human primary B cells (Fig. 5A). Electroporation in combination with precomplexed muIgH367 gRNA and Cas9 resulted in indels in ~80% of target alleles (Fig. 5B). Delivery of a murine RSV-emAb cassette encoded by AAV resulted in 8–24% of murine B cells binding RSV F antigen two days later (Fig. 5C, D). RSV F antigen-binding of 1–7% of B cells could be detected when an RSV-emAb cassette was delivered during the electroporation as a double stranded DNA (dsDNA) containing short 36 nucleotide homology regions (Fig. 5C, D), offering a potential to produce emAb B cells without the use of an AAV. Similar to human cells, murine B cells secreted engineered antibodies when induced to expand and differentiate into CD19LOW CD138+ antibody-secreting cells through additional culture (Fig. 5E–H). Most of the cells in this culture also lost the expression of IgM and IgD between day 3 and 7 (Fig. 5I, J), indicating the ability of these cells to undergo isotype switching.
Figure 5. Engineering of primary mouse B cells to express RSV-emAb.
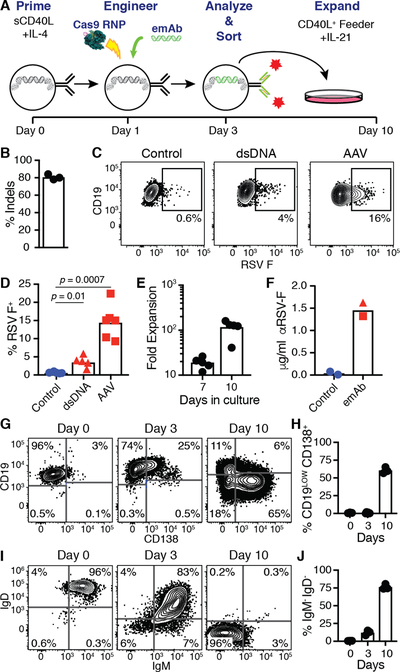
(A) Representation of the mouse B cell engineering protocol. (B) Frequency of indels detected in genomic heavy chain sequences from B cells two days after electroporation with muIgH367 gRNA/Cas9 in three independent experiments. (C) Representative flow cytometric analysis and (D) quantitation of RSV F antigen binding to mouse B cells engineered to express RSV-emAb using cassettes delivered using purified dsDNA or AAV versus control B cells that were mock electroporated. Data points in D represent the frequency of antigen-binding B cells from independent experiments (n=5–6) and p values were determined using an unpaired two-tailed t-test with Welch’s correction. (E) Number of RSV-emAb B cells in cultures from five experiments at day seven and ten displayed as a fold expansion over the number of cells sorted at day three. (F) ELISA-mediated quantitation of RSV F-specific antibodies in day 10 supernatants from RSV-emAb B cells engineered using dsDNA (triangle) or AAV (circle) versus control B cells. (G-J) Representative flow cytometric analysis and quantitation of (G, H) CD19 and CD138 or (I, J) IgM and IgD expression by RSV-emAb B cells from individual mice (n=3) at the start of culture, day three, and day ten. Representative of three similar experiments.
To assess the protective ability of RSV-emAb B cells, 0.5 – 1.5 × 107 Balb/c mouse B cells binding RSV antigen were transferred into wild-type Balb/c recipients (Fig. 6A). Six days following transfer, 3–29 μg/mL of RSV-specific antibodies were present in the serum from mice that received RSV-emAb B cells, but in those that received control B cells (Fig. 6B). These titers were not maintained, however, as antibody levels returned to baseline levels 25 days after transfer (Fig. 6C). We next assessed whether RSV-emAb B cells provided protection against infection while antibody levels were still high. For this, animals were challenged intranasally with 106 plaque-forming units (PFU) of RSV seven days following cell transfer and viral titers were measured in the lungs five days later. Around 5000 PFU of RSV was detected in lungs from control mice that did not receive cells, and recipients of control B cells (Fig. 6D). In contrast, RSV was nearly undetectable in mice that received RSV-emAb B cells (Fig. 6D). This protection was comparable to the protection afforded by the injection of a clinical dose, 15 mg/kg, of palivizumab two days before infection.
Figure 6. Immunocompetent mice are protected from RSV infection by RSV-emAb B cells.

(A) Schematic representation of experiment to test acute antiviral protection by RSV-emAb B cells seven days following transfer into Balb/cByJ recipient mice. (B) RSV F antigen-specific antibodies in serum by ELISA from individual control mice that did not receive transfer, or from mice six days after the transfer of control B cells or RSV-emAb B cells (n=4–9). Data combined from six independent experiments.(C) RSV F antigen-specific antibodies in serum from individual mice six, fifteen, and twenty-five days after the transfer of RSV-emAb B cells with or without infection with 106 PFU of RSV at day seven (n=3). The dashed line represents the mean level of RSV F-specific antibodies in uninfected control mice that did not receive cells. Data is combined from two independent experiments. (D) RSV PFU in the lungs of mice who received no B cell transfer, control B cells, RSV-emAb B cells, or 15 mg/kg palivizumab followed by intranasal infection with 106 PFU of RSV (n=2–9). The p values were determined using an unpaired two-tailed t-test with Welch’s correction.
Since hematopoietic stem cell transplant recipients are one of the most susceptible groups for RSV infection, we next assessed whether protective antibody levels would be sustained in immunodeficient hosts. Due to the profound immunodeficiency resulting from transplant, RSV infection in the three month period post-transplant carries a significant risk of lower respiratory tract infection, pneumonia, and death (34). For this reason, we tested the capacity of RSV-emAb B cells to provide long-term protection in immunodeficient RAG1−/− mice, which lack B and T cells (Fig. 7A). Transfer of 1.5 × 107 CD45.1+ C57Bl/6 RSV-emAb B cells into RAG1−/− mice led to a rapid accumulation of over 40 μg/mL RSV-specific antibodies in serum, which was maintained at this level for 40 days (Fig. 7B). Beginning at day 40, antibody levels declined to ~3 μg/mL 72 days after cell transfer (Fig. 7B, C). Despite this decline in titers, intranasal challenge of mice with RSV 82 days after RSV-emAb B cell transfer revealed near complete protection similar to mice challenged at day 7 when RSV PFU in the lung were assessed at day 87 and 12, respectively (Fig. 7D). Similar to wild-type recipients, protection did not appear to be mediated by a boosting effect, since serum from infected mice did not contain increased levels of serum antibody compared to their uninfected counterparts (Fig. 7E).
Figure 7. Long-term protection of immunocompromised mice from RSV infection by RSV-emAb B cells.

