

Summary
Cells have evolved complex mechanisms to maintain protein homeostasis, such as the UPRER, which are strongly associated with several diseases and the aging process. We performed a whole genome CRISPR-based knockout screen to identify genes important for cells to survive ER-based protein misfolding stress. We identified the cell-surface hyaluronidase, Transmembrane Protein 2 (TMEM2), as a potent modulator of ER-stress resistance. The breakdown of the glycosaminoglycan, hyaluronan (HA), by TMEM2 within the extracellular matrix (ECM) altered ER stress resistance independent of canonical UPRER pathways, but dependent upon the cell surface receptor, CD44, a putative HA receptor, and the MAPK cell-signaling components, ERK and p38. Lastly, and most surprisingly, ectopic expression of human TMEM2 in C. elegans protected animals from ER stress and increased both longevity and pathogen resistance independent of canonical UPRER activation, but dependent on the ERK ortholog, mpk-1, and the p38 ortholog, pmk-1.
ETOC
Intracellular ER stress resistance is impacted by changes in extracellular matrix metabolism
Graphical Abstract
Introduction
In order to ensure the integrity of the proteome, eukaryotic organisms evolved distinct subcellular stress response pathways, such as the unfolded protein response of the endoplasmic reticulum (UPRER), the mitochondrial unfolded protein response (UPRMT), and the cytosolic heat shock response (HSR). These pathways induce transcriptional programs that allow cells to adapt to subcellular stress and promote cell survival. However, under severe and unmitigated proteotoxic stress, they are also central in triggering cellular senescence or programmed cell death through apoptosis (Hetz, 2012; Walter and Ron, 2011). These challenges to the proteome can have a multitude of physiological and pathological causes. The ability to survive ER stress, for instance, is essential during the course of immune or inflammatory responses, during development, and cellular differentiation (Hetz, 2012; Wu and Kaufman, 2006). Additionally, ER stress is induced during intracellular pathogen replication, malignant cell growth, and the aging process(Hetz, 2009; Taylor and Dillin, 2013).
The induction of ER stress in both normal and pathophysiological contexts requires stress response pathways to respond appropriately and flexibly based on the circumstances involved: the cell type affected, the nature of the challenge, and the severity and persistence of the assault (Chen and Brandizzi, 2013; Hetz, 2012; Sano and Reed, 2013; Xu et al., 2005). Central to UPRER activation are ER-localized transmembrane proteins, IRE1, PERK1, and ATF6 (Gardner et al., 2013; Ron and Walter, 2007). These proteins serve as stress receptors, able to detect the load of unfolded proteins in the lumen of the organelle. In the presence of excessive levels of unfolded proteins, they signal to the nucleus to elicit a cellular response that results in a reduction in protein synthesis and expansion of the capacity of the ER. These changes are mediated through downstream signaling components, such as XBP1 or eIF2α. If there is no resolution to ER stress, the UPRER is also central to trigger cell death/senescence by influencing several MAPK signaling-mediated cell fate decisions (Darling and Cook, 2014; Hotamisligil and Davis, 2016). The MAPK-signaling components, p38, ERK, and JNK, integrate signals from the UPRER, from other subcellular stress response pathways, as well as other cell-signaling pathways, to initiate regulated cell death/senescence. This mechanism provides the cell with a certain flexibility in response to ER stress, allowing the modulation of cell fate decisions based on internal and extracellular cues. While much research has focused on the UPRER and its interaction with other stress response pathways, little is known how changes in the cellular microenvironment influence cell fate decisions or the aging process in the presence of ER stress. For example, it is not well understood how or which signals p38 and JNK receive to modulate ER stress responses.
Alongside a chronic activation of the UPRER, many pathologies present with significant changes in their cellular microenvironment, such as an altered glycosaminoglycan composition of the extracellular matrix (ECM). For example, an altered response to ER stress along with changes to the composition of the glycosaminoglycan, hyaluronan (HA), in the ECM has been observed in a variety of chronic-inflammatory and autoimmune diseases, in a subset of neurodegenerative diseases, and several malignancies (Brown and Naidoo, 2012; Chanmee et al., 2016; Hull et al., 2015; Majors et al., 2003; Nagy et al., 2015b, 2015a; Papy-Garcia et al., 2011). The polymer HA is a central component of the ECM and serves a variety of functions, such as basic structure, receptor protein attachment, and cell-to-cell communication (Cyphert et al., 2015; Laurent et al., 1996). The degradation of HA by enzymes, oxidative stress, and mechanical forces creates a continuum of different-sized HA-fragments ranging from several oligosaccharides to molecules of over 1 million Daltons in size. Different sizes of HA possess distinct biological effects and changes in the size distribution of HA in the ECM have been found to induce cell-signaling pathways (Cyphert et al., 2015). Recently, transmembrane protein 2 (TMEM2) has been identified as a cell-surface hyaluronidase (HAase) able to break down HMW-HA into low molecular weight HA (LMW-HA) (De Angelis et al., 2017; Yamamoto et al., 2017).
This complexity and significance of the cellular stress response pathways in the context of health and aging of an organism make protein quality control mechanisms central to our understanding of several diseases (Hetz, 2012; Wang and Kaufman, 2012). While these pathways have been extensively studied in yeast and other model organisms, studies in the mammalian background have been more limited (Adamson et al., 2016; Horlbeck et al., 2018; Wang et al., 2014). Moreover, large scale approaches almost always involved the use of cell lines derived from malignant cell growth, which often exhibit a severe dysregulation of stress response pathways, thus impeding our understanding of biological function under non-cancerous conditions (Hanahan and Weinberg, 2011; Hetz, 2009). We therefore turned to the karyotypical stable human fibroblast for our research. Within this experimental model, we used pooled CRISPR libraries to perform whole-genome functional knockout screens in order to identify novel candidate genes and pathways that influence the response of cells to ER stress. Once identified, we tested their relevance in-vivo using the nematode, C. elegans.
Results
We performed a whole-genome CRISPR knockout screen using the AVANA pooled sgRNA library, with the aim of identifying genes, that when inactivated, sensitized cells to ER stress (Figure 1A) (Doench et al., 2016; Shalem et al., 2014). Human immortalized fibroblasts were exposed to Tunicamycin, a drug that generates ER stress by inhibiting N-linked glycosylation, at a concentration that still maintained cell proliferation (Figure S1A). As non-transformed fibroblasts are not commonly used for CRISPR screens, we verified our experimental system by testing the extent of essential gene depletions compared to other published data sets, and found comparable quantitative depletion of essential genes (Figure S1B). Next, to identify gene knock-outs (KOs) that selectively sensitize cells to ER stress, we compared gene-based depletion p-values (see STAR methods) between the control and treatment arms and restricted our analysis to genes that do not show any depletion in the control arm (Figure 1B). This approach identified the main components of the UPRER, IRE1, XBP-1, and PERK, as some of the most selectively depleted genes. The approach was further validated by identifying the gene, MFSD2A, a putative transporter required for Tunicamycin entry into the cells, as the most significantly enriched genetic ablation in the presence of Tunicamycin (Reiling et al., 2011). A second independent screen replicate showed highly reproducible results (Figure S1C). We then combined the two replicates to produce a final list of genes, which are either selectively enriched or depleted in response to Tunicamycin induced ER stress (Supplemental Table 1). An enrichment analysis of the significantly depleted genes revealed functions associated with ER protein processing, peroxisome, and additional related pathways (Figure 1C) (Chen et al., 2013; Kuleshov et al., 2016).
Figure 1:

Whole genome CRISPR-KO library screen identifies TMEM2 as a potent modulator of ER stress sensitivity. A) Screen outline: Human immortalized fibroblasts were transduced with a genome-wide sgRNA lentiviral library and cultured for two weeks to maximize genome editing and target protein depletion. Cells were then split into control and Tunicamycin treatment and harvested after 3 weeks for sequencing (as described in detail in STAR METHODS) B) Comparison of gene depletion p-values between control and Tunicamycin-treated cells (individual depletion/enrichment are available in Supplemental Table 1) C) Enrichment analysis of the top differentially depleted genes (using 10% FDR as a cutoff) using the EnrichR online tool (https://amp.pharm.mssm.edu/Enrichr/). D) Viability and proliferation of Wildtype and clonal TMEM2-KO human immortalized fibroblast in the presence of Tunicamycin-induced ER stress with or without CMV-TMEM2 overexpression. Results are relative to untreated control to adjust for variability in initial cell number between cell lines. Cell density at the endpoint of a 5 day treatment period was determined via CellTiter-Glo analysis (CTG) (as described in detail in STAR METHODS); (n=3). Statistical Analysis: One-way ANOVA analysis with post-hoc Bonferroni-Holm analysis
Among the most significantly depleted genes, in addition to the known-members of the ER stress pathways, was Transmembrane Protein 2 (TMEM2) (Figure 1B). TMEM2 is localized to the plasma membrane, functions as a hyaluronidase within the ECM, and has not been previously implicated in ER stress, making it a prime candidate for further investigation.
TMEM2 is necessary and sufficient for regulating ER stress resistance
To verify the role of TMEM2 in ER stress regulation, we generated a clonal TMEM2-KO cell line (Figure S1D). TMEM2-KO cells had a significant decrease in resistance towards ER stress induced by either Tunicamycin or dithiothreitol (DTT) (Figure 1D, S1E). The re-introduction of Wildtype TMEM2 expressed under a strong constitutive promotor (Cytomegalovirus – CMV) was able to rescue the ER stress sensitive phenotype of the TMEM2 KO cells (Figure 1D). In addition, ectopic expression of TMEM2 in non-mutated, Wildtype cells also improved ER stress resistance (Figure 1D, S1E). We found no changes in cell proliferation in the absence of ER stress, resistance to mitochondrial stress (FCCP), actin destabilizing compounds (Cytochalasin D), and cytoplasmic protein misfolding (sodium arsenite) between Wildtype and TMEM2-KO cells (Figure S2A–D), suggesting that TMEM2’s role in ER stress was specific. More importantly, these results indicate that TMEM2 is both necessary for protection towards ER stress and sufficient for protection towards ER stress when overexpressed.
The hyaluronidase activity of TMEM2, and its products, are responsible for ER stress protection in human fibroblasts
TMEM2 functions as a cell-surface hyaluronidase (HAase) (De Angelis et al., 2017; Yamamoto et al., 2017). To determine if the enzymatic breakdown of HA is responsible for the change in ER stress resistance, we generated several CMV-TMEM2 expression vectors in which the hyaluronidase function of TMEM2 was disrupted. Cells expressing enzymatic dead versions of TMEM2 failed to rescue the increased sensitivity to ER stress of TMEM2-KO cells (Figure 2A). However, a construct carrying a neutral mutation was able to rescue the phenotype, similar to the functional CMV-TMEM2 overexpression construct. Moreover, supplementation of the growth media with HAase enzyme derived from Streptomyces hyalurolyticus was also sufficient to rescue the stress sensitivity phenotype of the TMEM2 KO cells to Wildtype levels and improved the resistance of Wildtype cells, phenocopying the effect of TMEM2 overexpression (Figure 2B).
Figure 2:

TMEM2’s enzymatic breakdown of HMW-HA to LMW-HA is responsible for the ER stress phenotype. A) The CMV-TMEM2 plasmid was altered through site-directed mutagenesis in order to disrupt HAase enzymatic activity of the gene. ER stress resistance was then measured through CTG analysis. The ER stress resistance was then compared between the constructs with no or a neutral mutation (ΔD275N) of the gene, and two lines in which the HAase function of TMEM2 was diminished (ΔP265C; ΔD273N); (n=3). B) Resistance to Tunicamycin-induced ER stress was measured in Wildtype and TMEM2-KO human fibroblasts in the presence and absence of supplemented hyaluronidase (HAase) (Concentration in bar graph: HAase 5U/ml). All HAase concentrations tested (0.6U/ml – 160U/ml) were equally able to evoke this phenotype. C-D) Wildtype and TMEM2-KO cells were exposed to low molecular weight hyaluronan (LMW-HA; <20kDa in molecular weight) medium molecular weight hyaluronan (MMW-HA; 200–1000kDa) or high molecular weight hyaluronan (HMW-HA; >1000kDa). The concentration of LMW-HA, MMW-HA and HMW-HA in the bar graphs was limited to 600ng/ml, since HMW-HA did not go fully into solution at higher concentrations (marked with #).
The HAase activity of TMEM2 is responsible for the breakdown of high molecular weight HA (HMW-HA – above 1000kDA) and moderate molecular weight HA (MMW – above 200kDA, below 1000kDa) to low molecular weight HA (LMW-HA – around 20kDa) in the ECM (De Angelis et al., 2017; Gialeli et al., 2014). Since different sizes of HA have been shown to possess distinct biological effects, we reasoned that the change in ER stress sensitivity of TMEM2 KO cells could be explained in two ways: either by the buildup of HA with a moderate to high molecular weight, or the lack of LMW-HA (Cyphert et al., 2015). In order to explore these two possibilities, we supplemented HA of varying sizes to the growth media. We found that LMW-HA, but not MMW-HA or HMW-HA, was able to rescue the Tunicamycin sensitivity of TMEM2-KO cells in a concentration dependent manner (Figure 2C–D). Therefore, the ER stress sensitivity in TMEM2-KO cells is not caused by a buildup of HMW-HA, but rather by the lack of LMW-HA products produced by the HAase activity of TMEM2.
TMEM2-mediated ER stress resistance is independent of the UPRER pathways
A central response and driver of ER stress resistance is the activation of one of the three branches of the UPRER regulated by PERK1, ATF6, or IRE1. In order to test if one of the three canonical UPRER branches was involved in the ER stress resistance conferred by TMEM2, we reduced the function of each branch of the UPRER and assessed ER stress resistance. We found that pharmacological inhibition of IRE1-dependent XBP1 splicing by the compounds, 4μ8C and STF-083010, did not impact the increased ER stress resistance observed in TMEM2 overexpressing cells, nor did it influence the decreased ER stress sensitivity in TMEM2-KO cells (Figure 3A, S3A). Similarly, we observed no changes to stress resistance in the presence of the PERK1 inhibitor, GSK-2656157, or the downstream eIF2a inhibitor, Salubrinal (Figure 3B, S3B). We next targeted the three canonical UPRER branches, PERK1, ATF6, and IRE1, through CRISPR-mediated KO. Mutations within PERK1, ATF6, or IRE1 were not able to alter the TMEM2-KO phenotype or influence the ability to respond to HAase supplementation (Figure 3C). Lastly, we investigated if TMEM2-KO human fibroblasts had an increased capacity to induce the UPRER when exposed to Tunicamycin-induced ER stress by performing RNAseq analysis. Compared to Wildtype cells, TMEM2-KO fibroblasts showed no significant difference in gene expression in response to ER stress both globally and specifically in UPRER target genes (Figure S4). Taken together, our data suggest that TMEM2 plays a unique role in ER stress resistance that is independent of the canonical pathways of PERK1, ATF6, and IRE1.
