

Abstract
The deluge of sequence information in the recent times provide us with an excellent opportunity to compare organisms on a large genomic scale. In this study we have tried to decipher the variation in the gene organization and structuring of a vital bacterial gene called ftsZ which codes for an integral component of the bacterial cell division, the FtsZ protein. FtsZ is homologous to tubulin protein and has been found to be ubiquitous in eubacteria. FtsZ is showing increasing promise as a target for antibacterial drug discovery. Our study of ftsZ protein from 143 different bacterial species spanning a wider range of morphological and physiological type demonstrates that the ftsZ gene of about ninety three percent of the organisms show relatively biased codon usage profile and significant GC deviation from their genomic GC content. Comparative codon usage analysis of ftsZ and a core housekeeping gene rpoB demonstrated that codon usage pattern of ftsZ CDS is shaped by natural selection to a large extent and mimics that of a housekeeping gene. We have also detected a tendency among the different organisms to utilize a core set of codons in structuring the ftsZ coding sequence. We observed that the compositional frequency of the amino acid serine in the FtsZ protein appears to be a indicator of the bacterial lifestyle. Our meticulous analysis of the ftsZ gene linked with the corresponding FtsZ protein show that there is a bias towards the use of specific synonymous codons particularly in the helix and strand regions of the multi-domain FtsZ protein. Overall our findings suggest that in an indispensable and vital protein such as FtsZ, there is an inherent tendency to maintain form for optimized performance in spite of the extrinsic variability in coding features.
Introduction
Codon usage bias (CUB) or the preference of an organism for a certain subset of codons coding for the different amino acids of polypeptides is an important evolutionary feature that has intrigued molecular biologists and evolutionists for decades [1,2]. It is a universal phenomenon observed in prokaryotes, eukaryotes [3] as well as viruses [4], and is predominantly dependent on selection, mutation, and genetic drift [5]. CUB exists as a balance between selective and neutral processes [6–10] and is shaped by several factors some of which include GC content, compositional bias, gene length, hydropathy, function, mutational bias and protein structure [11–15]. It has been found to be an important factor contributing to gene and genome evolution [15,16], and is a significant determinant of gene expression levels at the transcription level [3]. CUB has been reported to be responsible in controlling a variety of cellular processes such as translational efficiency, differential protein production and folding [17–19], and is a means to fine tune the expression of genes [17,20]. A strong CUB has been reported to be a characteristic feature of highly expressed genes [21–25]. The codon usage pattern of genes and genomes of a large number of organisms including bacteria, archaea, eukaryotes as well as viruses have been studied since the last five decades with the earliest record in PubMed [26] (https://www.ncbi.nlm.nih.gov/pubmed/) dating back to 1979 by Hasegawa et al. [27]. It has been found that codon usage pattern vary not only between genomes but also between coding sequences or genes within an organism [5,6]. In this study we have tried to decipher the variation in the gene organization and structuring of a vital bacterial gene called ftsZ which codes for an integral component of the bacterial cell division, the FtsZ protein. The process of bacterial cytokinesis is initiated by the assembly of the tubulin-like GTPase called FtsZ which is essential for bacterial cell division [28]. During bacterial cytokinesis, the FtsZ protein is recruited initially at the future division site which then polymerizes itself into a ring like structure called the Z ring [29,30] consisting of short, 30 subunits long, FtsZ protofilaments [31–33]. The Z ring provides a stage for assembly of the cell division apparatus and constricts at the leading edge of the invaginating septum [28]. The Z ring appears as a smooth, closed circular assembly under fluorescence light microscope [30,34]. The polymerization of FtsZ is dependent on GTP hydrolysis [35] and the remaining GDP bound FtsZ favours depolymerisation [36]. FtsZ is homologous to tubulin protein which acts as the building block of the microtubule cytoskeleton in eukaryotes FtsZ [37]. During cell division, FtsZ interacts with other membrane associated proteins like FtsW, FtsK, FtsQ and FtsI and helps in anchoring FtsZ to the bacterial cytoplasmic membrane [38]. FtsZ is reported to be a highly conserved protein [37] with a relative molecular mass of 40,000 and is ubiquitous in eubacteria. It is also found in the members of Euryarchaea, chloroplasts of plants and some mitochondria [39]. Higher plants have also been found to contain two distinct families of FtsZ homologues that seem to have diverged early in the evolution of plants [40]. Mutant bacteria which lacks FtsZ protein cannot divide but elongate into filamentous form. FtsZ is a vital cell-division protein in prokaryotes and is showing increasing promise as a target for antibacterial drug discovery [41]. The FtsZ protein has been projected as a potent target and has been studied extensively [42] for the discovery of next-generation antibacterial agents that can be used to counter drug-resistances to the commonly used drugs for methicillin resistant Staphylococcus aureus (MRSA), tuberculosis, and other microorganism mediated infections [43]. The ftsZ gene is regarded as an essential cell division gene in many bacteria including E. coli [44] and it has been found that the C-terminal domain for FtsZ is highly variable both in size and alignment among the different bacterial species [45]. The higher-order structure of FtsZ protein in vitro includes ribbons, sheets and bundles [46–48].