(A) Schematic representation of experiment to test long-term antiviral protection by transferred RSV-emAb B cells in RAG1−/− mice. (B) RSV F-specific antibodies were measured in serum from individual RAG1−/− mice at various time points after the transfer of 1.5 × 107 control or RSV-emAb B cells (n=2–9). Data is combined from three independent experiments. (C) Data from serum samples assessed 72 days after cell transfer shown on a different scale (n=2–4). (D) Combined data from two experiments in which RSV PFU were measured in the lungs of control mice which received no cell transfer compared to mice that received 1.5 × 107 RSV-emAb or control B cells 7 or 82 days prior to infection (n=2–4). The p values were determined using an unpaired two-tailed t-test with Welch’s correction. (E) RSV F-specific antibodies were measured in serum samples from individual RAG1−/− mice eleven days after the transfer of 0.5 × 107 control or RSV-emAb B cells with and without infection at day seven (n=3–5).
Analysis of transferred CD45.1+ cells revealed thousands of RSV-emAb B cells in the spleen and bone marrow of recipient mice (Fig. 8A–B). Many of the RSV-emAb B cells in the bone marrow expressed CD138 and low levels of CD19, a phenotype consistent with long-lived antibody-secreting plasma cells (Fig. 8C, D). In contrast, most of the RSV-emAb B cells in the spleen expressed CD19 and CD38, but not IgM, IgD, or CD138 (Fig. 8C–H), which would be consistent with the phenotype of an isotype switched memory B cell. The expression of these markers was indistinguishable in infected mice compared to their uninfected counterparts (Fig. 8F, H). CD38 expression was low on the CD19+ CD138− emAb B cells at the time of transfer (Fig. 8E, F), suggesting that cells re-expressed this molecule after transfer. Together, these results demonstrate that B cells can be efficiently engineered to provide robust and durable protection against infection.
Figure 8. Phenotype of RSV-emAb B cells in immunocompromised mice.

(A) Representative flow cytometric analysis and (B) quantitation of CD45.1+ RSV F+ CD3− F4/80− Gr-1− Fixable viability dye (FVD)− donor B cells in spleen and bone marrow from individual RAG1−/− mice from two combined experiments 12 or 87 days after transfer of 1.5 × 107 control or RSV-emAb B cells (n=2–4). Samples were enriched for CD45.1+ cells prior to analysis and all mice were infected with RSV five days prior to analysis. The limit of detection (LOD) was established in samples from mice that did not receive cell transfer (n=7). (C) Representative flow cytometric analysis and (D) quantitation of the % of CD19LOW CD138+ RSV-emAb B cells in the spleen and bone marrow from individual mice (n=2–4) from two combined experiments. (E) Representative flow cytometric analysis and (F) quantitation of CD38 expression by CD19+ CD138− RSV-emAb B cells in the spleen of recipient mice compared to B cells at start of culture and at the time of cell transfer (n=2–5) from three combined experiments. (G) Representative flow cytometric analysis and (H) quantitation of % of CD19+ CD138− RSV-emAb B cells in the spleen of individual animals that were IgM− IgD− twelve days after transfer (n=3–5).
Discussion
The isolation of monoclonal antibodies has transformed medicine as therapeutics (35–37). However, while the use of antibody-producing primary B cells in adoptive cellular therapy has lagged behind that of other cell types, there has recently been a wave of innovation in this area that paves the way for future clinical trials. There has been some recent genetic engineering work focused on taking advantage of the potent protein secretion capabilities of B cells to produce non-antibody therapeutic proteins (38, 39). Strategies to reprogram B cells to produce therapeutic antibodies have also been developed (17, 18), and several more will likely be published in the coming year. Our approach targets an intronic region, allowing for universal B cell engineering without a priori knowledge of the endogenous VDJ sequences. Engineered emAb receptors are not limited by the gene segments and recombination events that generate the endogenous B cell repertoire. This could be important in situations where current vaccines have failed. For example, many of the broadly neutralizing antibodies which have been identified for HIV-1 contain features which are rare in the naive B cell repertoire (40). As an alternative to isolation of rare antibodies, targeting domains could be designed in silico based either on the backbone of antibody variable domains, or entirely novel high affinity binding domains (41, 42). Combining these approaches with the emAb platform could allow targeting of pathogens for which no protective antibody has been isolated.
Insertion of emAb cassettes into this region was complicated by the ability of B cells to simultaneously express productive heavy chains from both alleles, which allowed the emAb to be expressed from one allele and an endogenous heavy chain from the other. Endogenous antibody/emAb co-expression can be circumvented by selecting against emAb B cells that express both kappa and lambda light chains when the endogenous antibody and emAb antibodies express one or the other. Alternatively, B cells can be engineered and selected based upon the simultaneous expression of two emAbs. The latter strategy is intriguing since complimentary epitopes on the same pathogen or escape mutations could be simultaneously targeted by the same engineered cells.
While we favor co-expression of two antibodies, there may be downsides to this approach. Normal B cells do not express multiple heavy and light chains, because this would result in numerous antibodies formed by different heavy and light chain parings. We have eliminated this possibility for dual-emAb B cells since the light and heavy chains are physically linked. However, dual-emAb expression could result in each cell expressing much less of each antibody compared to a cell that only expressed one antibody. For example, if one antibody is expressed to higher levels due to increased stability or a more active promoter, the expression of the second antibody could be greatly reduced compared to B cells only expressing a single antibody. This is particularly true of surface expressed antibody, where the level of CD79a and CD79b expressed by B cells is limiting. Another confounding factor is that the two antibodies in the cell could utilize different heavy chain constant regions unless class-switching was carefully controlled. Future work is necessary to probe these issues.
Our results indicate that serum antibody levels produced by emAb B cells were not stable long-term in wild-type recipients. One explanation is that cell fitness is decreased due to in vitro culture and we are exploring alternative culture methodologies to increase emAb B cell persistence in wild-type recipients. Another explanation is that emAb B cells are rejected by T or B cell responses targeting the emAb protein, and/or decreased cell fitness due to off-target cutting by Cas9. Future work is necessary to assess these mechanisms, and the strategy may need to be altered if cell rejection or off-target cutting is problematic.
Off-target cutting by Cas9 is a major concern of any engineering strategies utilizing gene cutting approaches. We have attempted to minimize the potential for off-target mutations by confining our strategy to a single gRNA and cut site for expression of both heavy and light chains and by using pre-complexed gRNA and Cas9 (43). Nevertheless, before emAb B cells could be utilized in the clinic, a thorough and deep analysis of off-target mutations must be conducted. If mutations are detected, the other gene cutting approaches or Cas9 variants with higher fidelity could be utilized (44).
