

Abstract
The cell death response to DNA damage is discussed in this Perspectives piece with cancer as the backdrop because DNA damaging agents (DDA) are widely used to treat cancer. From decades of clinical results, we learn that DDA have cured some cancers but their toxicity is temporary in most cancers due to emergence of DDA-resistant cancer cells. Investigation of DDA-activated genes, proteins, and pathways, known collectively as the DNA damage response (DDR), has uncovered the inner workings of DDR that protect the genome to sustain life. Paradoxically, however, DDR can also activate death. Current knowledge on DDA-activated death and hypotheses for how DDR may determine when and where to execute death are discussed. Given that cancer cells suffer from DDR defects, which account for their initial sensitivity to DDA, future therapeutic development may exploit those cancer-specific DDR defects to selectively create death-inducing DNA lesions, without using DDA, to kill DDA-resistant cancers.
Keywords: apoptosis, cancer, chemotherapy, mitochondria, mitotic catastrophe, necroptosis, radiation therapy
Introduction
DNA is the blueprint of life. When that blueprint is irrevocably damaged, life ceases. To stay alive, therefore, requires a continuous effort to protect that blueprint. By its chemical nature, DNA is not inert but highly reactive to elements that are prevalent in the macro- and the micro- environment, e.g., oxygen free radicals or UV light [1-3]. Hence, living organisms are endowed with a sophisticated toolbox to monitor and repair damages to their DNA. In the evolutionarily conserved parts of this toolbox, we have found proteins, enzymes, and gene products that detect DNA lesions, assemble the appropriate repair machineries, and coordinate a concerted effort from virtually every biological process to achieve the goal of survival, that is, to sustain life. Collectively, these proteins, enzymes and gene products constitute a biological network known as the DNA Damage Response (DDR) [4]. Since DNA is constantly damaged, DDR is constantly deployed. DDR defects are detrimental, for they either cause outright lethality or mutation accumulation to drive diseases, most notably, cancer [4-8].
The oncology practice of applying DNA damaging agents (DDA) to treat cancer began empirically in the early 20th century soon after physicists discovered X-rays [9,10]. To date, radiation and DNA-damaging drugs have remained the mainstay modality of cancer therapy. The clinical observation that DDA can kill cancer cells supports the idea that DNA damage is detrimental. However, we have now come to appreciate the reason for how oncologists can empirically determine a dose of DDA that kills cancer cells without destroying the body, and that is because cancer cells tend to suffer repair defects and are therefore more likely to be killed by DDA [11]. After a century of advancements and successes in curing a significant number of childhood cancers [12], DDA have failed to cure the majority of adult cancers because their toxicity is temporary. After the initial destruction of cancer cells by DDA, some malignant cells that survive the insult can resume proliferation and pass the know-how on surviving DDA to their progenies, and thereby neutralizing the toxic effect of radiation and chemotherapy on the recurring cancers. The conclusions that DDR defects drive cancer development and that DDR-defective cells are hypersensitive to DDA are irrefutable [11]; however, the consistent failure of DDA to cure adult cancer suggests that we may not have fully comprehended the capacity of DDR to sustain life. In the laboratories, biologists have observed cells to adapt to damage by resuming proliferation despite the presence of an irreversible lesion in their DNA [13-17]. Given that survival is perhaps the strongest selective pressure, it makes sense for DDR to have evolved strategies to sustain life even when DNA damage persists. The fact that we have not been very successful in either preventing or reversing the resistance of recurring cancers to DDA exposes the gaps in our knowledge on the life-sustaining capacity of DDR.
Paradoxically, this life-sustaining DDR also has the capability to actively kill cells [18-22]. In our body, the genetic blueprint assembled by the fusion of a sperm and an egg is repeatedly copied, i.e., replicated, during development and throughout adult life such that each human DNA blueprint is collectively held by tens of trillions of cells. This multiplicity of the blueprint affords multicellular organisms with the luxury to actively kill off excess or damaged cells [23], including those with damaged DNA [18-22]. The cell-killing programs are embedded in the DNA blueprint in that the genomes of multicellular organisms encode proteins and enzymes that when activated can kill cells [23,24]. Although the death programs are available to every cell of an organism, they are not always activated by DNA damage. For example, in the nematode Caenorhabditis elegans, DNA damage activates death in germ cells but not somatic cells [25]; and in the developing nervous system of the mouse, DNA damage activates death in neuroblasts but not differentiated neurons [26]. In other words, the death option in DDR is not universal – not all living organisms choose to die in response to DNA damage, and not all cell types in an organism choose to die, either. Obviously, the death programs could not have evolved without the co-evolution of suppressors to keep the killing-machines under wrap [27]. Generally speaking, the death option in DDR is more prevalently activated in cells that are either destined to die or are readily replaceable, such as those in highly regenerative tissues [28]. In this context, active elimination of damaged cells is not only affordable but may also be beneficial to the survival of the organisms [23]. On the whole, however, it is important to emphasize that the death option in DDR is suppressed in most mature tissues of our body.
In this Perspectives piece, I offer what I consider to be well-established death programs that can be activated by DNA damage, and discuss the current knowledge on how cancer cells may suppress these programs to survive radiation and chemotherapy.