Considering the critical nature and ubiquity of the FtsZ protein encoded by the ftsZ gene, it presents a fitting proposition to decipher the codon usage pattern of the ftsZ gene across the entire eubacterial domain to find out if there exist any differential codon usage bias in the structuring of the ftsZ coding sequence among different types of bacteria. The conservedness, ubiquity and imperative nature of the FtsZ protein in a critical bacterial cellular process like cytokinesis also portrays the ftsZ gene as a perfect candidate for housekeeping gene. The term ‘housekeeping genes’ refer to those genes that are required for the upkeep of basic cellular processes for the survival of a cell [49,50]. In this study, we have extensively analysed the codon usage pattern of the ftsZ gene from a wide range of bacteria differing in terms of their lifestyle and Gram nature, and spread across about 70 families belonging to 40 orders under 20 different classes encompassing the entire eubacterial domain to detect the factors shaping codon usage bias in ftsZ, and find out if the codon usage pattern of the ftsZ gene within different types of bacteria is a random phenomenon or has it been influenced by traits such as the lifestyle of the organism [51–55]. This includes their free-living behaviour or pathogenic association with specific host organisms and ecological traits. We have also tried to unravel whether the codon structuring of the ftsZ gene is influenced by the Gram nature of the organism to a certain extent. The Gram nature of a bacterium, although primarily attributed to the cell wall construction of the bacterium, have been found to manifest a host of comparative features in the organisms ranging from simple morphology [56] to advanced physiological, biochemical, ecological [57–59] and molecular characteristics such as GC content [60,61]. We have also compared the codon usage pattern of the ftsZ gene with a known bacterial housekeeping gene rpoB, coding for the β subunit of the bacterial RNA polymerase, to examine to what extent the codon usage pattern of the ftsZ gene deviate or resemble that of a core and conserved housekeeping gene such as rpoB. We have also attempted to estimate the compositional divergence of the ftsZ coding sequences. The FtsZ protein is a very vital component of bacterial cell division that demonstrates promiscuous variability both in terms of gene sequence and amino acid composition. This compositional variability in a conserved protein such as FtsZ has been our impetus to decipher and track whether there exists the preference for a ‘core’ set of codons in coding the gene sequence across a diverse group of bacteria. In our study we have tried to explore the codon usage tendency based on the positioning of the different amino acids in the different types of structural elements of the FtsZ protein. It has been reported that codon usage can play an important role in the translation process as well as the folding behaviour of nascent polypeptides [62,63]. We have adopted a unique approach to further explore the codon usage bias profile of the ftsZ sequence by linking the codon utilization profile with the secondary structural components of the protein. Thus, we have strived to correlate the coding pattern of the ftsZ gene with the structural attributes of the FtsZ protein. We have meticulously analysed the 61 sense codons coding for the twenty standard amino acids to find out the preference of disposition of specific codons in specific secondary structural elements of the FtsZ protein.
