

Abstract
Colloidal quantum dots (QDs) allow great flexibility in the design of optoelectronic devices, thanks to their size-dependent optical and electronic properties and the possibility to fabricate thin films with solution-based processing. In particular, in QD-based heterojunctions, the band gap of both components can be controlled by varying the size of the QDs. However, control over the band alignment between the two materials is required to tune the dynamics of carrier transfer across a heterostructure. We demonstrate that ligand exchange strategies can be used to control the band alignment of PbSe and CdSe QDs in a mixed QD solid, shifting it from a type-I to a type-II alignment. The change in alignment is observed in both spectroelectrochemical and transient absorption measurements, leading to a change in the energy of the conduction band edges in the two materials and in the direction of electron transfer upon photoexcitation. Our work demonstrates the possibility to tune the band offset of QD heterostructures via control of the chemical species passivating the QD surface, allowing full control over the energetics of the heterostructure without requiring changes in the QD composition.
Introduction
Semiconductor heterojunctions provide control over the distribution and the motion of charge carriers and thus are at the core of many optoelectronic devices. Semiconductor quantum dots (QDs) offer great flexibility in designing heterojunctions. The band gap of QDs can be readily tuned via a change in the QD size, without requiring changes in material composition.1−4 Colloidal solutions of QDs can be cast into thin films via solution-based processing, leading to densely packed films of QDs.5−8 During film fabrication, the long, insulating ligands capping the surface of as-synthesized QDs can be exchanged for shorter, more conductive ligands,8−15 allowing a high degree of electronic coupling between QDs while maintaining the quantum-confined nature of QD states. These properties make QD solids excellent candidates for use in solar cells,16−19 transistors,10,20−22 LEDs,23,24 lasers,25−28 and photodetectors.29−31 In contrast to bulk semiconductor heterojunctions, junctions formed between different QDs do not require epitaxial connections, which allows the fabrication of QD films composed of different, non-lattice-matched QD components, without building up strain in the system.
Beside the tunability of QD band gaps, numerous studies have demonstrated that the ligands on the QD surface significantly affect the position of the QD band edges with respect to the vacuum level.15,32,33,36−40 This effect arises from changes in the surface dipoles induced by the different ligands: inward-pointing surface dipoles increase the electron affinity, i.e., the energy difference between the vacuum level and the conduction band (CB), while outward-pointing surface dipoles decrease it.32 The possibility to shift the energy structure of QDs via control of the ligand-induced surface dipoles, depicted schematically in Figure 1a, can be used to control the band offset in a junction without affecting the QD band gap, as conduction- and valence-band edges are shifted by the same amount. Chuang et al.36 and Santra et al.37 showed the effect of the ligand-induced shift on working devices, by fabricating a homojunction with two different ligand treatments on PbS QDs, resulting in an increase of the photovoltaic performance of a QD-based solar cell.
Figure 1.

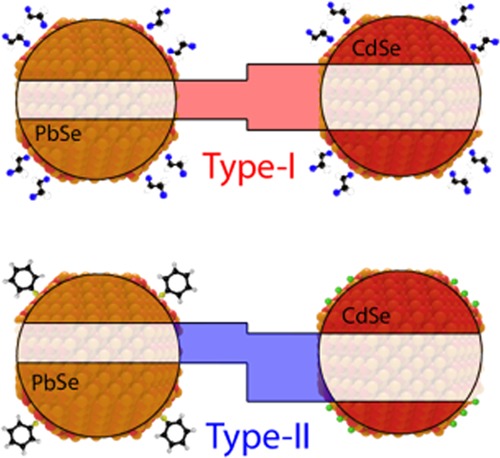
Schematics of QD heterojunction films. (a) Band edges of a semiconductor QD can be shifted by the presence of passivating ligands with different dipole moments.15,32,33 (b) Structure of a PbSe–CdSe heterojunction QD film, fabricated by dip-coating alternating layers of PbSe and CdSe QDs, and ligand-exchanging the original ligands of the as-synthesized QDs for shorter linker molecules. The inset shows the heterojunction formed by CdSe and PbSe QDs treated with ethanedithiol (EDT), characterized by a type-I band alignment.34,35 (c) Energy shift induced by the benzenethiol (BT, top left) and tetrabutylammonium chloride (TBACl, top right) ligand treatment on the conduction- and valence-band-edge positions in an EDT-capped QD, as reported by Brown et al.,32 together with the possible type-II heterojunction resulting from BT treatment of PbSe QDs and TBACl treatment of CdSe QDs.
In this work, we use ligand exchange treatments to control the band alignment of QD heterojunctions. We fabricated films composed of alternating layers of PbSe and CdSe QDs via layer-by-layer dip-coating. The film structure is shown schematically in Figure 1b. While this layer-by-layer geometry is not optimal for charge extraction in devices requiring transverse transport (such as solar cells), it allows for a facile and controlled formation of the heterostructure. Assembling the QDs in a bulk heterojunction or binary superlattice would enable efficient charge transport while maintaining the validity of the results.41−43 During the fabrication step, the oleate ligands, capping the as-synthesized QDs, are exchanged for shorter ligands. In earlier work performed in our group, we characterized the band alignment of PbSe–CdSe heterojunction films capped with ethanedithiolate (EDT) ligands. We found a type-I alignment between the two materials, in which the band edges of the smaller band-gap component (PbSe) are located within the band gap of the larger band-gap component (CdSe), as represented in Figure 1b.34,35
Although a type-I alignment is beneficial for optoelectronic applications involving radiative electron–hole recombination, such as LEDs or lasers, for many devices other band alignment configurations are preferred. Type-II heterojunctions lead to a separation of photoexcited electrons and holes, decreasing their recombination rate and thus increasing their lifetime. Such a configuration is preferred for light detection and photovoltaic applications. Alternatively, it could be beneficial to have a so-called quasi-type-II band alignment, where the CB levels of both materials are resonant. This would be ideal in the context of optimizing carrier multiplication, where, upon photon absorption in the CdSe QDs, impact ionization of the hole can lead to the generation of multiple electron–hole pairs, as has been shown for CdSe/PbSe core–shell QDs44 and Janus particles.45 In general, it is clear that the ability to control the relative alignment of the energy levels increases the possibilities to form functional QD heterojunctions.
We demonstrate that the type-I band alignment between CdSe and PbSe QDs can be altered into a type-II alignment (see Figure 1c) by the functionalization of the two QD components with appropriate ligands. Considering the changes in electron affinity reported by Brown et al.,32 we treated the PbSe QDs with benzenethiol (BT) ligands, leading to lower electron affinities than the EDT treatment. The CdSe QDs were treated with tetrabutylammonium chloride (TBACl), leading to higher electron affinities than the EDT treatment.
