

Abstract
Genome-wide association and whole exome sequencing studies from Autism Spectrum Disorder (ASD) patient populations have implicated numerous risk factor genes whose mutation or deletion results in significantly increased incidence of ASD. Behavioral studies of monogenic mutant mouse models of ASD-associated genes have been useful for identifying aberrant neural circuitry. However, behavioral results often differ from lab to lab, and studies incorporating both males and females are often not performed despite the significant sex-bias of ASD. In this study, we sought to investigate the simple, passive behavior of home-cage activity monitoring across multiple 24-h days in four different monogenic mouse models of ASD: Shank3b−/−, Cntnap2−/−, Pcdh10+/−, and Fmr1 knockout mice. Relative to sex-matched wildtype (WT) littermates, we discovered significant home-cage hypoactivity, particularly in the dark (active) phase of the light/dark cycle, in male mice of all four ASD-associated transgenic models. For Cntnap2−/− and Pcdh10+/− mice, these activity alterations were sex-specific, as female mice did not exhibit home-cage activity differences relative to sex-matched WT controls. These home-cage hypoactivity alterations differ from activity findings previously reported using short-term activity measurements in a novel open field. Despite circadian problems reported in human ASD patients, none of the mouse models studied had alterations in free-running circadian period. Together, these findings highlight a shared phenotype across several monogenic mouse models of ASD, outline the importance of methodology on behavioral interpretation, and in some genetic lines parallel the male-enhanced phenotypic presentation observed in human ASDs.
Keywords: Autism, Hypoactivity, Home-cage activity, Sex differences, Mouse models, Circadian rhythms
1. Introduction
Autism spectrum disorder (ASD) has an estimated prevalence of 1 in 59 children and is substantially sex-biased, with males 4 times more likely than females to receive an ASD diagnosis (Baio et al., 2018). Core features of ASD include repetitive behaviors, abnormal sensory processing, and deficits in communication, social interaction, and cognition (American Psychiatric Association, 2013). Mouse genetic models have been an invaluable tool for studying electrophysiological, circuit, and molecular mechanisms contributing to ASD (see Bey & Jiang, 2014; Ellegood & Crawley, 2015; Hulbert & Jiang, 2016 for recent reviews). In rodent models of ASD, behavioral assays have been developed to assess social interaction, repetitive behaviors, communication, and cognition (Chang, Cole, & Costa, 2017; Silverman, Yang, Lord, & Crawley, 2010). However, behavioral methodologies often vary between labs, and even when behavioral procedures are systematized as rigorously as possible, results within the same genetic line may differ from lab to lab, confounding interpretation (Crabbe, Wahlsten, & Dudek, 1999; Wahlsten et al., 2003).
Genome-wide association and whole exome sequencing studies have identified numerous monogenic risk factors for ASD (De Rubeis & Buxbaum, 2015; Persico & Napolioni, 2013; Willsey & State, 2015), and monogenic mouse models displaying both face and construct validity have been created (Bey & Jiang, 2014; Ellegood & Crawley, 2015; Hulbert & Jiang, 2016). In the present study, we investigated four monogenic mouse models of ASD: Shank3b−/−, Cntnap2−/−, Pcdh10+/−, and Fmr1 knockout (KO). SH3 and multiple Ankyrin repeat domains 3 (Shank3) is a scaffolding protein in the postsynaptic density whose deletion contributes to Phelan-McDermid syndrome, intellectual disability, and ASD-like behaviors in humans (Durand et al., 2007), and social deficits and repetitive behaviors in mice (Peça et al., 2011). Contactin associated protein-like 2 (CNTNAP2) is a member of the neurexin superfamily and is associated with potassium channels, myelination, and neuron-glia interaction (Poliak et al., 1999). Its deletion results in cortical dysplasia-focal epilepsy, seizures, and ASD in humans (Strauss et al., 2006), and seizures, social deficits, motor stereotypies, communication abnormalities, and interneuron reductions in mice (Peñagarikano et al., 2011). Protocadherin-10 is a member of the cadherin superfamily of cell-adhesion molecules whose reduction has been associated with ASD in human genome-wide association studies (Bucan et al., 2009; Morrow et al., 2008), and altered communication, male-specific social deficits, and dendritic spine morphology alterations of the lateral/basolateral amygdala in Pcdh10+/− mice (Schoch et al., 2017). Fmr1 encodes for the Fragile X mental retardation protein and is one of the leading single-gene causes of autism and intellectual disability (Hagerman, Rivera, & Hagerman, 2008; Turner, Webb, Wake, & Robinson, 1996). Fragile X patients display intellectual impairment, social deficits, communication problems, anxiety, hyperarousal, and hyperactivity (Garber, Visootsak, & Warren, 2008; Yu & Berry-Kravis, 2014), and Fmr1 KO mice have learning deficits, abnormal social behavior, anxiety, altered dendritic spine development, and aberrant cortical synchrony (The Dutch-Belgium Fragile X Consortium, 1994; Comery et al., 1997; Spencer, Alekseyenko, Serysheva, Yuva-Paylor, & Paylor, 2005; Gonçalves, Anstey, Golshani, & Portera-Cailliau, 2013).
In this study, we quantified home-cage activity and circadian rhythms across multiple 24-h days in Shank3b−/−, Cntnap2−/−, Pcdh10+/−, and Fmr1 KO mice. These behaviors were chosen because they are relatively high-throughput, they are objective and easily quantifiable, and they are passive, allowing for consistent, controlled, and comparable data between groups. Moreover, activity and circadian problems are commonly reported in ASD. An estimated 30–70% of ASD patients exhibit ADHD symptomology (Antshel, Zhang-James, Wagner, Ledesma, & Faraone, 2016; Davis & Kollins, 2012), with one-third exhibiting hyperactivity (Carlsson et al., 2013). Other studies have reported significantly reduced physical activity or hypoactivity in ASD children (Pan, 2008; Posserud, Hysing, Helland, Gillberg, & Lundervold, 2016). Sleep problems such as increased latency to sleep, early morning waking, and altered melatonin profiles are frequently reported in ASD, suggesting possible circadian rhythm abnormalities (Glickman, 2010; Kulman et al., 2000; Melke et al., 2008; Mulder et al., 2010; Ritvo et al., 1993; Tordjman, Anderson, Pichard, Charbuy, & Touitou, 2005).
Previous studies investigating activity levels in these monogenic mouse models of ASD have utilized brief (1 h or less) open field tests. Cntnap2−/− mice have been reported as hyperactive in short (< 1 h) activity monitoring sessions (Brunner et al., 2015; Peñagarikano et al., 2011). Likewise, Fmr1 KO mice have been described as hyperactive in the open field in numerous publications (The Dutch-Belgium Fragile X Consortium, 1994; Peier et al., 2000; Mineur, Sluyter, de Wit, Oostra, & Crusio, 2002; Restivo et al., 2005; Spencer et al., 2005; Dahlhaus & El-Husseini, 2010; Yuskaitis et al., 2010; Liu, Chuang, & Smith, 2011; Gholizadeh, Arsenault, Xuan, Pacey, & Hampson, 2014; Uutela et al., 2014; Ding et al., 2014). Shank3b−/− mice have previously been reported to be hypoactive (Bidinosti et al., 2016; Copping et al., 2017; Dhamne et al., 2017; Kouser et al., 2013; Lee et al., 2015; Mei et al., 2016), however numerous other labs have reported no open field activity differences between Shank3b−/− males and WT controls (Drapeau, Dorr, Elder, & Buxbaum, 2014; Duffney et al., 2015; Jaramillo et al., 2016; Peça et al., 2011). In some instances investigating both Shank3b−/− males and females in the open field, males were found to be hypoactive, but females were not (Wang et al., 2011; Yang et al., 2012). Activity has not previously been reported in Pcdh10+/− mice except during social behavior (Schoch et al., 2017), but we have not observed activity differences in the open field (data not shown). We speculated that activity results recorded in the home-cage might differ from, or even contradict, previous activity reports acquired from the novel open field area.
