

Abstract
Cell death can occur through numerous regulated mechanisms, from apoptosis to necrosis, entosis, and others. Each has a distinct mode of regulation and effect on tissue homeostasis. While the elimination of individual cells is typically considered the relevant physiologic endpoint of cell death, in some cases the remnants left behind by death can also function to support tissue homeostasis. Here we discuss specific functions of the end products of cell death, and how “after-death” functions may contribute to the roles of programmed cell death in physiology.
Keywords: cell death, cornification, entosis, phagocytosis, linker cell, lobe
Introduction
To choose to die instead of live is one of numerous cell fate decisions that individual cells make, along with entering quiescence or senescence, or undergoing differentiation, that contributes to maintaining proper tissue function. Individual cells undergo death and are removed from tissues when they are damaged, aged, or infected, and their dysfunction presents a threat to the organism. Some cells also die as part of a normal cycle of rapid cell turnover that supports the function of specific tissues, or are removed to eliminate specialized structures during development [1,2].
The idea that death is a regulated cell fate originated from observations of insect metamorphosis, where developmental tissue structures undergo hormone-induced regression [3]. Tissue regression occurs as a result of the programmed death of individual cells, typically through a mechanism called apoptosis that is executed by the activation of caspase proteases [4]. Cells undergoing apoptosis shrink and fragment into pieces, called apoptotic bodies, that are cleared by neighboring cells or immune cells through phagocytosis [5]. Because apoptotic bodies typically remain intact and do not release intracellular contents that can be pro-inflammatory, apoptotic death generally occurs in an immunologically silent manner, a feature that avoids potentially harmful immune responses when cells undergo death as part of normal physiology [6].
Research over the last two decades has revealed numerous additional regulated mechanisms of cell death [2]. Notably, several forms of necrosis can be induced by cell damage or infection that differ from apoptosis because they involve rapid rupture of the plasma membrane. By releasing pro-inflammatory cytokines and intracellular damage-associated molecular pattern molecules (DAMPs) that signal to activate immune cells, necrotic deaths promote immune responses that protect the organism from infection [7]. Numerous regulated mechanisms of necrosis, including necroptosis [8], pyroptosis [9], and ferroptosis [10], have been discovered that eliminate damaged or infected cells. Whereas apoptosis removes cells during development and under normal physiologic conditions, necrotic mechanisms are not utilized in normal tissues, likely due to their potent pro-inflammatory activity.
While differences in the crosstalk between apoptosis and necrosis and the immune system may underlie, at least in part, the different physiologic functions of these mechanisms, there are now numerous additional forms of cell death, up to at least 12 in total, that have been discovered [2]. That numerous mechanisms have emerged to eliminate cells suggests the presence of additional distinguishing features that may underlie the utilization of particular forms of cell death in different contexts. Here we consider one particular feature, that the remnants that are left behind by death may have specific “after-death” functions. We discuss how these may be shared or distinct between different mechanisms, and how after-death functions may contribute to the many different roles of cell death in physiology.
After-Death Functions
Nutrient Transfer From Cell Corpses Through Engulfment
One after-death function of cell death that may be shared among different mechanisms is the transfer of nutrients from dead cells to the engulfing cells that ingest them (Figure 1) [11]. The phagocytic clearance of apoptotic cells, for example, has been shown to lead to macrophage utilization of amino acids derived from ingested corpses, demonstrating that nutrients recovered from engulfed cells can be used to support metabolism [12]. The metabolic load from apoptotic cell digestion may be significant in some contexts, as individual macrophages can contain up to 10 to 20 phagocytosed corpses [13]. Similarly, pancreatic cancer cells have been shown to utilize amino acids from macropinocytosed necrotic cell debris to fuel the accumulation of biomass in support of proliferation [14]. In cancers where vascularization is often poor and nutrients can be scarce, the scavenging of extracellular protein through macropinocytosis is known to supply amino acids in support of cancer growth [11,14-16]. Nutrient transfer from dead cell corpses to living cells could also contribute significantly to disease progression in this context.
Figure 1.

After-death functions of cell death. Apoptosis and entosis both eliminate cells, and also have an after-death function to transfer nutrients from dead cells to engulfing cells. For apoptosis, an engulfing macrophage (white) is depicted next to an apoptotic cell (gray); for entosis, the engulfing cells are neighboring cells. Cornification does not function to eliminate cells, but has a structural function to support tissue formation, as dead cells called corneocytes compose the outer layers of skin. Entosis generates a subcellular lobe that may also have a structural after-death function, to support gonad development and fertility in C. elegans.