We performed spectroelectrochemical measurements on heterojunction QD films, confirming a type-I alignment for EDT-capped heterojunctions, which is altered into a type-II band alignment following a BT-TBACl treatment. Transient absorption (TA) measurements confirm that this type-II offset leads to charge separation and longer charge carrier lifetimes, while revealing the presence of a high density of interfacial trap states. Our results demonstrate the possibility to engineer the band alignment of QD heterojunctions via control of the passivating ligands on the QD surface, enabling full control over the energetics of the heterojunction. At the same time, a high degree of electron trapping at interfacial states is observed, which hinders the usage of PbSe–CdSe QD heterojunctions. Further improvement in the understanding and passivation of the heterojunction surface is needed to bridge the gap with device-grade heterojunctions.
Methods
CdSe QD Synthesis
The CdSe QDs were synthesized following a recipe reported in our previous work,35 in turn adapted from van Embden et al.46 The synthesis was performed by injecting a Se precursor into a flask containing a hot Cd precursor, followed by repeated injection of the Cd and Se precursors to further grow the QDs.
The Cd growth solution was prepared by adding 0.22 g of CdO (99.999%), 0.970 g of oleic acid (OA, 90%), and 6.23 g of 1-octadecene (ODE) to a three-neck round-bottom flask (BPF). The flask was attached to a Schlenk line, degassed for 1 h under vacuum (<1 mbar) at 80 °C. The solution was then heated to 260 °C under a nitrogen atmosphere, until it turned clear, indicating the formation of the Cd–oleate complex. The solution was then allowed to reach room temperature, and oleylamine (1.13 mL, tech grade 70%) was added during cooling.
The Se growth solution was obtained by dissolving 0.25 g of Se powder in 1.55 g of trioctylphosphine (TOP, tech grade 90%) in a nitrogen-filled glovebox.
The Se injection precursor was prepared by dissolving 0.327 g of Se powder in a solution of 2.5 g of TOP, 2.5 g of 1-octadecene (ODE, tech grade 90%), and 6 g of oleylamine (OAm, tech grade 70%) in a nitrogen-filled glovebox.
The Cd precursor for the hot injection was prepared by adding 0.22 g of CdO, 3 g of OA, and 30 g of ODE to a three-neck BPF flask. The solution was degassed under vacuum for 1 h at 80 °C and then heated to 260 °C under a nitrogen atmosphere until it turned clear.
The hot injection was performed by loading the Se injection precursor in a syringe equipped with a 16G needle and quickly injecting it into the three-neck BPF flask containing the Cd precursor, kept at a temperature of 260 °C. After injection, the temperature was allowed to decrease to 250 °C, where it was kept. After 20 min, 2 mL of Cd and Se growth precursors was added dropwise to the reaction solution. The growth precursor addition was repeated three times, waiting 10 min between each addition. After the last addition, the reaction was allowed to proceed at 250 °C for 10 min and was subsequently stopped by cooling to room temperature. The reaction product was washed three times by QD precipitation, achieved by adding an antisolvent (ethanol) to the reaction solution and centrifuging the mixture (3500 rpm), followed by re-dispersion in hexane.
PbSe QD Synthesis
The PbSe QDs were synthesized following a recipe reported in our previous work,35 in turn adapted from Steckel et al.47 The synthesis was performed by injecting a Se precursor into a flask containing a hot Pb precursor. The Se injection precursor was prepared by dissolving 0.553 g of Se powder in 19 mL of TOP and 0.13 mL of diphenylphosphine (DPP, 98%). The Pb reaction solution was prepared by adding 1.35 g of PbO (99.999%), 17 mL of ODE, and 4 mL of OA to a three-neck BPF flask. The flask was connected to a Schlenk line, and the solution was degassed under vacuum for 1 h. The solution was then heated to 125 °C under a nitrogen atmosphere, until it turned clear, and then cooled to 100 °C and degassed under vacuum for 30 min. To perform the injection reaction, we heated the Pb precursor to 180 °C and placed it under a nitrogen atmosphere. The injection was performed by loading the Se injection precursor into a 20 mL syringe equipped with a 16G needle and then quickly injecting the content into the Pb precursor. The reaction was allowed to proceed for 10 s, after which it was quenched by immersing the flask in cold water. The reaction product was washed three times by precipitation, achieved by adding an antisolvent (ethanol) to the reaction solution and centrifuging the mixture (3500 rpm), and re-dispersion in hexane.
Film Fabrication
The heterojunction QD films were fabricated by layer-by-layer deposition. For every QD layer, the substrate [quartz or indium tin oxide (ITO)] was dipped in a QD solution for 30 s, then was raised out of the solution, left to dry for 30 s, and subsequently immersed for 30 s in a solution of the new ligands dissolved in a polar solvent. After 30 s of drying, the substrate was immersed in the pure polar solvent, to wash away unbound residues of the new ligands, and finally left to dry for 30 s before starting again the same cycle for the other QD material. The QD solutions were prepared with a QD concentration in hexane of 0.2 mM, determined from linear absorption measurements and from the values of the extinction coefficient provided in the literature.48,49 For the EDT-capped films, the ligand exchange solution was a 10 mM solution of EDT in acetonitrile, and in the final washing step, the substrate was immersed in acetonitrile. For the TBACl layers, the ligand exchange solution consisted of a 30 mM solution of TBACl in methanol, and in the final washing step, the substrate was immersed in methanol. For the BT layers, the ligand exchange solution consisted of a 50 mM solution of BT in methanol, and in the final washing step, the substrate was immersed in methanol. The TBACl-BT-capped films were obtained by depositing 12 alternating layers of each material, resulting in a total film thickness of 80 ± 30 nm. The EDT-capped films were obtained by depositing four alternating layers of each material, resulting in a total film thickness of 37 ± 4 nm.
Transient Absorption
The fundamental laser beam is generated by a Yb:KGW oscillator (Light Conversion, Pharos SP), producing a 1028 nm wavelength beam with 180 fs pulse width and 5 kHz repetition rate. The fundamental laser beam is sent to a beam-splitter, separating the beam into a low-intensity and a high-intensity component. The high-intensity component (pump beam) is sent through an optical parametric amplifier (OPA, Light Conversion, Orpheus), allowing to tune the wavelength of the beam between 312 and 1330 nm. The pump beam is then sent through a mechanical chopper rotating at 2.5 kHz, blocking one out of two laser pulses. After transmission through the chopper, the pump beam is sent to the sample, photoexciting it. The low-intensity beam (probe beam) is converted into a broad-band continuum (500–1600 nm) by transmission through a sapphire crystal and then focused on the sample, where it overlaps spatially with the pump beam. After transmission through the sample, the probe beam is collected by a detector (Ultrafast Systems, Helios), measuring the spectrum of the probe pulse transmitted by the sample in the presence of a pump pulse (Ion) and the probe pulse transmitted when the pump beam was blocked (Ioff). The differential absorbance is calculated via ΔA = −ln(Ion/Ioff). The chirp of the probe spectrum, produced by the different group velocities of different probe wavelengths, is corrected via a polynomial fit to the coherent artifact response.50 The QD films were loaded into an air-tight sample holder inside a nitrogen-filled glovebox.