Materials and methods
The ftsZ and rpoB CDS of 143 bacterial species spanning the entire eubacterial domain and their whole genome sequences were retrieved from the NCBI GenBank [64] sequence database. The ftsZ and rpoB coding sequences (CDS), and their corresponding amino acid sequences were extracted from the whole genome sequences of the bacteria. Details regarding the NCBI accession number of the genomes, links pertaining to the genome information page, reference to the information regarding the organism in the corresponding volumes of Bergey’s Manual of Systematic Bacteriology [65–69], and locus tag/protein id of the ftsZ and rpoB CDS is provided in S1 Table. Analysis of the different codon usage bias parameters like effective number of codons (Nc) [70], GC content, guanine and cytosine content at the first (GC1), second (GC2) and third position of the codon (GC3) [70] and hydrophobicity were also estimated using CodonW [71] and INCA 2.1 [72].
The Nc determines the degree of bias for the use of codons [2] with value ranging from 20 to 61, where lower value indicates higher codon usage bias and vice versa. It is a commonly used measure to quantify how far a gene departs from the equal usage of synonymous codons [73,74]. The GC content plays a critical role in genome evolution [75], and it has been found to range from 13% to 75% in cellular organisms [76,77]. The GC content does not remain constant throughout the genome of an organism but varies based on different regions and coding sequences of the genome. The measurement of different GC based attributes like GC content, GC1, GC2 and GC3 content thus play a significant role in analysing the genomic as well as genic organization [18,78]. The percentage of genes in the genome of an organism with Nc and GC3 less than that of both ftsZ and rpoB along with their standard score (z-score) [79,80] was also calculated to determine how far the codon usage bias of ftsZ and rpoB differ with respect to each other, and the genome of the individual organism. The standard score (z) of a raw score (x; where x could be Nc/GC3 score of a CDS) was calculated as z = (x-μ)/δ where μ is the mean of the population (or genome) and δ is the standard deviation of the population (or genome of a bacterium). The Nc–plot [70], which is a parabolic curve used to measure and explore codon usage bias, and detect the effect of base content on CUB [81] was constructed using Microsoft Office Excel 2013 version.
Statistical analysis such as non-parametric One way ANOVA on Ranks [82] was used to find out whether there is a preferred set of codon for each of the amino acid that is used in the structuring of the ftsZ coding sequences. Two factor ANOVA on codon usage of ftsZ CDS was also performed to study the frequency of the individual 61 sense codons and their interrelation with lifestyle and Gram nature of the organisms. Furthermore, a two factor ANOVA was designed to study the interrelationship of the twenty different amino acids with lifestyle and Gram nature of the bacteria. Data analysis pack of Microsoft Office Excel 2013 version was used to perform all the statistical analyses.
The degree of identity in FtsZ protein sequences among the 142 organisms considered for this study was analysed using the Windows 64-bit version of Clustal Omega 1.2.2. This application employs HMM profile-profile techniques along with seeded guide trees to produce multiple alignments [83]. For clustering of similar proteins based on their sequence similarities, the web server version of the program CD-HIT [84] (http://weizhong-lab.ucsd.edu/cdhit-web-server/cgi-bin/index.cgi) was used. Multiple sequence alignment (MSA) of the 142 ftsZ CDS was also constructed using Clustal Omega [83] and a phylogenetic tree depicting the relationship between all the considered ftsZ CDS was generated using the Molecular Evolutionary Genetics Analysis or MEGA X software graphical interface based 64-bit version for Windows [85]. The phylogenetic tree was inferred using the neighbour-joining method [86] with 1000 bootstrap replicates [87], and the evolutionary distances, measured in terms of number of base substitutions per site, were computed using the Tajima-Nei method [88]. The rate variation among sites was modeled with a gamma distribution. Visualization of the phylogenetic tree was carried out using Interactive Tree of Life (iTOL) ver. 4.4.2, a web based tool hosted at https://itol.embl.de/ [89,90]. The MSA data in FASTA format and the raw phylogenetic inference data along with bootstrap support in Newick format is provided in the supplementary files S1 MSA and S1 Phylogeny respectively. All the 143 ftsZ CDS were further subjected to clustering using CD-HIT with a similarity threshold of 50%.