Topics: Modalities of DNA Damage-Induced Cell Death
Passive vs Active Death to DNA Damage
The death response to DNA damage falls into two categories: death-from-failure-to-repair vs death-by-choice. The death-from-failure-to-repair is passive; an unintended consequence brought on by detrimental DNA-lesions that interfere with the copying (replication), reading (transcription), and proper functioning (condensation and segregation) of the blueprint. The death-by-choice, in contrast, is an active response, an action brought on by the inner workings of the DDR [22]. Although the passive vs the active death responses are easy to discern on paper, they can be difficult to distinguish experimentally because the death-by-choice is not, and obviously cannot, be immediately activated upon the detection of any DNA lesion. Rather, the active death is a delayed response in DDR and coordinated with the status of DNA repair [20]. Further confounding the experimental categorization of passive vs active death is the finding that death-from-repair-failure, rather than an unintended consequence, may involve necroptosis, which is a killing-machine activated by inflammatory cytokines, oxidative stress, and other metabolic triggers [29]. Given these findings, it would be prudent, for the time being, to consider none of the DNA damage-induced death response to be entirely passive.
Coordination between DNA Repair and Death in DDR
A widely accepted theory posits that DDA activates the death-programs only when DNA-lesions become irreparable. Underlying this theory is the assumption that DDR has the ability to distinguish between a reparable DNA-lesion from one that is irreparable [20,21]. Although DDR can distinguish between different types of DNA lesions to stimulate the proper repairs of those lesions [20,30,31], there is no evidence that the lesion-detectors can distinguish between those that are reparable from those that are irreparable. In the absence of evidence for the existence of detectors that can recognize irreparable DNA-lesions, we must consider alternative hypotheses for how DNA repair may be coordinated with the execution of cell death.
Since the killing-machines must not be activated immediately upon the detection of a DNA lesion, because lesions do occur under normal physiological conditions, there should exist in DDR mechanisms to delay death execution. One such delay mechanism is the built-in requirement for transcription induction and new protein synthesis, which take time, to activate cell death [20,24]. The requirement for new gene expression to execute death not only provides a safeguard but could also allow for DDR to activate counter-measures to further control the death machines. It is conceivable that DNA lesion-detectors may simultaneously activate DNA-repair, a death-program, and suppressors of that death program (Figure 1). By activating both death and death-suppressors, it would become possible to set a time window for DNA repair (Figure 1). In this scenario, it is the combination of repair-efficiency and the death suppressor lifespan that determines the timing of death execution. In this scheme, if DNA-lesions are repaired within the lifespan of death-suppressors, cells do not die (Figure 1). However, if DNA-lesions are not repaired within the lifespan of death-suppressors, death is executed (Figure 1). In cells where DDR does not induce death-suppressors, or in cells where the lifespan of DDR-induced death-suppressors is very short, we would observe a rapid onset of death following exposure to a low dose of DDA, such as the case of embryonic neuroblasts [26,32]. On the other hand, in cells with constitutively expressed death-suppressors such as mature neurons [33], death can be prevented despite persistence of DNA-lesions. This idea that DDR can set a window-for-repair, either short or infinite, forgoes the need for detectors of irreparable lesions and negates the requirement for DDR to make judgments on the progress of repair [20]. Rather, in this model, the killing-machines can be pre-set, by the inner workings of DDR in conjunction with the context of a given cell type, for activation after a given window-of-time. While plausible, this theory of a time-dependent regulation of DNA damage-induced death is still pending experimental validation.
Figure 1.
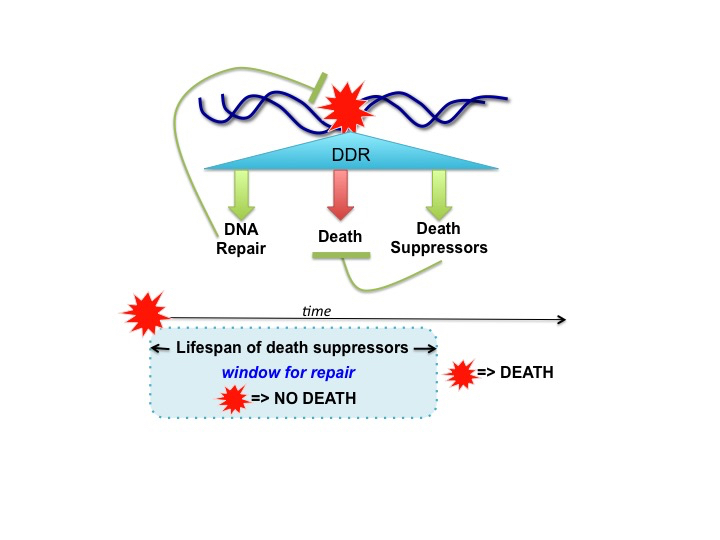
Regulation of Death Execution in DNA Damage Response: a Window-for-Repair Hypothesis. In DNA damage response (DDR), the detection of DNA-lesion (red star) must not immediately activate cell death because DNA lesions are continuously generated under physiological conditions. Instead, DNA damage-induced cell death is a delayed response, observed after hours and even days following exposure to DNA damaging agents (DDA) [20,22]. This delay in executing DDA-induced death is often explained by the theory that death is activated only when the DNA-lesions are irreparable. Since there is no evidence for any cellular mechanisms that can distinguish between reparable vs irreparable DNA-lesions [21], I propose an alternative hypothesis for how DDR can determine when and where to execute cell death. In this hypothesis, DNA damage simultaneously activates DNA repair, death, and death-suppressors (upper panel). Successful repair eliminates the damage and terminates DDR to prevent death, whereas death-suppressors also prevent death execution. By setting the lifespan of death-suppressors, this design of parallel pathway activations pre-sets a window of time for repair (lower panel). During this window of time, DNA damage cannot activate death because of the death suppressors. However, after the decay of death-suppressors, death is activated if the damage is not repaired (lower panel). Of course, if the damaged is repaired before the decay of death-suppressors, death is avoided because DDR is terminated. Cells with a short window-for-repair would be very sensitive to DDA-induced death. On the other hand, with an infinite window-for-repair, cells would become resistant to DDA-induced death.