Serum antibody levels were maintained for 40 days when emAb B cells were transferred into immunocompromised RAG1−/− mice that lack endogenous T and B cells. While titers declined after this time point, mice were protected for at least another 82 days due to persistence of emAb B cells. Notably, emAb B cells did not appear to respond to infection. We speculate that this poor response is the result of the absence of T cell help in RAG1−/− mice. While some memory B cells can respond independent of T cell help (45–49), in vitro cultured cells may not have gained this property and may be more functionally similar to the naive B cells that comprised the vast majority of the murine cells we utilized at the start of culture. In fact, it may not be appropriate to describe in vitro differentiated B cells that only encountered antigen at the time of cell sorting as “memory” B cells even though many of these cells are isotype switched. In the future, pre-selection of B cell subsets with reduced dependence on T cell help may improve the response to infection.
In situations where T cell help is present, emAb B cells may be able to enter the germinal center reaction and undergo affinity maturation. This could be beneficial in that the affinity of the engineered protective antibody was enhanced or mutation allowed for control of escape variants. We have not assessed whether activation-induced deaminase (AID) will effectively mutate inserted emAb genes. Using a different approach to replace the heavy chain VDJ, Voss et. al. demonstrated successful AID targeting and somatic hypermutation within the insert (17). This suggests that the emAb insert would be effectively targeted for somatic hypermutation, but it is possible that the inclusion of the light chain and insertion into the intronic region eliminates this targeting.
Our RAG1−/− experiments model immunodeficiencies where common viral infections frequently lead to hospitalization, disability, and death. Hematopoietic stem cell recipients are a particularly relevant group since they are vulnerable to infection post-transplant and are already receiving a cellular product as part of treatment for an underlying disease. If donor B cells were engineered and infused as emAb B cells targeting RSV, HMPV, EBV and CMV, then thousands of hospital visits, disabilities, and deaths could be prevented each year. For lower risk populations, in vitro culture and infusion of engineered cells could pose a barrier to the clinical translation of emAb B cells. However, new technology is being developed to bypass patient-specific in vitro preparation of adoptive cellular therapies such as the production of universal donor cells (50), as well as nanocarrier driven in vivo transduction of primary cells (51).
In summary, we have demonstrated specific and efficient engineering of primary mouse and human cells to produce multiple potent antiviral antibodies. Modified heavy chain loci in these engineered B cells retain the ability to undergo alternative splicing to generate both cell surface BCR and secreted antibodies at protective levels following adoptive transfer. This technique offers the possibility of engineering humoral immunity to produce sterilizing immunity to diseases for which no current therapy exists.
Materials and Methods
Study design
The aim of this study was to use CRISPR/Cas9 to replace the endogenous antibody expressed by human and murine B cells with antibodies known to be protective against RSV, influenza, HIV-1 or EBV. In the murine system, we also aimed to demonstrate that CRISPR/Cas9 engineered B cells could protect mice from infection. The size of the experimental groups is specified in figure legends. For RSV infection experiments, mice were randomly selected into infected versus uninfected groups. For most experiments the analysis was conducted unblinded, with the exception of quantitation of RSV PFU in the lung five days after infection.
Cell lines
3T3-msCD40L were obtained from Dr. Mark Connors at the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH (Cat#12535) and cultured in DMEM medium with 10% fetal calf serum (Gibco), 100 U/ml penicillin plus 100 μg/mL streptomycin (Gibco), and G418 (350 μg/mL). RAMOS cells were obtained from ATCC (CRL-1596™) and cultured in RPMI medium with 10% fetal calf serum (Gibco) and 100 U/mL penicillin plus 100 μg/mL streptomycin (Gibco). HEK293 cells were obtained from ATCC (CRL-1573™) cultured in DMEM medium with 10% fetal calf serum (Gibco), 100 U/mL penicillin plus 100 μg/mL streptomycin (Gibco). Vero cells were obtained from ATCC (CCL-8™) and cultured in DMEM medium with 10% fetal calf serum (Gibco), 100 U/mL penicillin plus 100 μg/mL streptomycin (Gibco).
Design of single-chain antibody template sequences
Human emAb cassettes consisted of a 450 base pair homology arm, the IgVH1–69 heavy chain promoter region, the full-length antibody light chain gene, a segment encoding a 57 amino acid glycine-serine linker containing three tandem copies of the Streptag-II motif, the variable region of the heavy chain, and a splice junction with 60 base pairs of flanking sequence derived from matching IgHJ variable regions followed by a 450 base pair homology arm. Antibody variable domain sequences were derived from the humanized monoclonal antibody MEDI-493/palivizumab (6), and the human monoclonal antibodies AMM01, VRC01, and MEDI8852 (27–29).
Murine emAb cassettes consisted of an upstream homology arm, the J5558H10 heavy chain promoter region (20), full length codon optimized antibody light chain, a segment encoding a 57 amino acid glycine-serine linker containing three tandem copies of the Streptag-II sequence, codon optimized variable region of the heavy antibody chain, and a splice junction with 60 base pairs of flanking sequence derived from the mouse IGHJ3 gene segment followed by a downstream homology arm. Antibody variable domains were derived from mouse Mab 1129 (6). The AAV emAb cassette included a 503 base pair upstream homology arm and a 968 base pair downstream homology arm while the dsDNA emAb cassette included a 36 base pair upstream and downstream homology arms.
Production of recombinant emAb-AAV
To generate AAV plasmids for homologous recombination, linearized AAV backbone was isolated from pAAV-GFP (Addgene, Plasmid #32395) by digestion with SnaBI (New England Biolabs), and homology arms for mouse and human heavy chain flanking an EcoRV restriction site inserted using NEB builder HiFi DNA assembly master mix (New England Biolabs). EmAb constructs were then synthesized as gene fragments (Integrated DNA Technologies) and cloned into the EcoRV site using Gibson HiFi master mix.
AAVs were generated by triple transfection of AAV emAb plasmid, serotype 6 capsid, and adenoviral helper plasmids into HEK293 cells using polyethylenimine (Polysciences). Eighteen hours after transfection, the media was changed to serum-free DMEM and the cells incubated for 48 hours cells prior to being lysed by freeze-thaw, treated with 20U of benzonase (Thermo Fisher Scientific) per 1 mL of viral lysate for 30 minutes at 37° C, then purified over iodixanol gradient. Purified AAV was concentrated into 1x DPBS using an Amicon Ultra-15 column (EMD Millipore) (52) prior to viral titer determination by qPCR of AAV genomes (53), which ranged from 1–7 × 1010 per microliter.