Representative amino acid sequences of ftsZ of the four main clusters as identified by CD-HIT were subjected to secondary structure (helix, strands and other elements) prediction using the SSpro 5.2 module of SCRATCH Protein Predictor (http://scratch.proteomics.ics.uci.edu) [91]. Accurately predicting protein secondary structure is important for the study of protein evolution, structure and function. The ftsZ CDS were further aligned with their corresponding amino acid sequences and secondary structure mark-up sequence was generated using SSpro 5.2. With the help of this triple alignment, we have identified each of the synonymous codons that are used for coding the amino acids, and we have linked those codons with the amino acids of the predicted secondary structural elements (SSE). The relative synonymous codon usage (RSCU) value which is measured by the ratio between the actual observed values of the codon and the theoretical expectations was also calculated. RSCU reflects the relative usage preference for the specific codons encoding the same amino acid [92]. If RSCU value equals to 1, codon usage is supposed to be unbiased but if RSCU>1, specific codon frequency is higher than other synonymous codons and codon usage is considered to be biased [93]. The RSCU values of the ftsZ CDS were calculated after splitting the sequences based on their propensity in constituting the different secondary structural classes as predicted by SSpro 5.2 [91].
Results and discussion
A comprehensive codon usage analysis of the ftsZ gene and its corresponding protein (FtsZ), along with the housekeeping gene rpoB was carried out in 143 spp. of bacteria of which 75 are non-pathogenic and 68 are pathogenic in nature. On the basis of the nature of cell wall, 43 are Gram positive, 99 organisms are Gram negative and one organism called Gardnerella vaginalis 409–05 is Gram variable in nature. A list of the organisms considered in this study along with their Gram nature and lifestyle is given in the file S2 Table.
After analysing the codon usage data of ftsZ given in S2 Table from the 142 species we found that Kocuria kristinae, which is a pathogenic, Gram-positive bacteria exhibits the lowest Nc value (24.56) among all the organisms. On the other hand, Chlamydophila pneumoniae CWL 029, a pathogenic, Gram-negative bacteria exhibited the highest Nc value of 61. A higher Nc value indicates poor codon bias of the gene [94]. Analysing the mean genomic Nc value of all the organisms studied, it was observed that the lowest mean genomic Nc value (29.198) is depicted by the organism Kocuria kristinae, a pathogenic, Gram-positive bacteria whereas the maximum mean genomic Nc value (55.68) is depicted by a pathogenic, Gram-negative bacteria called Anaplasma marginale str. Florida. Our observations primarily suggest that the degree of codon bias in the pathogenic organisms span a wider range. Analysis of the codon usage pattern of the rpoB gene (S3 Table) demonstrated Kocuria kristinae to exhibit the lowest Nc value (25.39) which is in line with its ftsZ Nc value.
Following the trend in Nc values, we clearly observed that the mean genomic Nc in majority of the organisms is higher than the genic Nc of ftsZ as well as rpoB. This suggests that the ftsZ gene is subjected to greater codon bias in comparison to the genomic Nc, a trend resembling the housekeeping gene rpoB. But in case of the ftsZ CDS of eight organisms, exceptions were evident where the genic Nc of ftsZ was found to be greater than that of the mean genomic Nc. These organisms include Haemophilus influenzae Rd KW20, Halomonas boliviensis LC1, Geobacillus subterraneus, Anaplasma marginale str. Florida, Coxiella burnetii RSA 493, Caldicellulosiruptor bescii DSM 6725, Brucella melitensis bv. 1 str. 16M and Chlamydophila pneumoniae CWL029. Most of these organisms are Gram negative and pathogenic in nature. When compared to ftsZ, the Nc score of rpoB was however found to lesser than that of ftsZ in a majority of the bacterial species including the eight species mentioned above suggesting comparatively greater codon bias in the housekeeping gene. About 30% of the 143 bacterial species considered in this study demonstrated lower Nc values for ftsZ in comparison to rpoB. The pathogenic bacterial species such as Corynebacterium diphtheria, Chlamydophila pneumoniae CWL029 and Brucella melitensis bv. 1 str. 16M demonstrated ftsZ Nc much higher than that of rpoB. A large number of Gram negative bacterial species were found to display genic Nc of ftsZ lower than that of rpoB. These organisms include Treponema denticola ATCC 35405, Porphyromonas gingivalis ATCC 33277, Geobacter sulfurreducens PCA, Francisella philomiragia subsp. philomiragia ATCC 25017, Eikenella corrodens ATCC 23834, Salinibacter ruber DSM 13855, Prochlorococcus marinus str. AS9601, Xylella fastidiosa 9a5c, etc.