Spectroelectrochemistry
The spectroelectrochemical measurements were performed inside a nitrogen-filled glovebox. The samples were placed in an electrochemical cell, having the ITO substrate coated with the QD film as the working electrode, a Ag wire as the quasireference electrode, and a Pt plate as the counter electrode. The cell was placed inside a cuvette, immersed in an electrolyte (0.1 M lithium perchlorate solution in acetonitrile), and connected to a potentiostat (Autolab, PGSTAT128N). Acetonitrile was dried before use in an Innovative Technology PureSolv Micro column. The quasireference electrode was calibrated by measuring the redox potential of the ferrocene/ferrocenium redox couple, with a scan speed of the applied potential of 50 mV/s. Absorbance measurements were performed by measuring the light transmitted through the sample for different applied potentials, using a deuterium halogen source (DH-2000, Ocean Optics) and measuring it on a USB2000+ spectrometer (Ocean Optics). Cyclic voltammograms measured on the PbSe–CdSe QD films were recorded with a scan speed of 10 mV/s.
Results and Discussion
Film Fabrication
We fabricated QD heterojunction films composed of alternating layers of PbSe and CdSe QDs via layer-by-layer dip-coating of quartz or ITO substrates in QD solutions. Two solutions of 2.3 nm PbSe QDs and 4.5 nm CdSe QDs were used for dip-coating, which is a combination of sizes that we previously characterized to have a type-I alignment with a small CB offset.34,35 Each QD layer is ligand-exchanged by dipping the substrate in a solution of the new ligand in a polar solvent (see Methods). For the TBACl-BT treatment, the layer-by-layer deposition technique used, which allows intimate contact between the CdSe and PbSe QDs, could lead to exposure of already ligand-exchanged QDs to the wrong ligand type, leading to the presence of benzenethiolate ligands on CdSe QDs and Cl– ligands on PbSe QDs. However, the electron affinity shifts observed in the spectroelectrochemical measurements are consistent with a predominant benzenethiolate passivation of PbSe QDs and Cl– passivation of CdSe QDs.32
Spectroelectrochemical Determination of Band Alignment
To determine the relative position of the conduction bands in the PbSe and CdSe QDs, we performed spectroelectrochemical measurements on the heterojunction QD films assembled on a conductive ITO substrate. In these measurements, the sample is immersed in an electrolyte solution (0.1 M lithium perchlorate in acetonitrile) and a potential is applied between the substrate and a Ag wire quasireference electrode, while the absorbance of the sample is recorded. The potential of the quasireference electrode was calibrated with the ferrocene/ferrocenium redox couple (see the Supporting Information, Figure S1). All potentials are reported with respect to vacuum.
When the Fermi level of the system is raised above the CB edge of one of the QD components, charges are injected into the CB of the QDs, leading to a reduction of the band-edge absorption, an effect called absorption bleach.51−58 The measurement of the development of the absorption bleach of the two QD components as a function of the applied potential allows the band alignment in the heterojunction to be to reconstructed: if the PbSe bleach develops at more positive potentials than the CdSe bleach, the PbSe band edges lie within the CdSe band gap and the system is in a type-I alignment; vice versa, if the CdSe bleach develops first, the system is in a type-II alignment.34
Figure 2 shows the results of spectroelectrochemical measurements performed on an EDT-treated sample and on a TBACl-BT-treated sample. Figure 2a shows the differential absorbance of the EDT-treated film for different applied potentials. As the potential is decreased, a negative bleach feature appears at the PbSe band-gap wavelength (1030 nm), followed at lower potential by a bleach feature at the CdSe band gap (603 nm). In contrast, for the TBACl-BT sample (Figure 2b), the first absorption bleach is observed at the CdSe QD band gap, while a PbSe absorption bleach becomes apparent at lower potential.
Figure 2.

Spectroelectrochemical determination of band alignment in EDT and TBACl/BT heterojunction films. (a, b) Differential absorbance spectra of the (a) EDT-treated and (b) TBACl/BT films for different applied potentials. Dotted lines indicate fits of the data around the band-gap energies of the two QD components. For the (a) EDT-treated film, the PbSe bleach appears at higher potential vs vacuum than the CdSe bleach, while the order of appearance is reversed in the (b) TBACl/BT film. (c, d) Amplitude of the bleach component extracted from a fit to the data (see the Supporting Information), plotted as a function of the applied potential. Dotted lines show error function fits to the experimental data (see Table 1). (e, f) Schemes representing the band alignment of the QD heterojunctions, highlighting that PbSe is the first QD material to bleach in the (e) type-I film, while CdSe QD bleach first in the (f) type-II film.
Beside the presence of the band-edge bleaches, the measurement shows a broad induced absorption background, which is often observed in similar measurements on QD films34,57 and is likely related to increased scattering and reflection due to the reduction of surface cations.59 To disentangle the potential dependence of the bleach features from the increase in the background, we performed a fit of the signal in a spectral range around each bleach feature, employing a Gaussian shape for the bleach and describing the slowly varying background with a straight line (dotted lines in Figure 2a,b). Figure 2c,d shows the amplitude of the two bleach features as a function of the potential in the EDT (Figure 2c) and in the TBACl-BT sample (Figure 2d), clearly demonstrating that the PbSe bleach appears at higher potential in the EDT sample, while the CdSe bleach appears at higher potential in the TBACl-BT sample.
Figure 2e,f displays the energy alignment inferred from the spectroelectrochemical results. In the EDT film, the PbSe CB edge lies at lower energy (higher potential) than the CdSe CB edge. Since the PbSe band gap is smaller than the CdSe band gap, the PbSe CB edges fall within the CdSe band gap, as shown in Figure 2e. Vice versa, the fact that in the TBACl-BT film the CdSe bleach appears at lower energy (higher potential) than the PbSe bleach implies a type-II band alignment, as shown in Figure 2f. These results demonstrate that the band alignment in CdSe–PbSe QD heterojunction films can be switched between type-I and type-II via ligand treatments.
As shown in Figure 2d, the CdSe bleach amplitude in the TBACl-BT film shows a second increase in the bleach amplitude below 3.7 V, occurring in the same potential range where the PbSe bleach develops. The second bleach increase could be caused by pockets of CdSe QDs isolated from the rest of the CdSe QDs network by surrounding PbSe QDs. When electrons are present only in the CdSe CB, electron injection in the isolated CdSe QD is slow and does not occur efficiently within the time scale of our experiment. When the Fermi level is raised above the PbSe CB, electrons can be injected into the PbSe QDs, increasing the conductivity of the entire film and allowing facile injection of electrons into the isolated CdSe QDs.
The potential dependence of the bleach amplitudes is well described by an error function (dotted black lines in Figure 2c,d), describing progressive filling of a Gaussian density of states centered at the CB energy (see the Supporting Information). Fitting the potential dependence of the bleach leads to a quantitative determination of the CB energy for the two ligand treatments, listed in Table 1.