Activation of Apoptosis by DNA Damaging Agents (DDA)
The evolutionarily conserved program of apoptosis, when activated, causes cells to condense and fragment their DNA and other content into apoptotic bodies that are recognized and engulfed by neighboring cells or professional phagocytes [34]. During embryonic development, the apoptosis program is deployed to eliminate cells in order to form digits [35]. In the development of immune tolerance, apoptosis is deployed to eliminate self-reactive lymphocytes [36]. Upon infection, apoptosis can be deployed to eliminate infected cells to prevent the spread of pathogens [37]. Biologists have found several different routes to executing the apoptotic form of death [38]. In DDR, activation of apoptosis requires mitochondrial release of cytochrome C to stimulate caspases (Figure 2), which cleave hundreds of proteins to cause the distinct features of apoptotic death [39].
Figure 2.
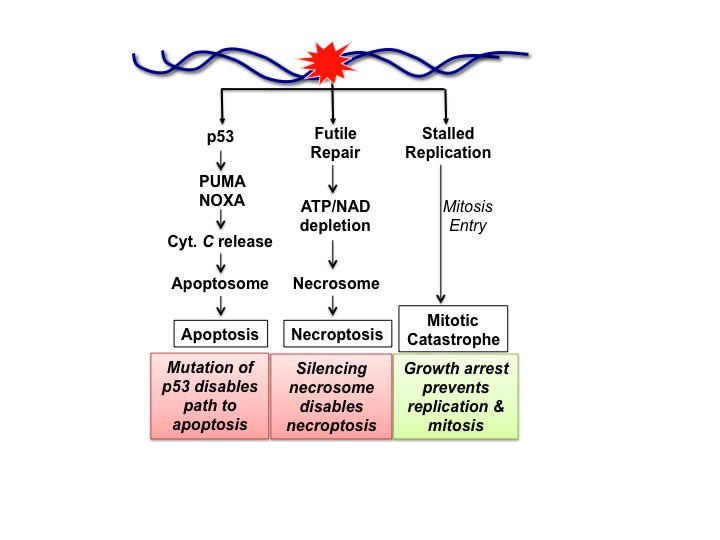
DNA Damage-Induced Cell Death Modalities and Their Suppression. The DDR master kinases, ATM, ATR, DNAPK, that are activated by DNA damage, phosphorylate a transcription factor p53 itself, encoded by the Tp53 tumor suppressor gene, and its inhibitors (MDM2) to stabilize and activate p53 [40]. The activated p53 stimulates the expression of hundreds of genes, including those encoding PUMA and NOXA, two pro-apoptotic proteins [44]. PUMA and NOXA stimulate apoptosis by causing the release of mitochondrial cytochrome C (Cyt. C), which then stimulates the assembly of apoptosome to activate apoptosis [24]. Tp53 is one of the most frequently mutated genes in cancers, and the loss of p53 disconnects DNA damage to the activation of apoptosis. Futile repair describes the process where a continuous and non-productive repair progress depletes ATP and NAD to cause oxidative stress, which activates the necrosome to stimulate necroptosis [29,52]. The necrosome can be silenced by the suppression of RIPK3 expression in cancers [51]. In actively proliferating cells, DNA lesions can interfere with DNA synthesis by stalling the replication forks. If cells with incompletely replicated DNA break through G2-arrest to enter mitosis, the condensation of partially replicated sister-chromatids will shatter the DNA to cause mitotic catastrophe [57]. DNA damage-induced growth arrest can sustain life by blocking DNA replication and mitosis to avoid mitotic catastrophe.