A comparative study of the genomic GC content with that of the ftsZ and rpoB genic GC content illustrated in Fig 1 clearly demonstrates that in a significant majority of the bacterial species the genic GC content of ftsZ CDS is greater than that of rpoB CDS. In comparison to the genomic GC content, nearly 54% of the studied bacterial species were found to have genic ftsZ GC content greater than the genomic GC content, whereas in the case of rpoB, about 47% of the organisms were found to have a genic GC content greater than that of the genomic one. In terms of GC3 content, Arcobacter butzleri RM 4018, a pathogenic Gram negative strain depicted the lowest GC3 value for ftsZ gene (0.01974). The maximum GC3 content for ftsZ was shown by Methylobacterium aquaticum (0.9819), a non-pathogenic Gram negative bacteria. When compared to rpoB, the GC3 of nearly 40% bacterial species considered in this study was found to be less than that of ftsZ, and majority of these are non-pathogenic in nature.
Fig 1. A superimposed radar plot comparing the genomic GC content with that of the ftsZ and rpoB CDS GC content of the 143 bacterial species considered in this study.
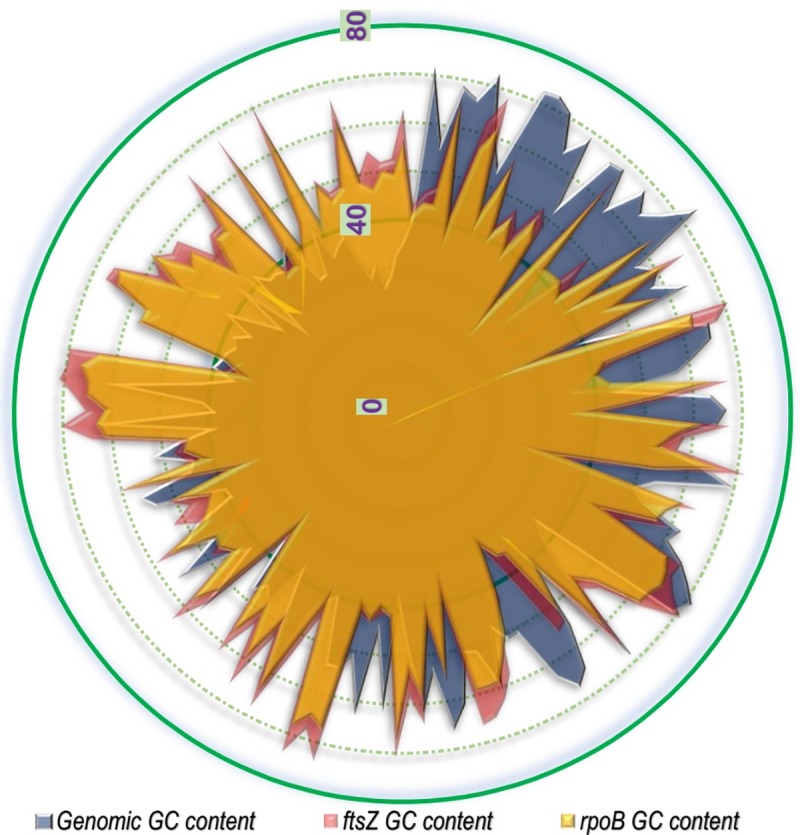
From the centre to the periphery each concentric ring represent increase in 10% GC content.
The study of the relation between Nc and GC3 is an important analytical tool for examining codon bias. In order to explore the relationship between the Nc and GC3 attributes of both the ftsZ and rpoB CDS, a Spearman’s rank correlation analysis was performed. This detected a negative correlation between the Nc and GC3 of ftsZ (ρ = -0.491, p<0.01) as well as rpoB (ρ = -0.562, p<0.01). To further clarify the degree of association between Nc and GC3 of ftsZ and rpoB CDS, non-linear regression analysis was carried out and we observed that a polynomial cubic equation was best fit to describe the relationship between Nc and GC3 (R2 = 0.71; standard error of estimate = 3.72) compared to a linear (R2 = 0.18; standard error of estimate = 6.22) or a polynomial quadratic (R2 = 0.69; standard error of estimate = 3.79) equation. A similar trend was also observed in case of rpoB where a polynomial cubic equation was best fit to describe the relationship between Nc and GC3 (R2 = 0.63; standard error of estimate = 3.78). The R2 value of ftsZ was found to greater than that of rpoB suggesting a relatively stronger association between Nc and GC3 of the ftsZ CDS in comparison to rpoB.