The mobilization of corpse-derived nutrients to other cells could similarly influence disease progression during stroke or myocardial infarction, conditions that also involve ischemia and the induction of cell death due to a loss of nutrients from vasculature [17]. Cardiac myofibroblasts are known to phagocytose dead cell corpses after myocardial infarction, and their participation in corpse clearance is linked to an anti-inflammatory response that promotes tissue recovery [18]. Nutrient uptake could conceivably contribute to supporting myofibroblast viability and function in tissue repair, although this possibility has not been explored. Similarly in the brain, microglia and astrocytes engulf dead cells that result from stroke, an activity that lasts from days to weeks following injury, and nutrient transfer from corpses could contribute to tissue recovery that is also known to be promoted by these cell types [19].
In some tissues, the ingestion of dead cells could support metabolism as part of normal physiology when nutrients from vasculature are limited. One example of this is in the testes, where Sertoli cells, which function as nurse cells to support sperm cell differentiation, are localized in seminiferous tubules behind an extracellular matrix, and form a blood-testis barrier that limits nutrient diffusion. Sertoli cells continuously ingest residual cytoplasm from developing spermatids and also phagocytose whole apoptotic germ cells, as more than 75 percent of all developing sperm undergo cell death [20,21]. Lipids that are scavenged from ingested cytoplasm and apoptotic cells are proposed to act as a nutrient source, generating ATP through β-oxidation in support of the function of Sertoli cells that otherwise have limited access to nutrients from blood [22].
In nutrient-replete conditions in well-vascularized tissues, macrophages, or other engulfing cells may not need to utilize corpse-derived nutrients when they are present in excess of cellular demand. In this case, nutrients may instead be exported to the extracellular environment. One example of export linked to phagocytosis occurs in the eye, where cells of the retinal pigment epithelium engulf the outer segments of photoreceptor cells on a daily basis. After lysosomal degradation of ingested outer segments, retinoids are recycled and exported back to photoreceptors to support their continued function, in what is called the visual cycle [23]. It is conceivable that when dying cells are ingested in other tissues, nutrient export could occur in a similar manner and large amounts of metabolites could be recycled back to the microenvironment. Macrophages have been shown to export cholesterol in response to the phagocytosis of apoptotic cells [24], but whether the export of cell corpse-derived nutrients from engulfing cells is a general feature of dead cell clearance has not been well explored in mammalian systems. In ancestral organisms, phagocytosis is used to support tissue metabolism, for example in sponges, where specialized phagocytic cells called choanocytes digest microorganisms and pass the scavenged nutrients to other cells [25]. Future studies may establish whether macrophages could function similarly to supply nutrients to cells in the microenvironment in mammalian tissues.
Nutrient Transfer Through a Specialized Mechanism Entosis
The direct transfer of nutrients from corpses to living cells also occurs with a specialized form of cell death called entosis, a competitive mechanism where certain cells within a population, called “winners,” ingest, kill, and degrade neighboring “loser” cells (Figure 1). Loser cells are digested within the lysosomes of winners, leading to direct nutrient transfer that supports winner cell survival and proliferation [12,26,27]. Intriguingly, loser cells play an active role, through Rho-GTPase and contractile myosin, to control their uptake by invading into winners [28-31], suggesting that nutrients are redistributed to winner cells by an altruistic activity of losers through this mechanism.
Entosis was recently shown to be induced in cancer cell populations by long-term starvation for glucose. In this context, high levels of activation of the starvation-induced, energy-sensing kinase AMPK control entosis by acting specifically within loser cells [26]. In starved cell populations, entosis can therefore mobilize and transfer cell-derived nutrients from the most-starved cells, or those with the highest levels of activation of AMPK, to the least-starved, an activity that was shown to promote the survival and outgrowth of cancer cell populations undergoing long-term starvation stress [26].
While entosis is a recently described form of cell competition between cancer cells [32], other forms of competition between cells in developing tissues may instead involve the induction of apoptosis in loser cells, followed by either loser cell engulfment by winners [33], or extrusion and engulfment by macrophages [34]. Winner cells may still benefit from direct nutrient transfer when they engulf apoptotic cells, and are also thought to receive additional signals from dying cells in the form of mechanical cues or secreted factors that promote compensatory proliferation to maintain tissue homeostasis [35,36]. Engulfing macrophages may also accumulate large quantities of metabolites derived from the lysosomal digestion of extruded loser cells, but the impact of nutrient scavenging on macrophage function or tissue homeostasis is not known. Finally, other mechanisms of live cell engulfment called cannibalism are also known to occur in cancer cell populations, where nutrient transfer from ingested neighboring cells or other cells in the microenvironment can contribute to supporting the metabolism of particularly aggressive cells within a cancer [37,38].