Without a doubt, the most important player that enables the apoptosis option in DDR is the transcription factor p53, encoded by the human TP53 gene, with orthologs in other vertebrate species. The p53 protein is a member of the rapid-response-team activated by the master kinases (ATM, ATR, DKN-PK) in DDR [40-42]. In keeping with the life-sustaining goal of DDR, activated p53 induces the transcription of hundreds of genes to promote survival by: (a) stimulating DNA repair, (b) installing cell cycle checkpoints, and (c) reprograming metabolism [43,44]. However, the activated p53 also induces the transcription of pro-apoptotic genes to produce PUMA and NOXA proteins [44-47]. PUMA (product of the human BBC3 gene) and NOXA (product of the human PMAIP1 gene) activate apoptosis by breaking down the anti-apoptosis defense mechanism to cause mitochondrial release of cytochrome C, which activates the apoptosome to stimulate caspases [24,39]. In mice, knockout of Tp53, or Bbc3 plus Pmaip1, abolished DDA-induced apoptosis in neuroblasts, thymocytes, and other cell types in which apoptosis is an option of DDR [48-50]. Therefore, the pathway from p53 to PUMA and NOXA is essential to DDA-induced apoptosis (Figure 2).
Futile Repair Activates Necroptosis
Necroptosis is a genetic program that, when activated, causes a necrotic form of cell death [29]. Necrotic death, or necrosis, describes the morphology of cell swelling, membrane rupture and the spilling of cell content, which are easily distinguishable from the morphological features of apoptosis [29]. Pathogens or blunt force injuries can rupture the cell membrane to cause necrosis. However, necrotic death can also result from activation of necroptosis, or from the opening of the mitochondrial permeability pore (PTP) [38]. The execution of necroptosis is stimulated by the necrosome, consisting of the RIPK1 and RPIK3 protein kinases, acting at the plasma membrane and in the mitochondria [29,38]. DNA damaging agents (DDA) can cause necrotic death that requires RIPK1 and RIPK3 [22,29,51]. Whereas the master kinases of DDR (ATM, ATR, DNA-PK) directly phosphorylate and activate p53 [40-42], there is no evidence as yet for a direct link between the DDR master kinases and the RIPKs. Instead, activation of necroptosis by DDA may result from futile-repair, which describes a continuous but non-productive repair process that can lead to the unintended depletion of ATP and NAD [52]. In other words, activation of necroptosis is an unintended consequence of repair failure (Figure 2).
Replication Failure and Mitotic Catastrophe
DNA replication is essential to cell proliferation, tissue regeneration and tumor growth. To ensure the complete and faithful replication of cellular DNA, DDR has a cadre of redundant and robust mechanisms to repair lesions ahead of replication and to re-start replication that is stalled by DNA-lesions [53,54]. In proliferative cells, upon detection of DNA damage, the DDR master kinases phosphorylate key proteins to prevent the initiation of DNA replication in G1-cells that have not yet committed to DNA synthesis, and this DDR action is referred to as the G1-checkpoint or G1-arrest [20,55,56]. In cells that have committed to but not yet completed replication, DDR master kinases phosphorylate other key proteins to prevent the onset of mitosis [20,55,56]. This G2-arrest reduces the risk of mitotic catastrophe, where chromosomes are shattered when incompletely replicated sister chromatids become condensed [57]. In the event where DDR fails to enforce G2-arrest while DNA replication forks are stalled, mitotic death would ensue [58,59]. In other words, the normal process of mitotic chromatin condensation can cause unintended death when DNA lesions prevent the completion of DNA replication (Figure 2).
Suppression of DNA Damage-Induced Cell Death
Quiescence, Senescence, or Terminal Differentiation
Generally speaking, DDA are more likely to kill proliferative than non-proliferative cells, including those in quiescence, senescence, or those that have undergone terminal differentiation [20,21]. The fact that it is possible for oncologists to empirically determine a DDA dosage that kills cancer cells but does not destroy the brain or the heart is because terminally differentiated neurons and cardiac muscle cells are more resistant to DDA [21,60]. The therapeutic doses of DDA do kill off normal proliferative cells in the bone marrow, the intestines, or the hair follicles, but these tissues contain sufficient numbers of quiescent stem cells for regeneration after cessation of DDA treatment.
The common characteristic among quiescent, senescent and terminally differentiated cells is the withdrawal from DNA replication. In quiescent cells, the withdrawal is reversible, that is, quiescent cells retain the capacity to initiate DNA replication when the conditions become favorable. In senescent cells, the withdrawal from DNA replication is irreversible and no longer stimulated by growth factors [61]. Because non-proliferative cells do not replicate DNA or enter mitosis, they are less likely to die by mitotic catastrophe (Figure 2). Terminally differentiated neurons and muscle cells also express higher levels of apoptosis suppressors [33,62]. It is of interest to note that necroptosis can still be activated by oxidative stress in aging neurons or cardiac muscle cells [63,64], and may account for neural and cardiac toxicity associated with chemotherapy [65,66].
Tp53 Mutation in Cancer
The human Tp53 is one of the most frequently mutated genes in cancers; as a result, p53-driven apoptotic response to DNA damage is lost in cancer cells. While p53 induces PUMA and NOXA to activate apoptosis, it is important to point out that p53 also inhibits DNA replication. According to a meta-analysis of published data on p53-target genes, CDKN1A is the most consistently, or universally, induced when p53 becomes activated [44]. CDKN1A encodes the p21CIP1 protein that inhibits Cdk/Cyclin to enforce G1-arrest, promote senescence and reduce death [67]. The fact that the p53-CDKN1A pathway is also a part of DDR shows that p53-activation alone is not sufficient to determine whether a damaged cell will undergo growth arrest or apoptosis. Despite decades of research and numerous hypotheses [22,68], we have not yet arrived at a consensus on how the conflict between the pro-arrest and the pro-apoptosis functions of p53 is resolved in DDR. Obviously, resolution of this conflict is irrelevant to cancer development because Tp53 is mutated in most cancer cells.