In order to better comprehend the codon usage bias profile of the ftsZ genes Nc-plots were constructed, delineating the ftsZ gene sequences based on the Gram nature and lifestyle of the organisms (Fig 2). Analysis of Fig 2(A) clearly demonstrates the ftsZ genes of majority of Gram positive bacterial species to lie well below the null hypothesis curve compared to the Gram negative ones. In terms of lifestyle, the ftsZ gene sequences of the non-pathogenic organisms were all found to scatter below the null hypothesis curve on the Nc-plot shown in Fig 2(B), suggesting translational selection as a major mechanistic force in shaping codon usage pattern in these bacterial species [70]. Analysis of the Nc-plot shows that the ftsZ genes of three pathogenic organisms—Anaplasma marginale str. Florida, Brucella melitensis bv. 1 str. 16M and Chlamydophila pneumoniae CWL029 occupy distinct positions on the Nc-plot (Fig 2). The common features shared by these three organisms are that they are Gram negative and pathogenic in nature. The bacteria Anaplasma marginale is a member of the order Rickettsiales. It is a small, obligate intracellular bacteria that typically have short genomes due to reductive evolution and survive as endosymbionts. It is also responsible for an infectious, non-contagious disease called bovine anaplasmosis in cattle and other ruminants [95]. The other organism Brucella melitensis is responsible for brucellosis, a common health hazard in people living in close vicinity of cattle [96]. The third organism called Chlamydophila pneumoniae represents an intracellular pathogen instigating different acute and chronic infections and has been found to be associated with chronic neurological disorders such as Alzheimer's disease and multiple sclerosis. Infection by C. pneumoniae which is a common cause of human respiratory disease [97] has also been suspected to cause chronic fatigue syndrome and the linked syndrome polymyalgia rheumatic in some patients [98]. A composite Nc-plot shown in Fig 3, consisting of both the ftsZ and rpoB CDS clearly depicts all the rpoB sequences to lie well below the null hypothesis curve suggesting selectional pressure as a major determinant of CUB [70]. In case of both rpoB and ftsZ, we observed that the gene sequences of some of the pathogenic species were found to be located much closer to the null hypothesis curve.
Fig 2. Nc-plots depicting the correlation between Nc and GC3 of the 143 ftsZ CDS selected for this study based on Gram nature and lifestyle.

(a) Nc-plot constructed by demarcating the ftsZ CDS on the basis of Gram nature where GP = CDS of Gram positive bacterial species, GN = CDS of Gram negative species and GV = CDS of Gram variable bacteria. (b) Nc-plot constructed by demarcating the ftsZ CDS on the basis of lifestyle where NP = CDS of non-pathogenic and P = CDS of pathogenic bacterial species. The green dashed curve in both (a) and (b) depicts the null hypothesis that the GC bias at the synonymous site is solely due to mutation but not selection.
Fig 3. A composite Nc-plot depicting the correlation between Nc and GC3 of the ftsZ and rpoB CDS selected for this study based on lifestyle of the bacterial species where NP = CDS of non-pathogenic and P = CDS of pathogenic bacterial species.

The orange dashed curve represents the null hypothesis that the GC bias at the synonymous site is solely due to mutation but not selection.
In order to study the compositional divergence of the gene sequences coding for FtsZ protein in the selected organisms with respect to their whole genome, the difference between the mean genomic Nc with Nc of ftsZ, the difference between average whole genome GC3 content with that of the ftsZ CDS and genomic GC content with the GC content of ftsZ gene was analysed. The relationship between GC1, GC2 and GC3 was also analysed to have a better understanding of the forces shaping codon usage pattern in both the rpoB and ftsZ gene sequences.