Figure 3:

The TMEM2 ER stress phenotype is independent the three canonical UPRER pathways. A-B) The resistance to Tunicamycin-induced ER stress of Wildtype, TMEM2-KO, and CMV-TMEM2-overexpressing cells was determined in the presence of A) an inhibitor of IRE1-mediated XBP1 splicing, 4μ8C (Concentration in bar graphs: 4μ8C 50μM; HAase 5U/ml) or B) an inhibitor of eIF2α phosphorylation, Salubrinal (Concentration in bar graphs: Salubrinal 200μM; HAase 5U/ml) through CTG analysis; (n=3). C) In Wildtype and TMEM2-KO cells, the UPRER pathway components IRE1, PERK1 and ATF6 were each targeted via CRISPR/CAS9-mediated gene disruption (see STAR METHODS for details). Each cell line was then cultured in the presence and absence of Tunicamycin (200ng/ml) and HAase (5U/ml) for 5 days, and the cell density at the endpoint of the experiment was measured by CTG analysis; (n=3). The additional graphs highlight the on the TMEM2-KO ER stress phenotype and the response to HAase due to the targeting of the UPRER pathways. Statistical Analysis: One-way ANOVA analysis with post-hoc Bonferroni-Holm analysis
TMEM2 mediates ER stress resistance through the CD44/ERK/p38 pathway
In the presence of severe or unmitigated ER stress, mitotic cells respond by inducing apoptosis or cellular senescence. Previous work identified three MAPK pathway components as central in cell fate decisions in the presence of ER stress: ERK, p38, and JNK (Darling and Cook, 2014; Sui et al., 2014). We therefore tested if any of these MAPK pathways were involved in the TMEM2-associated changes in ER stress resistance. For this, we exposed the cells to the ERK inhibitor SCH772984, the p38 inhibitor SB202190, or the JNK inhibitor SP600125, and measured their impact on the TMEM2-mediated ER stress phenotype. Inhibition of either p38 or ERK suppressed the ER stress resistance of TMEM2-overexpressing cells or cells treated with HAase to Wildtype levels (Figure 4A–B). In contrast, JNK inhibition had no effect (Figure 4C). While this set of inhibitors are commonly used in studying MAPK signaling, their specificity has been questioned (Bain et al., 2007, 2003). We therefore included an additional set of small molecule inhibitors for each pathway: DEL-22379 (ERK inhibitor), SB239063 (p38 inhibitor) and AEG 3482 (JNK inhibitor), which confirmed our original findings (Figure S5A–C).
Figure 4:

TMEM2 mediates ER stress resistance through the MAPK pathway components, ERK and p38, and the cell surface receptor CD44. ER stress resistance was measured in the presence of A) the ERK inhibitor SCH772984 (Concentration in bar graphs: SCH770984 5nM, HAase 5U/ml), B) the p38 MAPK pathway inhibitor SB202190 (Concentration in bar graphs: SB202190 10μM, HAase 5U/ml) or the C) JNK MAPK pathway inhibitor SP600125 (Concentration in bar graphs: SP600125 5μM, HAase 5U/ml); (n=3) D) In Wildtype and TMEM2-KO cells, the cell-surface receptors CD44, RHAMM, and ICAM-1 were targeted via CRISPR/CAS9-mediated gene disruption (see STAR METHODS for details). Each cell line was then cultured in the presence and absence of Tunicamycin (200ng/ml) and HAase. ER stress resistance was measured through CTG analysis (n=3). The additional graphs highlight the impact on the TMEM2-KO ER stress phenotype and the response to HAase due to the targeting of the receptors. Statistical Analysis: One-way ANOVA analysis with post-hoc Bonferroni-Holm analysis.
These results suggest that altered ER stress resistance in TMEM2 overexpressing cells is mediated through ERK and p38, but not JNK MAPK signaling. Intriguingly, similar to the overexpression phenotype, the decreased resistance of TMEM2-KO cells is dependent on ERK and p38, but not JNK signaling (Figure 4A–C). We therefore concluded that both the sensitivity to ER stress of TMEM2-KO cells and the ER stress resistance of TMEM2 overexpressing cells, are mediated through ERK/p38 MAPK signaling pathways.
Phenotypes associated with TMEM2 activity have been shown to depend upon the VEGF-VEGFR-ERK signaling pathway (De Angelis et al., 2017). However, neither VEGF supplementation (up to 200ng/ml) nor VEGFR receptor inhibition, using the compound SU5416, had an effect on ER stress resistance by either HAase supplementation or TMEM2 KO (Figure S3C–D). Besides VEGFR, LMW-HA fragments (around 5–20kDA) have been shown to interact with several other cell-surface receptors (De Angelis et al., 2017; Joy et al., 2018; Misra et al., 2015; Taylor et al., 2014; Tolg et al., 2014). Three of these have additionally been associated with changes to MAPK signaling: CD44, RHAMM, and ICAM-1 (Joy et al., 2018; Vigetti et al., 2014, 2008). We performed targeted disruption of the genomic locus of each of the receptors and measured potential changes to the ER stress resistance phenotype. We found no significant difference in response to RHAMM or ICAM-1 deletion. However, deletion of CD44 reduced the differential stress resistance we found in TMEM2-KO cells and greatly reduced the response of the TMEM2-KO cells to HAase treatment (Figure 4D). Therefore, CD44 appears to be the likely receptor responsible for the changes in ER stress resistance caused by changes to HAase activity, consistent with CD44 being a possible receptor for HA.
Ectopic expression of human TMEM2 in C. elegans results in increased lifespan, ER stress resistance and pathogen resistance
Altered ER stress resistance impacts longevity in several animal models, including the nematode, C. elegans. Specifically, an animal’s capacity to deal with ER stress decreases as a function of age, and hyperactivation of the ER stress response can ameliorate these defects, resulting in lifespan extension. The genetic activation of the UPRER through overexpression of the IRE1 pathway component, XBP-1s is able to increase the lifespan and resistance to proteotoxic stress in several model organisms, such as C. elegans and M. musculus (Taylor et al., 2014; Taylor and Dillin, 2013; Williams et al., 2014).
To determine whether TMEM2 can play a similar role in abrogating age-associated decline in an animal’s capacity to deal with ER stress, we used C. elegans as a model system to monitor the impact of TMEM2 expression upon aging. We introduced human TMEM2 (referred to as hTMEM2 in the context of C. elegans for clarity) into the worm using a pan-tissue promoter, sur-5p, and found that these animals were long-lived and protected from ER stress caused by Tunicamycin (Figure 5A, S6A). In an effort to determine whether hTMEM2 was necessary for these phenotypes in C. elegans, we identified the two closest nematode orthologues: R07C12.1, sharing a mere 30% identity based on amino acid sequence alignment, and chhy-1, the closest functional homologue (Csoka and Stern, 2013; Kaneiwa et al., 2008). We find that CRISPR-Cas9 knockout of R07C12.1 had no impact on longevity or stress resistance to Tunicamycin (Figure S6B). However, RNAi-knockdown of chhy-1 resulted in a significant decrease in lifespan and a very mild, but statistically significant, decrease in stress resistance to Tunicamycin, suggesting that there does exist some functional overlap between hTMEM2 and CHHY-1, and that chhy-1 is necessary for a normal lifespan in C. elegans (Figure S6C). Finally, to test whether hTMEM2-mediated lifespan extension and ER stress resistance is dependent on hTMEM2’s enzymatic function, we overexpressed an enzymatic dead version of hTMEM2 (R265C, D273N, D286N, referred to as hTMEM2-ED), and found that hTMEM2-ED was not sufficient to extend lifespan or promote ER stress resistance (Fig. 5B).
Figure 5:

hTMEM2 overexpression extends lifespan in C. elegans independent of canonical UPRER pathways. A) Lifespans were measured in Wildtype (N2) and sur-5p::hTMEM2 worms grown on EV RNAi on 1% DMSO and 25μg/ml Tunicamycin (Tm) from Day 1 (D1) as described in STAR METHODS. Data is representative of four independent trials. B) Lifespans were measured in Wildtype (N2), sur-5p::hTMEM2, and an enzymatic dead version of hTMEM2 (sur-5p::hTMEM2-ED, carrying R265C, D273N, D286N mutations; this overexpression line is an extrachromosomal array) using similar methods as A. Data is representative of three independent trials. C) Fluorescent micrographs of Wildtype (N2) and sur-5p::hTMEM2 animals expressing the UPRER reporter, hsp-4p::GFP. Animals were treated with DMSO or 25μg/ml Tunicamycin (Tm) at L4, and imaged at D1 as described in STAR METHODs. Data is representative of four independent trials. D) Lifespans were measured in Wildtype and sur-5p::hTMEM2 animals carrying either Wildtype alleles of xbp-1 and ire-1 or mutant alleles, xbp-1(zc12) or ire-1(v33), on EV RNAi. Data is representative of three independent trials. E) Lifespans were measured in Wildtype and sur-5p::hTMEM2 animals grown on EV, xbp-1, or ire-1 RNAi from hatch. Data is representative of three independent trials. F) Lifespans were measured in Wildtype and sur-5p::hTMEM2 animals grown on EV, atf-6, or pek-1 RNAi from hatch. Data is representative of two independent trials. All statistics for lifespans were performed using Log-Rank (Mantel-Cox) test using PRISM, and are available in Supplemental Table 2.
In our previous UPRER paradigm of longevity, xbp-1s overexpression in neurons (heretofore referred to as neuronal xbp-1s) was sufficient to induce UPRER in distal tissue and extend lifespan (Taylor and Dillin, 2013). Therefore, we tested whether ectopic expression of hTMEM2 affected lifespan by activation of the UPRER, similar to overexpression of xbp-1s. In contrast to our previous paradigm of longevity, overexpression of hTMEM2 throughout the animal did not activate the canonical UPRER in the absence of stress. However, it did result in increased UPRER induction in the presence of Tunicamycin (Figure 5C). To determine whether the physiological phenotypes of lifespan extension and ER stress resistance were dependent on canonical UPRER, similar to neuronal xbp-1s, we performed lifespan experiments in the presence of RNAi against the major regulators of the UPRER. We find that hTMEM2 extended lifespan in worms harboring null mutations or RNAi knockdown of either ire-1 or xbp-1 (Figure 5D–E). Similarly, hTMEM2 extended lifespan in worms with RNAi knockdown of pek-1 (the C. elegans ortholog of human PERK1) or atf-6, providing further evidence that the beneficial effects of hTMEM2 overexpression is not mediated through canonical UPRER (Figure 5F). Next, we tested the survival of animals with hTMEM2 overexpression when exposed to Tunicamycin at late age. Our previous work has reported that UPRER induction is lost at late age, resulting in increased sensitivity to ER stress (Taylor and Dillin, 2013). However, we find that hTMEM2 overexpressing animals still exhibit increased resistance to Tunicamycin even at late age when UPRER induction is completely abrogated (Figure S6D–E).
Next, we found that unlike the xbp-1s paradigm, neuronal overexpression of hTMEM2 alone was not sufficient to extend lifespan or increase resistance to ER stress (Figure S6F). Lastly, we compared the transcriptome of both strains using RNA-seq. Neuronal xbp-1s animals globally induce canonical UPRER genes, while hTMEM2 animals fail to do so. A closer look at specific targets of xbp-1s, such as crt-1, clearly show differences between these two lifespan extension paradigms (Figure S7). These data provide direct evidence that this longevity paradigm is distinct from previous ER stress response paradigm involving xbp-1s’ role in UPRER and further confirms our findings in human cells.
TMEM2 plays a potent role in innate immunity
During aging, C. elegans become increasingly susceptible to bacterial infection, which is considered an important cause of death of the nematodes in old age (Zhao et al., 2017). A decline in pmk-1/p38 activity, as well as a decrease in UPRER activation, are central in this age-associated decline in innate immunity (Youngman et al., 2011). Moreover, we found in our human studies that loss of either ERK or p38 suppressed the beneficial effects of TMEM2. Therefore, we tested what role, if any, the ERK/p38 homologues, mpk-1 or pmk-1, had on the beneficial physiological effects of hTMEM2 in C. elegans. Interestingly, we find that RNAi knockdown of mpk-1 or pmk-1 greatly suppressed the increased longevity of hTMEM2 overexpressing animals. Much like human cell studies, loss of jnk-1 had no effect on hTMEM2 overexpression (Figure 6A). To directly test whether hTMEM2 activates immune response, we determined whether hTMEM2 can induce a reporter for the PMK-1 transcriptional target, T24B8.5p::GFP (Shivers et al., 2009). Indeed, we find that hTMEM2 induces this immune response reporter in a pmk-1-dependent manner (Fig. 6B).
Figure 6:

hTMEM2 overexpression promotes immunity and extends lifespan through mpk-1/pmk-1. A) Lifespans were measured in Wildtype and sur-5p::hsf-1 animals on EV, jnk-1, mpk-1, or pmk-1 RNAi from hatch. Data is representative of three independent trials. B) Fluorescent micrographs of Wildtype (N2) and sur-5p::hTMEM2 animals expressing the immune response reporter, T24B8.5p::GFP. Animals were grown on EV or pmk-1 RNAi as described in STAR METHODS. Data is representative of three independent trials. C) Survival was scored in Wildtype (N2) and sur-5p::hTMEM2 animals exposed to PA14 infection at L4. Survival was scored every 6 hours as described in STAR METHODS. Data is representative of two independent trials. D) Lifespans were measured in Wildtype (N2) and sur-5p::hTMEM2 animals grown on dead EV RNAi from hatch. Bacteria were killed by UV irradiation, as described in STAR METHODS. All statistics for C-D were performed using Log-Rank (Mantel-Cox) test using PRISM, and are available in Supplemental Table 2. E) Wildtype and CMV-TMEM2 overexpressing human fibroblasts were exposed to lipopolysaccharides (LPS) derived from the E. coli bacteria strain (O111:B4). Resistance to the presence of LPS was measured through CTG to determine the cell density after 5 days of exposure; (n=3). Statistical analysis: One-way ANOVA test with a post-hoc Bonferroni-Holm analysis
Next, we tested what role, if any, hTMEM2 could play in the survival of animals to the natural challenges of bacterial pathogens. We exposed hTMEM2 animals to the pathogenic bacteria, Pseudomonas aeruginosa, and measured their survival compared to control animals. hTMEM2 animals had a significantly higher resistance to the pathogen compared to controls (Figure 6C). Furthermore, when the worms were grown on E. coli bacteria previously killed through UV exposure, thus preventing bacterial infection, the difference in lifespan due to hTMEM2 overexpression was lost (Figure 6D). Therefore, hTMEM2 plays an important role in the survival of cells and animals to pathogens.