Given that basal activity alterations can confound many behavioral assays utilized to investigate ASD-like phenotypes in rodents, including social interaction, repetitive behaviors, and cognitive tasks, it is important to investigate and consider the discrepancies between undisturbed home-cage activity and open field activity measurements, which may be impacted by anxiogenic or stimulating elements induced by either handling, or the novelty and brevity of the open field arena. The purposes of the present study were to investigate and compare home-cage activity and free-running circadian periods in multiple ASD models for the first time using consistent methodologies in the same lab environment, to examine both males and females given the pronounced sex-bias in ASD diagnoses (Baio et al., 2018), and to compare findings with published activity results— specifically highlighting the disparities between home-cage activity across multiple diurnal days and brief activity monitoring in a novel environment.
2. Methods
2.1. Animals
B6.129-Shank3tm2Gfng/J (Stock #017688) heterozygous males and females with exons 13–16 of Shank3 replaced with a neo cassette were purchased from The Jackson Laboratory and mated together to generate Shank3b−/− offspring and Wildtype (WT) controls as previously described (Peça et al., 2011). Heterozygous B6.129(Cg)-Cntnap2tm1Pele/ J males and females with exon 1 of Caspr2 replaced with a neo cassette were backcrossed to C57BL/6J for 10–12 generations, purchased from The Jackson Laboratory (Stock #017482), and mated together as previously described to produce experimental Cntnap2−/− males and females and WT controls (Peñagarikano et al., 2011; Poliak et al., 2003). Pcdh10+/− mice in which the first exon of Pcdh10 was replaced with a lacZ-neo cassette were created by Lexicon Pharmaceuticals, Inc. (Basking Ridge, NJ) and backcrossed for more than 15 generations as previously described (Schoch et al., 2017; Uemura, Nakao, Suzuki, Takeichi, & Hirano, 2007). Male Pcdh10+/− mice were mated with C57BL/6J female mice purchased from the Jackson Laboratory (Stock #000664) to produce heterozygous Pcdh10+/− experimental animals and Pcdh10+/+ wildtype controls. B6.129P2-Fmr1tm1Cgr/J female mice with exon 5 of Fmr1 replaced with a neo cassette were generated as previously described (The Dutch-Belgium Fragile X Consortium, 1994), backcrossed with C57BL/6J for many generations, and purchased from The Jackson Laboratory (Stock #003025) and mated with C57BL/6J (stock #000664) males to generate Fmr1 KO mice. Because Fragile X is an X-linked condition, and females display much milder and more variable symptoms than males due to random X-inactivation (Loesch, Huggins, & Hagerman, 2004; Yu & Berry-Kravis, 2014), only male Fmr1 KO mice were studied. Mice for all lines were weaned at 21 days old in cages of 3–5 sex-matched littermates. All mice were between 2 and 4.5 months old at the beginning of activity monitoring experimentation. Cohorts comprised of sex-matched littermates were used for all experimental groups. For all seven comparisons made in this study (male and female Shank3b−/−, Cntnap2−/−, and Pcdh10+/− mice and male Fmr1 KO mice), there were no differences in age between WT and sex-matched mutants (all p > 0.15). Age did not correlate with activity for any of the individual groups, with the lone exception of a negative correlation between age and activity for Shank3b WT males in the horizontal (Pearson’s r = −0.716, p = 0.020), but not vertical axis (p = 0.790). For the group comparison involving both WT and Shank3b−/− male mice, there was no correlation between age and activity (p = 0.134). All animals were maintained on a 12-h light: 12-h dark cycle (lights on at 7:00 am), except where indicated otherwise for circadian studies in constant darkness, with food and water provided ad libitum. All experiments were approved by the University of Pennsyl-vania Institutional Care and Use Committee (IACUC protocol 804407) and conducted in accordance to National Institute of Health guidelines.
2.2. Activity monitoring
Activity monitoring was performed as previously described (Angelakos et al., 2016). Mice were single housed within individual noise- and light-attenuating chambers (22″ × 16″ × 19″, Med Associates, St. Albans, VT) equipped with a 250 lx light source (80 lx at cage floor), ventilation fan, and an infrared beam break system which surrounded the mouse cage on all four sides and provided a high-resolution scaffold of infrared beams and detectors (Opto M3, Columbus Instruments, Columbus, OH). Infrared beams were spaced 0.5″ apart and provided two horizontal grids at 0.75″ and 2.75″ from the cage floor to quantify horizontal and vertical (rearing) activity, respectively. Mice were allowed to acclimate to the chambers for one week before data collection. Following acclimation, activity counts (beam breaks) were tabulated in 10-second intervals in both the XY (horizontal) and Z (vertical) direction continuously for 7 days in 12-h light: 12-h dark (12 h:12 h LD). Activity counts were pooled into 1-h bins across the entire diurnal cycle and averaged over the course of the 7 days. After 7 days of activity monitoring in 12 h:12 h LD, lights were switched off for 2 consecutive weeks of constant darkness (DD) and activity counts were compiled in 1-min intervals over the course of DD. Actogram generation and circadian period (tau) estimation were automated by Clocklab software (Actimetrics). Tau was calculated by Clocklab software as the slope of activity onset from day 2 to day 14 of the DD actogram. Day 1 of DD was discarded from circadian and activity analyses as all mice received a cage change on this day. Peak activity during DD was calculated by binning activity counts into every possible 1-h bin on a sliding scale and then averaging the bins with maximum activity count from each calendar day during the DD period. For Pcdh10+/− and Fmr1 KO groups, only a subset of mice underwent circadian monitoring in constant darkness (see Fig. 4 for numbers).
2.3. Statistics
All statistical analysis was performed using SPSS for Windows (V.24.0). To analyze home-cage activity (1-h bins), Mixed Design ANOVAs were utilized with genotype as the between-subjects factor (WT or transgenic) and time as the within-subjects factor. Post hoc multiple comparisons were performed using Bonferroni’s adjustment for multiple comparisons. Where the assumption of sphericity was violated, Greenhouse-Geisser corrected F values are given. Multivariate ANOVAs (MANOVAs) were performed to analyze activity counts in the light cycle and dark cycle, with alphas corrected for multiple ANOVAs and set at α = 0.05/2, followed by post hoc Bonferroni’s adjustment for multiple comparisons. Student’s T-test was used to compare 24-h activity counts, activity counts during habituation, circadian tau values, and peak activity counts during DD between WT and transgenic mice. One-way ANOVAs compared activity levels of transgenic animals to “reference” activity levels averaged across all sex-matched WT animals. Pearson’s r was used to correlate animal age with activity levels.
3. Results
3.1. Home-cage hypoactivity in male Shank3b−/−, Cntnap2−/−, Pcdh10+/−, and Fmr1 KO mice
Home-cage beam break counts were tabulated over 7 days of continuous monitoring in 12 h:12 h LD in male experimental mice and sex-matched WT littermate controls of the four transgenic models of autism spectrum disorder: Shank3b−/−, Cntnap2−/−, Pcdh10+/−, and Fmr1 KO. In the horizontal (XY) direction, compared to WT, ASD-associated genetic deletion resulted in decreased home-cage activity for male Shank3b−/− (main effect of genotype; F(1,18) = 14.466, p = 0.001, Fig. 1a), male Cntnap2−/− (time*genotype interaction: F (23,467) = 4.409, p = 0.003, Fig. 1c), male Pcdh10+/− (main effect of genotype: F(1,20) = 10.562, p = 0.004, Fig. 1e), and male Fmr1 KO mice (main effect of genotype: F(1,25) = 8.125, p = 0.009, Fig. 1g). Analysis by light/dark cycle revealed significantly reduced activity specifically in the dark (active) phase for Shank3b−/− (MANOVA, F (2,17) = 8.093, p = 0.003, Fig. 1b), Pcdh10+/− (MANOVA, F (2,19) = 6.331, p = 0.008, Fig. 1f), and Fmr1 KO males (MANOVA: F (2,24) = 26.801, p < 0.000001, Fig. 1h), relative to WT littermates. The magnitude of activity differences between male WT and ASD model littermates was even larger in the vertical (rearing) direction for all lines studied (Supplementary Fig. 1). Thus, mutation in four different ASD-associated genes resulted in decreased home-cage ambulatory and rearing activity in male mutant mice relative to sex-matched controls.