Production of murine double stranded DNA emAb templates
dsDNA templates containing short homology regions were generated from RSV-emAb AAV plasmids through PCR amplification using Platinum PCR Supermix High Fidelity (Thermo Fisher Scientific) and modified DNA oligos. PCR product was purified and concentrated using minElute PCR cleanup columns (Qiagen). The following primers were used for amplification, with mouse genomic homology region in bold and phosphorothioate stabilized DNA bonds denoted by *:
Forward primer:
5’Phosphate/ACCACCTCTGTGACAGCATTTATACAGTATCCGATGGACAAGTGAGTGTCTCAGGTTAGGATTCT
Reverse primer:
T*A*A*AGAAAGTGCCCCACTCCACTCTTTGTCCCTATGCTTGACCACAATGAATACTCCCACC
Mouse B cell culture and electroporation
Mouse B cell medium consisted of RPMI supplemented with 10% fetal calf serum (Gemini Biosciences), 10mM HEPES (Gibco), 55 μM Beta-mercaptoethanol (Sigma), and 100 U/ml penicillin plus 100 μg/mL streptomycin (Gibco) except in antibiotic free steps as noted. B cells were isolated from spleen and lymph nodes via negative selection with magnetic beads (Miltenyi Biotec) and 2×106 cells/mL were cultured for 24 hours at 37°C in a tissue culture incubator in B cell medium supplemented with 100 ng/mL recombinant carrier free HA-tagged mouse CD40L (R&D systems), 100 ng/mL αHA antibody (clone 543851, R&D systems), and 4 ng/mL mouse IL-4 (R&D systems). Next, the B cells were electroporated using the Neon transfection system as follows: Cas9 protein (Invitrogen) and synthetic gRNA (Synthego) were precomplexed at a 1 to 3 molar ratio in Neon Buffer T at room temperature for 20 minutes. The muIgH367 gRNA sequence with the PAM site in bold is TTATACAGTATCCGATGCATAGG. B cells were washed with 1xDPBS and suspended in Neon Buffer T at a final density of 2.5×107 cells/mL with 12 μg of Cas9 per 106 cells. When dsDNA emAb cassettes were used, 7.5 μg dsDNA template per 106 cells was included in the electroporation. Cells were electroporated with three 10 millisecond pulses at 1675 volts and immediately dispensed into pre-warmed antibiotic-free mouse B cell medium. For AAV experiments, after electroporation concentrated AAV in 1xDPBS was added at an up to 20% of final culture volume at a final multiplicity of infection (MOI) of 105–106 genome copies per cell and incubated for 1 hour. After AAV infection, B cells were expanded for an additional 48 hours with B cell medium supplemented with 100 ng/mL recombinant carrier free HA-tagged mouse CD40L, 100 ng/mL αHA antibody, 4 ng/mL mouse IL-4 (R&D systems), and 20 ng/mL mouse IL-21 (BioLegend). For additional expansion, B cells were co-cultured with irradiated (80 gy) NIH 3T3-CD40L feeder cells in the presence of 20 ng/mL mouse IL-21 for 6–8 days, with passage onto fresh irradiated 3T3-CD40L feeder cells every 4 days.
Human B cell culture and electroporation
Human B cell medium was IMDM supplemented with 10% FBS (Gemini Biosciences), 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco), except in antibiotic free steps as noted. Blood was obtained from healthy, HIV-seronegative adult volunteers as a part of the General Quality Control study in Seattle, WA by venipuncture and was approved by the Fred Hutch Institutional Review Board. Informed consent was obtained before enrollment. PBMCs were isolated from whole blood using Accuspin System Histopaque-1077 (Sigma-Aldrich) resuspended in 10% dimethylsulfoxide in heat-inactivated fetal bovine serum and cryopreserved in liquid nitrogen before use. PBMCs were thawed and B cells isolated using negative selection using the Human B Cell Isolation Kit II (Miltenyi Biotec) according to the manufacturer’s recommendations. Isolated B cells were resuspended at 0.5–1.0×106 cells/mL in stimulation media, which consisted of human B cell medium supplemented with 100 ng/mL MEGACD40L (Enzo Life Sciences), 50 ng/mL recombinant IL-2 (BioLegend), 50 ng/mL IL-10 (Shenandoah Biotech), 10 ng/mL IL-15 (Shenandoah Biotech), 1 μg/mL CpG ODN 2006 (IDT). After 48 hours, cells were electroporated using the Neon Transfection System. Cas9 protein (Invitrogen) and gRNA (Synthego) were precomplexed at a 1 to 2 molar ratio in Neon Buffer T for 20 minutes at room temperature. Cells were washed with 1xDPBS and resuspended to 2.5×107 cells/mL in Neon Buffer T containing 12 μg of pre-complexed gRNA/Cas9 per 106 cells. The huIgH296 gRNA sequence with the PAM site in bold is GTCTCAGGAGCGGTGTCTGTAGG. The cell/gRNA/Cas9 mixture was electroporated with one 20 millisecond pulse at 1750V and immediately plated into stimulation media as described above, without antibiotics. After 30 minutes, AAV was added to a final concentration of up to 20% culture volume amounting to a MOI of 105–106 genome copies per cell and incubated for 2–4 hours. Cells were next transferred to a larger culture dish to allow for further expansion. Two days after electroporation, cells were labeled with fluorochrome labeled antigen and/or streptactin and engineered cells FACS-purified. For secondary expansion, B cells were co-cultured for 4–8 days with irradiated (80 gy) NIH 3T3-CD40L feeder cells in human B cell medium containing 5 μg/mL human recombinant insulin (Sigma), 50 μg/mL transferrin (Sigma), 50 ng/mL human IL-2 (BioLegend), 20 ng/mL human IL-21 (BioLegend), and 10 ng/mL human IL-15 (Shenandoah Biotech). Cells were passaged onto fresh 3T3-CD40L feeder cells every 4 days. In order to promote differentiation to plasma cells, cells were washed, and transferred from expansion conditions into fresh feeder-free culture conditions containing human B cell medium supplemented with 5 μg /mL human recombinant insulin (Sigma), 50 μg/mL transferrin (Sigma), 500 U/mL Universal Type I IFN Protein (R&D Systems), 50 ng/mL IL-6 (Shenandoah Biotech), and 10 ng/mL IL-15 (Shenandoah Biotech).
Assessment of gRNA activity by Sanger sequencing
Total genomic DNA was isolated from 0.5–2 × 106 mock and Cas9/gRNA treated cells two-five days following electroporation using the DNeasy kit (Qiagen). The genomic DNA region flanking the gRNA target site was amplified by PCR using the following primers:
Mouse IgH Forward, GGCTCCACCAGACCTCTCTA
Mouse IgH Reverse, AACCTCAGTCACCGTCTCCT
Human IgH Forward, ACAGTAAGCATGCCTCCTAAG
Human IgH Reverse, GCCACTCTAGGGCCTTTGTT
The resulting PCR product was purified using minElute reaction cleanup kit (Qiagen) and Sanger sequenced (Genewiz). The frequency of indels in Cas9/gRNA electroporated cells relative to control cells was determined using the ICE algorithm (54).