Table 1. Conduction Band (CB) Energies for PbSe and CdSe QDs in the EDT and TBACl-BT Films, Determined via an Error Function Fit to the Bleach Amplitude, Shown in Figure 2c,d.
| PbSe CB (eV) | CdSe CB (eV) | ΔE (eV) | |
|---|---|---|---|
| EDT film | –4.2 | –3.9 | 0.3 |
| TBACl-BT film | –3.6 | –3.9 | –0.3 |
The CB energies extracted from the fit show that the band offset, ΔE = ECB (CdSe) – ECB (PbSe), changes sign between the two ligand treatments, shifting by 0.6 eV. The change is mostly due to the shift of the PbSe CB edge, while the CdSe CB edge is the same in the two films. The shift of the PbSe CB edge is twice as large as the shift observed by Brown et al. for PbS QDs.32 The difference is perhaps associated with the difference in material (PbSe vs PbS). Furthermore, the energy required to inject an electron inside a material with the higher CB edge can be greater than the electron affinity of the material, e.g., due to electrostatic repulsion between the injected electrons.34 While Boehme et al. have estimated that these effects contribute negligibly to the energy of the first electrons electrochemically injected in the system,34 they could lead to an overestimation of the offset between the conduction band-edge energies of the two QD components. However, the relative position of the CBs, and hence the type-I vs type-II band alignment, can be trusted.
To summarize, spectroelectrochemical measurements on PbSe–CdSe heterojunction QD films demonstrate that the band alignment in PbSe–CdSe heterojunctions can be changed from a type-I to a type-II alignment via ligand exchange with a BT treatment in the PbSe layers and with a TBACl treatment in the CdSe layers. The absolute energy position of the CB edges suggests that the shift is dominated by a change in the PbSe CB energy.
Electron Dynamics in Type-II QD Heterojunctions Measured by Transient Absorption Spectroscopy
In a type-II band alignment, photoexcited electrons and holes should transfer to opposite sides of the heterojunction. In particular, in the type-II structure assigned to the TBACl-BT-treated film, photoexcited electrons are expected to minimize their energy by occupying the CB edge of the CdSe QD component. Thus, photoexcitation of either the PbSe QDs or the CdSe QDs should lead to electrons bleaching the CdSe band edge. Furthermore, the CdSe QD band-edge bleach induced by the electron should persist long after the initial thermalization, as electron–hole recombination should be slowed down by the spatial separation of the charges. To test the energy alignment determined above and to verify that charge separation takes place, we performed transient absorption (TA) measurements on a BT-TBACl film on a quartz substrate, photoexciting the system either below or above the band gap of the CdSe QDs.
Figure 3a shows the results of a TA measurement of the TBACl-BT-treated type-II film, excited at 700 nm to obtain selective photoexcitation of the PbSe QD component, with an absorbed photon fluence of 1.96 × 1013 photons/cm2 per pulse. At early times after photoexcitation, the signal shows a bleach at the CdSe band gap (blue in the color map), indicating the presence of electrons at the CdSe CB edge. This bleach feature decays rapidly and is replaced by a negative–positive antisymmetric signal, resembling the first derivative of the first absorption peak (the red trace in Figure 3d). The derivative-like feature is associated with an electrostatic shift of the QD absorption60,61 and can be linked to the presence of charge carriers close to the CdSe QDs but not directly populating the CdSe CB edge. We associate the rapid decay of the bleach in the type-II system with the presence of a high density of electronic trap states. The presence of sub-ns trapping was also shown in our previous TA study on type-I CdSe–PbSe QD heterojunction films.35 Very fast electron trapping was also observed in CdSe-CdTe type-II QD heterojunction films. In that case, electrochemical trap filling was required to observe efficient electron transfer from CdTe to CdSe QDs.62
Figure 3.

Transient absorption (TA) measurements of electron dynamics in CdSe–PbSe QD heterojunction films upon selective PbSe excitation. (a) TA color map of the TBACl/BT film, excited at 700 nm with an absorbed fluence of 1.96 × 101 3 photons/cm2 per pulse. The CdSe band-edge signal is characterized by a bleach feature that decays rapidly and is replaced by a derivative-like feature. (b) TA color map of the TBACl/BT film, excited at 700 nm with an absorbed fluence of 8.58 × 1014 photons/cm2 per pulse. A long-lived bleach is present at the CdSe band edge. (c) TA color map of the EDT film, excited at 700 nm with an absorbed fluence of 4.24 × 1014 photons/cm2 per pulse. The CdSe bleach is short-lived. (d) Absorbed fluence-normalized TA signal of the TBACl/BT film excited at 700 nm with different photon fluences, obtained at a delay time of 200 ps. With increasing photon fluence, the signal evolves from a derivative-like feature to a Gaussian bleach. (e) Absorbed fluence-normalized TA signal of the EDT film excited at 700 nm with different photon fluences, obtained at a delay time of 200 ps. The shape of the signal at the CdSe band gap is weakly influenced by the absorbed fluence. (f) Time dependence of the energy-integrated band gap signal (see the Supporting Information) of the EDT sample (red) and TBACl-BT sample (blue), for low (dotted-line) and high (continuous line) fluence. As the fluence increases, the bleach lifetime in the TBACl-BT film increases dramatically, while in the EDT sample, it is hardly affected. (g–i) Schemes representing electron transfer processes occurring in the (g, h) type-II film and in the (i) type-I film. For the type-II film, at low fluence, electrons are rapidly trapped at interfacial states (g), and higher fluence is needed to saturate the trap states and allow long-lived electron occupation of the CdSe CB edge (h). In the (i) type-I film, the electron dynamics is described by hot-electron transfer to the CdSe QDs, followed by back-transfer to the PbSe QDs.35
In analogy with the approach used for the CdSe-CdTe QD heterojunction films, in this work, we sought to fill the traps by increasing the excitation fluence. We find that the fast decay dynamics of the CdSe band-edge bleach changes significantly as a function of excitation fluence. A TA measurement at a high absorbed photon fluence of 8.58 × 1014 photons/cm2 per pulse is shown in Figure 3b. In this case, the CdSe band-edge response is characterized by a long-lived bleach feature, eventually decaying with a half-life of 400 ps, comparable with the band-edge bleach lifetime of a CdSe-only film (see also Figure 4a,b and the Supporting Information).
Figure 4.

Transient absorption (TA) measurements of electron dynamics upon direct CdSe excitation. (a) TA color maps of a TBACl/BT heterojunction film (left), a CdSe-only film treated with TBACl (middle), and an EDT heterojunction film (right) excited at 570 nm, above the onset of CdSe absorption. The minimum of each color scale is set to the lowest differential absorbance value. The TBACl/BT film shows the longest bleach lifetime, while the bleach of the EDT film is the shortest-lived. (b) Normalized bleach amplitude for the TA measurements displayed in (a), quantitatively showing the difference in bleach decay between the films. (c) Proposed model for the dynamics of bleach decay upon direct CdSe excitation. The increased bleach lifetime in the TBACl/BT film is attributed to spatial separation between electrons and holes across the type-II heterojunction. In the type-I EDT-treated film, instead, electron transfer contributes to the depopulation of the CdSe band edges, increasing the rate of bleach decay.