Fig. 1.

Male Shank3b−/−, Cntnap2−/−, Pcdh10+/−, and Fmr1 KO mice are hypoactive in the home-cage relative to sex-matched WT controls. (A,B) Shank3b−/− males exhibit significantly reduced horizontal activity compared to WT littermates. Expressed in 1-h bins (A) and by light/dark cycle (B). (C,D) Cntnap2−/− males have decreased horizontal activity compared to WT controls. Expressed in 1-h bins (C) and by light/dark cycle (D). (E,F) Pcdh10+/− males display significantly less horizontal activity than WT littermates. Expressed in 1-h bins (E) and by light/dark cycle (F). (G,H) Fmr1 KO males demonstrate significantly lower horizontal activity than WT controls. Expressed in 1-h bins (G) and by light/dark cycle (H). Mean ± standard error of the mean (s.e.m.) *p < 0.05, **p < 0.01, ***p < 0.001.
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.nlm.2019.02.010.
3.2. Shank3b−/− female mice exhibit home-cage hypoactivity, but female Cntnap2−/− and Pcdh10+/− have normal activity levels
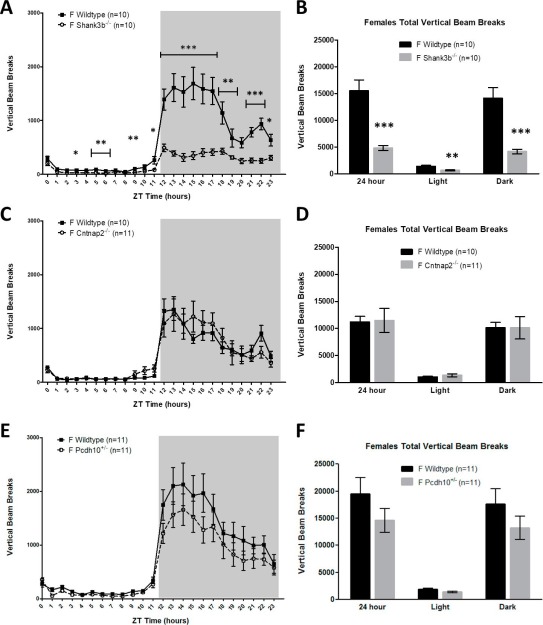
Because ASD is a male-biased disorder and females often exhibit less severe symptomology than males, even under the same genetic insult (Jacquemont et al., 2014; Robinson, Lichtenstein, Anckarsäter, Happé, & Ronald, 2013; Werling & Geschwind, 2013), we also quantified home-cage activity behavior in female mice and sex-matched littermates to look for potential sex-specific deficits. Female Fmr1 KO mice were not studied because Fragile X is an X-linked condition and phenotypes in females are less severe, more variable, and less predictable than males due to random X-inactivation (Loesch et al., 2004; Yu & Berry-Kravis, 2014). Similar to Shank3b−/− male mice, Shank3b−/− females exhibited robust home-cage hypoactivity relative to WT (main effect of genotype: F(1,18) = 20.447, p < 0.001, Fig. 2a). Analysis by light/dark cycle indicated that Shank3b−/− females were less active than WT females in both the light and dark phases (MANOVA, F (2,17) = 9.699, p = 0.002, Fig. 2b). Interestingly, unlike male Cntnap2−/− and Pcdh10+/− mice, there were no activity differences relative to sex-matched WT littermates in female Cntnap2−/− mice (F (1,19) = 0.033, p = 0.86, Fig. 2d) or female Pch10+/− mice (F (1,20) = 1.960, p = 0.18, Fig. 2f). These female activity findings were similar in rearing behavior (Supplementary Fig. 2). Altogether, activity alterations were male-specific for two of the four ASD-associated mouse models studied, consistent with heightened ASD symptomology in males compared to females given the same genetic insult (Jacquemont et al., 2014; Robinson et al., 2013).
Fig. 2.

Female Shank3b−/− mice are hypoactive, but female Cntnap2+/− and Pcdh10+/− display no home-cage activity differences, relative to sex-matched WT controls. (A,B) Shank3b−/− females exhibit significantly reduced horizontal activity compared to WT littermates. Expressed in 1-h bins (A) and by light/dark cycle (B). (C,D) Cntnap2−/− females have no differences in horizontal activity compared to WT controls. Expressed in 1-h bins (C) and by light/dark cycle (D). (E,F) Pcdh10+/− females display no differences in horizontal activity in comparison to WT littermates. Expressed in 1-h bins (E) and by light/dark cycle (F). Mean ± standard error of the mean (s.e.m.) *p < 0.05, **p < 0.01, ***p < 0.001.
3.3. Cntnap2−/−, Pcdh10+/−, and Fmr1 KO mice are not hypoactive during the habituation phase
Activity monitoring experiments are typically performed in a novel open field arena and are brief in nature (usually 1 h or less). Because our activity monitoring cages differ in dimension, environment, and number of cohabitants from the group-housed home-cages that the mice are reared in, we can capture some of this novelty by quantifying activity levels during the initial habituation phase in our activity monitoring chambers. Activity measurements were made during the first 10 minutes, 30 minutes, and 1 h in the activity monitoring chambers, coinciding with common time intervals used for open field experiments. In comparison to sex-matched wildtype littermates, Shank3b−/− male and female mice were hypoactive in the XY-axis during the first 30 min (Student’s t-test, male: p = 0.017, female: p = 0.013; Fig. 3b) and 1 h (Student’s t-test, male: p = 0.011, female: p = 0.007; Fig. 3c) in the activity monitoring system. For male Cntnap2−/−, Pcdh10+/− and Fmr1 KO mice, however, there were no activity differences during the habituation phase between transgenic mice and WT controls (p > 0.180 for all comparisons; Fig. 3d–l), in contrast to the hypoactivity observed following acclimation to the home-cage (Fig. 1). There were also no activity differences between Cntnap2−/− and Pcdh10+/− female mice and WT littermates during habituation (p > 0.131 for all comparisons; Fig. 3d–i). Activity findings in the vertical direction during habituation were similar to those observed in the horizontal direction (Supplementary Fig. 3). The lack of activity differences between male Cntnap2−/−, Pcdh10+/− and Fmr1 KO mice and their WT counterparts during habituation highlights the differences between home-cage activity monitoring and short-term activity monitoring in a novel environment.
Fig. 3.

Shank3b−/− mice are hypoactive during habituation to the activity monitors, but Cntnap2−/−, Pcdh10+/−, and Fmr1 KO mice exhibit no activity differences in the habituation phase relative to sex-matched WT littermates. (A–C) Shank3b−/− males and females have reduced horizontal activity in the first 30 min (B) and 1h (C) of activity monitoring in comparison to sex-matched WT controls. D-F) There are no differences in activity counts between Cntnap2−/− mice and WT during the first 10 min (D), 30 min (E), or 1h (F) of habituation. (G–I) Pcdh10+/− male and female mice display similar activity levels during the first hour of habituation compared to sex-matched WT littermates. (J–L) There are no differences in activity counts during the habituation phase between Fmr1 KO males and WT controls. Mean ± standard error of the mean (s.e.m.) *p < 0.05, **p < 0.01.
3.4. Circadian rhythms are unaltered in the four ASD mouse models
Circadian issues are common in ASD patients, who often have difficulties falling asleep and have more frequent night awakenings than the control population (Glickman, 2010; Richdale & Prior, 1995). Because of this, we investigated intrinsic circadian period of the ASD-associated transgenic mouse lines. Following two weeks of continuous activity monitoring, lights were shut off and mice were allowed to free-run in continuous darkness for two weeks. Despite activity alterations found in the ASD mouse models, there were no differences in free-running circadian period between male and female transgenic mice and sex-matched littermates for any of the four ASD-associated lines studied (Student’s t-test, p > 0.25 for all comparisons, Fig. 4a–d). Moreover, activity patterns found in 12 h:12 h LD remained consistent in constant darkness (DD), with the exception of Fmr1 KO mice not being hypoactive in the horizontal axis relative to WT controls. Male Shank3b−/ − (Student’s t-test, horizontal: p = 0.0005; Fig. 4e, vertical: p = 0.00003; Fig. 4i), Cntnap2−/− (horizontal: p = 0.26; Fig. 4f, vertical: p = 0.014; Fig. 4j), Pcdh10+/− (horizontal: p = 0.018; Fig. 4g, vertical: p = 0.030; Fig. 4k), and Fmr1 KO (horizontal: p = 0.99; Fig. 4h, vertical: p = 0.024; Fig. 4l) mice displayed lower daily peak activity in DD than sex-matched WT controls. Female Shank3b−/− mice were hypoactive in DD relative to sex-matched WT (horizontal: p = 0.0007; Fig. 4e, vertical: p = 0.0006; Fig. 4i), but female Cntnap2−/− (horizontal: p = 0.41; Fig. 4f, vertical: p = 0.55; Fig. 4j) and Pcdh10+/− (horizontal: p = 0.58; Fig. 4g, vertical: p = 0.61; Fig. 4k) mice displayed no peak activity differences compared to WT littermates.