In order to test if a similar change in the response to pathogen play a role in human fibroblasts overexpressing TMEM2, we exposed cells to lipopolysaccharides (LPS) derived from the enteropathogenic E. coli strain (O111:B4). We found that overexpressing TMEM2 in fibroblasts abrogated the detrimental impact of LPS exposure compared to Wildtype cells. The impact of LPS on both cell lines was entirely mediated through p38 MAPK signaling (Figure 6E).
Finally, we sought to determine whether the lifespan extension of neuronal xbp-1s animals was also dependent on mpk-1 and pmk-1 signaling, similar to hTMEM2 overexpression. In contrast to hTMEM2-mediated lifespan extension, lifespan extension by neuronal xbp-1s is completely independent of jnk-1, mpk-1, or pmk-1 (Figure 7A). Moreover, neuronal xbp-1s animals promotes lifespan extension when grown on bacteria previously killed through UV exposure (Figure 7B). Lastly, we find that neuronal xbp-1s and hTMEM2 overexpression exhibit synergistic effects when combined (Figure 7C–D). Taken together, these data provide further evidence that neuronal xbp-1s and hTMEM2 overexpression play independent and non-overlapping roles in lifespan extension where neuronal xbp-1s modulates canonical UPRER and hTMEM2 mediates innate immunity through mpk-1 and pmk-1.
Figure 7:

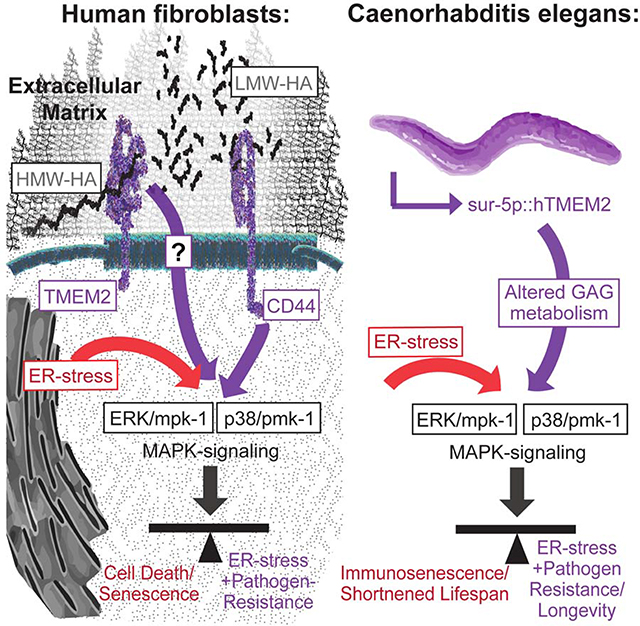
Lifespan extension through canonical xbp-1s signaling is not dependent on mpk-1/pmk-1 and is distinct from hTMEM2. A) Lifespans were measured in Wildtype (N2) and rab-3p::xbp-1s animals on EV, jnk-1, mpk-1, or pmk-1 RNAi from hatch. Data is representative of three independent trials. B) Lifespans were measured in Wildtype (N2) and rab-3p::xbp-1s animals grown on dead EV RNAi from hatch as per 5D. Data is representative of two independent trials. C-D) Lifespans were measured in Wildtype (N2), sur-5p::hTMEM2, rab-3p::xbp-1s, and sur-5p::hTMEM2/rab-3p::xbp-1s animals in the absence (C) and presence (D) of FUDR. Animals were grown on EV RNAi from hatch, and the assay was either performed on standard EV plates (C) or moved to FUDR containing plates at L4 for (D) - see STAR Methods for details. E) Graphical representation of the key insight generated by this work. In human fibroblasts, TMEM2 breaks down HMW-HA into LMW-HA within the ECM. Through interaction with CD44, LMW-HA influences ERK/mpk1- and p38/pmk-1-mediated MAPK-signaling. This in turn alters ER stress resistance and pathogen resistance in human fibroblasts. In C. elegans, TMEM2-mediated changes to glycosaminoglycan (GAG) metabolism cause a shift in MAPK signaling. This in turn alters the response to ER stress and with it, changes pathogen resistance and the lifespan of the animals.
Discussion
We performed a genome-wide knockout screen using pooled CRISPR libraries in karyotypical stable human fibroblasts to identify genes that mediate cell survival in the presence of ER-stress. Through this approach, we were able to establish Transmembrane Protein 2 (TMEM2), a cell-surface hyaluronidase, as a potent modulator of ER-stress resistance. TMEM2 activity is able to modulate ER stress resistance by altering the composition of the glycosaminoglycan hyaluronan in the extracellular matrix through enzymatic breakdown of HMW-HA to LMW-HA. The increase of the small metabolite LMW-HA ultimately alters the cell-fate decision in the presence of ER stress, through changes in p38/ERK MAPK signaling, mediated by the cell-surface receptor, CD44.
The ER stress response is thought to involve two stages. The first stage is characterized by a protective response that involves the expansion of the ER, induction of ER localized chaperones, and the reduction in protein load to the ER through decreased translation (Hetz, 2012; Wu and Kaufman, 2006). In the case of a persistent and unmitigated assault on the ER, the second phase involves the induction of apoptotic cell death or cellular senescence in mitotic cells (Chanmee et al., 2016; Chen and Brandizzi, 2013; Gerakis and Hetz, 2018; Hetz, 2012). Since the vast majority of cells in the nematode, C. elegans, are post-mitotic after development, the lifespan extension is likely to be independent of an adjustment of programmed cell-death. The central role of pmk-1/p38 activity in C. elegans involves innate immunity (Youngman et al., 2011). During aging, C. elegans exhibits tissue deterioration and an increased intestinal proliferation of bacteria, along with an increased susceptibility to bacterial infection. This susceptibility has been associated with a decline in pmk-1/p38 activity with increased age. The decline in innate immunity, generally referred to as innate immunosensescence, is considered an important cause of death of the nematodes in old age (Youngman et al., 2011; Zhao et al., 2017). Activation of ER stress responses have been shown to suppress immunity against bacterial pathogens and to contribute to immunosenescence (Singh and Aballay, 2006). The importance of cell survival under ER stress in the context of pathogen infection, is also supported by the results of the whole genome KO screen we performed in human fibroblast (Figure 1A–B). The enrichment analysis performed on the top, differentially deleted genes (sensitizers to ER stress), associated many of the genes as important factors in several different forms of pathogen infection.
The expression of hTMEM2 in C. elegans was able to significantly increase the lifespan of animals and resistance to ER stress. This lifespan extension seems to follow different rules than the previously characterized C. elegans ER-stress longevity model (Taylor and Dillin, 2013). Here, the selective expression of xbp-1s in neurons of the nematode, extended the lifespan in an xbp-1s and ire-1 dependent manner. Expression of xbp-1s throughout the animal did not impact longevity. With hTMEM2, expression restricted to neurons is insufficient for the lifespan extension; rather expression of hTMEM2 in all tissues is necessary to evoke the longevity phenotype. Furthermore, the lifespan extension is independent of xbp-1 and ire-1 in contrast to the xbp1s UPRER model. Lastly, in the UPRER longevity model, the expression of the ER localized chaperone, HSP-4, is significantly increased throughout the life of the animal. In the context of hTMEM2 expression, only changes to stress-induced hsp-4 were observed in early adulthood.
One of the most surprising findings of this work is that TMEM2 regulates ER stress resistance independent of the UPRER. This was found in several ways. First, overexpression of TMEM2 protected Wildtype human fibroblasts from ER stress in the presence of pharmacological inhibitors of XBP1, PERK1, and eIF2α. Second, genetic ablation of IRE1, PERK1, or ATF6 had no effect on the stress resistance phenotype mediated by TMEM2 in human cells or worms. Third, while overexpression of TMEM2 protected cells and worms from ER stress, it did not result in induction of the canonical UPRER target, HSPA5/hsp-4. Instead, TMEM2 links the small metabolite LMW-HA to CD44 and MAPK signaling through ERK and p38 signaling to protect cells from the damages of ER stress.
Why then has TMEM2 been missed in the plethora of discoveries surrounding ER stress resistance? The main reason for the lack of insight into TMEM2’s role in ER stress could be that most studies in this field have focused on the transcriptional output of the response, in which TMEM2 has no role in. Indeed, the RNA-sequencing analysis of TMEM2 KO cells exposed to Tunicamycin, showed little difference in the gene expression response compared to Wildtype cells (Figure S4A–D). This indicates that TMEM2-KO cells are perfectly capable of inducing the full extent of the UPRER.
Similarly, our RNA-sequencing data comparing hTMEM2 overexpressing animals and neuronal xbp-1s animals showed that, unlike neuronal xbp-1s animals, hTMEM2 overexpression does not activate canonical UPRER targets. Moreover, xbp-1s-mediated lifespan extension is independent of pmk-1/p38 and mpk-1/ERK. Finally, these two lifespan extension paradigms have synergistic effects, as animals with simultaneous neuronal xbp-1s and hTMEM2 overexpression, exhibit more than double the lifespan extension of either paradigm independently. Based on the results presented here, we propose that hTMEM2 overexpression in C. elegans, rather than by altering the UPRER directly, conveys longevity through changes to the pmk-1/p38 signaling pathway. This in turn alters how they adapt their cell fate in the presence of ER stress, ultimately prolonging the lifespan of the animals by delaying immunosenescence (Youngman et al., 2011). This suggests that rather than by altering the initial, protective stage of the response to ER stress, the longevity phenotype we observe in the presence of hTMEM2 overexpression would be due to changes of the second stage of the response, through the avoidance of the detrimental consequences of ER stress, specifically its impact on innate immunity.
The effects of hTMEM2 on C. elegans ER stress resistance and longevity are also surprising since the main structural glycosaminoglycan utilized by the animal is Chondroitin and the presence of HA is disputed (Csoka and Stern, 2013; Yamada et al., 2011). We provide evidence that hTMEM2’s enzymatic function is critical for its effect on ER stress resistance and longevity and that there is a striking conservation of the phenotypes and cellular signaling mechanisms involved across species. Furthermore, supplementation of HA to the animals caused a substantial developmental perturbation and alterations in sex determination in C. elegans (data not shown). While these results point to the role of HA metabolism in the nematode biology, alternative explanations are plausible. Most prominently, hTMEM2 could potentially serve as a Chondroitinase in the nematode. Supporting this reasoning is the structural similarity of Chondroitin and HA, the conservation of the functional domains of both enzyme families, and the previously observed considerable overlap of substrate specificities of both HAase and Chondroitinase enzymes (Csoka and Stern, 2013; Wang et al., 2017). Unfortunately, due to a range of experimental constraints, our attempts to distinguish between these two possibilities were not successful. We can therefore only come to the more general conclusion that the effects of hTMEM2 in C. elegans is likely due to changes in glycosaminoglycan metabolism. However, the impact of hTMEM2 expression on mpk-1/pmk-1 MAPK signaling solidify an important contribution for this enzyme and the downstream signaling pathway in ER stress resistance and longevity.
In human fibroblast, the results of our experiments strongly implicate a shift of HA metabolism as the driver of the TMEM2-mediated shift in ER stress resistance. HAase supplementation phenocopies the effect of TMEM2 overexpression while LMW-HA supplementation rescues defects in TMEM2-KO. Our data strongly implicate CD44/LMW-HA interaction as an important factor for the shift in ER stress resistance in human cells. These experiments nonetheless leave room for the possibility that CD44 is not the sole LMW-HA-receptor responsible, nor can they completely rule out a role for Chondroitin metabolism in the phenotypes we observed. Furthermore, since both Chondroitin and HA is thought to serves as an attachment point for a variety of cell-surface receptors, changes to glycosaminoglycan composition in the ECM might also impact receptor abundance on the cell surface more generally and additional confounding interactions are plausible.
There are several physiological conditions that are known to cause a degradation of glycosaminoglycans within the cellular microenvironment, including an exposure to oxidative stress, mechanical forces, and enzymatic breakdown through bacterial pathogens and leukocytes. The involvement of the ECM and cell surface receptors in the modulation of ER stress resistance we described here, would therefore allow the cell to integrate extracellular signals from the prevailing microenvironment to the response to intracellular protein-folding perturbations. The fragmentation of glycosaminoglycans in the ECM in this context can be seen as an extracellular cue that adjust the cell-fate decision of cells experiencing cellular stress, such as the presence of pathogens or the activation of an immune response.
An increase in ER stress, changes to the ECM, as well an altered MAPK-signaling have all been identified as characteristic cellular phenotypes in tissues undergoing age-associated decline (Brown and Naidoo, 2012; Kurz and Tan, 2004; Morawski et al., 2014; Robert and Labat-Robert, 2015; Tigges et al., 2014). Similarly, the pathology of multiple diseases, ranging from a subset of neurodegenerative diseases, autoimmune and inflammatory diseases, and several malignancies show similar characteristic pathophysiological changes to the ECM, MAPK signaling, and signs of a prolonged exposure to ER stress (Gerakis and Hetz, 2018; Kim and Choi, 2010; Morawski et al., 2014; Robert and Labat-Robert, 2015; Robertson, 2016; Sherman et al., 2015). In many of these diseases, age is an important risk factor. Taken together, a loosely framed network of TMEM2, HA, cell surface receptors, and p38/ERK can be created that could serve as an explanatory model in how these factors contribute to age-associated decline and disease etiology. Furthermore, our work introduces an additional mechanisms of how age-associated changes to ER stress levels and ECM composition, influence pathogenesis.
Besides an opportunity to illuminate new disease mechanisms, our work might serve to inform therapeutic interventions, be it through changes in ECM composition or cellular signaling. That a modification of this network is in principle possible, is suggested by experimental results in another longevity model organisms in which HA plays an important part, the Naked Mole Rat (NMR). The NMR is the longest living rodent species and seems almost completely resistant to cancer. This resistance is thought to be caused by a high abundance of HA with up to 5x higher molecular weight, partly due to an increase in HA synthesis through HAS2 (HA synthase) and due to lowered activity of HA enzymatic breakdown (HYAL2). The abundance and composition of HA seems to be at the root of the resistance to malignant transformation, since either knocking down HA synthesis, or overexpression of the HA-degrading enzyme, HYAL2, caused NMR fibroblast to become susceptible to malignant transformation (Tian et al., 2013). These results, along with the insights presented here, strongly suggest that at least part of the cancer resistance is due to a decreased resistance to ER stress. This line of reasoning is supported by the observation that fibroblasts from NMR show an increase in ER stress sensitivity, further highlighting the interconnection of ER stress resistance to the abundance and enzymatic breakdown of HA (Salmon et al., 2008). However, in contrast to our hTMEM2-overexpression model in C. elegans, the NMR is long-lived while at the same time exhibiting generally reduced HAase activity. This might be explained by the observation that cells of the NMR are more responsive to some types of HA signaling, compared to other mammalian cells (Tigges et al., 2014). Understanding the unique molecular mechanism underlying the different animal and human cell-culture models could therefore provide the opportunity to manipulate these pathways independently, thereby guiding the development of novel therapeutic interventions.