Structural Functions: Cornification
While the transfer of nutrients to engulfing cells through corpse digestion is one after-death function of cell death, in some circumstances cell corpses may not be cleared, but may instead persist and perform specific functions for the organism. One clear example of this is a mechanism of cell death called cornification that generates the outer protective layer of skin, the stratum corneum, as well as nails and hair in mammals (Figure 1) [39].
Cornification begins when progenitor cells located in the basal skin layer, the matrix of hair follicles, or the root of nails undergo asymmetric divisions, generating cells that detach from the basement membrane and move toward the exterior surface of the organism. Matrix detachment initiates the process of cornification that is induced by loss of β1-integrin engagement [40] and activation of the TAp63 transcription factor [41], and involves successive stages of intermediate filament protein expression and upregulation of protein crosslinking activity that generates keratin bundles. The plasma membrane also becomes replaced by a corneocyte lipid envelope (CLE), which is supported by an underlying protein-rich cornified envelope composed of proteins such as involucrin and loricrin that become crosslinked to lipids in the CLE [42].
To make space for an increased number of intermediate filament protein bundles, all major cellular organelles, including the nucleus and mitochondria, endoplasmic reticulum, and endosomes, are eventually degraded [39]. Nuclei are degraded in part by the DNAse1L2 and DNAse2 enzymes that control DNA degradation [43-45], and by upregulation of the lysosomal degradative pathway autophagy that clears nuclear fragments through nucleophagy [46]. Autophagy also participates in clearing mitochondria and the endoplasmic reticulum [47,48], and contributes to remodeling cytoplasmic protein composition [49]. The resulting terminally differentiated corneocytes are rendered functionally dead but remain connected by desmosomal adhesions that link the keratin bundles of adjacent cells to generate structural support for skin, hair, and nails. While dead corneocytes in skin are eventually shed from the body after the cleavage of intercellular junctions [50], hair and nails maintain persistent cell junctions, linking cornified cell corpses into architectures that support specialized tissue function.
Structural Functions: Sebocyte Cell Death
Another form of cell death that occurs in skin and performs a structural function is a mechanism that controls the death of specialized cells called sebocytes within sebaceous glands. These cells undergo death through a process of holocrine secretion that involves the degradation of cellular organelles, generation of lipid droplets, and ultimately secretion of lipid-rich sebum from dead cells that provides a waterproofing function for skin and hair [51-53]. Nuclear degradation during sebocyte cell death is controlled by DNAse2 that is released from degrading lysosomes, suggesting that lysosomal damage, a known trigger of cell death in other contexts [54], is linked to this death mechanism that generates an important structural component of healthy hair and skin [55]. The degradation of DNA during sebocyte cell death also contributes purines that are utilized to make uric acid, a component of sebum that is proposed to serve a protective function for skin by acting as an antioxidant [55].
Structural Functions: The Entotic and Linker Cell Lobe
A recently discovered function for entosis suggests that in addition to promoting competition between cells, entosis may also play a structural role to support fertility in Caenorhabditis elegans (Figure 1). While most cell deaths in C. elegans development occur by apoptosis, one particular cell, called the linker cell, dies in a non-apoptotic manner [56,57]. The linker cell has a unique function in development, to shape the male gonad by leading a collective migration that involves movement toward the head, dorsal, and ventral turns, and migration back to the tail in the last larval stage. After the completion of migration, the linker cell undergoes death and is removed in order to facilitate the joining of the gonad to the digestive opening, called the cloaca, through which sperm are released from adults during mating. A failure to kill and remove the linker cell disrupts gonad-to-cloaca fusion and renders adult male worms sterile [56].
While genetic screens identified a pathway involved in promoting linker cell death [58-60], the mechanism underlying linker cell engulfment had remained elusive since the discovery that clearance occurs in a manner distinct from phagocytosis [56]. A recent report identified entosis as the mechanism that clears the linker cell [61]. Both entosis and linker cell clearance were shown to involve the formation of cell adhesions between the ingested cells and their engulfers, an active role for actin within the ingested cells to promote uptake, and both processes resulted in the formation and separation of a subcellular structure, called a lobe, from the ingested cells (Figures 1, 2) [61]. The linker cell lobe, which ranged from 2 to 3 microns in size, was deposited at the site of gonad to cloaca fusion, and persisted for long periods of time as the linker cell body containing the nucleus was engulfed and degraded (Figure 2). The long-term persistence of the lobe structure identified in this study [61], although differing from another report that suggested the lobe can also become engulfed [62], may indicate that this subcellular structure has a specialized function.
Figure 2.