Figure 3c shows the differential absorbance of an EDT-treated type-I film obtained with measurement conditions similar to those in Figure 3b (700 nm excitation with an absorbed photon fluence of 4.24 × 1014 photons/cm2 per pulse). As previously reported for EDT-treated PbSe–CdSe QD films, excitation below the CdSe QD absorption onset results in a short-lived CdSe QD bleach, originating from hot-electron transfer from the PbSe QDs to the CdSe QDs, followed by back-transfer to the PbSe QDs.35 While the CdSe QD absorption bleach in the type-II film decays with a half-life of 400 ps (Figure 3b), the CdSe QD bleach in this type-I film decays with a short half-lifetime of 3.8 ps. This short decay time is attributed to fast electron back-transfer in the type-I heterojunction, confirming the difference in band alignment between the two films.
Figure 3d shows the fluence-normalized TA spectra of the TBACl-BT sample obtained 200 ps after photoexcitation at the CdSe QD band gap for varying absorbed photon fluences. While at low fluence the signal is dominated by the derivative-like feature, due to the trapping of photoexcited carriers, as the fluence increases, the signal becomes dominated by a band-edge bleach, indicating the presence of electrons at the CdSe CB edge. In contrast to the type-II TBACl-BT sample, the type-I EDT sample shows little variation of the fluence-normalized band-edge TA signal with fluence, as shown in Figure 3e.
Figure 3f shows the time dependence of the bleach signal for the TBACl-BT sample (blue lines) and EDT sample (red lines), measured in the low fluence (dotted lines) and high fluence (continuous lines) regimes. The TA signal, here labeled ΔA*, is integrated over a spectral range centered at the CdSe QD band gap (581–652 nm), to remove the contribution of the antisymmetric positive–negative shift feature from the time dependence of the bleach. The increase in absorbed photon fluence causes only a minor variation in the bleach lifetime for the EDT sample, while the dynamics of the bleach in the TBACl-BT film changes dramatically, going from a bleach decay half-life of 2.7 to 400 ps.
Our interpretation of the TA measurements on the type-II TBACl-BT sample is summarized by the schemes in Figure 3g,h. Figure 3g shows the dynamics of the photoexcited electron in the low-fluence regime, in which carrier trapping and electron transfer both occur on a picosecond time scale, leading to a short-lived CdSe QD bleach and the development of a pronounced derivative-like feature at later times. However, in the high-fluence regime (Figure 3h), the increased number of trapped electrons can saturate the density of trap states, allowing additionally photogenerated electrons to escape the trapping process and remain at the CdSe band edge. The power dependence of the bleach lifetime distinguishes the type-II TBACl-BT sample from the type-I EDT sample (Figure 3i). In the latter case, the bleach dynamics is dominated by the back-transfer of hot electrons from CdSe QDs to PbSe QDs, which does not depend on the excitation fluence.
Since the fast trapping process is not observed in a PbSe-only or CdSe-only QD film (see the Supporting Information, Figures S4 and S5), we tentatively assign these trap states to interfacial states, occurring at the PbSe–CdSe heterojunction interface. The presence of localized states at the interface between Pb chalcogenides and Cd chalcogenides has been predicted by density functional theory (DFT) simulations of PbS–CdS Janus particles63 and of PbSe QDs embedded in a CdSe matrix,63 suggesting that epitaxial contact between PbSe and CdSe nanostructures leads to the formation of localization centers. However, we performed DFT calculations of PbSe–CdSe QD-dimers capped with thiolate ligands on the PbSe QD and Cl– ligands on the CdSe QD (see the Supporting Information, Figure S6), displaying a clean band gap, i.e., free of localized interfacial states. This suggests that CdSe–PbSe QD heterointerfaces do not necessarily lead to the formation of localized interfacial states and that the presence of fast electron trapping in the heterojunction films might be associated with insufficient ligand passivation.
To further demonstrate the effect of the band offset on the lifetime of photogenerated electrons, we excited mixed CdSe–PbSe and CdSe-only QD films above the CdSe QD band gap, at 570 nm. Figure 4a compares the resulting TA response of the TBACl/BT heterojunction film (Figure 4a, left) with the response of a CdSe-only film (Figure 4a, center) and of an EDT-treated heterojunction film (Figure 4a, right). In each color map, the blue edge of the color scale is set to the minimum ΔA value. Figure 4b shows the time dependence of the normalized bleach, highlighting the differences between the three films. It is evident that the bleach lifetime is longest in the TBACl/BT type-II heterojunction film, shortest in the EDT type-I heterojunction film, and intermediate in the CdSe-only film.
The differences in the bleach decay can be attributed to differences in the carrier recombination rate induced by charge transfer, as depicted in Figure 4c. For the type-II film (left panel), an electron–hole pair, generated on CdSe QD in contact with a PbSe QD, is separated, as the hole transfers to the PbSe QDs. Charge separation decreases the rate of recombination and increases the electron lifetime with respect to the CdSe-only film. On the contrary, in the type-I film, charge transfer between adjacent CdSe and PbSe QDs depopulates the band edges of the CdSe QDs, thus increasing the rate of band-edge bleach decay. These TA measurements, with excitation both above and below the CdSe QD band gap, confirm the type-I alignment of the EDT-treated QD heterojunction film and the type-II alignment of the TBACl-BT-treated film, in line with the results of the spectroelectrochemical measurements.
Conclusions
Using spectroelectrochemistry and ultrafast transient absorption spectroscopy, we have shown that the band alignment in CdSe/PbSe QD heterojunction films can be tuned from type-I, when the entire film is treated with ethanedithiol, to type-II alignment, when the PbSe QDs are treated with benzenethiol and the CdSe QDs with TBACl. Spectroelectrochemical measurements on TBACl-BT-treated and on EDT-treated films show a clear inversion in the order of the PbSe and CdSe CB edge levels. Transient absorption measurements performed with a high photon fluence on the TBACl-BT-treated film show the presence of a long-lived CdSe QD bleach developing upon selective PbSe QD excitation, indicative of electron transfer from the PbSe QD to the CdSe QD conduction band edge. Furthermore, direct CdSe QD photoexcitation in the TBACl/BT film results in a longer band-edge bleach lifetime compared to a CdSe-only QD film, characteristic of reduced recombination of spatially separated electrons and holes. Our results demonstrate the possibility to control the energy alignment of QD heterojunctions via ligand exchange, providing an additional handle to control the energetics of QD-based devices. On the other hand, low-fluence TA measurements reveal the presence of a high density of trap states. This highlights the importance of a greater understanding of the electronic passivation of QD heterojunctions to exploit their potential for optoelectronic applications.
Acknowledgments
This research is funded by the European Research Council Horizon 2020 ERC Grant agreement no. 678004 (Doping on Demand), STW (project no. 13903, Stable and Non-Toxic Nanocrystal Solar Cells), and NWO (Vidi grant, no. 723.013.002). This work was sponsored by NWO Exact and Natural Sciences for the use of supercomputer facilities. This work was carried out on the Dutch National e-Infrastructure with the support of the SURF Cooperative. We thank I. Infante for his assistance in the DFT simulations and for the helpful discussions on the interpretation of the DFT results.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcc.9b09470.