Lastly, a cautionary note. Hyaluronan is increasingly used as a growth matrix to grow cells in-vitro, and HAase enzymatic breakdown of the matrix is utilized to dissociate the cells from the culture dish. We would like to point out that the biological activity of the HA fragments generated in this process might interfere with experimental results, especially in the context of studying cellular stress.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
All strains and cell lines used in this study are available by direct request to the lead contact. Raw sequencing data are available in the following formats: C. elegans RNA-seq raw data is available at https://data.mendeley.com/datasets/dy97pwyf74/1. All human RNA-seq and screening raw data is available at http://dx.doi.org/10.17632/tmtkc8gcs8.1. Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Andrew Dillin (dillin@berkeley.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
For C. elegans work, all strains used are derivatives of N2 from Canorhabdities Genetics Center and specific genotypes of all strains used in this study are available in Key Resources Table. All worms are hermaphrodites for all studies. Specific growth conditions are specific under each experimental method detailed below. General growth and maintenance is as follows:
Key Resources Table.
| ANTIBODIES | ||
| None used | ||
| BACTERIAL AND VIRUS STRAINS | ||
| OP50 | CGC | N/A |
| HT115 | CGC | N/A |
| PA14 | CGC | N/A |
| DH5α | Invitrogen | 18258012 |
| Stbl3 | Thermo Fisher | C7373–03 |
| BIOLOGICAL SAMPLES | ||
| None used | ||
| CHEMICALS, PEPTIDES, AND RECOMBINANT PROTEINS | ||
| 4μ8C | EMD Millipore | 412512 |
| (+)-5-Fluorodeoxyuridine (FUDR) | Spectrum Chemical | 50–91–9 |
| A-(2→3.6.8.9) Neuraminidase | Sigma-Aldrich | N8271 |
| AEG 3482 | TOCRIS | 2651 |
| Agarose, low melting | Sigma-Aldrich | A9414–10G |
| Ammonium Acetate | Sigma-Aldrich | A1542 |
| Bacto Peptone | Fisher Scientific | DF0118072 |
| BD Difco granulated agar | VWR | 90000–782 |
| Blasticidin S HCL | Thermo Fisher | A11139–03 |
| BODIPY 493/503 | Thermo Fisher | D3922 |
| Calcium chloride dehydrate | VWR | 97061–904 |
| Carbenicillin | BioPioneer | C0051–25 |
| Cholesterol | Sigma-Aldrich | 57–88–5 |
| Cytochalasin D | Cayman Chemical | 11330 |
| DEL 22379 | TOCRIS | 5774 |
| DMEM media | Thermo Fisher | 11995–073 |
| DNase I | New England Biolabs | M03035 |
| DTT | Sigma Aldrich | C2920 |
| FBS, Premium Grade 500 ml (Lot# 190B14) | VWR | 97068–091 |
| FCCP | Cayman Chemical | CAS 370–86–5 |
| Gentamicin | VWR | 17–5182 |
| GlutaMAX supplement | Thermo Fisher | 35050–061 |
| GSK2606414 | Cayman Chemical | 17376 |
| Herculase II Fusion DNA Polymerase | Agilent | 600677 |
| Hyaluronan, various sizes | R&D Systems | GLR001 |
| Hyaluronidase, sheep | Abcam | ab208484 |
| IPTG dioxane free | Denville Scientific | CI8280–4 |
| Lipofectamine 2000 | Thermo Fisher | 1–3603 |
| LB Broth Miller | Fisher Scientific | BP1426500 |
| LPS (O55:B5) | Sigma Aldrich | L2880 |
| Magnesium sulfate heptahydrate | VWR | EM-MX0070–3 |
| Non-essential Amino Acids solution | Thermo Fisher | 11140–050 |
| Opti-MEM | Thermo Fisher | 4916 |
| PBS, pH 7.4 | Thermo Fisher | 10010–049 |
| Penicillin/Streptomycin solution | Thermo Fisher | 15070–063 |
| Polybrene | Fisher Scientific | TR-1003-G |
| Potassium Chloride | Fisher | P217–500 |
| Potassium phosphate dibasic | VWR | EM-PX1570–2 |
| Potassium phosphate monobasic | VWR | EM-PX1565–5 |
| Proteinase K | QIAGEN | 19131 |
| Puromycin dihydrochloride | Thermo Fisher | A11138–03 |
| RNase A | QIAGEN | 19101 |
| Rifampicin | Fisher | BP2679250 |
| Salubrinal | Chem Scene | CS-1012 |
| SB 202190 | Sigma Aldrich | S7067 |
| SB 239062 | TOCRIS | 1962 |
| SCH772984 | Selleck Chemicals | S7101 |
| Sodium Arsenite | Santa Cruz Biotechnology | Sc-301816 |
| Sodium Azide | Sigma-Aldrich | 71289–50G |
| Sodium Chloride | EMD Millipore | SX0420–5 |
| Sodium phosphate dibasic | VWR | 71003–472 |
| Sodium phosphate monobasic monohydrate | Sigma-Aldrich | S9638–1KG |
| SP600125 | Sigma-Aldrich | S5567 |
| STF-083010 | Cayman Chemical | 17370 |
| SU 5413 | Cayman Chemical | 13342 |
| TE buffer | Sigma-Aldrich | T9285 |
| Tetracycline hydrochloride | Sigma-Aldrich | T7660–5G |
| Trypsin-EDTA | Thermo Fisher | 25200–072 |
| Tunicamycin | Sigma-Aldrich | T7765–50MG |
| VEGF Recombinant Human Protein | Thermo Fisher | PHC9394 |
| CRITICAL COMMERCIAL ASSAYS | ||
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | G7571 |
| HA ELISA kit :: Human Hyaluronic Acid | MyBioSource | MBS262948 |
| Pierce™ Detergent Compatible Bradford Assay Kit | Fisher Scientific | 23246 |
| QIAquick Gel Extraction Kit | Qiagen | 28706 |
| QIAquick PCR Purification Kit | Qiagen | 28106 |
| Qiagen RNeasy Mini Kit | Qiagen | 74106 |
| QIAprep Spin Miniprep Kit | Qiagen | 27106 |
| DEPOSITED DATA | ||
| C. elegans RNAseq | Mendeley | https://data.mendeley.com/datasets/dy97pwyf74/1 |
| Human RNAseq & Screen Data | - | http://dx.doi.org/10.17632/tmtkc8gcs8.1. |
| EXPERIMENTAL MODELS: CELL LINES | ||
| HEK293T | ATCC | CRL-11268 |
| Human foreskin fibroblast, BJ | ATCC | CRL-2522 |
| Human BJ fibroblast, pLentiCas9-Blast; pSGR119puro TMEM2.1 polyclonal | This study | N/A |
| Human BJ fibroblast, pLentiCas9-Blast; pSGR120puro TMEM2.2 polyclonal | This study | N/A |
| Human BJ fibroblast, pLentiCas9-Blast; pSGR121puro TMEM2.3 polyclonal | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal: pLentiCas9-Blast; pSGR121puro clonal expansion | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGRscramble.1gent | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGRscramble.2gent | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGRscramble.3gent | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGR49gent ERN1.1 | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGR50gent ERN1.2 | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGR51gent ERN1.3 | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGR52gent EIF2AK3.1 | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGR53gent EIF2AK3.2 | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGR54gent EIF2AK3.3 | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGR58gent ATF6.1 | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGR59gent ATF6.2 | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGR60gent ATF6.3 | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGR199gent CD44.1 | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGR200gent CD44.2 | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGR201gent CD44.3 | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGR202gent RHAMM.1 | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGR203gent RHAMM.2 | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGR204gent RHAMM.3 | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGR205gent ICAM.1 | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGR206gent ICAM.2 | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal + sSGR207gent ICAM.3 | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal+CMV-TMEM2gent | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal+CMV-TMEM2 ΔP265Cgent | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal+CMV-TMEM2 ΔD273Ngent | This study | N/A |
| Human BJ fibroblast, TMEM2-KO clonal+CMV-TMEM2 ΔD275Ngent | This study | N/A |
| EXPERIMENTAL MODELS: ORGANISMS/STRAINS | ||
| C. elegans: Bristol (N2) strain as wild type (WT) | CGC | N2 |
| C. elegans: SJ4005: zcls4(hsp-4p::GFP)V | (Taylor and Dillin, 2013) | SJ4005 |
| C. elegans: RE666: ire-1(v33)II | CGC | RE666 |
| C. elegans: AU78: agIs219 [T24B8.5p::GFP::unc-54–3′ UTR + ttx-3p::GFP::unc-54–3′ UTR] III | CGC | AU78 |
| C. elegans:AGD1049: xbp-1(zc12) III | (Taylor and Dillin, 2013) | N/A |
| C. elegans:AGD1952; N2, uthIs485(sur-5p::hTMEM2::unc-54 UTR, myo-2p::tdtomato) strain C4a | This study (Taylor and Dillin, 2013) | N/A |
| C. elegans:AGD1953; N2, uthIs486(sur-5p::hTMEM2::unc-54 UTR, myo-2p::tdtomato) strain G4e | This study | N/A |
| C. elegans: AGD1940: uthEx847(sur-5p::TMEM2::unc-54 UTR; myo-2p::tdtomato); zcIs4[hsp-4p::GFP] | This study | N/A |
| C. elegans: AGD1961; N2, uthEx852(rgef-1p::hTMEM::unc-54 UTR, myo-2p::tdtomato) | This study | N/A |
| C. elegans: AGD2004: N2, uthIs486(sur-5p::hTMEM::unc-54 UTR, myo-2p::tdtomato) strain G4e; xbp-1(zc12)III | This study | N/A |
| C. elegans: AGD2005: N2, uthIs486(sur-5p::hTMEM::unc-54 UTR, myo-2p::tdtomato) strain G4e; ire-1(v33)II | This study | N/A |
| C. elegans: AGD2121: N2, uthEx870(vha-6p::hTMEM2::unc-54 UTR; myo-2p::tdtomato) | This study | N/A |
| C. elegans: AGD2343: N2, uthIs486(sur-5p::hTMEM::unc-54 UTR, myo-2p::tdTomato) strain G4e; uthIs270[rab-3p::xbp-1s, myo-2p::tdTomato] | This study | N/A |
| C. elegans: AGD2357: N2, uthIs486(sur-5p::hTMEM::unc-54 UTR, myo-2p::tdTomato) strain G4e; zcls4[hsp-4p::GFP]V | This study | N/A |
| C. elegans: AGD2416: N2, agIs219 [T24B8.5p::GFP::unc-54–3′ UTR + ttx-3p::GFP::unc-54–3′ UTR] III; uthIs486(sur-5p::hTMEM::unc-54 UTR, myo-2p::tdt) | This study | N/A |
| C. elegans: AGD2484: N2, uthEx924(sur-5p::hTMEM2 (R265C, D273N, D286N)::unc-54 UTR, myo-2p::tdt) | This study | N/A |
| OLIGONUCLEOTIDES – ALL OLIGOS ORDERED FROM IDT – Please see Table S3 | ||
| RECOMBINANT DNA | ||
| pCMV-TMEM2gent: pCDH-CMV-TMEM2-MCS-EF1a-Gentamicin | This study | N/A |
| pCMV-TMEM2ΔP265Cgent: pCDH-CMV-TMEM2ΔP265C-MCS-EF1a-Gentamicin | This study | N/A |
| pCMV-TMEM2ΔD273Ngent: pCDH-CMV-TMEM2ΔD273N-MCS-EF1a-Gentamicin | This study | N/A |
| pCMV-TMEM2ΔD275Ngent: pCDH-CMV-TMEM2ΔD275N-MCS-EF1a-Gentamicin | This study | N/A |
| pEK2: myo-2p::tdtomato::unc-54 3’ UTR | This study | N/A |
| pFUDW LentiCas9-Blast | Addgene | 52962 |
| pRHS47: sur-5p::hTMEM2::unc-54 3’ UTR | This study | N/A |
| pRHS48: rgef-1p::hTMEM2::unc-54 3’ UTR | This study | N/A |
| pRHS49: vha-6p::hTMEM2::unc-54 3’ UTR | This study | N/A |
| pRHS54 sur-5p::hTMEM2(R265C, D273N, D286N)::unc-54 3’ UTR | This study | N/A |
| pSGRscramble.1puro; pHKO_42-pLKO-(N)_10-U6-sgScramble.1-EF1a-Puro | This study | N/A |
| pSGRscramble.1gent; pPF12-pLKO-(N)_10-U6-sgScramble.1-EF1a-Gentamycin | This study | N/A |
| pSGRscramble.2puro; pHKO_42-pLKO-(N)_10-U6-sgScramble.2-EF1a-Puro | This study | N/A |
| pSGRscramble.2gent; pPF12-pLKO-(N)_10-U6-sgScramble.2-EF1a-Gentamycin | This study | N/A |
| pSGRscramble.3puro; pHKO_42-pLKO-(N)_10-U6-sgScramble.3-EF1a-Puro | This study | N/A |
| pSGRscramble.3gent; pPF12-pLKO-(N)_10-U6-sgScramble.1-EF1a-Gentamycin | This study | N/A |
| pSGR49puro; pHKO_42-pLKO-(N)_10-U6-sgERN1.1-EF1a-Puro | This study | N/A |
| pSGR49gent; pPF12-pLKO-(N)_10-U6-sgERN1.1-EF1a-Gentamycin | This study | N/A |
| pSGR50puro; pHKO_42-pLKO-(N)_10-U6-sgERN1.2-EF1a-Puro | This study | N/A |
| pSGR50gent; pPF12-pLKO-(N)_10-U6-sgERN1.2-EF1a-Gentamycin | This study | N/A |
| pSGR51puro; pHKO_42-pLKO-(N)_10-U6-sgERN1.3-EF1a-Puro | This study | N/A |
| pSGR51gent; pPF12-pLKO-(N)_10-U6-sgERN1.3-EF1a-Gentamycin | This study | N/A |
| pSGR52puro; pHKO_42-pLKO-(N)_10-U6-sgEIF2AK3.1-EF1a-Puro | This study | N/A |
| pSGR52gent; pPF12-pLKO-(N)_10-U6-sgEIF2AK3.1-EF1a-Gentamycin | This study | N/A |
| pSGR53puro; pHKO_42-pLKO-(N)_10-U6-sgEIF2AK3.2-EF1a-Puro | This study | N/A |
| pSGR53gent; pPF12-pLKO-(N)_10-U6-sgEIF2AK3.2-EF1a-Gentamycin | This study | N/A |
| pSGR54puro; pHKO_42-pLKO-(N)_10-U6-sgEIF2AK3.3-EF1a-Puro | This study | N/A |
| pSGR54gent; pPF12-pLKO-(N)_10-U6-sgEIF2AK3.3-EF1a-Gentamycin | This study | N/A |
| pSGR58puro; pHKO_42-pLKO-(N)_10-U6-sgATF6.1-EF1a-Puro | This study | N/A |
| pSGR58gent; pPF12-pLKO-(N)_10-U6-sgATF6.1-EF1a-Gentamycin | This study | N/A |
| pSGR59puro; pHKO_42-pLKO-(N)_10-U6-sgATF6.2-EF1a-Puro | This study | N/A |
| pSGR59gent; pPF12-pLKO-(N)_10-U6-sgATF6.2-EF1a-Gentamycin | This study | N/A |
| pSGR60puro; pHKO_42-pLKO-(N)_10-U6-sgATF6.3-EF1a-Puro | This study | N/A |
| pSGR119gent; pPF12-pLKO-(N)_10-U6-sgTMEM2.1-EF1a-Gentamycin | This study | N/A |
| pSGR119puro; pHKO_42-pLKO-(N)_10-U6-sgTMEM2.1-EF1a-Puro | This study | N/A |
| pSGR120gent; pPF12-pLKO-(N)_10-U6-sgTMEM2.2-EF1a-Gentamycin | This study | N/A |
| pSGR120puro; pHKO_42-pLKO-(N)_10-U6-sgTMEM2.2-EF1a-Puro | This study | N/A |
| pSGR121gent; pPF12-pLKO-(N)_10-U6-sgTMEM2.3-EF1a-Gentamycin | This study | N/A |
| pSGR121puro; pHKO_42-pLKO-(N)_10-U6-sgTMEM2.3-EF1a-Puro | This study | N/A |