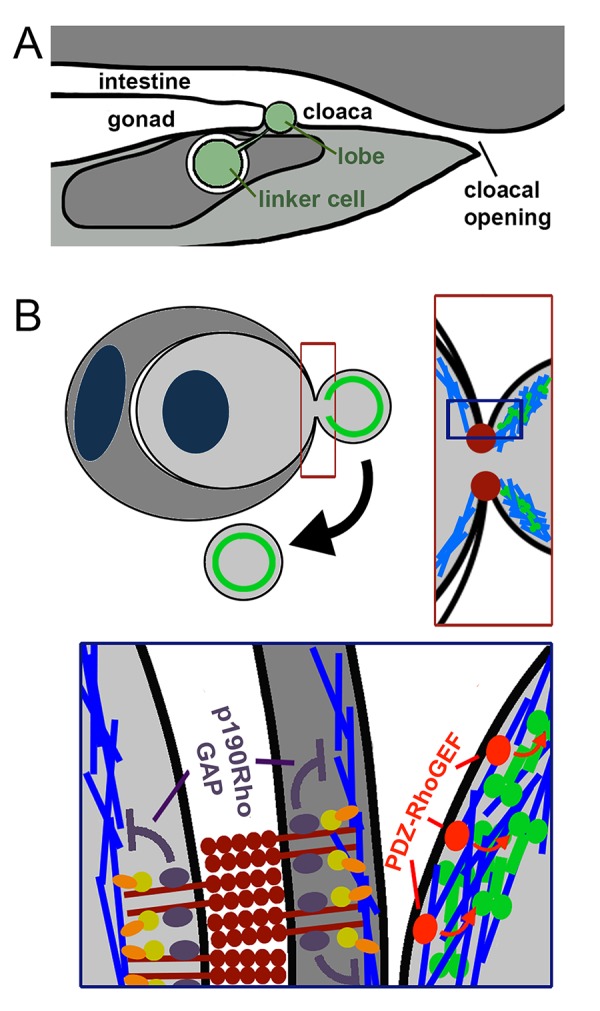
Lobe formation by entosis. a. The linker cell (green) is cleared by entosis in the late L4 stage of development and leaves behind a subcellular lobe that is deposited in between the developing gonad and cloaca, which will form the exit route for sperm. The gonad and intestine share this common exit channel in adult worms. b. Entotic lobes also detach from cells during entosis in culture. Left, top: contractile myosin is known to accumulate at the rear cortex, toward the back of invading cells, and likely accumulates in detaching lobes as well (green, circle). Right, top: entosis is mediated by cell-cell junctions that are formed by E- and P-cadherins (red) at the engulfment interface between internalizing and host cells. The cell adhesions form a ring-like structure, depicted in two dimensions by red foci. Contractile myosin (green) is predicted to accumulate in the lobe; actin is shown in blue. Bottom: E- and P-cadherin junctions (dark red) between internalizing and host cells inhibit the accumulation of contractile actomyosin, through p120 catenin (purple) – dependent recruitment of p190RhoGAP, and suppression of Rho-GTPase activity (inhibitory arrows, purple). Contractile myosin (green) may be activated in the lobe by PDZ-RhoGEF (red) – dependent activation of Rho-GTPase, which is known to occur at the rear cortex of invading cells during entosis.
Subcellular lobes have been shown to form and detach from other cells, including in C. elegans, where large lobes form and separate from migrating primordial germ cells. Germ cell lobes become engulfed and degraded by neighboring endothelial cells through a process that is proposed to promote germ cell maturation [63]. Lobes have also been shown to form and separate from leukocytes undergoing transendothelial migration in mice, and are left behind at blood vessels, where their function is unknown but could involve participation in junctional resealing of the endothelium [64]. For the linker cell, the separating lobe structure can be long-lived, in some cases persisting into adulthood, and then becoming cleared during mating [61]. The long-lived nature of the linker cell lobe and its localization suggest that it could have a specialized function, potentially to regulate the joining of the gonad and cloaca, or perhaps to serve as a protective barrier to shield the developing germline from the digestive opening and external environment. The formation and long-lived nature of the subcellular lobe generated by entosis suggests that this process could have been selected to clear the linker cell, in part, due to specialized function of this remnant that is left behind by cell death.
Conclusions and Outlook
Here we have discussed different ways that cell death mechanisms can contribute to physiology through the end products that they generate. While numerous mechanisms of cell death may lead to the redistribution of nutrients between cells, other after-death functions are more specialized, such as structural functions resulting from cornification in skin. For entosis, this mechanism appears to have two different after-death functions, including direct nutrient transfer that promotes cell competition, and lobe formation that may have a specialized role in supporting fertility during C. elegans development (Figure 1).