Proliferation Despite Persistent Damage
Besides the suppression of cell death, recurring cancer cells may also activate strategies to continue proliferation despite persistent DNA damage. Several such strategies discovered in unicellular organisms are worth considerations.
Adaptation to Cell Cycle Checkpoints
Cell cycle checkpoints describe DDR-mechanisms that prevent the initiation of DNA replication (G1-checkpoint) or the entry into mitosis (G2-checkpoint) [69]. The checkpoint mechanisms are reversible so that cells can resume proliferation after lesions are repaired. Genetic experiments conducted in the model eukaryotic yeast cells have found that checkpoints are also reversible even when DNA lesions are not repaired. By inducing a double-stranded break (DSB) to activate cell cycle checkpoints in yeast cells that cannot repair DSB, it was found that the repair-defective yeast cells broke through the checkpoints and resumed DNA replication despite the DSB in their DNA [13]. This process of overcoming checkpoints despite lesion-persistence is described as the adaption to DNA damage. In human cancer cells, Tp53 mutation weakens the checkpoints and can thus promote adaptation. Orthologs of Tp53 are not found in yeast, but the yeast genes required for adaptation are conserved through evolution [13]. Hence, DDA-resistant cancer cells could, beyond Tp53 mutation, additionally activate the evolutionarily conserved adaption genes to resume proliferation even when DNA lesions are not repaired.
Lesion-Bypass DNA Replication
In cells that have committed to DNA replication, DDA activates the S-phase checkpoint that inhibits the firing of replication origins, but only temporarily [53]. Thus, the commitment to replicating DNA is accompanied with ways to rapidly adapt to DNA lesions. One such way is to activate DNA polymerases that can synthesize DNA across lesions [70]. These lesion-bypass polymerases are conserved in prokaryotic and eukaryotic cells [70]. Although lesion-bypass DNA synthesis causes mutations, the evolutionary conservation of these enzymes suggests that error-prone replication is preferable to no replication. The human genome encodes over a dozen of lesion-bypass DNA polymerases, which cancer cells can explore to replicate damaged DNA. Such mutagenic DNA synthesis may further drive the emergence of DDA-resistant progenies.
Polyploidy
The bacteria Deinococcus radiodurans, named for extreme resistance to radiation, can withstand radiation doses that shatter their DNA into hundreds of fragments. By sequencing its genome, biologists found D. radiodurans to contain between 4 to 10 genome equivalent of sequences [71]. This redundancy in gene copies, together with a unique arrangement of these DNA [72] and non-unique but efficient repair machines [73], contribute to the ability of D. radiodurans to reassemble their genetic blueprint even after extensive breakage. One of the phenotypes of malignant cancer cells is aneuploidy, with losses and gains in chromosome copy numbers [74]. The lessons from D. radiodurans suggest that chromosome copy number gains may promote cancer resistance to radiation therapy. The idea that polyploidy can contribute to acquired radio-resistance in cancer is supported by experimental evidence [75,76]; however, the mechanisms that drive polyploidy-dependent resistance to radiation therapy have remained to be elucidated.
Conclusions and Outlooks
Our knowledge on the cell death response to DNA damage has advanced at a fast pace over the past few decades. Considering that pace, the current gaps in our knowledge on the life-sustaining capacity of DDR will certainly be filled in due course. The important question to ponder, at this time, is how we may apply the knowledge on DNA damage-induced cell death to efficiently and irreversibly kill off cancer cells.
Outlook-1: The field will continue to search for death-suppressors that are activated in recurring cancer cells to block DDA-induced death. Relevant to this line of investigation is the fact that execution of apoptosis or programmed necrosis requires leakage of killer-proteins or calcium ions from the outer or the inner mitochondrial membrane, respectively [38]. Although Tp53 is often mutated in cancer cells, PUMA, NOXA, and other mitochondrial killer-proteins are seldom mutated. Furthermore, cancer cells are dependent on mitochondrial metabolites to support nucleotide biosynthesis and proliferation [77]. Because tumor growth requires mitochondria, and because mitochondria execute death, it is reasonable to assume that mitochondrial protection is enhanced in DDA-resistant cancer cells. The best-known suppressors of mitochondria-dependent apoptosis are the BCL2-family of mitochondrial outer membrane protectors [78]. Drugs have been developed to neutralize the protective functions of BCL2-family members; however, disabling this family of death-suppressors has had limited success in reversing DDA-resistance [79]. Protection of the mitochondrial outer membrane, unfortunately, does not prevent mitochondria permeability transition (MPT), where the opening of the PTP (permeability transition pore) in the inner mitochondrial membrane causes massive leakage of calcium and other metabolites to kill cells [38]. After four decades of investigation, the molecular identify of PTP has remained mysterious [80], thus hindering the search for suppressors of MPT. While new insights on mitochondrial protection may reveal additional death suppressors in DDA-resistant cancer cells, we have to consider the possibility that non-proliferating neurons and cardiac muscle cells may also depend on those mitochondrial protectors to survive, as suggested by the fact that defects in mitochondrial quality control underline many forms of neural degenerative diseases [81]. Because DDA-resistant cancer cells may adopt mechanisms that protect neurons and cardiac muscles, there may exist a limit on how far we can push for mitochondria-dependent cell killing in cancer therapy.