| pSGR60gent; pPF12-pLKO-(N)_10-U6-sgATF6.3-EF1a-Gentamycin | This study | N/A |
| pSGR199puro; pHKO_42-pLKO-(N)_10-U6-sgCD44.1-EF1a-Puro | This study | N/A |
| pSGR199gent; pPF12-pLKO-(N)_10-U6-sgCD44.1-EF1a-Gentamycin | This study | N/A |
| pSGR199puro; pHKO_42-pLKO-(N)_10-U6-sgCD44.1-EF1a-Puro | This study | N/A |
| pSGR199gent; pPF12-pLKO-(N)_10-U6-sgCD44.1-EF1a-Gentamycin | This study | N/A |
| pSGR200puro; pHKO_42-pLKO-(N)_10-U6-sgCD44.2-EF1a-Puro | This study | N/A |
| pSGR200gent; pPF12-pLKO-(N)_10-U6-sgCD44.2-EF1a-Gentamycin | This study | N/A |
| pSGR201puro; pHKO_42-pLKO-(N)_10-U6-sgCD44.3-EF1a-Puro | This study | N/A |
| pSGR201gent; pPF12-pLKO-(N)_10-U6-sgCD44.3-EF1a-Gentamycin | This study | N/A |
| pSGR202puro; pHKO_42-pLKO-(N)_10-U6-sgRHAMM.1-EF1a-Puro | This study | N/A |
| pSGR202gent; pPF12-pLKO-(N)_10-U6-sgRHAMM.1-EF1a-Gentamycin | This study | N/A |
| pSGR203puro; pHKO_42-pLKO-(N)_10-U6-sgRHAMM.2-EF1a-Puro | This study | N/A |
| pSGR203gent; pPF12-pLKO-(N)_10-U6-sgRHAMM.2-EF1a-Gentamycin | This study | N/A |
| pSGR204puro; pHKO_42-pLKO-(N)_10-U6-sgRHAMM.3-EF1a-Puro | This study | N/A |
| pSGR204gent; pPF12-pLKO-(N)_10-U6-sgRHAMM.3-EF1a-Gentamycin | This study | N/A |
| pSGR205puro; pHKO_42-pLKO-(N)_10-U6-sgICAM-1.1-EF1a-Puro | This study | N/A |
| pSGR205gent; pPF12-pLKO-(N)_10-U6-sgICAM-1.1-EF1a-Gentamycin | This study | N/A |
| pSGR206puro; pHKO_42-pLKO-(N)_10-U6-sgICAM-1.2-EF1a-Puro | This study | N/A |
| pSGR206gent; pPF12-pLKO-(N)_10-U6-sgICAM-1.2-EF1a-Gentamycin | This study | N/A |
| pSGR207puro; pHKO_42-pLKO-(N)_10-U6-sgICAM-1.3-EF1a-Puro | This study | N/A |
| pSGR207gent; pPF12-pLKO-(N)_10-U6-sgICAM-1.3-EF1a-Gentamycin | This study | N/A |
| SOFTWARE AND ALGORITHMS | ||
| ImageJ | NIH | N/A- download available from imagej.nih.gov/ij/ |
| Prism7 | GraphPad | N/A |
| GOrilla | http://cbl-gorilla.cs.technion.ac.il/ | |
C. elegans growth and maintenance
All C. elegans strains used are derivatives of N2 from Caenorhabditis Genetics Center (CGC) and are listed in Key Resources Table. All worms are grown on NGM agar plates fed OP50 E. coli bacteria at 15°C. For experimentation, worms are s ynchronized via bleaching using a standard bleach solution (1.8% sodium hypochlorite, 0.375M KOH) until all carcasses are degraded and only eggs remain. The eggs are then washed 4x with M9 solution (22mM KH2PO4 monobasic, 42.3mM Na2HPO4, 85.6mM NaCl, 1mM MgSO4) and spun at 20°C in M9 solution until eggs hatch and L1 worms are arrested (~12–16hours). L1s are then plated onto NGM agar plates containing μM IPTG, 100μg/ml carbenicillin, and 10μg/ml tetracycline and kept at 20°C until the desired stage. All experimental worms are fed HT115 E. coli bacteria harboring either empty vector (EV – pL4440) plasmids or pL4440 plasmids expressing double-stranded RNA containing the sequence of the target gene. All RNAi vectors were isolated from the Vidal libraries and sequence-verified prior to use.
All transgenic worms were synthesized by injecting N2 worms with plasmids listed in Key Resources Table at 25μg/ml with co-injection marker pEK2 (myo-2p::tdtomato) at 2.5μg/ml and 100μg/ml of pD64 vehicle as filler DNA. Worms positive for myo-2p::tdtomato were selected for stable arrays. Integration of sur5p::hTMEM2 worms was performed by gamma irradiation. L4 worms were irradiated with 4400 rems of radiation and integrants were identified by selectin animals that maintained 100% myo-2p::tdtomato at 100% frequency past the F3 generation. Three independent lines were isolated, backcrossed to N2 animals 8x to eliminate mutations, and animals with the most similar phenotypes to the array animals were used for the experiment.
Cell culture and maintenance of human fibroblasts
We used the human foreskin fibroblast line BJ ATCC® CRL-2522™ (BJ fibroblasts). The cells were cultured at 37°C, 95% air and 5% CO 2 in a humidified incubator on gelatin-coated dishes in medium containing DMEM, 15% Fetal Bovine Serum (FBS), 1% Glutamax, 1% Non-Essential Amino Acids (NEAA) and 1% Penicillin/Streptomycin. When cells reached confluence, the cells were washed with PBS, trypsinized and replated in a 1:3 to 1:6 split ratio, or used for experimental purposes. Media was replaced every other day, if necessary cells were frozen in maintenance media +10% DMSO. Human fibroblast cells are expressing Cas9 and hTERT in all experiments performed and are labelled as Wildtype.
Cell Counting
Cells were counted using Countess FL II automated cell counter (Thermo Fisher; AMQAF1000) in 4x replicates (n=4) per cell line.
METHOD DETAILS
C. elegans induction of ER stress and immune response reporter
Animals were synchronized to the L4 stage and treated with 25μg/ml Tunicamycin in M9 buffer for 4 hours spinning at 20°C. Control animal s were treated with an equal concentration of DMSO vehicle in M9 buffer (1% for concentrations used in this study). After four hour incubation, animals were washed with M9 buffer 2x, then plated onto OP50 plates overnight (~16 hours) at 20°C to recove r and allow for hsp-4p::GFP expression. Animals were picked at random (under white light) from a population and immobilized in 100nM sodium azide and lined up with pharynx facing up on an NGM plate. Worms were imaged using a Leica M250FA automated fluorescent stereomicroscope equipped with a Hamamatsu ORCA-ER camera.
For aging experiments, age-synchronized animals were grown on EV RNAi from hatch at 20 °C and manually moved away from progeny onto new RNAi plates, similar to lifespans. Worms were moved onto 1% DMSO or 25μg/ml Tunicamycin plates containing EV RNAi 16 hours prior to imaging (L4 for D1 imaging, D3 for D4 imaging, etc.). Imaging was performed similar to above.
For the immune response reporter, transgenic animals carrying T24B8.5p::GFP were synchronized to the L4 stage and moved to either EV or pmk-1 RNAi. Animals were grown to adulthood and laid progeny, and the L4s from the second generation on RNAi were imaged using the similar protocol above.
C. elegans lifespan measurements
Lifespan measurements were performed on solid NGM agar plates spotted with RNAi bacteria (HT115 E. coli strain with either EV pL4440 or RNAi vectors). Worms were synchronized by bleaching, L1 arrested, and grown to adulthood at 20°C. Adult worms were moved away from progeny daily onto new RNAi plates until progeny were no longer visible (~7–10 days). Animals were then scored every 1–2 days for death until all animals were scored. Animals with bagging, vulval explosion, or other age-unrelated deaths were censored and removed from statistics. For Tunicamycin lifespans, either 1% DMSO, 5μg/ml Tunicamycin, 10μg/ml Tunicamycin, or 25μg/ml Tunicamycin was included in the NGM agar plates, and animals were moved onto DMSO or Tunicamycin plates at D1. All animals were grown on DMSO plates until D1 to allow proper development of animals, since tunicamycin results in developmental defects. Prism5 software was used for statistical analysis and log-rank (Mantel-Cox) method was used to determine significance. All lifespans are performed with the experimenter blinded to the identity of strains throughout the entirety of the experiment.
For Pseudomonas aeruginosa (PA14) lifespans, worms were synchronized by bleaching, L1 arrested, and grown to L4 at 20°C on NGM agar plates spotted with RNAi bacteria (HT115 E. coli strain with EV pL4440). Worms were then transferred onto solid agar plates (0.3% NaCl w/v; 0.35% peptone w/v; 1.7% agar w/v; 1mM CaCl2; 1mM MgSO4; 5ng/ml cholesterol; 1mM KPO4) seeded with 10μl of PA14 culture spread evenly along the surface of the plates 24 hours prior. Animals were then scored every 6 hours for death until all animals were scored. Bagged animals were scored as dead, and only animals crawling off the plates were considered censored in this assay.
For FUDR lifespans, worms were synchronized and grown on EV bacteria similar to above. Worms were then transferred onto agar plates seeded with EV and spotted with 100μl of 10mg/ml FUDR. Lifespans were scored similar to standard lifespans.
To kill bacteria for dead bacteria assays, NGM agar plates were spotted with RNAi bacteria (HT115 E. coli strain with EV pL4440) and allowed to dry overnight. Spotted plates were then put into a UV cross-linker (CL-1000 Ultraviolet Crosslinker; 254nm; Energy ×100 μ J/cm2) for ten minutes, where both the spotted plate and lids were exposed to UV treatment face up. Treated plates were left at room temperature overnight prior to using. Lifespans were carried out on these plates similar to standard lifespans described above.
C. elegans RNA-seq and analysis
Animals were synchronized and grown to D2 on EV RNAi plates. ~2,000 animals were harvested using M9. M9 was subsequently aspirated, replaced with trizol, and worms were freeze/thawed 3x with lipid nitrogen/37 °C wat er bath cycles. After the final thaw, chloroform was added at a 1:5 ratio of chloroform:trizol volume for aqueous separation of RNA, which was performed via centrifugation in heavy gel phase-lock tubes (VWR, 10847–802). The aqueous phase was mixed with isopropanol, then RNA purification was performed using a Qiagen RNeas Mini Kit as per manufacturer’s directions. Library preparation was performed using Kapa Biosystems mRNA Hyper Prep Kit. Sequencing was performed using Illumina HS4000, mode SR100 through the Vincent J. Coates Genomic Sequencing Core at University of California, Berkeley. Reads were aligned and quantified using Salmon (Patro et al., 2017), with WBcel235 as the worm reference genome. Fold changes were determined using R-package DESeq2. For analysis of UPRER genes, the GO term ER-UPR was used (GO 0030968). Enrichment was calculated using Gene Ontology enrichment analysis and visualization tool(Eden et al., 2009, 2007). It should be noted that both sur-5p::hTMEM2 animals and rab-3p::xbp-1s animals have a significant increase in aex-5 transcripts. This is because the unc-54 3’UTR used in these transgenes has a small part of the last exon of aex-5. This serves as a validation that our transgenes are highly expressed in these animals.
CellTiter-Glo® luminescence viability assay
We experienced significant variation in the cell number plated based on cell counting alone. We therefore generally seeded each cell line in two concentrations in order to control for any cell density effects that might be contributing to the stress phenotype. Furthermore, we generally focused on results that are relative to the no treatment control conditions of each cell line, in order to allow us a comparison between cell lines. Cells were seeded at 100k and 200k/plate on gelatin-coated 24/well (VWR; 29442–044) or 96well (Corning; 3904) plates. After 24h the compounds were added to the plated cells. After an additional 5days, the plates were washed 1x with PBS and CellTiter-Glo/media (1:3), was added to the wells, 100μl/96well, 250μl/24well with a multichannel pipette. After an incubation of 30min at 37°C, lumi nescence of the 96well was measured using Tecan M1000. In case of the 24well plates, 100μl of each well was removed and added to a 96well (Corning; 3904), using a multichannel pipette, for measurement, in order to avoid interference of the measurements through diffraction. Wells on the periphery of the plate were generally avoided, due to differences in oxygen exposure and evaporation during the culture of these cells. For the statistical analysis of these experiments, we utilized a one-way ANOVA test with a post-hoc Bonferroni-Holm analysis. All results presented here passed the equal variance test and normal distribution was assumed.
Clonal expansion
Single human fibroblast cells are often troublesome to expand. We therefore started by plating Wildtype fibroblast at 25k/plate density on a gelatin-coated 48well plate in order to support the initial stages of clonal fibroblast expansion. The cell line chosen for clonal expansion, was then added at a 0.5cells/well density to the Wildtype fibroblasts. As soon as the cells reached confluency, the initial transduction selection marker was used once more, this time to remove the supporting wildtype cells. The surviving cells were then left for expansion until the well was confluent (roughly 6–8 weeks). Part of the cells were then send for sequence validation, the rest frozen down until the clonal nature and successful genome editing was verified.