Fig. 4.

Normal free-running circadian periods and hypoactivity in Shank3b−/−, Cntnap2−/−, Pcdh10+/−, and Fmr1 KO mice during constant darkness. A-D) Relative to sex-matched WT controls, there are no differences in free-running circadian period (tau) in (A) Shank3b−/− males (23.60 ± 0.02 h, WT: 23.64 ± 0.02 h; p = 0.25) or females (23.62 ± 0.03 h, WT: 23.67 ± 0.02 h; p = 0.25), (B) Cntnap2−/− males (23.61 ± 0.02 h, WT: 23.61 ± 0.02 h; p =0.93) or females (23.65 ± 0.04 h, WT: 23.64 ± 0.03 h; p= 0.64), (C) Pcdh10+/− males (23.53 ± 0.04 h, WT: 23.54 ± 0.08 h; p= 0.87) or females (23.51 ± 0.05 h, WT: 23.50 ± 0.03 h; p =0.83), or (D) Fmr1 KO males (23.67 ± 0.02 h, WT: 23.64 ± 0.02 h; p =0.26). (E–L) Peak activity patterns during DD (1-h bins) are similar to activity patterns observed during 12h:12 h LD. (E,I) Shank3b−/− male and female mice have lower peak activity in DD than sex-matched WT controls. (F,J) Relative to sex-matched WT in DD, male Cntnap2−/− mice are not hypoactive in the (F) horizontal axis, but are hypoactive in the (J) vertical direction. Female Cntnap2−/− mice are not hypoactive relative to sex-matched WT in DD. (G,K) Pcdh10+/− males but not females have lower peak activity levels in DD compared to WT. (H,L) Fmr1 KO males do not have altered peak activity levels in the (H) XY-axis, but display reduced peak activity in the (L) Z-axis during DD in comparison to WT littermates. Mean ± standard error of the mean (s.e.m.) *p < 0.05, **p < 0.01, ***p < 0.001.
4. Discussion
In this study, we performed home-cage activity monitoring and circadian behavioral analysis in four monogenic mouse models of ASD. Male mice of all four lines studied— Shank3b−/−, Cntnap2−/−, Pcdh10+/−, and Fmr1 KO—exhibited home-cage hypoactivity, particularly in the dark (active) phase of the diurnal cycle, relative to sex-matched WT controls. Our findings present a shared phenotype across multiple mouse models of ASD, performed in one lab using consistent methodologies. Activity alterations (both hyperactivity and hypoactivity) are commonly reported in ASD (Carlsson et al., 2013; Posserud et al., 2016), and the mutual phenotype of hypoactivity observed in these monogenic mouse lines may suggest common neural underpinnings. One possibility may be aberrant corticostriatal circuitry, which plays a role in coordinated movement and is believed to be one of the major structural alterations contributing to repetitive behaviors and ASD (Delmonte, Gallagher, O’Hanlon, McGrath, & Balsters, 2013; Hollander et al., 2005; Langen et al., 2012; Mosconi et al., 2009; Shepherd, 2013). Altered corticostriatal connectivity and neural firing have previously been reported in Shank3b−/− mice (Peça et al., 2011; Peixoto, Wang, Croney, Kozorovitskiy, & Sabatini, 2016), Cntnap2−/− mice (Peñagarikano et al., 2011; Selimbeyoglu et al., 2017), Fmr1 KO mice (Centonze et al., 2008; Lai, Lerch, Doering, Foster, & Ellegood, 2016), and Fragile X humans (Dennis & Thompson, 2013; Haas et al., 2009). Pcdh10 has been shown to be involved in striatal axon growth (Uemura et al., 2007), and corticostriatal alterations have also been reported in other mouse models of ASD, including neuroligin-1 KO and BTBR mice (Blundell et al., 2010; Ellegood, Babineau, Henkelman, Lerch, & Crawley, 2013). In contrast to the hypoactivity observed in the present study, other mouse models of ASD including the 16p11.2 del/+ chromosomal copy number variation mouse model (Angelakos et al., 2016; Arbogast et al., 2016; Horev et al., 2011; Portmann et al., 2014) and the Ptchd1Y/- mouse model of ASD (Ung et al., 2017; Wells, Wimmer, Schmitt, Feng, & Halassa, 2016) exhibit robust hyperactivity in the home-cage. 16p11.2 del/+ mice display cellular and molecular alterations in the striatum and deficits in striatum-dependent behaviors (Grissom et al., 2017; Portmann et al., 2014), and PTCHD1 expression is enriched in the cortex and striatum (Ung et al., 2017). Together, these results suggest that hypoactivity is not a defining characteristic of ASD mouse models, but rather, activity alterations in both directions are commonly observed in various rodent models of ASD, as well as in humans with ASD. Future studies will need to determine if misregulated corticostriatal circuits underlie these activity alterations.
4.1. Cntnap2−/− and Pcdh10+/− mice may be useful models for studying sex differences in ASD
For Cntnap2−/− and Pcdh10+/− mice, home-cage hypoactivity was sex-specific, as female mice of these lines displayed activity levels similar to sex-matched WT littermates. Male-enhanced activity alterations in these lines parallel sex differences in phenotypic presentation in human ASD patients (Werling & Geschwind, 2013) and supports findings that females require a larger mutational burden than males to manifest similar ASD-related symptomology (Jacquemont et al., 2014; Robinson et al., 2013). Cntnap2−/− mice have previously been shown to have male-specific reductions in visually-evoked cortical activity (Townsend & Smith, 2017), and male but not female mice exposed to a Cntnap2-reactive antibody in utero display ASD-associated behavioral deficits and structural alterations in the cortex and hippocampus (Brimberg et al., 2016). Relative to sex-matched WT littermates, Pcdh10+/− male mice exhibit significantly reduced social approach behavior, whereas female Pcdh10+/− mice do not (Schoch et al., 2017). These findings, in concert with the male-specific activity reductions outlined in the present study, suggest that Cntnap2−/− and Pcdh10+/− mice may serve as informative genetic models for investigating sex differences in ASD.
4.2. Home-cage activity dissociates from activity observed in a novel arena
Another main conclusion from this study is the importance of dissociating continuous, undisturbed home-cage activity from novelty-induced, brief activity measurements that may contain anxiogenic elements, such as those obtained in an open field test. Activity findings reported in the open field and during the habituation phase of our study do not necessarily match our findings in the acclimated home-cage. Cntnap2−/− and Fmr1 KO mice have been reported to be hyperactive in the open field (The Dutch-Belgium Fragile X Consortium, 1994; Peier et al., 2000; Mineur et al., 2002; Restivo et al., 2005; Spencer et al., 2005; Dahlhaus & El-Husseini, 2010; Yuskaitis et al., 2010; Liu et al., 2011; Peñagarikano et al., 2011; Gholizadeh et al., 2014; Uutela et al., 2014; Brunner et al., 2015) and displayed no activity differences during the habituation phase in our task (Fig. 3), but longer activity studies across multiple undisturbed days corroborate our findings of hypoactivity in Cntnap2−/− (Thomas, Schwartz, Saxe, & Kilduff, 2016) and Fmr1 KO male mice (Bonasera, Chaudoin, Goulding, Mittek, & Dunaevsky, 2017). Although it is difficult to make direct comparisons between results in the open field, which contains no bedding and is 1.5–10 × larger in area than the 4.5″ × 8.5″ home-cages we use in our study, it is clear that activity patterns in a short-duration novel environment greatly differ from activity patterns in the undisturbed home-cage.