The discussion of after-death functions raises an interesting question: what defines when a cell dies? The point of no return for apoptosis was once considered to be any of numerous stages of execution, from the release of cytochrome c from mitochondria, to the activation of caspases, the degradation of DNA, or even the exposure of phagocytic eat-me signals and fragmentation of cells into apoptotic bodies, all of which have now been shown to be stages from which cells can recover, through a process called anastasis [65-71]. This leaves the phagocytic clearance of dying cells, or their lysosomal digestion, as possible points of no return. From studies of entosis, it is clear that engulfment can also be a reversible process [28], suggesting that it is the lysosomal degradation of engulfed cells that is the ultimate, irreversible endpoint.
For cells that are not engulfed, such as corneocytes, which ultimately die as a result of the cornification process, death could, by analogy, be considered to occur when they are sloughed off and removed from the body. Yet prior to removal, individual cells lose all organelles, including mitochondria, and as a result, the ability to generate energy. This irreversible aspect of cornification has been considered the point of death for the individual cells that make up the major structural components of skin, hair, and nails [47]. A loss of cellular organelles is also a defining feature of differentiation programs that generate other cell types, including red blood cells and fiber cells that make up the lens in the eye. Lens fiber cells, unlike corneocytes, are extremely long-lived after organelle removal, persisting perhaps for the lifespan of the organism [72]. Red blood cells are relatively short lived, persisting for 120 days, but they can be activated to undergo cell suicide even after losing organelles [73], and may ultimately die through engulfment by macrophages [74], providing evidence for continued cell viability in some contexts in the absence of major cellular organelles including mitochondria and the nucleus [75].
While entotic cells become degraded by the cells that engulf them, and are at that point irreversibly dead, they also leave behind a subcellular lobe structure that may function after degradation of the cell body. Entotic lobes contain cortical actin and do not contain nuclei [61], but their composition with respect to other organelles or specific proteins is unknown. While lobes have been shown to form from other cells in different contexts, what is unusual about the entotic or linker cell lobe is that it can persist long after the cell body from which it originates is degraded. By analogy, the entotic lobe may resemble platelets, which are 2 to 4 micron cell fragments formed by megakaryocytes in the bone marrow, through a process involving internal membrane formation and outward budding. After platelet release, the megakaryocyte cell body containing the nucleus is engulfed and degraded by macrophages, while platelets enter circulation and can survive and function for up to 10 days in support of blood clotting. Although platelets do not have nuclei, they do contain other organelles, including mitochondria, secretory granules, lysosomes, and peroxisomes, and they are rich actin and myosin, microtubules, and cell surface proteins involved in adhesion [76,77]. Interestingly, platelets can be induced to undergo cell death through apoptosis [78], demonstrating cell viability that extends beyond the lifespan of the original nucleated megakaryocyte precursor cells. If lobes that are formed by entosis perform a specific structural function, their formation may similarly represent the extension of lifespan of the entotic precursor cells from which they are generated.
Acknowledgments
We thank members of the Overholtzer lab for helpful discussions. This work was supported by a grant from the National Cancer Institute (RO1CA154649, to M.O.).
Glossary
- AMPK
Adenosine Monophosphate-Activated Protein Kinase
- CLE
corneocyte lipid envelope
- DAMPs
damage-associated molecular pattern molecules
- DNAse1L2
deoxyribonuclease-1-like-2
- E-cadherin
epithelial cadherin
- P-cadherin
placental cadherin
- P190RhoGAP
p190 Rho-GTPase activating protein
- PDZ-RhoGEF
PDZ-Rho-GTPase guanine exchange factor
- Rho-GTPase
Ras homologous-guanine triphosphatase
- TAp63
p63 transcription factor with the N-terminal transactivation domain
Author Contributions
Yongchan Lee and Michael Overholtzer both contributed to the writing and editing of the manuscript.