Clonal expansion of TMEM2 and control lines
For our initial attempt to characterize the impact of TMEM2 on ER stress resistance, we exposed a population of cells to CRISPR/Cas9 and sgRNA targeting TMEM2. This approach generates a complex mutant pool within this population of cells, some of which possess heterozygotic or homozygotic ablation of the targeted gene product, while others maintain two functional copies of the gene due to neutral modification of the targeted locus. We observed significant changes to ER stress resistance, due to the disruption of TMEM2 expression, when compared to Wildtype and scramble control conditions under this experimental paradigm (data not shown).
From this complex mutant pools we generated clonal lines of TMEM2. During the derivation of these clonal lines, we also generated Wildtype and scramble sgRNA clonal control lines, in order to test if the presence of scramble sgRNA or the clonal nature impacted ER stress resistance itself. We did not observe any significant differences between the clonal lines, between the pooled and clonal control lines, or due to the expression of scramble sgRNA, on ER stress resistance. However, ablation of TMEM2, be it of one or both alleles, significantly reduced ER stress resistance as described in the results section. While we continued to include these controls in our characterization of the TMEM2 phenotype, the additional controls show no significant differences to the Wildtype controls, and therefore do not provide additional insight or information. We therefore decided to exclude the clonal control results for clarity and ease of the presentation of the results.
CRISPR/CAS9-mediated gene disruption
In order to target a specific genomic locus we cloned single guide sgRNA vectors using the AVANA sgRNA library sequences, into the LENTI-viral expression vector pLKO.1. We then proceeded to transduce the fibroblasts with lenti-virus at an MOI of 0.3–0.5 (usually between 50–100μl of virus/well). We then added maintenance media to a total of 800μl/well. One well without viral transduction was included to the setup in order to serve as a selection control. Cells were incubated overnight and washed 2x with PBS and normal fibroblast culture media added. After 48h, the cells were then exposed to either Puromycin, Blasticidine or Gentamicin based on the selection cassette used. The selection was removed as soon as all the cells in the non-transduction control were dead, roughly after 5–7days. The cells were then cultured and expanded for an additional 7–14days in order to maximize genome editing efficacy and to allow for target protein depletion. Finally, the cells were then frozen down or immediately used for experiments.
Genomic DNA extraction and screen library preparation
For gDNA extraction 3×107 – 5×107 frozen cell pellet in a 15ml conical tube, 6ml of NK Lysis Buffer (50 mM Tris, 50 mM EDTA, 1% SDS, pH 8) and 30μl of 20mg/ml Proteinase K were added to the tissue/cell sample and incubated at 55°C overnight. The next day, 30μL of 10mg/ml RNase A, diluted in NK Lysis Buffer to 10mg/ml and then stored at 4°C, was added to the lysed sample, which was then inverted 25 times and incubated at 37°C for 30min. Samples were coole d on ice before addition of 2ml of pre-chilled 7.5M ammonium acetate to precipitate proteins. Stock solutions of 7.5M ammonium acetate was made in sterile dH2O and kept at 4°C until use. After adding ammonium acetate, the samples were vortexed at high speed for 20s and then centrifuged at ≥ 4,000×g for 10min. After the spin, a tight pellet was visible in each tube and the supernatant was carefully decanted into a new 15ml conical tube. Then 6ml 100% isopropanol was added to the tube, inverted 50 times and centrifuged at ≥ 4,000×g for 10min. Genomic DNA was visible as a small white pellet in each tube. The supernatant was discarded, 6ml of freshly prepared 70% ethanol was added to the tube and inverted 10times.he tube was then centrifuged at ≥4,000×g for 1min. The supernatant was discarded by pouring; the tube was briefly spun, and remaining ethanol was removed using a P200 pipette. After air drying for 10–30min, the DNA changed appearance from a milky white pellet to slightly translucent. At this stage, 500μL of 1xTE buffer was added, the tube was incubated at 65°C fo r 1h and at room temperature overnight to fully resuspend the DNA. The next day, the gDNA samples were vortexed briefly. The gDNA concentration was measured using a Nanodrop (Thermo Scientific).
To measure the distribution of the different library sgRNA within each screen arm we used Illumina Next Generation Sequencing applied to an amplicon generated from a single targeted PCR of the integrated sgRNA cassette (PMID: 24336571). Briefly, we use all the collected gDNA (1000x coverage) divided into 100μl PCR reactions with 5μg of DNA per reaction. We used Herculase Fusion II DNA polymerase using the default mix protocol only with double the amount of primers. PCR program: (950 2min, (980 10sec, 600 30sec, 720 30sec) × 24, 720 5min). As forward primer for all samples we used an equimolar mix of the following primers:
| Illumina sequences | Stagger | Priming site |
|---|---|---|
| AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT | t | Tcttgtggaaaggacgaaacaccg |
| AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT | at | Tcttgtggaaaggacgaaacaccg |
| AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT | gat | Tcttgtggaaaggacgaaacaccg |
| AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT | cgat | Tcttgtggaaaggacgaaacaccg |
| AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT | tcgat | Tcttgtggaaaggacgaaacaccg |
| AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT | atcgat | Tcttgtggaaaggacgaaacaccg |
| AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT | gatcgat | Tcttgtggaaaggacgaaacaccg |
One of the following reverse primers was used to each sample
| Illumina sequence | Sample specific barcode | Illumina sequence | Priming site |
|---|---|---|---|
| CAAGCAGAAGACGGCATACGAGAT | AAGTAGAG | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | TCTACTATTCTTTCCCCTGCACTGT |
| CAAGCAGAAGACGGCATACGAGAT | ACACGATC | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | TCTACTATTCTTTCCCCTGCACTGT |
| CAAGCAGAAGACGGCATACGAGAT | CGCGCGGT | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | TCTACTATTCTTTCCCCTGCACTGT |
| CAAGCAGAAGACGGCATACGAGAT | CATGATCG | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | TCTACTATTCTTTCCCCTGCACTGT |
| CAAGCAGAAGACGGCATACGAGAT | CGTTACCA | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | TCTACTATTCTTTCCCCTGCACTGT |
PCR products were gel purified and an equimolar mix was sequences of HiSeq2000 using standard Illumina primers.
Human Fibroblast RNAseq and analysis
RNA purification was performed using a Qiagen RNase Mini Kit as per manufacturer’s instructions. Library preparation was performed using Kapa Biosystems mRNA Hyper Prep Kit. Sequencing was performed using Illumina HS4000, mode SR100 through the Vincent J. Coates Genomic Sequencing Core at University of California, Berkeley. Trimmed fastq reads were then aligned to the human genome (GRCh38) using STAR RNAseq aligner (Dobin et al., 2013) version 2.5.2a using default parameters. Sam files were then converted to Bam files using Samtools (Li et al., 2009). Read counts per gene were calculated using the R bioconductor package GenomicAlignments with the function summarizeOverlaps with parameters: mode=“Union”, singleEnd=TRUE, ignore.strand=TRUE. Differential expression fold change and significance where calculated using DEseq package (Anders and Huber, 2010).
Lentiviral production and transduction of expression vectors in general
HEK293T cells were seeded at ~40% confluence the day before transfection in D10 media on gelatin-coated 6well plates (VWR; 29442–042). One hour prior to transfection, media was removed and 1ml of pre-warmed reduced serum OptiMEM media was added to each well. We then mixed for each 6well, 2μg of the plasmid of choice, and the packaging vectors pMDLg/pRRE (1.3μg), pRSV.Rev (500ng) and pVSVg (700ng) to a total of 100μl OptiMEM. In a separate tube, we mixed 5μl/well Lipofectamine 2000 reagent diluted to 100μl with OptiMEM, and after 5min, added to the mixture of DNA/OptiMEM. The complete mixture was incubated for 20min before being added to the HEK293T cells. After 6h, the media was changed to 1ml/well fibroblast culture media. After 48h, the supernatant was collected and centrifuged at 3,000 rpm for 10min to pellet cell debris. The supernatant was then filtered through a 0.45μm low protein binding membrane (Millipore Steriflip HV/PVDF). Aliquots were stored at −80°C.
Library lentiviral production
Four 15cm2 dishes (VWR; 430599) of HEK293T cells were seeded at ~40% confluency the day before transfection in D10 media (DMEM supplemented with 10% fetal bovine serum). One hour prior to transfection, media was removed and 13ml of pre-warmed reduced serum OptiMEM media was added to each dish. Transfection was performed using Lipofectamine 2000. For each dish, 20μg of plasmid library, 10μg of pVSVg, and 15μg of psPAX2 (Addgene) were diluted in 2ml OptiMEM (Life Technologies). 100μl of Lipofectamine 2000 was diluted in 2ml OptiMEM and, after 5min, it was added to the mixture of DNA. The complete mixture was incubated for 20min before being added to cells. After 6h, the media was changed to 15ml D10. Media containing lentiviral particles was removed after 48h and stored in 1ml aliquots at −80°C.
Screen cell culture
Human immortalized foreskin fibroblasts (BJ fibroblast) were cultured at 37°C, 95% air and 5% CO2 in a humidified incubator on gelatin-coated dishes in medium containing DMEM, 15% Fetal Bovine Serum (FBS), 1% Glutamax, 1% Non-Essential Amino Acids (NEAA) and 1% Penicillin/Streptomycin in all experiments. Cells were transduced with the AVANA library via spinfection. To find optimal virus volumes for achieving an MOI of 0.3–0.5, each new virus lot was tested by spinfecting 2×106 cells with several different volumes of virus. Briefly, 2×106 cells per well were plated into a 12-well plate in media supplemented with 8μg/ml polybrene (Sigma). Each well received a different titrated virus amount (usually between 5 and 200μl) along with a no-transduction control. The 12-well plate was centrifuged at 1,000g for 2h at 37°C and left in the incubator overnight. Media was aspirated the next morning and cells were enzymatically detached using trypsin. Cells were counted and each well was split into duplicate wells diluted 1:10. One replicate received 1μg/ml puromycin. After 3days cells were counted to calculate a percent transduction. Percent transduction is calculated as cell count from the replicate with puromycin divided by cell count from the replicate without puromycin multiplied. The virus volume yielding a MOI closest to 0.4 was chosen for large-scale screening.
For the screen itself we aimed to maintain a 500x coverage over library complexity (80,000). We transduced 250×106 cells (divided into 12 well plates) using spinfection and the predetermined virus and cell amount. Day following transduction all cells were mixed together and plated on 60 15cm plated with puromycin selection. One well was used to ensure that the screen MOI was indeed as aimed using the same method described above. Cells were cultured for two weeks to maximize genome-editing and target protein depletion while maintaining a minimum of 40M cells (500x coverage) at all times. After two weeks cells were spitted to a control and treatment arm and cultured for an additional three weeks in the presence of 200ng/ml Tunicamycin or 0.1% DMSO, before harvesting gDNA.
Screening data analysis
To count the number of reads associated with each sgRNA in each fastq file, we first extracted the sgRNA targeting sequencing using a regular expression containing the three nucleotides flanking each side of the sgRNA 20bp target. sgRNA spacer sequences were then aligned to a pre-indexed AVANA library using the short-read aligner ‘bowtie’ using parameters –v 0 –m 1. Data analysis was performed using custom R scripts. Gene level p-values were calculated using the mean of the four sgRNA compared to an empirical distribution of means generated by random data permutations.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses are specifically described in figure legends and in the experimental methods above. All graphical representations and sample sizes are also provided in the figure legends. PRISM software is used to perform all statistical tests.
DATA AND CODE AVAILABILITY
All necessary data to draw conclusions presented are available within the manuscript. Raw sequencing data are available in the following formats: C. elegans RNA-seq raw data is available at https://data.mendeley.com/datasets/dy97pwyf7471. All human RNA-seq and screening raw data is available at http://dx.doi.org/10.17632/tmtkc8gcs8.1. Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Andrew Dillin (dillin@berkeley.edu). No program code has been used in this manuscript.
Supplementary Material
Figure S1: →Related to Figure 1. A) Proliferation of human immortalized fibroblasts exposed to varying levels of Tunicamycin, as determined through cell counting (n=3). 200ng/ml Tunicamycin was used for the whole genome KO screen. B) Quantitative comparison of the depletion of essential genes in the control arm of the two screen replicates, compared to the mean depletion of >400 screens that used the AVANA library (data downloaded from https://depmap.org/portal/). C) Comparison of gene depletion p-values (see Methods) between Control and Tunicamycin-treated fibroblast in the second screen replicate. D) Results of the sequence validation of the TMEM2 locus in TMEM2-KO clonal fibroblast line. E) CellTiter-Glo (CTG) analysis to determine cell density of Wildtype, TMEM2-KO and CMV-TMEM2 overexpressing fibroblasts exposed to varying levels of DTT-induced ER stress, at the endpoint of a 5 day treatment. Results are presented relative to the untreated control conditions in order to adjust for cell number variability between the cell lines at the start of the experiment; (n=3). Statistical Analysis: One-way ANOVA analysis with post-hoc Bonferroni-Holm analysis
Table S3. Oligonucleotides used in this study.
Figure S2: →Related to Figure 2. TMEM2-KO fibroblast showed no changes in proliferation or resistance to mitochondrial, cytoplasmic protein-misfolding or actin destabilization stress. A) Wildtype and TMEM2-KO fibroblast proliferation speed was determined through cell counting in the absence of cellular stress; (n=3). B) Resistance to FCCP-induced mitochondrial stress was measured in Wildtype and TMEM2-KO human fibroblasts. The cells were exposed to FCCP for 5 days and cell density measured at the endpoint of the experiment using CTG analysis; (n=3). C) Wildtype and TMEM2-KO cells were exposed to the actin destabilization compound Cytochalasin D for 5 days. Cell density was then determined via CTG analysis; (n=3). D) CTG analysis of TMEM2-KO to Wildtype fibroblast grown for 5 days in the presence and absence of Sodium Arsenite, a compound which induces cytoplasmic protein misfolding; (n=3).