4.3. Limitations of circadian analysis in C57BL/6J mice
Some limitations exist in our study. Circadian alterations have been reported in ASD patients (Glickman, 2010), but none of the ASD mouse models studied showed changes in circadian periodicity or activity in DD (Fig. 4). These circadian abnormalities have been speculated to be related to altered melatonin synthesis and concentration in those with ASD compared to typically developing controls (Bourgeron, 2007; Kulman et al., 2000; Melke et al., 2008; Mulder et al., 2010; Nir et al., 1995; Ritvo et al., 1993; Tordjman et al., 2005). However, due to a mutation in the melatonin synthesis pathway, C57BL/6J mice have significantly attenuated melatonin production (Roseboom et al., 1998). Although augmentation of melatonin protein in C57BL/6J mice via outcrossing to a melatonin-proficient strain results in no alterations in the circadian rhythm (Kasahara, Abe, Mekada, Yoshiki, & Kato, 2010), given the link between altered melatonin production and ASD, future circadian experiments should be performed using transgenic ASD model mice on a melatonin-proficient strain, such as CBA mice (Kennaway, Voultsios, Varcoe, & Moyer, 2002).
4.4. Additional limitations
Although home-cage hypoactivity was demonstrated across the various monogenic ASD mouse models utilized in this study, the nature of the hypoactive behavior was not observable. Future studies video tracking the movement patterns of these mouse models may prove useful for identifying potential stereotypies or behavioral abnormalities underlying the hypoactive phenotypes. Another possible limitation of our study is the variance in wildtype activity between sex-matched mice of different genetic lines, confounding direct comparison between transgenic groups. One possible explanation for the inconsistencies in wildtype activity across groups may be that this simply reflects inherent variability within C57BL/6J mice. However, other possibilities are likely to play a role. Although none of the ASD-associated genes studied are known to be imprinted (http://www.geneimprint.com/), the possibility remains that parental origin of the mutant allele impacts behavioral phenotypes of the progeny. Pcdh10 was transmitted via the paternal allele in accordance with previous studies using this mouse model (Uemura et al., 2007; Schoch et al., 2017) so that offspring would be reared by WT mothers. Out of necessity, however, experimental animals for the other three lines were generated as the offspring of transgenic mothers. It is entirely possible that mutations in Shank3b, Caspr2, or Fmr1 impact maternal behavior, which is known to affect both development and subsequent behavior of the offspring (Branchi & Cirulli, 2014), but these would have to act in the heterozygous state. For these reasons, investigating how maternally-transmitted Pcdh10 impacts the development and behavior of Pcdh10+/− mice, which is hitherto unknown, would be an interesting future avenue of research. Another possible explanation for the variance in WT activity levels is that age/body weight was not strictly controlled between groups. Importantly, cohorts of sex-matched littermates were used for all within-genotype direct comparisons performed in this study. Slight variations in age or body weight between genotypes, however, may contribute to the between-group discrepancies in WT activity levels. Although comparisons utilizing sex-matched littermates, which eliminate environmental variables, are strongly preferred, we also re-analyzed activity counts after collapsing wildtype activity levels across the different genotypes to create “reference” wildtype values. Relative to the “reference” WT value, male Fmr1 KO mice were hypoactive in the vertical axis, but were no longer hypoactive in the horizontal axis. Apart from this, all activity findings utilizing “reference” WT values were the same as the findings directly comparing the transgenic animals to sex-matched WT littermates (Supplementary Fig. 4).
4.5. Conclusions and future directions
This study revealed a common home-cage hypoactivity phenotype, relative to WT, in male mice of four different monogenic mouse models of ASD. While human Fragile X patients and those with CNTNAP2 mutation are hyperactive (Baumgardner, Reiss, Freund, & Abrams, 1995; Strauss et al., 2006; Sullivan et al., 2006; Tranfaglia, 2011), the consistent phenotype of activity alterations in ASD patients and mouse models may point to common aberrant neural circuity. Future studies including other activity-dependent behavioral tasks such as tests of motivation/depression, or involving brain region-specific genetic rescue in our current activity setup, may help elucidate the neural circuitry underlying the hypoactivity observed in these mice. Given that ASD is a neurodevelopmental disorder, future investigation tracking home-cage activity patterns across development may reveal critical developmental timepoints and may shed further light onto potential mechanisms underlying the activity alterations. Together, this shared phenotype between Shank3b−/−, Cntnap2−/−, Pcdh10+/−, and Fmr1 KO mice helps validate these monogenic models of ASD and may assist in pinpointing structural and mechanistic deficits common between humans with ASD and rodent genetic models.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was funded by a grant from the Simons Foundation Autism Research Initiative, 248429, the Department of Defense/U.S. Army grant, AR110189, and government support under and awarded by DoD, Air Force Office of Scientific Research, National Defense Science and Engineering Graduate (NDSEG) Fellowship, 32 CFR 168a. T.A. is the Roy J. Carver Chair in Neuroscience and Director of the Iowa Neuroscience Institute at the University of Iowa. Previously T.A. was the Brush Family Professor of Biology at the University of Pennsylvania.
Footnotes
Conflicts of interest
The authors declare no competing financial interests.
References
- American Psychiatric Association (2013). Diagnostic and statistical manual of mental disorders-5. Washington, DC: American Psychiatric Association. [Google Scholar]
- Angelakos CC, Watson AJ, O’Brien WT, Krainock KS, Nickl-Jockschat T, & Abel T. (2016). Hyperactivity and male-specific sleep deficits in the 16p11.2 deletion mouse model of autism. Autism Research. 10.1002/aur.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antshel KM, Zhang-James Y, Wagner KE, Ledesma A, & Faraone SV (2016). An update on the comorbidity of ADHD and ASD: A focus on clinical management. Expert Review of Neurotherapeutics, 16(3), 279–293. 10.1586/14737175.2016.1146591. [DOI] [PubMed] [Google Scholar]
- Arbogast T, Ouagazzal A-M, Chevalier C, Kopanitsa M, Afinowi N, Migliavacca E, … Herault Y. (2016). Reciprocal effects on neurocognitive and metabolic phenotypes in mouse models of 16p11.2 deletion and duplication syndromes. PLoS Genetics, 12(2), e1005709. 10.1371/journal.pgen.1005709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baio J, Wiggins L, Christensen DL, Maenner MJ, Daniels J, Warren Z, … Dowling NF (2018). Prevalence of autism spectrum disorder among children aged 8 years — Autism and developmental disabilities monitoring network, 11 sites, United States, 2014. Morbidity and Mortality Weekly Report. Surveillance Summaries (Washington, D.C.: 2002), 67(6), 1–23. 10.15585/mmwr.ss6706a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgardner TL, Reiss AL, Freund LS, & Abrams MT (1995). Specification of the neurobehavioral phenotype in males with fragile X syndrome. Pediatrics, 95(5) http://pediatrics.aappublications.org/content/95/5/744.long. [PubMed] [Google Scholar]
- Bey AL, & Jiang Y. (2014). Overview of mouse models of autism spectrum disorders. Current Protocols in Pharmacology, 66, 5.66.1–26 10.1002/0471141755.ph0566s66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidinosti M, Botta P, Krüttner S, Proenca CC, Stoehr N, Bernhard M, … Galimberti I. (2016). CLK2 inhibition ameliorates autistic features associated with SHANK3 deficiency. Science, 351(6278)http://science.sciencemag.org/content/351/6278/1199.full. [DOI] [PubMed] [Google Scholar]