Protein antigens
RSV prefusion F antigen trimer, EBV gH/gL complex, and a modified HIV Envelope GP140 trimer (426c TM4ΔV1–3) were produced as described (29, 55, 56). Stabilized influenza HA stem was produced from VRC clone 3925, derived from strain H1 1999 NC as described (57). All antigens were conjugated to Biotin NHS ester (Thermo Fisher Scientific) followed by tetramerization with streptavidin-PE, streptavidin-APC or streptavidin (all from Prozyme) as described previously (58). RSV F antigen was conjugated to Alexa Fluor 488 NHS ester (Thermo Fisher Scientific) according to the manufacturer’s recommendations and used for flow cytometry.
Flow Cytometry
Cells were incubated in 50 μL of FACS buffer containing a cocktail of antibodies for 30 minutes on ice prior to washing and analysis on a FACSymphony (BD Bioscience) or sorted on FACS Aria II (BD Bioscience). FACS buffer consisted of 1xDPBS containing 5 mM EDTA and either 1% newborn calf serum (Life Technologies) or 1% bovine serum albumin (Sigma) for experiments including streptactin staining. For murine experiments, cells were labeled with a cocktail including combinations of streptactin PE (IBA Lifesciences), anti-CD45.1 APC (A20, BioLegend), anti-IgM PerCP-eFluor710 (II/41, eBioscience), anti-IgD PE-Cy7 (11–26c, eBioscience) anti-CD138 BV421 (281–2, BioLegend), anti-CD38 AF700 (90, eBioscience), anti-CD19 BUV395 (ID3, BD Biosciences), anti-CD3e BV510 (145–2C11, BD Biosciences), anti-Gr- BV510 (RB6–8C5, BioLegend), anti-F4/80 BV510 (BM8, BioLegend), and a fixable viability dye (eBioscience) prior to analysis. For human experiments, cells were labeled with a cocktail including a combinations of streptactin PE, anti-Igλ PE (MHL-38, BioLegend), anti-Igλ V450 (JDC-12, BD Biosciences), anti-CD19 APC (GSJ25C1, BD Biosciences), anti-CD20 BUV395 (2H7, BD Biosciences), anti-CD27 PE-Cy7 (LG.7F9, eBioscience), anti-CD38 BV650 (HB-7, BioLegend), anti-CD45, (HI30, BD Biosciences), anti-CD79b APC/Fire750 (3B3–1, BioLegend), and a fixable viability dye.
For ex vivo analysis of transferred cells after RSV infection in mice, single cell suspensions of spleen and bone marrow from the femurs were generated by manual disassociation and filtration. Cells were stained with anti-CD45.1 APC (A20, BioLegend) and purified anti-CD16/32 (2.4G2, BioXcell) for 30 minutes on ice, washed with FACS buffer and then incubated with 25 μL of anti-APC conjugated magnetic microbeads (Miltenyi Biotec). Following a 15–30 minute incubation on ice, 3 mL of FACS buffer was added and the sample was passed over a magnetized LS column (Miltenyi Biotec). The tube and column were washed once with 5 mL of FACS buffer and then removed from the magnetic field. Five mL of FACS buffer was pushed through the column with a plunger twice to elute column-bound cells. Cells from the column-bound and 1/40 of the column flow through fractions were stained as described above. 20,000 AccuCheck counting beads (Invitrogen) were added to the samples to calculate total cell numbers. To account for cells in bones that were not harvested, the number of cells detected in the pooled femurs was multiplied by ten (59).
Calcium flux was measured by flow cytometry with the Fluo-4 Direct kit (Thermo Fisher Scientific). Briefly, 1.5 × 106 cells were labelled with 1 mL Fluo-4 according to manufacturer instructions for 30 minutes at 37°C. Fluo-4 baseline florescence was measured for 60 seconds, then cells were stimulated with 1 μg tetramerized RSV F antigen, followed by 180 seconds of measurement, and finally cells were simulated with 1 μg ionomycin in DMSO, and florescence was measured for an additional 60 seconds. Florescence intensity data was binned by time and displayed as fold change over baseline measurement. Flow cytometry data was analyzed using FlowJo X software (Tree Star).
Animals
Animal studies were approved and conducted in accordance with the Fred Hutchinson Cancer Center Institutional Animal Care and Use Committee. Six- to ten-week old male and female BALB/cByJ, BALB/cByJ-CD45.1, C57bl/6-RAG1−/− (RAG1−/−), and C57bl/6-CD45.1 mice were obtained from the Jackson Laboratory. For transfer of emAb B cells, age matched BALB/cByJ mice or RAG1−/− mice received a single intraperitoneal (IP) injection of emAb or control B cells derived from CD45.1+ congenic donor mice, or a single IP injection of 15 mg/kg palivizumab at the indicated two days prior to RSV challenge.
RSV infections and titer measurement
In RSV challenge experiments, mice were inoculated intranasally with 106 PFU of sucrose purified RSV expressing eGFP (60) in 40 μL 1xDPBS. Lungs were harvested five days post-infection and the titer was determined using a plaque assay (61). In brief, lungs were homogenized in 2 mL of DMEM using a GentleMACS M Dissociator using preset program lung_02 (Miltenyi Biotec) followed by centrifugation at 400xg for 10 minutes. Supernatant was flash frozen and stored at −80°C. The supernatant was diluted 1:10 and 1:20 in DMEM and 100 μL of each dilution was added duplicate to confluent Vero cells in 24 well flat-bottoms tissue-culture plates and incubated for 2 hours at 37°C. An overlay of 0.8% methylcellulose was then added and plates incubated for five days prior to imaging on a Typhoon imager (GE Healthcare) with filter settings for eGFP. The titer in pfu/lung was calculated by counting the number of eGFP+ plaques with ImageJ software in the highest positive dilution and correcting for the dilution factor.
ELISA
Nunc maxsorp 96-well plates (Thermo Fisher) were coated with 1 μg/mL of RSV F, HIV Env, or influenza HA in 1xDPBS overnight at 4°C. Plates were washed three times with 1xDPBS containing 0.05% TWEEN-20 (PBST) and blocked with 150 μL/well PBST+3% bovine serum albumin (BSA) (Sigma-Aldrich) for 1 hour at room temperature. Alternatively, 1 μg/mL of EBV gH/gL in 1x DPBS was coated on pre-blocked 96-well Ni-NTA plates (Qiagen) for 1 hour at room temperature, then washed three times with PBST. Antigen coated plates were incubated with culture supernatant or mouse plasma samples in duplicate diluted in PBST+3%BSA and a standard curve generated using the purified recombinant mouse RSV-specific antibody Mab 1127, or purified recombinant human RSV-specific palivizumab (Synagis clinical grade, MedImmune), influenza HA-specific Medi8852, EBV gH/gL-specific AMM01, or HIV-1 Env-specific VRC01 positive control antibodies for 90 minutes at room temperature. Plates were washed five times with PBST prior to 1 hour incubation with horseradish peroxidase-conjugated goat anti-mouse or anti-human total Ig (Southern Biotech) diluted 1:4000 in PBST+3%BSA. Plates were then washed three times with PBST prior to a 2–15 minute incubation with 100 uL/well of ELISA 1xTMB substrate (Thermo Fisher) and absorbance was measured at 405 nm using a Softmax Pro plate reader (Molecular Devices). The concentration of antigen-specific antibody in each sample was determined by reference to the standard curve and dilution factor.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 7. Pairwise statistical comparisons were performed using unpaired two-tailed t-test with Welch’s correction. P < 0.05 was considered statistically significant. Data points from individual samples are typically displayed and raw values can be found in Table S1.