Figure S3: →Related to Figure 3 and 4. The impact of TMEM2 on stress resistance is independent of the canonical UPRER pathway components IRE1 and PERK1, nor does it involve VEGF signaling. A-B) Wildtype, TMEM2-KO and CMV-TMEM2 overexpressing human fibroblasts were exposed to varying concentrations of A) the IRE-1-mediated XBP1-splicing inhibitor STF-083010 (Concentration in bar graphs: 10μM) (n=3) or the B) PERK1 inhibitor GSK-2606414 (Concentration in bar graphs: 10nM). Tunicamycin-induced ER stress resistance was monitored through CTG analysis after a 5 day treatment with 200ng/ml Tunicamycin; (n=3). C) CTG analysis of Wildtype and TMEM2-KO human fibroblasts was performed to determine the cell density after the exposure to Tunicamycin-induced ER-stress (5 days; 200ng/ml Tunicamycin), and in the presence or absence of the growth factor VEGF (n=3). D) CTG analysis of Wildtype, TMEM2-KO and CMV-TMEM2 overexpressing cells in the presence and absence of the VEGF receptor inhibitor SU5416 and Tunicamycin-induced ER stress (5 day exposure, Tunicamycin 200ng/ml) (n=3). Statistical Analysis: One-way ANOVA analysis with post-hoc Bonferroni-Holm analysis
Figure S4: →Related to Figure 3. RNAseq analysis of Wildtype and TMEM2-KO human fibroblasts reveals that TMEM2-KO cells are capable to induce the UPRER in the presence of ER stress. Volcano plot of A) Wildtype and B) TMEM2-KO cells, and their shift in gene expression in response to an 8h, 200ng/ml Tunicamycin treatment. Genes highlighted in red are known UPRER target genes, HSPA5 is highlighted in blue. C) Dot plot comparing the response between the two cell lines, known UPRER target genes are highlighted in red; D) Analysis of the expression changes in response to Tunicamycin treatment, of gene targets associated with each of the canonical UPRER pathways, ATF6, IRE1 and PERK1. Each point represents a gene mean expression across three replicate measurements (see STAR METHODS for details of the analysis).
Figure S5: →Related to Figure 4. The impact of TMEM2 on stress resistance depends on p38 and ERK MAPK signaling, and is independent JNK MAPK signaling. ER stress resistance of Wildtype, TMEM2-KO and CMV-TMEM2 overexpressing human fibroblast was measured in the presence of Tunicamycin (200ng/ml) and A) the ERK inhibitor DEL-22379 (Concentration in bar graphs: DEL-22379 2μM), B) the p38 MAPK pathway inhibitor SB239063 (Concentration in bar graphs: SB239063 50μM) or the C) JNK MAPK pathway inhibitor AEG 3482 (Concentration in bar graphs: AEG 3482 100μM). Changes to Tunicamycin-induced ER stress resistance was determined through CTG analysis, after a 5 day treatment with 200ng/ml Tunicamycin; (n=3) Statistical Analysis: One-way ANOVA analysis with post-hoc Bonferroni-Holm analysis
Figure S6: →Related to Figure 5 The predicted C. elegans TMEM2 homologue, chhy-1, but not R07C12.1, is required for ER stress resistance and whole animal overexpression of hTMEM2 is necessary for lifespan extension and stress resistance. A) Lifespans were measured in Wildtype (N2) and sur-5p::hTMEM2 worms grown on EV RNAi on 1% DMSO, 5μg/ml, 10μg/ml, and 25μg/ml of Tunicamycin (Tm) from Day 1 (D1) as described in STAR METHODS. Animals were developed on standard EV RNAi plates from hatch until D1. Data is representative of two independent trials. B) Lifespan measurements of Wildtype (N2) or CRISPR-Cas9 null mutant R07C12.1(uth37) animals grown on EV RNAi on 1% DMSO and 25μg/ml Tunicamycin (Tm) from Day 1 (D1). Animals were developed on standard EV RNAi plates from hatch until D1. Data is representative of two independent trials. C) Lifespan measurements of Wildtype (N2) animals grown on EV or chhy-1 RNAi on 1% DMSO and 25μg/ml Tunicamycin (Tm) from Day 1 (D1). Animals were developed on standard EV RNAi plates from hatch until D1. Data is representative of two independent trials. D) Lifespan measurements of Wildtype (N2) and sur-5p::hTMEM2 worms grown on EV RNAi from hatch until D7. Animals were moved to either 1% DMSO or 25μg/ml Tunicamycin (Tm) at D7. Data is representative of three independent trials. E) Fluorescent micrographs of Wildtype (N2) and sur-5p::hTMEM2 animals expressing the UPRER reporter, hsp-4p::GFP. Animals were moved onto standard EV plates containing 1% DMSO or 25μg/ml Tunicamycin (Tm) 16 hours prior to imaging – see STAR Methods for details. Data is representative of 3 independent trials. F) Lifespan measurements of Wildtype (N2) or rgef-1p::hTMEM2 animals grown on EV RNAi on 1% DMSO and 25μg/ml Tunicamycin (Tm) from Day 1 (D1). Animals were developed on standard EV RNAi plates from hatch until D1. Data is representative of three independent trials. All statistics for lifespans were performed using Log-Rank (Mantel-Cox) test using PRISM, and are available in Supplemental Table 2.
Figure S7: →Related to Figure 5 Overexpression of hTMEM2 in C. elegans does not promote the expression of canonical UPRER targets. A) RNA-sequencing was performed on Wildtype (N2), rab-3p::xbp-1s, and sur-5p::hTMEM2 animals. The log2-fold change in expression as a function of mean expression level is shown as compared to Wildtype. Genes are highlighted according to the significance of fold change (blue, p-value < 0.01; yellow p-value < 0.001). UPRER transcription factor, xbp-1s, is highlighted in green. B) Fold changes of the most significant changes (p-value < 0.001) are plotted individually for sur-5p::hTMEM2 (left) and rab-3p::xbp-1s (right) animals compared to N2. Genes are non-overlapping. C) Fold change of genes annotated as UPRER (GO: 0030968) is shown. The median fold change is noted in a red line. The log2-fold change is also shown separately for each gene in (D). These data show that the UPRER is generally not activated in sur-5p::hTMEM2 in comparison to rab-3p::xbp-1s animals. E) Venn diagram of significantly changing genes (p-value < 0.05) (top). Fold-change of the 27 differentially expressed genes that are shared between the two strains (bottom). These data show that hTMEM2 and xbp-1s overexpression result in distinct transcriptional changes.
Table S1. CRISPR-Cas9 Screen Data. →Related to Figure 1.
Highlights.
CRISPR-Cas9 screening in human fibroblasts reveals TMEM2 as a regulator of ER stress
TMEM2 alters intracellular ER stress resistance through changes in ECM metabolism
TMEM2 promotes ER stress resistance independent of UPRER through MAPK signaling
Ectopic expression of human TMEM2 promotes lifespan and immunity in C. elegans
Acknowledgements
We thank the members of the Dillin lab for feedback, discussion, and technical assistance. This work was supported by grant 5R01AG02679-01A1 and 5R01AG059566 from the National Institute of Aging (NIA) and Howard Hughes Medical Institute to AD, by the California Institute of Regenerative Medicine and by the Siebel Stem Cell Institute for RTS, Jane Coffins Child to EAM, EMBO Long-Term Fellowship (462-2017) to RBZ, grant 5F32AG053023-02 from NIA and the Glenn Foundation for Medical Research Postdoctoral Fellowship to RHS, and a postdoctoral fellowship from the Klarman family foundation to OS.
Footnotes
Competing Financial Interests/Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, Pak RA, Gray AN, Gross CA, Dixit A, Parnas O, Regev A, Weissman JS, 2016. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 167, 1867–1882.e21. 10.1016/j.cell.2016.11.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P, 2003. The specificities of protein kinase inhibitors: an update. Biochem. J 371, 199–204. 10.1042/BJ20021535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JSC, Alessi DR, Cohen P, 2007. The selectivity of protein kinase inhibitors: a further update. Biochem. J 408, 297–315. 10.1042/BJ20070797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MK, Naidoo N, 2012. The endoplasmic reticulum stress response in aging and age-related diseases. Front. Physiol 3, 263 10.3389/fphys.2012.00263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanmee T, Ontong P, Itano N, 2016. Hyaluronan: A modulator of the tumor microenvironment. Cancer Lett. 375, 20–30. 10.1016/j.canlet.2016.02.031 [DOI] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A, 2013. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 10.1186/1471-2105-14-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Brandizzi F, 2013. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 23, 547–555. 10.1016/j.tcb.2013.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csoka AB, Stern R, 2013. Hypotheses on the evolution of hyaluronan: a highly ironic acid. Glycobiology 23, 398–411. 10.1093/glycob/cws218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyphert JM, Trempus CS, Garantziotis S, 2015. Size Matters: Molecular Weight Specificity of Hyaluronan Effects in Cell Biology. Int. J. Cell Biol 2015, 563818 10.1155/2015/563818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling NJ, Cook SJ, 2014. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta 1843, 2150–2163. 10.1016/j.bbamcr.2014.01.009 [DOI] [PubMed] [Google Scholar]
- De Angelis JE, Lagendijk AK, Chen H, Tromp A, Bower NI, Tunny KA, Brooks AJ, Bakkers J, Francois M, Yap AS, Simons C, Wicking C, Hogan BM, Smith KA, 2017. Tmem2 Regulates Embryonic Vegf Signaling by Controlling Hyaluronic Acid Turnover. Dev. Cell 40, 421 10.1016/j.devcel.2017.02.005 [DOI] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, Virgin HW, Listgarten J, Root DE, 2016. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191. 10.1038/nbt.3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Lipson D, Yogev S, Yakhini Z, 2007. Discovering motifs in ranked lists of DNA sequences. PLoS Comput. Biol. 3, e39 10.1371/journal.pcbi.0030039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z, 2009. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48 10.1186/1471-2105-10-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P, 2013. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol 5, a013169 10.1101/cshperspect.a013169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerakis Y, Hetz C, 2018. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 285, 995–1011. 10.1111/febs.14332 [DOI] [PubMed] [Google Scholar]
- Gialeli C, Viola M, Barbouri D, Kletsas D, Passi A, Karamanos NK, 2014. Dynamic interplay between breast cancer cells and normal endothelium mediates the expression of matrix macromolecules, proteasome activity and functional properties of endothelial cells. Biochim. Biophys. Acta 1840, 2549–2559. 10.1016/j.bbagen.2014.02.019 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA, 2011. Hallmarks of cancer: the next generation. Cell 144, 646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Hetz C, 2012. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol 13, 89–102. 10.1038/nrm3270 [DOI] [PubMed] [Google Scholar]
- Hetz C, 2009. The UPR as a survival factor of cancer cells: More than folding proteins? Leuk. Res 33, 880–882. 10.1016/j.leukres.2009.02.017 [DOI] [PubMed] [Google Scholar]
- Horlbeck MA, Xu A, Wang M, Bennett NK, Park CY, Bogdanoff D, Adamson B, Chow ED, Kampmann M, Peterson TR, Nakamura K, Fischbach MA, Weissman JS, Gilbert LA, 2018. Mapping the Genetic Landscape of Human Cells. Cell 174, 953–967.e22. 10.1016/j.cell.2018.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Davis RJ, 2016. Cell Signaling and Stress Responses. Cold Spring Harb. Perspect. Biol 8 10.1101/cshperspect.a006072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull RL, Bogdani M, Nagy N, Johnson PY, Wight TN, 2015. Hyaluronan: A Mediator of Islet Dysfunction and Destruction in Diabetes? J. Histochem. Cytochem. Off. J. Histochem. Soc 63, 592–603. 10.1369/0022155415576542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joy RA, Vikkath N, Ariyannur PS, 2018. Metabolism and mechanisms of action of hyaluronan in human biology. Drug Metab. Pers. Ther 33, 15–32. 10.1515/dmpt-2017-0031 [DOI] [PubMed] [Google Scholar]
- Kaneiwa T, Yamada S, Mizumoto S, Montaño AM, Mitani S, Sugahara K, 2008. Identification of a novel chondroitin hydrolase in Caenorhabditis elegans. J. Biol. Chem 283, 14971–14979. 10.1074/jbc.M709236200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EK, Choi E-J, 2010. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 1802, 396–405. 10.1016/j.bbadis.2009.12.009 [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma’ayan A, 2016. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–97. 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz CL, Tan M-W, 2004. Regulation of aging and innate immunity in C. elegans. Aging Cell 3, 185–193. 10.1111/j.1474-9728.2004.00108.x [DOI] [PubMed] [Google Scholar]
- Laurent TC, Laurent UB, Fraser JR, 1996. The structure and function of hyaluronan: An overview. Immunol. Cell Biol 74, A1–7. 10.1038/icb.1996.32 [DOI] [PubMed] [Google Scholar]
- Majors AK, Austin RC, de la Motte CA, Pyeritz RE, Hascall VC, Kessler SP, Sen G, Strong SA, 2003. Endoplasmic reticulum stress induces hyaluronan deposition and leukocyte adhesion. J. Biol. Chem 278, 47223–47231. 10.1074/jbc.M304871200 [DOI] [PubMed] [Google Scholar]
- Misra S, Hascall VC, Markwald RR, Ghatak S, 2015. Interactions between Hyaluronan and Its Receptors (CD44, RHAMM) Regulate the Activities of Inflammation and Cancer. Front. Immunol 6, 201 10.3389/fimmu.2015.00201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morawski M, Filippov M, Tzinia A, Tsilibary E, Vargova L, 2014. ECM in brain aging and dementia. Prog. Brain Res 214, 207–227. 10.1016/B978-0-444-63486-3.00010-4 [DOI] [PubMed] [Google Scholar]
- Nagy N, Kaber G, Johnson PY, Gebe JA, Preisinger A, Falk BA, Sunkari VG, Gooden MD, Vernon RB, Bogdani M, Kuipers HF, Day AJ, Campbell DJ, Wight TN, Bollyky PL, 2015a. Inhibition of hyaluronan synthesis restores immune tolerance during autoimmune insulitis. J. Clin. Invest 125, 3928–3940. 10.1172/JCI79271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy N, Kuipers HF, Frymoyer AR, Ishak HD, Bollyky JB, Wight TN, Bollyky PL, 2015b. 4-methylumbelliferone treatment and hyaluronan inhibition as a therapeutic strategy in inflammation, autoimmunity, and cancer. Front. Immunol 6, 123 10.3389/fimmu.2015.00123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papy-Garcia D, Christophe M, Huynh MB, Fernando S, Ludmilla S, Sepulveda-Diaz JE, Raisman-Vozari R, 2011. Glycosaminoglycans, protein aggregation and neurodegeneration. Curr. Protein Pept. Sci 12, 258–268. [DOI] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C, 2017. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. 10.1038/nmeth.4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert L, Labat-Robert J, 2015. Longevity and aging: role of genes and of the extracellular matrix. Biogerontology 16, 125–129. 10.1007/s10522-014-9544-x [DOI] [PubMed] [Google Scholar]