Outlook-2: As an alternative to enforcing mitochondria-dependent cell killing, cancer researchers are exploiting the DDR defects in cancer cells to induce cancer-specific DNA-lesions that can activate mitochondria-independent death. Since the cell cycle checkpoints are weakened and the repair machines are compromised by DDR-defects in cancer, it is plausible to devise strategies, without using DDA, to selectively create death-inducing DNA lesions only in cancer cells [82]. As an example, cancer cells with defects in homologous recombination (HR) repair, due to mutations of BRCA1 or BRCA2, are hypersensitive to DDA [83]. Interestingly, BRCA1- or BRCA2-mutant cells are also sensitive to inhibitors of PARP (poly-ADP-ribose polymerase), which is a DNA-repair enzyme [84]. In patients with HR-defective cancers, PARP inhibitors have delayed cancer progression but they have not improved overall survival [85]. Nevertheless, the success with PARP inhibitors demonstrates that it is possible to design enzyme inhibitors to selectively cause DNA damage in repair-defective cancer cells [86]. Moving forward, it is likely that other enzyme inhibitors will be developed to irreversibly block the replication forks in DDR-defective cancer cells. By combining replication blockade with induction of chromatin condensation, it is possible to kill cancer cells through mitotic catastrophe without the need for apoptosis or necroptosis. Judging from the recent flurry of investigations on replication stress [87-89], we look forward to new insights on how to selectively activate replication stress and chromosome fragmentation in DDR-defective and DDA-resistant cancer cells.
Acknowledgments
Research in the author’s laboratory is supported by a grant (R01CA043054) from the National Cancer Institute, USA.
Glossary
- DDA
DNA damaging agents
- DDR
DNA damage response
- DSB
double-stranded break
- HR
homologous recombination
Author Contribution
JYJW reviewed the literature, wrote the article, designed and drew the Figures.
References
- Adelman R, Saul RL, Ames BN. Oxidative damage to DNA: relation to species metabolic rate and life span. Proc Natl Acad Sci USA. 1988;85(8):2706–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setlow RB. Cyclobutane-type pyrimidine dimers in polynucleotides. Science. 1966;153(3734):379–86. [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA. DNA repair and mutagenesis. 2nd ed. Washington, D.C.: ASM Press; 2006. xxix, 1118 p. p. [Google Scholar]
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28(5):739–45. [DOI] [PubMed] [Google Scholar]
- Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene. 2007;26(56):7773–9. [DOI] [PubMed] [Google Scholar]
- Lobrich M, Jeggo PA. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat Rev Cancer. 2007;7(11):861–9. [DOI] [PubMed] [Google Scholar]
- Jeggo PA, Pearl LH, Carr AM. DNA repair, genome stability and cancer: a historical perspective. Nat Rev Cancer. 2016;16(1):35–42. [DOI] [PubMed] [Google Scholar]
- Tubbs A, Nussenzweig A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell. 2017;168(4):644–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor GG. Rodent Cancer Treated with X Rays. BMJ. 1904;1(2260):946–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVita VT, Jr, Chu E. A history of cancer chemotherapy. Cancer Res. 2008;68(21):8643–53. [DOI] [PubMed] [Google Scholar]
- Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer. 2012;12(9):587–98. [DOI] [PubMed] [Google Scholar]
- Frei E., 3rd Curative cancer chemotherapy. Cancer Res. 1985;45(12 Pt 1):6523–37. [PubMed] [Google Scholar]
- Toczyski DP, Galgoczy DJ, Hartwell LH. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell. 1997;90(6):1097–106. [DOI] [PubMed] [Google Scholar]
- Galgoczy DJ, Toczyski DP. Checkpoint adaptation precedes spontaneous and damage-induced genomic instability in yeast. Mol Cell Biol. 2001;21(5):1710–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol. 2007;19(2):238–45. [DOI] [PubMed] [Google Scholar]
- Syljuasen RG. Checkpoint adaptation in human cells. Oncogene. 2007;26(40):5833–9. [DOI] [PubMed] [Google Scholar]
- Shaltiel IA, Krenning L, Bruinsma W, Medema RH. The same, only different - DNA damage checkpoints and their reversal throughout the cell cycle. J Cell Sci. 2015;128(4):607–20. [DOI] [PubMed] [Google Scholar]
- Gobe GC, Axelsen RA, Harmon BV, Allan DJ. Cell death by apoptosis following X-irradiation of the foetal and neonatal rat kidney. Int J Radiat Biol. 1988;54(4):567–76. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362(6423):847–9. [DOI] [PubMed] [Google Scholar]
- Wang JY, Cho SK. Coordination of repair, checkpoint, and cell death responses to DNA damage. Adv Protein Chem. 2004;69:101–35. [DOI] [PubMed] [Google Scholar]
- Borges HL, Linden R, Wang JY. DNA damage-induced cell death: lessons from the central nervous system. Cell Res. 2008;18(1):17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matt S, Hofmann TG. The DNA damage-induced cell death response: a roadmap to kill cancer cells. Cell Mol Life Sci. 2016;73(15):2829–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis RE, Yuan JY, Horvitz HR. Mechanisms and functions of cell death. Annu Rev Cell Biol. 1991;7:663–98. [DOI] [PubMed] [Google Scholar]
- Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011;30(18):3667–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermezovic J, Stergiou L, Hengartner MO, d’Adda di Fagagna F. Differential regulation of DNA damage response activation between somatic and germline cells in Caenorhabditis elegans. Cell Death Differ. 2012;19(11):1847–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges HL, Chao C, Xu Y, Linden R, Wang JY. Radiation-induced apoptosis in developing mouse retina exhibits dose-dependent requirement for ATM phosphorylation of p53. Cell Death Differ. 2004;11(5):494–502. [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Coulson EJ, Vaux DL. Inhibitor of apoptosis proteins and their relatives: IAPs and other BIRPs. Genome Biol. 2001;2(7):REVIEWS3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova EA, Chernov MV, Franks R, Wang K, Armin G, Zelnick CR, et al. Transgenic mice with p53-responsive lacZ: p53 activity varies dramatically during normal development and determines radiation and drug sensitivity in vivo. EMBO J. 1997;16(6):1391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11(10):700–14. [DOI] [PubMed] [Google Scholar]
- Friedberg EC. G. C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellengerberg, T. DNA Repair and Mutagenesis. 2nd ed. Washington (DC): ASM Press; 2006. 1118 pp. [Google Scholar]
- Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12(12):801–17. [DOI] [PubMed] [Google Scholar]
- Herzog KH, Chong MJ, Kapsetaki M, Morgan JI, McKinnon PJ. Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science. 1998;280(5366):1089–91. [DOI] [PubMed] [Google Scholar]
- Hollville E, Romero SE, Deshmukh M. Apoptotic Cell Death Regulation in Neurons. FEBS J. 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori C, Nakamura N, Kimura S, Irie H, Takigawa T, Shiota K. Programmed cell death in the interdigital tissue of the fetal mouse limb is apoptosis with DNA fragmentation. Anat Rec. 1995;242(1):103–10. [DOI] [PubMed] [Google Scholar]
- Owen JJ, Jenkinson EJ. Apoptosis and T-cell repertoire selection in the thymus. Ann N Y Acad Sci. 1992;663:305–10. [DOI] [PubMed] [Google Scholar]
- Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358(6382):167–9. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler M, Green DR. Mechanisms of p53-dependent apoptosis. Biochem Soc Trans. 2001;29(Pt 6):684–8. [DOI] [PubMed] [Google Scholar]
- Lakin ND, Jackson SP. Regulation of p53 in response to DNA damage. Oncogene. 1999;18(53):7644–55. [DOI] [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281(5383):1677–9. [DOI] [PubMed] [Google Scholar]
- Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell. 2017;66(6):801–17. [DOI] [PubMed] [Google Scholar]
- Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16(7):393–405. [DOI] [PubMed] [Google Scholar]
- Fischer M. Census and evaluation of p53 target genes. Oncogene. 2017;36(28):3943–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288(5468):1053–8. [DOI] [PubMed] [Google Scholar]
- Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7(3):673–82. [DOI] [PubMed] [Google Scholar]
- Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7(3):683–94. [DOI] [PubMed] [Google Scholar]
- Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302(5647):1036–8. [DOI] [PubMed] [Google Scholar]
- Michalak EM, Villunger A, Adams JM, Strasser A. In several cell types tumour suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ. 2008;15(6):1019–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JB, Hutt KJ, Michalak EM, Cook M, Vandenberg CJ, Liew SH, et al. DNA damage-induced primordial follicle oocyte apoptosis and loss of fertility require TAp63-mediated induction of Puma and Noxa. Mol Cell. 2012;48(3):343–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysko O, Aaes TL, Kagan VE, D’Herde K, Bachert C, Leybaert L, et al. Necroptotic cell death in anti-cancer therapy. Immunol Rev. 2017;280(1):207–19. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297(5579):259–63. [DOI] [PubMed] [Google Scholar]
- Yekezare M, Gomez-Gonzalez B, Diffley JF. Controlling DNA replication origins in response to DNA damage - inhibit globally, activate locally. J Cell Sci. 2013;126(Pt 6):1297–306. [DOI] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, Cimprich KA. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol. 2017;18(10):622–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15(17):2177–96. [DOI] [PubMed] [Google Scholar]
- Lanz MC, Dibitetto D, Smolka MB. DNA damage kinase signaling: checkpoint and repair at 30 years. EMBO J. 2019;•••:e101801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23(16):2825–37. [DOI] [PubMed] [Google Scholar]
- Canman CE. Replication checkpoint: preventing mitotic catastrophe. Curr Biol. 2001;11(4):R121–4. [DOI] [PubMed] [Google Scholar]