References
- Baehrecke EH. How death shapes life during development. Nat Rev Mol Cell Biol. 2002;3(10):779–87. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockshin RA, Williams CM. Programmed Cell Death—I. Cytology of Degeneration in the Intersegmental Muscles of the Pernyi Silkmoth. J Insect Physiol. 1965;11:123–33. [DOI] [PubMed] [Google Scholar]
- Miura M, Zhu H, Rotello R, Hartwieg EA, Yuan J. Induction of apoptosis in fibroblasts by IL-1 beta-converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. Cell. 1993;75(4):653–60. [DOI] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran KS. “Recruitment signals” from apoptotic cells: invitation to a quiet meal. Cell. 2003;113(7):817–20. [DOI] [PubMed] [Google Scholar]
- Tait SW, Ichim G, Green DR. Die another way—non-apoptotic mechanisms of cell death. J Cell Sci. 2014;127(Pt 10):2135–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–9. [DOI] [PubMed] [Google Scholar]
- Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9(3):113–4. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna S, Palm W, Lee Y, Yang W, Bandyopadhyay U, Xu H, et al. PIKfyve Regulates Vacuole Maturation and Nutrient Recovery following Engulfment. Dev Cell. 2016;38(5):536–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajcovic M, Krishna S, Akkari L, Joyce JA, Overholtzer M. mTOR regulates phagosome and entotic vacuole fission. Mol Biol Cell. 2013;24(23):3736–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zent CS, Elliott MR. Maxed out macs: physiologic cell clearance as a function of macrophage phagocytic capacity. FEBS J. 2017;284(7):1021–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SM, Nguyen TT, Ravi A, Kubiniok P, Finicle BT, Jayashankar V, et al. PTEN Deficiency and AMPK Activation Promote Nutrient Scavenging and Anabolism in Prostate Cancer Cells. Cancer Discov. 2018;8(7):866–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florey O, Overholtzer M. Macropinocytosis and autophagy crosstalk in nutrient scavenging. Philos Trans R Soc Lond B Biol Sci. 2019;374(1765):20180154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497(7451):633–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieweke MH, Allen JE. Beyond stem cells: self-renewal of differentiated macrophages. Science. 2013;342(6161):1242974. [DOI] [PubMed] [Google Scholar]
- Nakaya M, Watari K, Tajima M, Nakaya T, Matsuda S, Ohara H, et al. Cardiac myofibroblast engulfment of dead cells facilitates recovery after myocardial infarction. J Clin Invest. 2017;127(1):383–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morizawa YM, Hirayama Y, Ohno N, Shibata S, Shigetomi E, Sui Y, et al. Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat Commun. 2017;8(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakberg EF. A description of spermiogenesis in the mouse and its use in analysis of the cycle of the seminiferous epithelium and germ cell renewal. Am J Anat. 1956;99(3):391–413. [DOI] [PubMed] [Google Scholar]
- Huckins C. The morphology and kinetics of spermatogonial degeneration in normal adult rats: an analysis using a simplified classification of the germinal epithelium. Anat Rec. 1978;190(4):905–26. [DOI] [PubMed] [Google Scholar]
- Xiong W, Wang H, Wu H, Chen Y, Han D. Apoptotic spermatogenic cells can be energy sources for Sertoli cells. Reproduction. 2009;137(3):469–79. [DOI] [PubMed] [Google Scholar]
- Wright CB, Redmond TM, Nickerson JM. A History of the Classical Visual Cycle. Prog Mol Biol Transl Sci. 2015;134:433–48. [DOI] [PubMed] [Google Scholar]
- Fond AM, Lee CS, Schulman IG, Kiss RS, Ravichandran KS. Apoptotic cells trigger a membrane-initiated pathway to increase ABCA1. J Clin Invest. 2015;125(7):2748–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godefroy N, Le Goff E, Martinand-Mari C, Belkhir K, Vacelet J, Baghdiguian S. Sponge digestive system diversity and evolution: filter feeding to carnivory. Cell Tissue Res. 201;377(3):341–351. [DOI] [PubMed] [Google Scholar]
- Hamann JC, Surcel A, Chen R, Teragawa C, Albeck JG, Robinson DN, et al. Entosis Is Induced by Glucose Starvation. Cell Rep. 2017;20(1):201–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Luo T, Ren Y, Florey O, Shirasawa S, Sasazuki T, et al. Competition between human cells by entosis. Cell Res. 2014;24(11):1299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overholtzer M, Mailleux AA, Mouneimne G, Normand G, Schnitt SJ, King RW, et al. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell. 2007;131(5):966–79. [DOI] [PubMed] [Google Scholar]