Supplementary Material
Acknowledgments:
We thank M. D. Grey and L. Stamatatos for providing HIV-1 Env, plasmids containing the VRC01 sequence and purified VRC01 antibody, A. T. McGuire for EBV GH/GL, plasmids containing AMM01 and purified AMM01 antibody., B. Graham for plasmids encoding RSV prefusion F antigen, P. Kwong for plasmids encoding influenza HA stem, D. Stone for help producing AAVs, P. Collins and U. Buchholz for RSV expressing eGFP, M. J. McElrath for PBMCs from Seattle Area Control cohort, and K. G. Anderson for critical reading of the manuscript.
Funding: Supported by a donation from E. Averett, an Individual Biomedical Research Award from The Hartwell Foundation (J.J.T.), a Sponsored Research Agreement from Vir Biotechnology (J.J.T.), and the National Institutes of Health under award numbers T32AI118690 (J. B.) and T32GM095421 (K.S.F.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing interests: Work described in this manuscript has been included in patent application PCT/US2018/056789.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper. All constructs and reagents are available for research purposes by contacting J.J.T. The sequences of human RSV-emAb (MK689353), human Flu-emAb (MK689354), human HIV-emAb (MK689352), human EBV-emAb (MK689357) and murine RSV-emAb AAV (MK689356) and murine RSV-emAb dsDNA (MK689355) have been deposited in GenBank.
References and Notes:
- 1.Chemaly RF, Shah DP, Boeckh MJ, Management of respiratory viral infections in hematopoietic cell transplant recipients and patients with hematologic malignancies. Clin Infect Dis 59 Suppl 5, S344–351 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shi T, McAllister DA, O’Brien KL, Simoes EAF, Madhi SA, Gessner BD, Polack FP, Balsells E, Acacio S, Aguayo C, Alassani I, Ali A, Antonio M, Awasthi S, Awori JO, Azziz-Baumgartner E, Baggett HC, Baillie VL, Balmaseda A, Barahona A, Basnet S, Bassat Q, Basualdo W, Bigogo G, Bont L, Breiman RF, Brooks WA, Broor S, Bruce N, Bruden D, Buchy P, Campbell S, Carosone-Link P, Chadha M, Chipeta J, Chou M, Clara W, Cohen C, de Cuellar E, Dang DA, Dash-Yandag B, Deloria-Knoll M, Dherani M, Eap T, Ebruke BE, Echavarria M, de Freitas Lazaro Emediato CC, Fasce RA, Feikin DR, Feng L, Gentile A, Gordon A, Goswami D, Goyet S, Groome M, Halasa N, Hirve S, Homaira N, Howie SRC, Jara J, Jroundi I, Kartasasmita CB, Khuri-Bulos N, Kotloff KL, Krishnan A, Libster R, Lopez O, Lucero MG, Lucion F, Lupisan SP, Marcone DN, McCracken JP, Mejia M, Moisi JC, Montgomery JM, Moore DP, Moraleda C, Moyes J, Munywoki P, Mutyara K, Nicol MP, Nokes DJ, Nymadawa P, da Costa Oliveira MT, Oshitani H, Pandey N, Paranhos-Baccala G, Phillips LN, Picot VS, Rahman M, Rakoto-Andrianarivelo M, Rasmussen ZA, Rath BA, Robinson A, Romero C, Russomando G, Salimi V, Sawatwong P, Scheltema N, Schweiger B, Scott JAG, Seidenberg P, Shen K, Singleton R, Sotomayor V, Strand TA, Sutanto A, Sylla M, Tapia MD, Thamthitiwat S, Thomas ED, Tokarz R, Turner C, Venter M, Waicharoen S, Wang J, Watthanaworawit W, Yoshida LM, Yu H, Zar HJ, Campbell H, Nair H, R. S. V. G. E. Network, Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: a systematic review and modelling study. Lancet 390, 946–958 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE, Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med 352, 1749–1759 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Kim HW, Canchola JG, Brandt CD, Pyles G, Chanock RM, Jensen K, Parrott RH, Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am J Epidemiol 89, 422–434 (1969). [DOI] [PubMed] [Google Scholar]
- 5.Mazur NI, Higgins D, Nunes MC, Melero JA, Langedijk AC, Horsley N, Buchholz UJ, Openshaw PJ, McLellan JS, Englund JA, Mejias A, Karron RA, Simoes EA, Knezevic I, Ramilo O, Piedra PA, Chu HY, Falsey AR, Nair H, Kragten-Tabatabaie L, Greenough A, Baraldi E, Papadopoulos NG, Vekemans J, Polack FP, Powell M, Satav A, Walsh EE, Stein RT, Graham BS, Bont LJ, Respiratory F Syncytial Virus Network, The respiratory syncytial virus vaccine landscape: lessons from the graveyard and promising candidates. Lancet Infect Dis 18, e295–e311 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Johnson S, Oliver C, Prince GA, Hemming VG, Pfarr DS, Wang SC, Dormitzer M, O’Grady J, Koenig S, Tamura JK, Woods R, Bansal G, Couchenour D, Tsao E, Hall WC, Young JF, Development of a humanized monoclonal antibody (MEDI-493) with potent in vitro and in vivo activity against respiratory syncytial virus. J Infect Dis 176, 1215–1224 (1997). [DOI] [PubMed] [Google Scholar]
- 7.Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. The IMpact-RSV Study Group. Pediatrics 102, 531–537 (1998). [PubMed] [Google Scholar]
- 8.Walker LM, Burton DR, Passive immunotherapy of viral infections: ‘super-antibodies’ enter the fray. Nat Rev Immunol 18, 297–308 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fonseca MHG, Furtado GP, Bezerra MRL, Pontes LQ, Fernandes CFC, Boosting half-life and effector functions of therapeutic antibodies by Fc-engineering: An interaction-function review. Int J Biol Macromol 119, 306–311 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Skaricic D, Traube C, De B, Joh J, Boyer J, Crystal RG, Worgall S, Genetic delivery of an anti-RSV antibody to protect against pulmonary infection with RSV. Virology 378, 79–85 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Schnepp BC, Johnson PR, Adeno-associated virus delivery of broadly neutralizing antibodies. Curr Opin HIV AIDS 9, 250–256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo XM, Maarschalk E, O’Connell RM, Wang P, Yang L, Baltimore D, Engineering human hematopoietic stem/progenitor cells to produce a broadly neutralizing anti-HIV antibody after in vitro maturation to human B lymphocytes. Blood 113, 1422–1431 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Kuhlmann AS, Haworth KG, Barber-Axthelm IM, Ironside C, Giese MA, Peterson CW, Kiem HP, Long-Term Persistence of Anti-HIV Broadly Neutralizing Antibody-Secreting Hematopoietic Cells in Humanized Mice. Mol Ther 27, 164–177 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martincic K, Alkan SA, Cheatle A, Borghesi L, Milcarek C, Transcription elongation factor ELL2 directs immunoglobulin