- Robertson C, 2016. The extracellular matrix in breast cancer predicts prognosis through composition, splicing, and crosslinking. Exp. Cell Res 343, 73–81. 10.1016/j.yexcr.2015.11.009 [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P, 2007. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol 8, 519–529. 10.1038/nrm2199 [DOI] [PubMed] [Google Scholar]
- Salmon AB, Sadighi Akha AA, Buffenstein R, Miller RA, 2008. Fibroblasts from naked mole-rats are resistant to multiple forms of cell injury, but sensitive to peroxide, ultraviolet light, and endoplasmic reticulum stress. J. Gerontol. A. Biol. Sci. Med. Sci 63, 232–241. 10.1093/gerona/63.3.232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano R, Reed JC, 2013. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 1833, 3460–3470. 10.1016/j.bbamcr.2013.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F, 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. 10.1126/science.1247005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman LS, Matsumoto S, Su W, Srivastava T, Back SA, 2015. Hyaluronan Synthesis, Catabolism, and Signaling in Neurodegenerative Diseases. Int. J. Cell Biol 2015, 368584 10.1155/2015/368584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivers RP, Kooistra T, Chu SW, Pagano DJ, Kim DH, 2009. Tissue-specific activities of an immune signaling module regulate physiological responses to pathogenic and nutritional bacteria in C. elegans. Cell Host Microbe 6, 321–330. 10.1016/j.chom.2009.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V, Aballay A, 2006. Heat-shock transcription factor (HSF)-1 pathway required for Caenorhabditis elegans immunity. Proc. Natl. Acad. Sci. U. S. A 103, 13092–13097. 10.1073/pnas.0604050103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui X, Kong N, Ye L, Han W, Zhou J, Zhang Q, He C, Pan H, 2014. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 344, 174–179. 10.1016/j.canlet.2013.11.019 [DOI] [PubMed] [Google Scholar]
- Taylor RC, Berendzen KM, Dillin A, 2014. Systemic stress signalling: understanding the cell non-autonomous control of proteostasis. Nat. Rev. Mol. Cell Biol 15, 211–217. 10.1038/nrm3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC, Dillin A, 2013. XBP-1 Is a Cell-Nonautonomous Regulator of Stress Resistance and Longevity. Cell 153, 1435–1447. 10.1016/j.cell.2013.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Azpurua J, Hine C, Vaidya A, Myakishev-Rempel M, Ablaeva J, Mao Z, Nevo E, Gorbunova V, Seluanov A, 2013. High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature 499, 346–349. 10.1038/nature12234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tigges J, Krutmann J, Fritsche E, Haendeler J, Schaal H, Fischer JW, Kalfalah F, Reinke H, Reifenberger G, Stühler K, Ventura N, Gundermann S, Boukamp P, Boege F, 2014. The hallmarks of fibroblast ageing. Mech. Ageing Dev 138, 26–44. 10.1016/j.mad.2014.03.004 [DOI] [PubMed] [Google Scholar]
- Tolg C, McCarthy JB, Yazdani A, Turley EA, 2014. Hyaluronan and RHAMM in wound repair and the “cancerization” of stromal tissues. BioMed Res. Int 2014, 103923 10.1155/2014/103923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigetti D, Karousou E, Viola M, Deleonibus S, De Luca G, Passi A, 2014. Hyaluronan: biosynthesis and signaling. Biochim. Biophys. Acta 1840, 2452–2459. 10.1016/j.bbagen.2014.02.001 [DOI] [PubMed] [Google Scholar]
- Vigetti D, Viola M, Karousou E, Rizzi M, Moretto P, Genasetti A, Clerici M, Hascall VC, De Luca G, Passi A, 2008. Hyaluronan-CD44-ERK1/2 regulate human aortic smooth muscle cell motility during aging. J. Biol. Chem 283, 4448–4458. 10.1074/jbc.M709051200 [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D, 2011. The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- Wang S, Kaufman RJ, 2012. The impact of the unfolded protein response on human disease. J. Cell Biol. 197, 857–867. 10.1083/jcb.201110131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES, 2014. Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84. 10.1126/science.1246981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wang J, Li F, 2017. Hyaluronidase and Chondroitinase. Adv. Exp. Med. Biol 925, 75–87. 10.1007/5584_2016_54 [DOI] [PubMed] [Google Scholar]
- Williams KW, Tiemin Liu, Kong X, Fukuda M, Deng Y, Berglund ED, Deng Z, Gao Y, Tianya Liu, Sohn J-W, Jia L, Fujikawa T, Kohno D, Scott MM, Lee S, Lee CE, Sun K, Chang Y, Scherer PE, Elmquist JK, 2014. Xbp1s in Pomc neurons connects ER stress with energy balance and glucose homeostasis. Cell Metab. 20, 471–482. 10.1016/j.cmet.2014.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Kaufman RJ, 2006. From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ. 13, 374–384. 10.1038/sj.cdd.4401840 [DOI] [PubMed] [Google Scholar]
- Xu C, Bailly-Maitre B, Reed JC, 2005. Endoplasmic reticulum stress: cell life and death decisions. J. Clin. Invest 115, 2656–2664. 10.1172/JCI26373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S, Sugahara K, Ozbek S, 2011. Evolution of glycosaminoglycans: Comparative biochemical study. Commun. Integr. Biol 4, 150–158. 10.4161/cib.4.2.14547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Tobisawa Y, Inubushi T, Irie F, Ohyama C, Yamaguchi Y, 2017. A mammalian homolog of the zebrafish transmembrane protein 2 (TMEM2) is the long-sought-after cell-surface hyaluronidase. J. Biol. Chem 292, 7304–7313. 10.1074/jbc.M116.770149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman MJ, Rogers ZN, Kim DH, 2011. A decline in p38 MAPK signaling underlies immunosenescence in Caenorhabditis elegans. PLoS Genet. 7, e1002082 10.1371/journal.pgen.1002082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Gilliat AF, Ziehm M, Turmaine M, Wang H, Ezcurra M, Yang C, Phillips G, McBay D, Zhang WB, Partridge L, Pincus Z, Gems D, 2017. Two forms of death in ageing Caenorhabditis elegans. Nat. Commun 8, 15458 10.1038/ncomms15458 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: →Related to Figure 1. A) Proliferation of human immortalized fibroblasts exposed to varying levels of Tunicamycin, as determined through cell counting (n=3). 200ng/ml Tunicamycin was used for the whole genome KO screen. B) Quantitative comparison of the depletion of essential genes in the control arm of the two screen replicates, compared to the mean depletion of >400 screens that used the AVANA library (data downloaded from https://depmap.org/portal/). C) Comparison of gene depletion p-values (see Methods) between Control and Tunicamycin-treated fibroblast in the second screen replicate. D) Results of the sequence validation of the TMEM2 locus in TMEM2-KO clonal fibroblast line. E) CellTiter-Glo (CTG) analysis to determine cell density of Wildtype, TMEM2-KO and CMV-TMEM2 overexpressing fibroblasts exposed to varying levels of DTT-induced ER stress, at the endpoint of a 5 day treatment. Results are presented relative to the untreated control conditions in order to adjust for cell number variability between the cell lines at the start of the experiment; (n=3). Statistical Analysis: One-way ANOVA analysis with post-hoc Bonferroni-Holm analysis
Table S3. Oligonucleotides used in this study.
Figure S2: →Related to Figure 2. TMEM2-KO fibroblast showed no changes in proliferation or resistance to mitochondrial, cytoplasmic protein-misfolding or actin destabilization stress. A) Wildtype and TMEM2-KO fibroblast proliferation speed was determined through cell counting in the absence of cellular stress; (n=3). B) Resistance to FCCP-induced mitochondrial stress was measured in Wildtype and TMEM2-KO human fibroblasts. The cells were exposed to FCCP for 5 days and cell density measured at the endpoint of the experiment using CTG analysis; (n=3). C) Wildtype and TMEM2-KO cells were exposed to the actin destabilization compound Cytochalasin D for 5 days. Cell density was then determined via CTG analysis; (n=3). D) CTG analysis of TMEM2-KO to Wildtype fibroblast grown for 5 days in the presence and absence of Sodium Arsenite, a compound which induces cytoplasmic protein misfolding; (n=3).
Figure S3: →Related to Figure 3 and 4. The impact of TMEM2 on stress resistance is independent of the canonical UPRER pathway components IRE1 and PERK1, nor does it involve VEGF signaling. A-B) Wildtype, TMEM2-KO and CMV-TMEM2 overexpressing human fibroblasts were exposed to varying concentrations of A) the IRE-1-mediated XBP1-splicing inhibitor STF-083010 (Concentration in bar graphs: 10μM) (n=3) or the B) PERK1 inhibitor GSK-2606414 (Concentration in bar graphs: 10nM). Tunicamycin-induced ER stress resistance was monitored through CTG analysis after a 5 day treatment with 200ng/ml Tunicamycin; (n=3). C) CTG analysis of Wildtype and TMEM2-KO human fibroblasts was performed to determine the cell density after the exposure to Tunicamycin-induced ER-stress (5 days; 200ng/ml Tunicamycin), and in the presence or absence of the growth factor VEGF (n=3). D) CTG analysis of Wildtype, TMEM2-KO and CMV-TMEM2 overexpressing cells in the presence and absence of the VEGF receptor inhibitor SU5416 and Tunicamycin-induced ER stress (5 day exposure, Tunicamycin 200ng/ml) (n=3). Statistical Analysis: One-way ANOVA analysis with post-hoc Bonferroni-Holm analysis
Figure S4: →Related to Figure 3. RNAseq analysis of Wildtype and TMEM2-KO human fibroblasts reveals that TMEM2-KO cells are capable to induce the UPRER in the presence of ER stress. Volcano plot of A) Wildtype and B) TMEM2-KO cells, and their shift in gene expression in response to an 8h, 200ng/ml Tunicamycin treatment. Genes highlighted in red are known UPRER target genes, HSPA5 is highlighted in blue. C) Dot plot comparing the response between the two cell lines, known UPRER target genes are highlighted in red; D) Analysis of the expression changes in response to Tunicamycin treatment, of gene targets associated with each of the canonical UPRER pathways, ATF6, IRE1 and PERK1. Each point represents a gene mean expression across three replicate measurements (see STAR METHODS for details of the analysis).
Figure S5: →Related to Figure 4. The impact of TMEM2 on stress resistance depends on p38 and ERK MAPK signaling, and is independent JNK MAPK signaling. ER stress resistance of Wildtype, TMEM2-KO and CMV-TMEM2 overexpressing human fibroblast was measured in the presence of Tunicamycin (200ng/ml) and A) the ERK inhibitor DEL-22379 (Concentration in bar graphs: DEL-22379 2μM), B) the p38 MAPK pathway inhibitor SB239063 (Concentration in bar graphs: SB239063 50μM) or the C) JNK MAPK pathway inhibitor AEG 3482 (Concentration in bar graphs: AEG 3482 100μM). Changes to Tunicamycin-induced ER stress resistance was determined through CTG analysis, after a 5 day treatment with 200ng/ml Tunicamycin; (n=3) Statistical Analysis: One-way ANOVA analysis with post-hoc Bonferroni-Holm analysis
Figure S6: →Related to Figure 5 The predicted C. elegans TMEM2 homologue, chhy-1, but not R07C12.1, is required for ER stress resistance and whole animal overexpression of hTMEM2 is necessary for lifespan extension and stress resistance. A) Lifespans were measured in Wildtype (N2) and sur-5p::hTMEM2 worms grown on EV RNAi on 1% DMSO, 5μg/ml, 10μg/ml, and 25μg/ml of Tunicamycin (Tm) from Day 1 (D1) as described in STAR METHODS. Animals were developed on standard EV RNAi plates from hatch until D1. Data is representative of two independent trials. B) Lifespan measurements of Wildtype (N2) or CRISPR-Cas9 null mutant R07C12.1(uth37) animals grown on EV RNAi on 1% DMSO and 25μg/ml Tunicamycin (Tm) from Day 1 (D1). Animals were developed on standard EV RNAi plates from hatch until D1. Data is representative of two independent trials. C) Lifespan measurements of Wildtype (N2) animals grown on EV or chhy-1 RNAi on 1% DMSO and 25μg/ml Tunicamycin (Tm) from Day 1 (D1). Animals were developed on standard EV RNAi plates from hatch until D1. Data is representative of two independent trials. D) Lifespan measurements of Wildtype (N2) and sur-5p::hTMEM2 worms grown on EV RNAi from hatch until D7. Animals were moved to either 1% DMSO or 25μg/ml Tunicamycin (Tm) at D7. Data is representative of three independent trials. E) Fluorescent micrographs of Wildtype (N2) and sur-5p::hTMEM2 animals expressing the UPRER reporter, hsp-4p::GFP. Animals were moved onto standard EV plates containing 1% DMSO or 25μg/ml Tunicamycin (Tm) 16 hours prior to imaging – see STAR Methods for details. Data is representative of 3 independent trials. F) Lifespan measurements of Wildtype (N2) or rgef-1p::hTMEM2 animals grown on EV RNAi on 1% DMSO and 25μg/ml Tunicamycin (Tm) from Day 1 (D1). Animals were developed on standard EV RNAi plates from hatch until D1. Data is representative of three independent trials. All statistics for lifespans were performed using Log-Rank (Mantel-Cox) test using PRISM, and are available in Supplemental Table 2.
Figure S7: →Related to Figure 5 Overexpression of hTMEM2 in C. elegans does not promote the expression of canonical UPRER targets. A) RNA-sequencing was performed on Wildtype (N2), rab-3p::xbp-1s, and sur-5p::hTMEM2 animals. The log2-fold change in expression as a function of mean expression level is shown as compared to Wildtype. Genes are highlighted according to the significance of fold change (blue, p-value < 0.01; yellow p-value < 0.001). UPRER transcription factor, xbp-1s, is highlighted in green. B) Fold changes of the most significant changes (p-value < 0.001) are plotted individually for sur-5p::hTMEM2 (left) and rab-3p::xbp-1s (right) animals compared to N2. Genes are non-overlapping. C) Fold change of genes annotated as UPRER (GO: 0030968) is shown. The median fold change is noted in a red line. The log2-fold change is also shown separately for each gene in (D). These data show that the UPRER is generally not activated in sur-5p::hTMEM2 in comparison to rab-3p::xbp-1s animals. E) Venn diagram of significantly changing genes (p-value < 0.05) (top). Fold-change of the 27 differentially expressed genes that are shared between the two strains (bottom). These data show that hTMEM2 and xbp-1s overexpression result in distinct transcriptional changes.
Table S1. CRISPR-Cas9 Screen Data. →Related to Figure 1.
Data Availability Statement
All necessary data to draw conclusions presented are available within the manuscript. Raw sequencing data are available in the following formats: C. elegans RNA-seq raw data is available at https://data.mendeley.com/datasets/dy97pwyf7471. All human RNA-seq and screening raw data is available at http://dx.doi.org/10.17632/tmtkc8gcs8.1. Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Andrew Dillin (dillin@berkeley.edu). No program code has been used in this manuscript.