- Kojima R, Fussenegger M. Engineering Whole Mammalian Cells for Target-Cell-Specific Invasion/Fusion. Adv Sci (Weinh). 2018;5(7):1700971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purvanov V, Holst M, Khan J, Baarlink C, Grosse R. G-protein-coupled receptor signaling and polarized actin dynamics drive cell-in-cell invasion. eLife. 2014:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Cibas ES, Huang H, Hodgson L, Overholtzer M. Induction of entosis by epithelial cadherin expression. Cell Res. 2014;24(11):1288–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Huang H, Overholtzer M. Cell-in-cell structures are involved in the competition between cells in human tumors. Mol Cell Oncol. 2015;2(4):e1002707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis SJ, Gomez NC, Levorse J, Mertz AF, Ge Y, Fuchs E. Distinct modes of cell competition shape mammalian tissue morphogenesis. Nature. 2019;569(7757):497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolo FN, Casas-Tinto S, Moreno E. Cell competition time line: winners kill losers, which are extruded and engulfed by hemocytes. Cell Rep. 2012;2(3):526–39. [DOI] [PubMed] [Google Scholar]
- Madan E, Gogna R, Moreno E. Cell competition in development: information from flies and vertebrates. Curr Opin Cell Biol. 2018;55:150–7. [DOI] [PubMed] [Google Scholar]
- Bove A, Gradeci D, Fujita Y, Banerjee S, Charras G, Lowe AR. Local cellular neighborhood controls proliferation in cell competition. Mol Biol Cell. 2017;28(23):3215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fais S, Overholtzer M. Cell-in-cell phenomena in cancer. Nat Rev Cancer. 2018;18(12):758–66. [DOI] [PubMed] [Google Scholar]
- Lugini L, Matarrese P, Tinari A, Lozupone F, Federici C, Iessi E, et al. Cannibalism of live lymphocytes by human metastatic but not primary melanoma cells. Cancer Res. 2006;66(7):3629–38. [DOI] [PubMed] [Google Scholar]
- Eckhart L, Lippens S, Tschachler E, Declercq W. Cell death by cornification. Biochim Biophys Acta. 2013;1833(12):3471–80. [DOI] [PubMed] [Google Scholar]
- Levy L, Broad S, Diekmann D, Evans RD, Watt FM. beta1 integrins regulate keratinocyte adhesion and differentiation by distinct mechanisms. Mol Biol Cell. 2000;11(2):453–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candi E, Rufini A, Terrinoni A, Dinsdale D, Ranalli M, Paradisi A, et al. Differential roles of p63 isoforms in epidermal development: selective genetic complementation in p63 null mice. Cell Death Differ. 2006;13(6):1037–47. [DOI] [PubMed] [Google Scholar]
- Elias PM, Gruber R, Crumrine D, Menon G, Williams ML, Wakefield JS, et al. Formation and functions of the corneocyte lipid envelope (CLE). Biochim Biophys Acta. 2014;1841(3):314–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer H, Eckhart L, Mildner M, Jaeger K, Buchberger M, Ghannadan M, et al. DNase1L2 degrades nuclear DNA during corneocyte formation. J Invest Dermatol. 2007;127(1):24–30. [DOI] [PubMed] [Google Scholar]
- Fischer H, Szabo S, Scherz J, Jaeger K, Rossiter H, Buchberger M, et al. Essential role of the keratinocyte-specific endonuclease DNase1L2 in the removal of nuclear DNA from hair and nails. J Invest Dermatol. 2011;131(6):1208–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer H, Buchberger M, Napirei M, Tschachler E, Eckhart L. Inactivation of DNase1L2 and DNase2 in keratinocytes suppresses DNA degradation during epidermal cornification and results in constitutive parakeratosis. Sci Rep. 2017;7(1):6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinduro O, Sully K, Patel A, Robinson DJ, Chikh A, McPhail G, et al. Constitutive Autophagy and Nucleophagy during Epidermal Differentiation. J Invest Dermatol. 2016;136(7):1460–70. [DOI] [PubMed] [Google Scholar]
- Jones LA, Harland DP, Jarrold BB, Connolly JE, Davis MG. The walking dead: sequential nuclear and organelle destruction during hair development. Br J Dermatol. 2018;178(6):1341–52. [DOI] [PubMed] [Google Scholar]
- Koenig U, Robenek H, Barresi C, Brandstetter M, Resch GP, Groger M, et al. Cell death induced autophagy contributes to terminal differentiation of skin and skin appendages. Autophagy. 2019:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger K, Sukseree S, Zhong S, Phinney BS, Mlitz V, Buchberger M, et al. Cornification of nail keratinocytes requires autophagy for bulk degradation of intracellular proteins while sparing components of the cytoskeleton. Apoptosis. 2019;24(1-2):62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Has C. Peeling Skin Disorders: A Paradigm for Skin Desquamation. J Invest Dermatol. 2018;138(8):1689–91. [DOI] [PubMed] [Google Scholar]
- Plewig G, Christophers E. Renewal rate of human sebaceous glands. Acta Derm Venereol. 1974;54(3):177–82. [PubMed] [Google Scholar]
- Zouboulis CC. Acne and sebaceous gland function. Clin Dermatol. 2004;22(5):360–6. [DOI] [PubMed] [Google Scholar]
- Atsugi T, Yokouchi M, Hirano T, Hirabayashi A, Nagai T, Ohyama M, et al. Holocrine secretion occurs outside the tight junction barrier in multicellular glands: lessons from claudin-1-deficient mice. J Invest Dermatol. 2019. pii: S0022-202X(19)33139–2. [DOI] [PubMed] [Google Scholar]