secretion in plasma cells by stimulating altered RNA processing. Nat Immunol 10, 1102–1109 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fusil F, Calattini S, Amirache F, Mancip J, Costa C, Robbins JB, Douam F, Lavillette D, Law M, Defrance T, Verhoeyen E, Cosset FL, A Lentiviral Vector Allowing Physiologically Regulated Membrane-anchored and Secreted Antibody Expression Depending on B-cell Maturation Status. Mol Ther 23, 1734–1747 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watson CT, Glanville J, Marasco WA, The Individual and Population Genetics of Antibody Immunity. Trends Immunol 38, 459–470 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Voss JE, Gonzalez-Martin A, Andrabi R, Fuller RP, Murrell B, McCoy LE, Porter K, Huang D, Li W, Sok D, Le K, Briney B, Chateau M, Rogers G, Hangartner L, Feeney AJ, Nemazee D, Cannon P, Burton DR, Reprogramming the antigen specificity of B cells using genome-editing technologies. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greiner V, Bou Puerto R, Liu S, Herbel C, Carmona EM, Goldberg MS, CRISPR-Mediated Editing of the B Cell Receptor in Primary Human B Cells. iScience 12, 369–378 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gillies SD, Morrison SL, Oi VT, Tonegawa S, A tissue-specific transcription enhancer element is located in the major intron of a rearranged immunoglobulin heavy chain gene. Cell 33, 717–728 (1983). [DOI] [PubMed] [Google Scholar]
- 20.Love VA, Lugo G, Merz D, Feeney AJ, Individual V(H) promoters vary in strength, but the frequency of rearrangement of those V(H) genes does not correlate with promoter strength nor enhancer-independence. Mol Immunol 37, 29–39 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Delpy L, Decourt C, Le Bert M, Cogne M, B cell development arrest upon insertion of a neo gene between JH and Emu: promoter competition results in transcriptional silencing of germline JH and complete VDJ rearrangements. J Immunol 169, 6875–6882 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Koerber JT, Hornsby MJ, Wells JA, An improved single-chain Fab platform for efficient display and recombinant expression. J Mol Biol 427, 576–586 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmidt TG, Skerra A, The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat Protoc 2, 1528–1535 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Chu VT, Graf R, Wirtz T, Weber T, Favret J, Li X, Petsch K, Tran NT, Sieweke MH, Berek C, Kuhn R, Rajewsky K, Efficient CRISPR-mediated mutagenesis in primary immune cells using CrispRGold and a C57BL/6 Cas9 transgenic mouse line. Proc Natl Acad Sci U S A 113, 12514–12519 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K, dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29, 308–311 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korndorfer IP, Skerra A, Improved affinity of engineered streptavidin for the Strep-tag II peptide is due to a fixed open conformation of the lid-like loop at the binding site. Protein Sci 11, 883–893 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, Longo NS, McKee K, O’Dell S, Louder MK, Wycuff DL, Feng Y, Nason M, Doria-Rose N, Connors M, Kwong PD, Roederer M, Wyatt RT, Nabel GJ, Mascola JR, Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329, 856–861 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kallewaard NL, Corti D, Collins PJ, Neu U, McAuliffe JM, Benjamin E, Wachter-Rosati L, Palmer-Hill FJ, Yuan AQ, Walker PA, Vorlaender MK, Bianchi S, Guarino B, De Marco A, Vanzetta F, Agatic G, Foglierini M, Pinna D, Fernandez-Rodriguez B, Fruehwirth A, Silacci C, Ogrodowicz RW, Martin SR, Sallusto F, Suzich JA, Lanzavecchia A, Zhu Q, Gamblin SJ, Skehel JJ, Structure and Function Analysis of an Antibody Recognizing All Influenza A Subtypes. Cell 166, 596–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Snijder J, Ortego MS, Weidle C, Stuart AB, Gray MD, McElrath MJ, Pancera M, Veesler D, McGuire AT, An Antibody Targeting the Fusion Machinery Neutralizes Dual-Tropic Infection and Defines a Site of Vulnerability on Epstein-Barr Virus. Immunity 48, 799–811 e799 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sonoda E, Pewzner-Jung Y, Schwers S, Taki S, Jung S, Eilat D, Rajewsky K, B cell development under the condition of allelic inclusion. Immunity 6, 225–233 (1997). [DOI] [PubMed] [Google Scholar]
- 31.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC, Predominant autoantibody production by early human B cell precursors. Science 301, 1374–1377 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Bernard O, Cory S, Gerondakis S, Webb E, Adams JM, Sequence of the murine and human cellular myc oncogenes and two modes of myc transcription resulting from chromosome translocation in B lymphoid tumours. EMBO J 2, 2375–2383 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wiman KG, Clarkson B, Hayday AC, Saito H, Tonegawa S, Hayward WS, Activation of a translocated c-myc gene: role of structural alterations in the upstream region. Proc Natl Acad Sci U S A 81, 6798–6802 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shah JN, Chemaly RF, Management of RSV infections in adult recipients of hematopoietic stem cell transplantation. Blood 117, 2755–2763 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Kohler G, Milstein C, Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497 (1975). [DOI] [PubMed] [Google Scholar]