Calibration of the Ag wire used in the spectroelectrochemical measurement; fit of the QD bleach in the spectroelectrochemical data; effect of power on the decay lifetime of the bleach for the EDT and TBACl-BT samples; transient absorption measurements of PbSe-only and CdSe-only QD films; and density functional theory calculations on a PbSe/CdSe dimer (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Brus L. E. Electron-Electron and Electron-Hole Interactions in Small Semiconductor Crystallites: The Size Dependence of the Lowest Excited Electronic State. J. Chem. Phys. 1984, 80, 4403–4409. 10.1063/1.447218. [DOI] [Google Scholar]
- Ekimov A. I.; Efros A. L.; Onushchenko A. A. Quantum Size Effect in Semiconductor Microcrystals. Solid State Commun. 1985, 56, 921–924. 10.1016/S0038-1098(85)80025-9. [DOI] [Google Scholar]
- Colvin V. L.; Alivisatos A. P.; Tobin J. G. Valence-Band Photoemission from a Quantum-Dot System. Phys. Rev. Lett. 1991, 66, 2786–2789. 10.1103/PhysRevLett.66.2786. [DOI] [PubMed] [Google Scholar]
- Ekimov A. I.; Hache F.; Schanne-Klein M. C.; Ricard D.; Flytzanis C.; Kudryavtsev I. A.; Yazeva T. V.; Rodina A. V.; Efros A. L. Absorption and Intensity-Dependent Photoluminescence Measurements on CdSe Quantum Dots: Assignment of the First Electronic Transitions. J. Opt. Soc. Am. B 1993, 10, 100–107. 10.1364/JOSAB.10.000100. [DOI] [Google Scholar]
- Kagan C. R.; Lifshitz E.; Sargent E. H.; Talapin D. V. Building Devices from Colloidal Quantum Dots. Science 2016, 353, aac5523 10.1126/science.aac5523. [DOI] [PubMed] [Google Scholar]
- Colvin V. L.; Schlamp M. C.; Alivisatos A. P. Light-Emitting Diodes Made from Cadmium Selenide Nanocrystals and a Semiconducting Polymer. Nature 1994, 370, 354–357. 10.1038/370354a0. [DOI] [Google Scholar]
- Kagan C. R.; Murray C. B.; Nirmal M.; Bawendi M. Electronic Energy Transfer in CdSe Quantum Dot Solids. Phys. Rev. Lett. 1996, 76, 1517 10.1103/PhysRevLett.76.1517. [DOI] [PubMed] [Google Scholar]
- Yu D.; Wang C.; Guyot-Sionnest P. n-Type Conducting CdSe Nanocrystal Solids. Science 2003, 300, 1277–1280. 10.1126/science.1084424. [DOI] [PubMed] [Google Scholar]
- Law M.; Luther J. M.; Song Q.; Hughes B. K.; Perkins C. L.; Nozik A. J. Structural, Optical, and Electrical Properties of PbSe Nanocrystal Solids Treated Thermally or with Simple Amines. J. Am. Chem. Soc. 2008, 130, 5974–5985. 10.1021/ja800040c. [DOI] [PubMed] [Google Scholar]
- Talapin D. V.; Murray C. B. PbSe Nanocrystal Solids for n- and p-Channel Thin Film Field-Effect Transistors. Science 2005, 310, 86–89. 10.1126/science.1116703. [DOI] [PubMed] [Google Scholar]
- Jarosz M. V.; Porter V. J.; Fisher B. R.; Kastner M. A.; Bawendi M. G. Photoconductivity Studies of Treated CdSe Quantum Dot Films Exhibiting Increased Exciton Ionization Efficiency. Phys. Rev. B 2004, 70, 195327 10.1103/PhysRevB.70.195327. [DOI] [Google Scholar]
- Luther J. M.; Law M.; Song Q.; Perkins C. L.; Beard M. C.; Nozik A. J. Structural, Optical and Electrical Properties of Self-Assembled Films of PbSe Nanocrystals Treated with 1,2-Ethanedithiol. ACS Nano 2008, 2, 271–280. 10.1021/nn7003348. [DOI] [PubMed] [Google Scholar]
- Kovalenko M. V.; Scheele M.; Talapin D. V. Colloidal Nanocrystals with Molecular Metal Chalcogenide Surface Ligands. Science 2009, 324, 1417–1420. 10.1126/science.1170524. [DOI] [PubMed] [Google Scholar]
- Crisp R. W.; Callahan R.; Reid O. G.; Dolzhnikov D. S.; Talapin D. V.; Rumbles G.; Luther J. M.; Kopidakis N. Photoconductivity of CdTe Nanocrystal-Based Thin Films: Te2– Ligands Lead to Charge Carrier Diffusion Lengths over 2 μm. J. Phys. Chem. Lett. 2015, 6, 4815–4821. 10.1021/acs.jpclett.5b02252. [DOI] [PubMed] [Google Scholar]
- Crisp R. W.; Kroupa D. M.; Marshall A. R.; Miller E. M.; Zhang J.; Beard M. C.; Luther J. M. Metal Halide Solid-State Surface Treatment for High Efficiency PbS and PbSe QD Solar Cells. Sci. Rep. 2015, 5, 9945 10.1038/srep09945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan A.; Houtepen A.; Crisp R. Quantum Dot Solar Cells: Small Beginnings Have Large Impacts. Appl. Sci. 2018, 8, 1867 10.3390/app8101867. [DOI] [Google Scholar]
- Zhang H.; Kurley J. M.; Russell J. C.; Jang J.; Talapin D. V. Solution-Processed, Ultrathin Solar Cells from CdCl3–(-)-Capped CdTe Nanocrystals: The Multiple Roles of CdCl3– Ligands. J. Am. Chem. Soc. 2016, 138, 7464–7467. 10.1021/jacs.6b03240. [DOI] [PubMed] [Google Scholar]
- Semonin O. E.; Luther J. M.; Choi S.; Chen H.-Y.; Gao J.; Nozik A. J.; Beard M. C. Peak External Photocurrent Quantum Efficiency Exceeding 100% Via MEG in a Quantum Dot Solar Cell. Science 2011, 334, 1530–1533. 10.1126/science.1209845. [DOI] [PubMed] [Google Scholar]
- Sanehira E. M.; Marshall A. R.; Christians J. A.; Harvey S. P.; Ciesielski P. N.; Wheeler L. M.; Schulz P.; Lin L. Y.; Beard M. C.; Luther J. M. Enhanced Mobility CsPbI3 Quantum Dot Arrays for Record-Efficiency, High-Voltage Photovoltaic Cells. Sci. Adv. 2017, 3, eaao4204 10.1126/sciadv.aao4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung D. S.; Lee J.-S.; Huang J.; Nag A.; Ithurria S.; Talapin D. V. Low Voltage, Hysteresis Free, and High Mobility Transistors from All-Inorganic Colloidal Nanocrystals. Nano Lett. 2012, 12, 1813–1820. 10.1021/nl203949n. [DOI] [PubMed] [Google Scholar]