- Blundell J, Blaiss CA, Etherton MR, Espinosa F, Tabuchi K, Walz C, … Powell CM (2010). Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 30(6), 2115–2129. 10.1523/JNEUROSCI.4517-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonasera SJ, Chaudoin TR, Goulding EH, Mittek M, & Dunaevsky A. (2017). Decreased home cage movement and oromotor impairments in adult Fmr1 -KO mice. Genes, Brain and Behavior, 16(5), 564–573. 10.1111/gbb.12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeron T. (2007). The possible interplay of synaptic and clock genes in autism spectrum disorders. Cold Spring Harbor Symposia on Quantitative Biology, 72(1), 645–654. 10.1101/sqb.2007.72.020. [DOI] [PubMed] [Google Scholar]
- Branchi I, & Cirulli F. (2014). Early experiences: Building up the tools to face the challenges of adult life. Developmental Psychobiology, 56(8), 1661–1674. 10.1002/dev.21235. [DOI] [PubMed] [Google Scholar]
- Brimberg L, Mader S, Jeganathan V, Berlin R, Coleman TR, Gregersen PK, … Diamond B. (2016). Caspr2-reactive antibody cloned from a mother of an ASD child mediates an ASD-like phenotype in mice. Molecular Psychiatry, 21(12), 1663–1671. 10.1038/mp.2016.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner D, Kabitzke P, He D, Cox K, Thiede L, Hanania T, … Biemans B. (2015). Comprehensive analysis of the 16p11.2 deletion and null cntnap2 mouse models of autism spectrum disorder. PloS One, 10(8), e0134572. 10.1371/journal.pone.0134572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucan M, Abrahams BS, Wang K, Glessner JT, Herman EI, Sonnenblick LI, … Hakonarson H. (2009). Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genetics, 5(6), e1000536. 10.1371/journal.pgen.1000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson LH, Norrelgen F, Kjellmer L, Westerlund J, Gillberg C, & Fernell E. (2013). Coexisting disorders and problems in preschool children with autism spectrum disorders. The Scientific World Journal, 2013, 213979. 10.1155/2013/213979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Rossi S, Mercaldo V, Napoli I, Ciotti MT, De Chiara V, … Bagni C. (2008). Abnormal striatal GABA transmission in the mouse model for the fragile X syndrome. Biological Psychiatry, 63(10), 963–973. 10.1016/j.biopsych.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Chang Y-C, Cole TB, & Costa LG (2017). Behavioral phenotyping for autism spectrum disorders in mice Current protocols in toxicology: Vol. 72, (pp. 11.22.1–11.22.21). Hoboken, NJ, USA: John Wiley & Sons, Inc; 10.1002/cptx.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, & Greenough WT (1997). Abnormal dendritic spines in fragile X knockout mice: Maturation and pruning deficits. Proceedings of the National Academy of Sciences of the United States of America, 94(10), 5401–5404. http://www.ncbi.nlm.nih.gov/pubmed/9144249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copping NA, Berg EL, Foley GM, Schaffler MD, Onaga BL, Buscher N, … Yang M. (2017). Touchscreen learning deficits and normal social approach behavior in the Shank3B model of Phelan-McDermid Syndrome and autism. Neuroscience, 345, 155–165. 10.1016/j.neuroscience.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe JC, Wahlsten D, & Dudek BC (1999). Genetics of mouse behavior: Interactions with laboratory environment. Science (New York, N.Y.), 284(5420), 1670–1672. http://www.ncbi.nlm.nih.gov/pubmed/10356397. [DOI] [PubMed] [Google Scholar]
- Dahlhaus R, & El-Husseini A. (2010). Altered neuroligin expression is involved in social deficits in a mouse model of the fragile X syndrome. Behavioural Brain Research, 208(1), 96–105. 10.1016/j.bbr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- Davis NO, & Kollins SH (2012). Treatment for co-occurring attention deficit/hyperactivity disorder and autism spectrum disorder. Neurotherapeutics: The Journal of the American Society for Experimental NeuroTherapeutics, 9(3), 518–530. 10.1007/s13311-012-0126-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, & Buxbaum JD (2015). Recent advances in the genetics of autism spectrum disorder. Current Neurology and Neuroscience Reports, 15(6), 36 10.1007/s11910-015-0553-1. [DOI] [PubMed] [Google Scholar]
- Delmonte S, Gallagher L, O’Hanlon E, McGrath J, & Balsters JH (2013). Functional and structural connectivity of frontostriatal circuitry in Autism Spectrum Disorder. Frontiers in Human Neuroscience, 7, 430 10.3389/fnhum.2013.00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis EL, & Thompson PM (2013). Typical and atypical brain development: A review of neuroimaging studies. Dialogues in Clinical Neuroscience, 15(3), 359–384. http://www.ncbi.nlm.nih.gov/pubmed/24174907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhamne SC, Silverman JL, Super CE, Lammers SHT, Hameed MQ, Modi ME, … Sahin M. (2017). Replicable in vivo physiological and behavioral phenotypes of the Shank3B null mutant mouse model of autism. Molecular Autism, 8, 26 10.1186/s13229-017-0142-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Sethna F, & Wang H. (2014). Behavioral analysis of male and female Fmr1 knockout mice on C57BL/6 background. Behavioural Brain Research, 271, 72–78. 10.1016/j.bbr.2014.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drapeau E, Dorr NP, Elder GA, & Buxbaum JD (2014). Absence of strong strain effects in behavioral analyses of Shank3-deficient mice. Disease Models & Mechanisms, 7(6), 667–681. 10.1242/dmm.013821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffney LJ, Zhong P, Wei J, Matas E, Cheng J, Qin L, … Yan Z. (2015). Autismlike deficits in shank3-deficient mice are rescued by targeting actin regulators. Cell Reports, 11(9), 1400–1413. 10.1016/j.celrep.2015.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, … Bourgeron T. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nature Genetics, 39(1), 25–27. 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegood J, Babineau BA, Henkelman RM, Lerch JP, & Crawley JN (2013). Neuroanatomical analysis of the BTBR mouse model of autism using magnetic resonance imaging and diffusion tensor imaging. NeuroImage, 70, 288–300. 10.1016/j.neuroimage.2012.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Dutch-Belgian Fragile X Consortium, Bakker CE, Verheij C, Willemsen R, van der Helm R, Oerlemans F, Vermey M, Bygrave A, Hoogeveen A, Oostra BA, Reyniers E, 1994. Cell 78, 23–33. https://www.ncbi.nlm.nih.gov/pubmed/?term=8033209.8033209 [Google Scholar]
- Ellegood J, & Crawley JN (2015). Behavioral and neuroanatomical phenotypes in mouse models of autism. Neurotherapeutics: The Journal of the American Society for Experimental NeuroTherapeutics, 12(3), 521–533. 10.1007/s13311015-0360-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber KB, Visootsak J, & Warren ST (2008). Fragile X syndrome. European Journal of Human Genetics: EJHG, 16(6), 666–672. 10.1038/ejhg.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gholizadeh S, Arsenault J, Xuan ICY, Pacey LK, & Hampson DR (2014). Reduced phenotypic severity following adeno-associated virus-mediated Fmr1 gene delivery in fragile X mice. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 39(13), 3100–3111. 10.1038/npp.2014.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman G. (2010). Circadian rhythms and sleep in children with autism. Neuroscience & Biobehavioral Reviews, 34(5), 755–768. 10.1016/j.neubiorev.2009.11.017. [DOI] [PubMed] [Google Scholar]