- 36.Sadelain M, Riviere I, Riddell S, Therapeutic T cell engineering. Nature 545, 423–431 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carter PJ, Lazar GA, Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat Rev Drug Discov 17, 197–223 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Hung KL, Meitlis I, Hale M, Chen CY, Singh S, Jackson SW, Miao CH, Khan IF, Rawlings DJ, James RG, Engineering Protein-Secreting Plasma Cells by Homology-Directed Repair in Primary Human B Cells. Mol Ther 26, 456–467 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson MJ, Laoharawee K, Lahr WS, Webber BR, Moriarity BS, Engineering of Primary Human B cells with CRISPR/Cas9 Targeted Nuclease. Sci Rep 8, 12144 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou T, Georgiev I, Wu X, Yang ZY, Dai K, Finzi A, Kwon YD, Scheid JF, Shi W, Xu L, Yang Y, Zhu J, Nussenzweig MC, Sodroski J, Shapiro L, Nabel GJ, Mascola JR, Kwong PD, Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science 329, 811–817 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baran D, Pszolla MG, Lapidoth GD, Norn C, Dym O, Unger T, Albeck S, Tyka MD, Fleishman SJ, Principles for computational design of binding antibodies. Proc Natl Acad Sci U S A 114, 10900–10905 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chevalier A, Silva DA, Rocklin GJ, Hicks DR, Vergara R, Murapa P, Bernard SM, Zhang L, Lam KH, Yao G, Bahl CD, Miyashita SI, Goreshnik I, Fuller JT, Koday MT, Jenkins CM, Colvin T, Carter L, Bohn A, Bryan CM, Fernandez-Velasco DA, Stewart L, Dong M, Huang X, Jin R, Wilson IA, Fuller DH, Baker D, Massively parallel de novo protein design for targeted therapeutics. Nature 550, 74–79 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang X, Potter J, Kumar S, Zou Y, Quintanilla R, Sridharan M, Carte J, Chen W, Roark N, Ranganathan S, Ravinder N, Chesnut JD, Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J Biotechnol 208, 44–53 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Vakulskas CA, Dever DP, Rettig GR, Turk R, Jacobi AM, Collingwood MA, Bode NM, McNeill MS, Yan S, Camarena J, Lee CM, Park SH, Wiebking V, Bak RO, Gomez-Ospina N, Pavel-Dinu M, Sun W, Bao G, Porteus MH, Behlke MA, A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat Med 24, 1216–1224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Von Eschen KB, Rudbach JA, Immunological responses of mice to native protoplasmic polysaccharide and lipopolysaccharide: functional separation of the two signals required to stimulate a secondary antibody response. J Exp Med 140, 1604–1614 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernasconi NL, Traggiai E, Lanzavecchia A, Maintenance of serological memory by polyclonal activation of human memory B cells. Science 298, 2199–2202 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Richard K, Pierce SK, Song W, The agonists of TLR4 and 9 are sufficient to activate memory B cells to differentiate into plasma cells in vitro but not in vivo. J Immunol 181, 1746–1752 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, Good-Jacobson KL, Shlomchik MJ, CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol 15, 631–637 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krishnamurty AT, Thouvenel CD, Portugal S, Keitany GJ, Kim KS, Holder A, Crompton PD, Rawlings DJ, Pepper M, Somatically Hypermutated Plasmodium-Specific IgM(+) Memory B Cells Are Rapid, Plastic, Early Responders upon Malaria Rechallenge. Immunity 45, 402–414 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, Butler K, Rivat C, Wright G, Somana K, Ghorashian S, Pinner D, Ahsan G, Gilmour K, Lucchini G, Inglott S, Mifsud W, Chiesa R, Peggs KS, Chan L, Farzeneh F, Thrasher AJ, Vora A, Pule M, Veys P, Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med 9, (2017). [DOI] [PubMed] [Google Scholar]
- 51.Lee K, Conboy M, Park HM, Jiang F, Kim HJ, Dewitt MA, Mackley VA, Chang K, Rao A, Skinner C, Shobha T, Mehdipour M, Liu H, Huang WC, Lan F, Bray NL, Li S, Corn JE, Kataoka K, Doudna JA, Conboy I, Murthy N, Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat Biomed Eng 1, 889–901 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi VW, Asokan A, Haberman RA, Samulski RJ, Production of recombinant adeno-associated viral vectors. Curr Protoc Hum Genet Chapter 12, Unit 12 19 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Aurnhammer C, Haase M, Muether N, Hausl M, Rauschhuber C, Huber I, Nitschko H, Busch U, Sing A, Ehrhardt A, Baiker A, Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum Gene Ther Methods 23, 18–28 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Hsiau T, Maures T, Waite K, Yang J, Kelso R, Holden K, Stoner R, Inference of CRISPR Edits from Sanger Trace Data. bioRxiv, (2018). [DOI] [PubMed] [Google Scholar]
- 55.McLellan JS, Chen M, Joyce MG, Sastry M, Stewart-Jones GB, Yang Y, Zhang B, Chen L, Srivatsan S, Zheng A, Zhou T, Graepel KW, Kumar A, Moin S, Boyington JC, Chuang GY, Soto C, Baxa U, Bakker AQ, Spits H, Beaumont T, Zheng Z, Xia N, Ko SY, Todd JP, Rao S, Graham BS, Kwong PD, Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 342, 592–598 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McGuire AT, Gray MD, Dosenovic P, Gitlin AD, Freund NT, Petersen J, Correnti C, Johnsen W, Kegel R, Stuart AB, Glenn J, Seaman MS, Schief WR, Strong RK, Nussenzweig MC, Stamatatos L, Specifically modified Env immunogens activate B-cell precursors of broadly neutralizing HIV-1 antibodies in transgenic mice. Nat Commun 7, 10618 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yassine HM, Boyington JC, McTamney PM, Wei CJ, Kanekiyo M, Kong WP, Gallagher JR, Wang L, Zhang Y, Joyce MG, Lingwood D, Moin SM, Andersen H, Okuno Y, Rao SS, Harris AK, Kwong PD, Mascola JR, Nabel GJ, Graham BS, Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat Med 21, 1065–1070 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Taylor JJ, Martinez RJ, Titcombe PJ, Barsness LO, Thomas SR, Zhang N, Katzman SD, Jenkins MK, Mueller DL, Deletion and anergy of polyclonal B cells specific for ubiquitous membrane-bound self-antigen. J Exp Med 209, 2065–2077 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Colvin GA, Lambert JF, Abedi M, Hsieh CC, Carlson JE, Stewart FM, Quesenberry PJ, Murine marrow cellularity and the concept of stem cell competition: geographic and quantitative determinants in stem cell biology. Leukemia 18, 575–583 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Munir S, Le Nouen C, Luongo C, Buchholz UJ, Collins PL, Bukreyev A, Nonstructural proteins 1 and 2 of respiratory syncytial virus suppress maturation of human dendritic cells. J Virol 82, 8780–8796 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murphy BR, Sotnikov AV, Lawrence LA, Banks SM, Prince GA, Enhanced pulmonary histopathology is observed in cotton rats immunized with formalin-inactivated respiratory syncytial virus (RSV) or purified F glycoprotein and challenged with RSV 3–6 months after immunization. Vaccine 8, 497–502 (1990). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.