- Kagan C. R.; Mitzi D. B.; Dimitrakopoulos C. D. Organic-Inorganic Hybrid Materials as Semiconducting Channels in Thin-Film Field-Effect Transistors. Science 1999, 286, 945–947. 10.1126/science.286.5441.945. [DOI] [PubMed] [Google Scholar]
- Bisri S. Z.; Piliego C.; Yarema M.; Heiss W.; Loi M. A. Low Driving Voltage and High Mobility Ambipolar Field-Effect Transistors with PbS Colloidal Nanocrystals. Adv. Mater. 2013, 25, 4309–4314. 10.1002/adma.201205041. [DOI] [PubMed] [Google Scholar]
- Shirasaki Y.; Supran G. J.; Bawendi M. G.; Bulović V. Emergence of Colloidal Quantum-Dot Light-Emitting Technologies. Nat. Photonics 2013, 7, 13–23. 10.1038/nphoton.2012.328. [DOI] [Google Scholar]
- Lim J.; Park Y. S.; Wu K.; Yun H. J.; Klimov V. I. Droop-Free Colloidal Quantum Dot Light-Emitting Diodes. Nano Lett. 2018, 18, 6645–6653. 10.1021/acs.nanolett.8b03457. [DOI] [PubMed] [Google Scholar]
- Klimov V. I.; Mikhailovsky A. A.; Xu S.; Malko A.; Hollingsworth J. A.; Leatherdale C. A.; Eisler H.; Bawendi M. G. Optical Gain and Stimulated Emission in Nanocrystal Quantum Dots. Science 2000, 290, 314–317. 10.1126/science.290.5490.314. [DOI] [PubMed] [Google Scholar]
- Klimov V. I.; Ivanov S. A.; Nanda J.; Achermann M.; Bezel I.; McGuire J. A.; Piryatinski A. Single-Exciton Optical Gain in Semiconductor Nanocrystals. Nature 2007, 447, 441–446. 10.1038/nature05839. [DOI] [PubMed] [Google Scholar]
- Fan F.; et al. Continuous-Wave Lasing in Colloidal Quantum Dot Solids Enabled by Facet-Selective Epitaxy. Nature 2017, 544, 75–79. 10.1038/nature21424. [DOI] [PubMed] [Google Scholar]
- Lim J.; Park Y.-S.; Klimov V. I. Optical Gain in Colloidal Quantum Dots Achieved with Direct-Current Electrical Pumping. Nat. Mater. 2018, 17, 42–49. 10.1038/nmat5011. [DOI] [PubMed] [Google Scholar]
- Konstantatos G.; Howard I.; Fischer A.; Hoogland S.; Clifford J.; Klem E.; Levina L.; Sargent E. H. Ultrasensitive Solution-Cast Quantum Dot Photodetectors. Nature 2006, 442, 180–183. 10.1038/nature04855. [DOI] [PubMed] [Google Scholar]
- Keuleyan S.; Lhuillier E.; Brajuskovic V.; Guyot-Sionnest P. Mid-Infrared HgTe Colloidal Quantum Dot Photodetectors. Nat. Photonics 2011, 5, 489–493. 10.1038/nphoton.2011.142. [DOI] [Google Scholar]
- Deng Z.; Jeong K. S.; Guyot-Sionnest P. Colloidal Quantum Dots Intraband Photodetectors. ACS Nano 2014, 8, 11707–11714. 10.1021/nn505092a. [DOI] [PubMed] [Google Scholar]
- Brown P. R.; Kim D.; Lunt R. R.; Zhao N.; Bawendi M. G.; Grossman J. C.; Bulović V. Energy Level Modification in Lead Sulfide Quantum Dot Thin Films through Ligand Exchange. ACS Nano 2014, 8, 5863–5872. 10.1021/nn500897c. [DOI] [PubMed] [Google Scholar]
- Kroupa D. M.; Voros M.; Brawand N. P.; McNichols B. W.; Miller E. M.; Gu J.; Nozik A. J.; Sellinger A.; Galli G.; Beard M. C. Tuning Colloidal Quantum Dot Band Edge Positions through Solution-Phase Surface Chemistry Modification. Nat. Commun. 2017, 8, 15257 10.1038/ncomms15257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehme S. C.; Vanmaekelbergh D.; Evers W. H.; Siebbeles L. D. A.; Houtepen A. J. In Situ Spectroelectrochemical Determination of Energy Levels and Energy Level Offsets in Quantum-Dot Heterojunctions. J. Phys. Chem. C 2016, 120, 5164–5173. 10.1021/acs.jpcc.5b12016. [DOI] [Google Scholar]
- Grimaldi G.; et al. Hot-Electron Transfer in Quantum-Dot Heterojunction Films. Nat. Commun. 2018, 9, 2310 10.1038/s41467-018-04623-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang C.-H. M.; Brown P. R.; Bulović V.; Bawendi M. G. Improved Performance and Stability in Quantum dot Solar Cells through Band Alignment engineering. Nat. Mater. 2014, 13, 796–801. 10.1038/nmat3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santra P. K.; Palmstrom A. F.; Tanskanen J. T.; Yang N.; Bent S. F. Improving Performance in Colloidal Quantum Dot Solar Cells by Tuning Band Alignment through Surface Dipole Moments. J. Phys. Chem. C 2015, 119, 2996–3005. 10.1021/acs.jpcc.5b00341. [DOI] [Google Scholar]
- Soreni-Harari M.; Yaacobi-Gross N.; Steiner D.; Aharoni A.; Banin U.; Millo O.; Tessler N. Tuning Energetic Levels in Nanocrystal Quantum Dots through Surface Manipulations. Nano Lett. 2008, 8, 678–684. 10.1021/nl0732171. [DOI] [PubMed] [Google Scholar]
- Timp B. A.; Zhu X. Y. Electronic Energy Alignment at the PbSe Quantum Dots/Zno(1010̅) Interface. Surf. Sci. 2010, 604, 1335–1341. 10.1016/j.susc.2010.04.026. [DOI] [Google Scholar]
- Munro A. M.; Zacher B.; Graham A.; Armstrong N. R. Photoemission Spectroscopy of Tethered CdSe Nanocrystals: Shifts in Ionization Potential and Local Vacuum Level as a Function of Nanocrystal Capping Ligand. ACS Appl. Mater. Interfaces 2010, 2, 863–869. 10.1021/am900834y. [DOI] [PubMed] [Google Scholar]
- Barkhouse D. A. R.; Debnath R.; Kramer I. J.; Zhitomirsky D.; Pattantyus-Abraham A. G.; Levina L.; Etgar L.; Grätzel M.; Sargent E. H. Depleted Bulk Heterojunction Colloidal Quantum Dot Photovoltaics. Adv. Mater. 2011, 23, 3134–3138. 10.1002/adma.201101065. [DOI] [PubMed] [Google Scholar]