- Gonçalves JT, Anstey JE, Golshani P, & Portera-Cailliau C. (2013). Circuit level defects in the developing neocortex of Fragile X mice. Nature Neuroscience, 16(7), 903–909. 10.1038/nn.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissom NM, McKee SE, Schoch H, Bowman N, Havekes R, O’Brien WT, … Abel T. (2017). Male-specific deficits in natural reward learning in a mouse model of neurodevelopmental disorders. Molecular Psychiatry. 10.1038/mp.2017.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BW, Barnea-Goraly N, Lightbody AA, Patnaik SS, Hoeft F, Hazlett H, … Reiss AL (2009). Early white-matter abnormalities of the ventral frontostriatal pathway in fragile X syndrome. Developmental Medicine and Child Neurology, 51(8), 593–599. 10.1111/j.1469-8749.2009.03295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman R, Rivera S, & Hagerman P. (2008). The fragile X family of disorders: A model for autism and targeted treatments. Current Pediatric Reviews, 4(1), 40–52. 10.2174/157339608783565770. [DOI] [Google Scholar]
- Hollander E, Anagnostou E, Chaplin W, Esposito K, Haznedar MM, Licalzi E, … Buchsbaum M. (2005). Striatal volume on magnetic resonance imaging and repetitive behaviors in autism. Biological Psychiatry, 58(3), 226–232. 10.1016/j.biopsych.2005.03.040. [DOI] [PubMed] [Google Scholar]
- Horev G, Ellegood J, Lerch JP, Son Y-EE, Muthuswamy L, Vogel H, … Mills AA (2011). Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proceedings of the National Academy of Sciences, 108(41), 17076–17081. http://www.pnas.org/cgi/doi/10.1073/pnas.1114042108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulbert SW, & Jiang Y-H (2016). Monogenic mouse models of autism spectrum disorders: Common mechanisms and missing links. Neuroscience, 321, 3–23. 10.1016/j.neuroscience.2015.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S, Coe BP, Hersch M, Duyzend MH, Krumm N, Bergmann S, … Eichler EE (2014). A higher mutational burden in females supports a “Female Protective Model” in neurodevelopmental disorders. The American Journal of Human Genetics, 94(3), 415–425. 10.1016/j.ajhg.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo TC, Speed HE, Xuan Z, Reimers JM, Liu S, & Powell CM (2016). Altered striatal synaptic function and abnormal behaviour in shank3 exon4–9 deletion mouse model of autism. Autism Research: Official Journal of the International Society for Autism Research, 9(3), 350–375. 10.1002/aur.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara T, Abe K, Mekada K, Yoshiki A, & Kato T. (2010). Genetic variation of melatonin productivity in laboratory mice under domestication. Proceedings of the National Academy of Sciences of the United States of America, 107(14), 6412–6417. 10.1073/pnas.0914399107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennaway DJ, Voultsios A, Varcoe TJ, & Moyer RW (2002). Melatonin in mice: Rhythms, response to light, adrenergic stimulation, and metabolism. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, 282(2), R358–R365. 10.1152/ajpregu.00360.2001. [DOI] [PubMed] [Google Scholar]
- Kouser M, Speed HE, Dewey CM, Reimers JM, Widman AJ, Gupta N, … Powell CM (2013). Loss of predominant Shank3 isoforms results in hippocampus-dependent impairments in behavior and synaptic transmission. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 33(47), 18448–18468. 10.1523/JNEUROSCI.3017-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulman G, Lissoni P, Rovelli F, Roselli MG, Brivio F, & Sequeri P. (2000). Evidence of pineal endocrine hypofunction in autistic children. Neuro Endocrinology Letters, 21(1), 31–34. http://www.ncbi.nlm.nih.gov/pubmed/11455326. [PubMed] [Google Scholar]
- Lai JKY, Lerch JP, Doering LC, Foster JA, & Ellegood J. (2016). Regional brain volumes changes in adult male FMR1-KO mouse on the FVB strain. Neuroscience, 318, 12–21. 10.1016/j.neuroscience.2016.01.021. [DOI] [PubMed] [Google Scholar]
- Langen M, Leemans A, Johnston P, Ecker C, Daly E, Murphy CM, … Murphy DGM (2012). Fronto-striatal circuitry and inhibitory control in autism: Findings from diffusion tensor imaging tractography. Cortex, 48(2), 183–193. 10.1016/j.cortex.2011.05.018. [DOI] [PubMed] [Google Scholar]
- Lee J, Chung C, Ha S, Lee D, Kim D-Y, Kim H, & Kim E. (2015). Shank3-mutant mice lacking exon 9 show altered excitation/inhibition balance, enhanced rearing, and spatial memory deficit. Frontiers in Cellular Neuroscience, 9, 94 10.3389/fncel.2015.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z-H, Chuang D-M, & Smith CB (2011). Lithium ameliorates phenotypic deficits in a mouse model of fragile X syndrome. The International Journal of Neuropsychopharmacology, 14(5), 618–630. 10.1017/S1461145710000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loesch DZ, Huggins RM, & Hagerman RJ (2004). Phenotypic variation and FMRP levels in fragile X. Mental Retardation and Developmental Disabilities Research Reviews, 10(1), 31–41. 10.1002/mrdd.20006. [DOI] [PubMed] [Google Scholar]
- Mei Y, Monteiro P, Zhou Y, Kim J-A, Gao X, Fu Z, & Feng G. (2016). Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature. 10.1038/nature16971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melke J, Goubran Botros H, Chaste P, Betancur C, Nygren G, Anckarsäter H, … Bourgeron T. (2008). Abnormal melatonin synthesis in autism spectrum disorders. Molecular Psychiatry, 13(1), 90–98. 10.1038/sj.mp.4002016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineur YS, Sluyter F, de Wit S, Oostra BA, & Crusio WE (2002). Behavioral and neuroanatomical characterization of theFmr1 knockout mouse. Hippocampus, 12(1), 39–46. 10.1002/hipo.10005. [DOI] [PubMed] [Google Scholar]
- Morrow EM, Yoo S-Y, Flavell SW, Kim T-K, Lin Y, Hill RS, … Walsh CA (2008). Identifying autism loci and genes by tracing recent shared ancestry. Science, 321(5886), 218–223. 10.1126/science:1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi MW, Kay M, D’Cruz A-M, Seidenfeld A, Guter S, Stanford LD, & Sweeney JA (2009). Impaired inhibitory control is associated with higher-order repetitive behaviors in autism spectrum disorders. Psychological Medicine, 39(9), 1559–1566. 10.1017/S0033291708004984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder EJ, Anderson GM, Kemperman RFJ, Oosterloo-Duinkerken A, Minderaa RB, & Kema IP (2010). Urinary excretion of 5-hydroxyindoleacetic acid, serotonin and 6-sulphatoxymelatonin in normoserotonemic and hyperserotonemic autistic individuals. Neuropsychobiology, 61(1), 27–32. 10.1159/000258640. [DOI] [PubMed] [Google Scholar]
- Nir I, Meir D, Zilber N, Knobler H, Hadjez J, & Lerner Y. (1995). Brief report: Circadian melatonin, thyroid-stimulating hormone, prolactin, and cortisol levels in serum of young adults with autism. Journal of Autism and Developmental Disorders, 25(6), 641–654. http://www.ncbi.nlm.nih.gov/pubmed/8720032. [DOI] [PubMed] [Google Scholar]
- Pan C-Y (2008). Objectively measured physical activity between children with autism spectrum disorders and children without disabilities during inclusive recess settings in Taiwan. Journal of Autism and Developmental Disorders, 38(7), 1292–1301. 10.1007/s10803-007-0518-6. [DOI] [PubMed] [Google Scholar]
- Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, … Feng G. (2011). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature, 472(7344), 437–442. 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peier AM, McIlwain KL, Kenneson A, Warren ST, Paylor R, & Nelson DL (2000). (Over)correction of FMR1 deficiency with YAC transgenics: Behavioral and physical features. Human Molecular Genetics, 9(8), 1145–1159. http://www.ncbi.nlm.nih.gov/pubmed/10767339. [DOI] [PubMed] [Google Scholar]
- Peixoto RT, Wang W, Croney DM, Kozorovitskiy Y, & Sabatini BL (2016). Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B−/− mice. Nature Neuroscience, 19(5), 716–724. 10.1038/nn.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, … Geschwind DH (2011). Absence of CNTNAP2 Leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell, 147(1), 235–246. 10.1016/j.cell.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persico AM, & Napolioni V. (2013). Autism genetics. Behavioural Brain Research, 251, 95–112. 10.1016/j.bbr.2013.06.012. [DOI] [PubMed] [Google Scholar]
- Poliak S, Gollan L, Martinez R, Custer A, Einheber S, Salzer JL, … Peles E. (1999). Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron, 24(4), 1037–1047. 10.1016/S0896-6273(00)81049-1. [DOI] [PubMed] [Google Scholar]
- Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L, … Peles E. (2003). Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. The Journal of Cell Biology, 162(6), 1149–1160. 10.1083/jcb.200305018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portmann T, Yang M, Mao R, Panagiotakos G, Ellegood J, Dolen G, … Dolmetsch RE (2014). Behavioral abnormalities and circuit defects in the basal ganglia of a mouse model of 16p11.2 deletion syndrome. Cell Reports, 7(4), 1077–1092. 10.1016/j.celrep.2014.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posserud M, Hysing M, Helland W, Gillberg C, & Lundervold AJ (2016). Autism traits: The importance of ‘co-morbid’ problems for impairment and contact with services. Data from the Bergen Child Study. Research in Developmental Disabilities. 10.1016/j.ridd.2016.01.002. [DOI] [PubMed] [Google Scholar]
- Restivo L, Ferrari F, Passino E, Sgobio C, Bock J, Oostra BA, … Ammassari-Teule M. (2005). Enriched environment promotes behavioral and morphological recovery in a mouse model for the fragile X syndrome. Proceedings of the National Academy of Sciences of the United States of America, 102(32), 11557–11562. 10.1073/pnas.0504984102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richdale AL, & Prior MR (1995). The sleep/wake rhythm in children with autism. European Child & Adolescent Psychiatry, 4(3), 175–186. 10.1007/BF01980456. [DOI] [PubMed] [Google Scholar]
- Ritvo ER, Ritvo R, Yuwiler A, Brothers A, Freeman BJ, & Plotkin S. (1993). Elevated daytime melatonin concentrations in autism: A pilot study. European Child & Adolescent Psychiatry, 2(2), 75–78. 10.1007/BF02098862. [DOI] [PubMed] [Google Scholar]
- Robinson EB, Lichtenstein P, Anckarsäter H, Happé F, & Ronald A. (2013).Examining and interpreting the female protective effect against autistic behavior. Proceedings of the National Academy of Sciences of the United States of America, 110(13), 5258–5262. 10.1073/pnas.1211070110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseboom PH, Namboodiri MA, Zimonjic DB, Popescu NC, Rodriguez IR, Gastel JA, & Klein DC (1998). Natural melatonin “knockdown” in C57BL/6J mice: Rare mechanism truncates serotonin N-acetyltransferase. Brain Research. Molecular Brain Research, 63(1), 189–197. http://www.ncbi.nlm.nih.gov/pubmed/9838107. [DOI] [PubMed] [Google Scholar]
- Schoch H, Kreibich AS, Ferri SL, White RS, Bohorquez D, Banerjee A, … Brodkin ES (2017). Sociability deficits and altered amygdala circuits in mice lacking Pcdh10, an autism associated gene. Biological Psychiatry, 81(3), 193–202. 10.1016/j.biopsych.2016.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selimbeyoglu A, Kim CK, Inoue M, Lee SY, Hong ASO, Kauvar I, … Deisseroth K. (2017). Modulation of prefrontal cortex excitation/inhibition balance rescues social behavior in CNTNAP2-deficient mice. Science Translational Medicine, 9(401), eaah6733. 10.1126/scitranslmed.aah6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GMG (2013). Corticostriatal connectivity and its role in disease. Nature Reviews. Neuroscience, 14(4), 278–291. 10.1038/nrn3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JL, Yang M, Lord C, & Crawley JN (2010). Behavioural phenotyping assays for mouse models of autism. Nature Reviews. Neuroscience, 11(7), 490–502. 10.1038/nrn2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer CM, Alekseyenko O, Serysheva E, Yuva-Paylor LA, & Paylor R. (2005). Altered anxiety-related and social behaviors in the Fmr1 knockout mouse model of fragile X syndrome. Genes, Brain and Behavior, 4(7), 420–430. 10.1111/j.1601-183X.2005.00123.x. [DOI] [PubMed] [Google Scholar]
- Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE, Parod JM, … Morton DH (2006). Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. New England Journal of Medicine, 354(13), 1370–1377. 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- Sullivan K, Hatton D, Hammer J, Sideris J, Hooper S, Ornstein P, & Bailey D. (2006). ADHD symptoms in children with FXS. American Journal of Medical Genetics Part A, 140A(21), 2275–2288. 10.1002/ajmg.a.31388. [DOI] [PubMed] [Google Scholar]
- Thomas AM, Schwartz MD, Saxe MD, & Kilduff TS (2016). Cntnap2 knockout rats and mice exhibit epileptiform activity and abnormal sleep/wake physiology. Sleep. http://www.ncbi.nlm.nih.gov/pubmed/27692059. [DOI] [PubMed] [Google Scholar]
- Tordjman S, Anderson GM, Pichard N, Charbuy H, & Touitou Y. (2005). Nocturnal excretion of 6-sulphatoxymelatonin in children and adolescents with autistic disorder. Biological Psychiatry, 57(2), 134–138. 10.1016/j.biopsych.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Townsend LB, & Smith SL (2017). Genotype- and sex-dependent effects of altered Cntnap2 expression on the function of visual cortical areas. Journal of Neurodevelopmental Disorders, 9, 2 10.1186/s11689-016-9182-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tranfaglia MR (2011). The psychiatric presentation of fragile x: Evolution of the diagnosis and treatment of the psychiatric comorbidities of fragile X syndrome. Developmental Neuroscience, 33(5), 337–348. 10.1159/000329421. [DOI] [PubMed] [Google Scholar]
- Turner G, Webb T, Wake S, & Robinson H. (1996). Prevalence of fragile X syndrome. American Journal of Medical Genetics, 64(1), 196–197. 10.1002/(SICI)1096-8628(19960712)64:1<196::AID-AJMG35>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Uemura M, Nakao S, Suzuki ST, Takeichi M, & Hirano S. (2007). OL-protocadherin is essential for growth of striatal axons and thalamocortical projections. Nature Neuroscience, 10(9), 1151–1159. 10.1038/nn1960. [DOI] [PubMed] [Google Scholar]
- Ung DC, Iacono G, Méziane H, Blanchard E, Papon M-A, Selten M, … Laumonnier F. (2017). Ptchd1 deficiency induces excitatory synaptic and cognitive dysfunctions in mouse. Molecular Psychiatry. 10.1038/mp.2017.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uutela M, Lindholm J, Rantamäki T, Umemori J, Hunter K, Võikar V, & Castrén ML (2014). Distinctive behavioral and cellular responses to fluoxetine in the mouse model for Fragile X syndrome. Frontiers in Cellular Neuroscience, 8, 150 10.3389/fncel.2014.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlsten D, Metten P, Phillips TJ, Boehm SL, Burkhart-Kasch S, Dorow J, … Crabbe JC (2003). Different data from different labs: Lessons from studies of gene-environment interaction. Journal of Neurobiology, 54(1), 283–311. 10.1002/neu.10173. [DOI] [PubMed] [Google Scholar]
- Wang X, McCoy PA, Rodriguiz RM, Pan Y, Je HS, Roberts AC, … Jiang Y-H (2011). Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Human Molecular Genetics, 20(15), 3093–3108. 10.1093/hmg/ddr212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells MF, Wimmer RD, Schmitt LI, Feng G, & Halassa MM (2016). Thalamic reticular impairment underlies attention deficit in Ptchd1(Y/-) mice. Nature, 532(7597), 58–63. 10.1038/nature17427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werling DM, & Geschwind DH (2013). Sex differences in autism spectrum disorders. Current Opinion in Neurology, 26(2), 146–153. 10.1097/WCO.0b013e32835ee548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, & State MW (2015). Autism spectrum disorders: From genes to neurobiology. Current Opinion in Neurobiology, 30, 92–99. 10.1016/j.conb.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Bozdagi O, Scattoni ML, Wöhr M, Roullet FI, Katz AM, … Crawley JN (2012). Reduced excitatory neurotransmission and mild autism-relevant phenotypes in adolescent Shank3 null mutant mice. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 32(19), 6525–6541. 10.1523/JNEUROSCI.6107-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, & Berry-Kravis E. (2014). Autism and Fragile X Syndrome. Seminars in Neurology, 34(3), 258–265. 10.1055/s-0034-1386764. [DOI] [PubMed] [Google Scholar]
- Yuskaitis CJ, Mines MA, King MK, Sweatt JD, Miller CA, & Jope RS (2010). Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of fragile X syndrome. Biochemical Pharmacology, 79(4), 632–646. 10.1016/j.bcp.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.