- Aits S, Jaattela M. Lysosomal cell death at a glance. J Cell Sci. 2013;126(Pt 9):1905–12. [DOI] [PubMed] [Google Scholar]
- Fischer H, Fumicz J, Rossiter H, Napirei M, Buchberger M, Tschachler E, et al. Holocrine Secretion of Sebum Is a Unique DNase2-Dependent Mode of Programmed Cell Death. J Invest Dermatol. 2017;137(3):587–94. [DOI] [PubMed] [Google Scholar]
- Abraham MC, Lu Y, Shaham S. A morphologically conserved nonapoptotic program promotes linker cell death in Caenorhabditis elegans. Dev Cell. 2007;12(1):73–86. [DOI] [PubMed] [Google Scholar]
- Yuan J. Evolutionary conservation of a genetic pathway of programmed cell death. J Cell Biochem. 1996;60(1):4–11. [DOI] [PubMed] [Google Scholar]
- Kinet MJ, Malin JA, Abraham MC, Blum ES, Silverman MR, Lu Y, et al. HSF-1 activates the ubiquitin proteasome system to promote non-apoptotic developmental cell death in C. elegans. eLife. 2016:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin JA, Kinet MJ, Abraham MC, Blum ES, Shaham S. Transcriptional control of non-apoptotic developmental cell death in C. elegans. Cell Death Differ. 2016;23(12):1985–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum ES, Abraham MC, Yoshimura S, Lu Y, Shaham S. Control of nonapoptotic developmental cell death in Caenorhabditis elegans by a polyglutamine-repeat protein. Science. 2012;335(6071):970–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Hamann JC, Pellegrino M, Durgan J, Domart MC, Collinson LM, et al. Entosis Controls a Developmental Cell Clearance in C. elegans. Cell Rep. 2019;26(12):3212–20 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutscher LM, Keil W, Shaham S. RAB-35 and ARF-6 GTPases Mediate Engulfment and Clearance Following Linker Cell-Type Death. Dev Cell. 2018;47(2):222–38 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdu Y, Maniscalco C, Heddleston JM, Chew TL, Nance J. Developmentally programmed germ cell remodelling by endodermal cell cannibalism. Nat Cell Biol. 2016;18(12):1302–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun YM, Sumagin R, Sarangi PP, Lomakina E, Overstreet MG, Baker CM, et al. Uropod elongation is a common final step in leukocyte extravasation through inflamed vessels. J Exp Med. 2012;209(7):1349–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinou I, Desagher S, Eskes R, Antonsson B, Andre E, Fakan S, et al. The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J Cell Biol. 1999;144(5):883–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SW, Green DR. Mitochondrial regulation of cell death. Cold Spring Harb Perspect Biol. 2013;5(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SW, Parsons MJ, Llambi F, Bouchier-Hayes L, Connell S, Munoz-Pinedo C, et al. Resistance to caspase-independent cell death requires persistence of intact mitochondria. Dev Cell. 2010;18(5):802–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang HL, Tang HM, Mak KH, Hu S, Wang SS, Wong KM, et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol Biol Cell. 2012;23(12):2240–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang HM, Tang HL. Anastasis: recovery from the brink of cell death. R Soc Open Sci. 2018;5(9):180442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang HM, Fung MC, Tang HL. Detecting Anastasis In Vivo by CaspaseTracker Biosensor. J Vis Exp. 2018(132). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang HL, Tang HM, Hardwick JM, Fung MC. Strategies for tracking anastasis, a cell survival phenomenon that reverses apoptosis. J Vis Exp. 2015(96). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JR, Levchenko VA, Blanksby SJ, Mitchell TW, Williams A, Truscott RJ. No turnover in lens lipids for the entire human lifespan. eLife. 2015:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang KS, Lang PA, Bauer C, Duranton C, Wieder T, Huber SM, et al. Mechanisms of suicidal erythrocyte death. Cell Physiol Biochem. 2005;15(5):195–202. [DOI] [PubMed] [Google Scholar]
- Brown GC, Neher JJ. Eaten alive! Cell death by primary phagocytosis: ‘phagoptosis’. Trends Biochem Sci. 2012;37(8):325–32. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16(1):3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendu F, Brohard-Bohn B. The platelet release reaction: granules’ constituents, secretion and functions. Platelets. 2001;12(5):261–73. [DOI] [PubMed] [Google Scholar]
- White JG. Current concepts of platelet structure. Am J Clin Pathol. 1979;71(4):363–78. [DOI] [PubMed] [Google Scholar]
- Leytin V, Gyulkhandanyan AV, Freedman J. Platelet Apoptosis Can Be Triggered Bypassing the Death Receptors. Clin Appl Thromb Hemost. 2019;25:1076029619853641. [DOI] [PMC free article] [PubMed] [Google Scholar]