- Urban J. J.; Talapin D. V.; Shevchenko E. V.; Kagan C. R.; Murray C. B. Synergism in Binary Nanocrystal Superlattices Leads to Enhanced p-Type Conductivity in Self-Assembled PbTe/Ag2Te Thin Films. Nat. Mater. 2007, 6, 115–121. 10.1038/nmat1826. [DOI] [PubMed] [Google Scholar]
- Kramer I. J.; et al. Ordered Nanopillar Structured Electrodes for Depleted Bulk Heterojunction Colloidal Quantum Dot Solar Cells. Adv. Mater. 2012, 24, 2315–2319. 10.1002/adma.201104832. [DOI] [PubMed] [Google Scholar]
- Cirloganu C. M.; Padilha L. A.; Lin Q.; Makarov N. S.; Velizhanin K. A.; Luo H.; Robel I.; Pietryga J. M.; Klimov V. I. Enhanced Carrier Multiplication in Engineered Quasi-Type-II Quantum Dots. Nat. Commun. 2014, 5, 4148 10.1038/ncomms5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroupa D. M.; et al. Enhanced Multiple Exciton Generation in PbS|CdS Janus-Like Heterostructured Nanocrystals. ACS Nano 2018, 12, 10084–10094. 10.1021/acsnano.8b04850. [DOI] [PubMed] [Google Scholar]
- van Embden J.; Mulvaney P. Nucleation and Growth of CdSe Nanocrystals in a Binary Ligand System. Langmuir 2005, 21, 10226–10233. 10.1021/la051081l. [DOI] [PubMed] [Google Scholar]
- Steckel J. S.; Yen B. K.; Oertel D. C.; Bawendi M. G. On the Mechanism of Lead Chalcogenide Nanocrystal Formation. J. Am. Chem. Soc. 2006, 128, 13032–13033. 10.1021/ja062626g. [DOI] [PubMed] [Google Scholar]
- Moreels I.; Lambert K.; De Muynck D.; Vanhaecke F.; Poelman D.; Martins J. C.; Allan G.; Hens Z. Composition and Size-Dependent Extinction Coefficient of Colloidal PbSe Quantum Dots. Chem. Mater. 2007, 19, 6101–6106. 10.1021/cm071410q. [DOI] [Google Scholar]
- Jasieniak J.; Smith L.; van Embden J.; Mulvaney P.; Califano M. Re-Examination of the Size-Dependent Absorption Properties of CdSe Quantum Dots. J. Phys. Chem. C 2009, 113, 19468–19474. 10.1021/jp906827m. [DOI] [Google Scholar]
- Kunneman L. T.Exciton and Charge Carrier Dynamics in Semiconductor Nanorods and Nanoplatelets; Delft University of Technology, 2015. [Google Scholar]
- Bawendi M. G.; Wilson W. L.; Rothberg L.; Carroll P. J.; Jedju T. M.; Steigerwald M. L.; Brus L. E. Electronic Structure and Photoexcited-Carrier Dynamics in Nanometer-Size CdSe Clusters. Phys. Rev. Lett. 1990, 65, 1623–1626. 10.1103/PhysRevLett.65.1623. [DOI] [PubMed] [Google Scholar]
- Dneprovskii V. S.; Klimov V.; Okorokov D. K.; Vandyshev Y. V. Strong Optical Nonlinearities and Laser Emission of Semiconductor Microcystals. Solid State Commun. 1992, 81, 227–230. 10.1016/0038-1098(92)90504-3. [DOI] [Google Scholar]
- Woggon U.; Gaponenko S.; Langbein W.; Uhrig A.; Klingshirn C. Homogeneous Linewidth of Confined Electron-Hole-Pair States in II-VI Quantum Dots. Phys. Rev. B 1993, 47, 3684–3689. 10.1103/PhysRevB.47.3684. [DOI] [PubMed] [Google Scholar]
- Wang C.; Shim M.; Guyot-Sionnest P. Electrochromic Nanocrystal Quantum Dots. Science 2001, 291, 2390–2392. 10.1126/science.291.5512.2390. [DOI] [PubMed] [Google Scholar]
- Wehrenberg B. L.; Guyot-Sionnest P. Electron and Hole Injection in PbSe Quantum Dot Films. J. Am. Chem. Soc. 2003, 125, 7806–7807. 10.1021/ja035369d. [DOI] [PubMed] [Google Scholar]
- Guyot-Sionnest P.; Wehrenberg B.; Yu D. Intraband Relaxation in CdSe Nanocrystals and the Strong Influence of the Surface Ligands. J. Chem. Phys. 2005, 123, 074709 10.1063/1.2004818. [DOI] [PubMed] [Google Scholar]
- Houtepen A. J.; Vanmaekelbergh D. Orbital Occupation in Electron-Charged CdSe Quantum-Dot Solids. J. Phys. Chem. B 2005, 109, 19634–19642. 10.1021/jp053103i. [DOI] [PubMed] [Google Scholar]
- Koole R.; Allan G.; Delerue C.; Meijerink A.; Vanmaekelbergh D.; Houtepen A. J. Optical Investigation of Quantum Confinement in PbSe Nanocrystals at Different Points in the Brillouin Zone. Small 2008, 4, 127–133. 10.1002/smll.200700632. [DOI] [PubMed] [Google Scholar]
- du Fossé I.; ten Brinck S.; Infante I.; Houtepen A. J. Role of Surface Reduction in the Formation of Traps in n-Doped II–VI Semiconductor Nanocrystals: How to Charge without Reducing the Surface. Chem. Mater. 2019, 31, 4575–4583. 10.1021/acs.chemmater.9b01395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Empedocles S. A.; Bawendi M. G. Quantum-Confined Stark Effect in Single CdSe Nanocrystallite Quantum Dots. Science 1997, 278, 2114–2117. 10.1126/science.278.5346.2114. [DOI] [PubMed] [Google Scholar]
- Trinh M. T.; Houtepen A. J.; Schins J. M.; Piris J.; Siebbeles L. D. Nature of the Second Optical Transition in PbSe Nanocrystals. Nano Lett. 2008, 8, 2112–2117. 10.1021/nl8010963. [DOI] [PubMed] [Google Scholar]
- Boehme S. C.; Walvis T. A.; Infante I.; Grozema F. C.; Vanmaekelbergh D.; Siebbeles L. D. A.; Houtepen A. J. Electrochemical Control over Photoinduced Electron Transfer and Trapping in CdSe-CdTe Quantum-Dot Solids. ACS Nano 2014, 8, 7067–7077. 10.1021/nn501985e. [DOI] [PubMed] [Google Scholar]
- Giberti F.; Voros M.; Galli G. Design of Heterogeneous Chalcogenide Nanostructures with Pressure-Tunable Gaps and without Electronic Trap States. Nano Lett. 2017, 17, 2547–2553. 10.1021/acs.nanolett.7b00283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.