- Yazinski SA, Zou L. Functions, Regulation, and Therapeutic Implications of the ATR Checkpoint Pathway. Annu Rev Genet. 2016;50:155–73. [DOI] [PubMed] [Google Scholar]
- Latella L, Lukas J, Simone C, Puri PL, Bartek J. Differentiation-induced radioresistance in muscle cells. Mol Cell Biol. 2004;24(14):6350–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll B, Nelson G, Rabanal-Ruiz Y, Kucheryavenko O, Dunhill-Turner NA, Chesterman CC, et al. Persistent mTORC1 signaling in cell senescence results from defects in amino acid and growth factor sensing. J Cell Biol. 2017;216(7):1949–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson AB, Gottlieb RA. Bcl-2 family members and apoptosis, taken to heart. Am J Physiol Cell Physiol. 2007;292(1):C45–51. [DOI] [PubMed] [Google Scholar]
- Fricker M, Tolkovsky AM, Borutaite V, Coleman M, Brown GC. Neuronal Cell Death. Physiol Rev. 2018;98(2):813–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. 2016;22(2):175–82. [DOI] [PubMed] [Google Scholar]
- Waseem M, Kaushik P, Tabassum H, Parvez S. Role of Mitochondrial Mechanism in Chemotherapy-Induced Peripheral Neuropathy. Curr Drug Metab. 2018;19(1):47–54. [DOI] [PubMed] [Google Scholar]
- Piper S, McDonagh T. Heart failure and chemotherapeutic agents. Future Cardiol. 2015;11(4):453–70. [DOI] [PubMed] [Google Scholar]
- El-Deiry WS. p21(WAF1) Mediates Cell-Cycle Inhibition, Relevant to Cancer Suppression and Therapy. Cancer Res. 2016;76(18):5189–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner A, Bulyk ML, Jambhekar A, Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol. 2019;20(4):199–210. [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246(4930):629–34. [DOI] [PubMed] [Google Scholar]
- Yang W, Gao Y. Translesion and Repair DNA Polymerases: Diverse Structure and Mechanism. Annu Rev Biochem. 2018;87:239–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White O, Eisen JA, Heidelberg JF, Hickey EK, Peterson JD, Dodson RJ, et al. Genome sequence of the radioresistant bacterium Deinococcus radiodurans R1. Science. 1999;286(5444):1571–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin-Zaidman S, Englander J, Shimoni E, Sharma AK, Minton KW, Minsky A. Ringlike structure of the Deinococcus radiodurans genome: a key to radioresistance? Science. 2003;299(5604):254–6. [DOI] [PubMed] [Google Scholar]
- Zahradka K, Slade D, Bailone A, Sommer S, Averbeck D, Petranovic M, et al. Reassembly of shattered chromosomes in Deinococcus radiodurans. Nature. 2006;443(7111):569–73. [DOI] [PubMed] [Google Scholar]
- Tang YC, Amon A. Gene copy-number alterations: a cost-benefit analysis. Cell. 2013;152(3):394–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmukhanov SB, Yefimov ML, Kleinbock TS. Acquired radioresistance of tumour cells. Nature. 1967;216(5116):709–11. [DOI] [PubMed] [Google Scholar]
- Ivanov A, Cragg MS, Erenpreisa J, Emzinsh D, Lukman H, Illidge TM. Endopolyploid cells produced after severe genotoxic damage have the potential to repair DNA double strand breaks. J Cell Sci. 2003;116(Pt 20):4095–106. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between Metabolism and Cancer Biology. Cell. 2017;168(4):657–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton MI, Whysong D, O’Connor PM, Hockenbery D, Korsmeyer SJ, Kohn KW. Constitutive expression of human Bcl-2 modulates nitrogen mustard and camptothecin induced apoptosis. Cancer Res. 1993;53(8):1853–61. [PubMed] [Google Scholar]
- Delbridge AR, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016;16(2):99–109. [DOI] [PubMed] [Google Scholar]
- Baines CP, Gutierrez-Aguilar M. The still uncertain identity of the channel-forming unit(s) of the mitochondrial permeability transition pore. Cell Calcium. 2018;73:121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm A, Eckert A. Brain aging and neurodegeneration: from a mitochondrial point of view. J Neurochem. 2017;143(4):418–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilie PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol. 2019;16(2):81–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer. 2016;16(1):20–33. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21. [DOI] [PubMed] [Google Scholar]
- Ledermann JA. Front-line therapy of advanced ovarian cancer: new approaches. Ann Oncol. 2017;28(suppl_8):viii46-viii50. [DOI] [PubMed] [Google Scholar]
- Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA Repair in Cancer: beyond PARP Inhibitors. Cancer Discov. 2017;7(1):20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Techer H, Koundrioukoff S, Nicolas A, Debatisse M. The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat Rev Genet. 2017;18(9):535–50. [DOI] [PubMed] [Google Scholar]
- Rickman K, Smogorzewska A. Advances in understanding DNA processing and protection at stalled replication forks. J Cell Biol. 2019;218(4):1096–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley MP, Bocek M, Cimprich KA. R-Loops as Cellular Regulators and Genomic Threats. Mol Cell. 2019;73(3):398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]