Comparative analysis of mean genomic Nc and ftsZ Nc
Out of the 143 organisms, about 93% (134 species) demonstrated relatively biased codon usage configuration in terms of Nc value. Out of these, 21 species viz., Lactococcus garvieae Lg2, Ensifer adhaerens, Mycobacterium abscessus, Aerococcus viridans, Cupriavidus metallidurans CH34, Enterococcus avium ATCC 14025, Deinococcus radiodurans R1, Lactococcus lactis subsp. lactis Il1403, Butyrivibrio proteoclasticus B316, Neorhizobium galegae bv. orientalis str. HAMBI 540, Ochrobactrum anthropi ATCC 49188, Geobacter sulfurreducens PCA, Rhizobium etli CFN 42, Polynucleobacter asymbioticus QLW-P1DMWA-1, Burkholderia ubonensis MSMB22, Granulibacter bethesdensis CGDNIH1, Alteromonas macleodii ATCC 27126, Sinorhizobium fredii NGR234, Serratia fonticola and Streptococcus pneumoniae R6 demonstrated Nc values of ftsZ CDS that are 20% or less than their mean genomic Nc values. This is suggestive of a significant codon bias existing within the ftsZ CDS. On the other hand, the ftsZ CDS of two Gram negative and pathogenic species Chlamydophila pneumoniae CWL029 and Brucella melitensis bv. 1 str. 16M were found to display Nc values twenty units greater than their mean genomic Nc score. A comparative graphical representation of the genomic Nc and ftsZ Nc segregated on the basis of lifestyle is depicted in Fig 4. The percentage of genes with Nc less than that of ftsZ residing within the genome of each of the organism was also calculated (S5 Table) and compared with that of rpoB. From Fig 5 it is clearly evident that majority of the species considered in this study have relatively fewer percentage of genes with Nc score below that of ftsZ as well as rpoB, which is a core bacterial housekeeping gene. A common trend was visible in the non-pathogenic bacterial species where a majority depicted relatively fewer number of genes with Nc score below that of ftsZ and rpoB. Furthermore, the analysis of z-score as a relative statistical measure to normalize and compare the difference between mean genomic Nc with ftsZ and rpoB genic Nc (S6 Table) in the 143 bacterial species, shown in Fig 6, demonstrates that a significantly large number of bacterial species have ftsZ and rpoB genic Nc lower than that of the mean genomic Nc. This is a significant indicator of greater codon bias existing within the CDS of ftsZ, and a trend well in line expected of a housekeeping gene like rpoB. Organisms with exceptional Nc z-score for ftsZ and rpoB CDS indicating significantly reduced codon bias compared to the genome have been pointed out in Fig 6.
Fig 4.

A comparative graphical representation of the genomic Nc and Nc of ftsZ CDS from (a) non-pathogenic [NP] bacterial species and (b) pathogenic [P] bacterial species included in this study.
Fig 5. A scatter plot showing the distribution of the percentage of genes with Nc less than that of ftsZ and rpoB residing within the genome of each of the organism segregated on the basis of the non-pathogenic and pathogenic nature of the organism.

Fig 6. A scatter plot depicting the distribution of z-score for the Nc of ftsZ and rpoB CDS of all the bacterial species included in this study.

The name of the organisms with relatively higher and lower Nc z-score have been labelled in the plot.
Comparative analysis of genomic GC3 and genic ftsZ GC3
Out of the 143 organisms, the ftsZ CDS of organisms like Fusobacterium nucleatum, Arcobacter butzleri RM4018, Staphylococcus aureus, Aerococcus viridans, Lactobacillus crispatus ST1, Enterococcus avium ATCC 14025, Lactococcus lactis subsp. lactis Il1403, Bacillus mycoides, Butyrivibrio proteoclasticus B316 hve GC3 content which is substantially less (upto 66% lesser) than the mean genomic GC3 content. Barring Lactococcus lactis subsp. lactis Il1403, Bacillus mycoides and Butyrivibrio proteoclasticus B316, the remaining organisms are pathogenic in nature. This is an interesting observation which shows that the ftsZ CDS of these pathogenic bacteria are structured without significant bias towards G/C ending codons. Organisms like Prochlorococcus marinus str. AS9601, Piscirickettsia salmonis LF-89 and Bacteroides cellulosilyticus on the other hand, had significantly greater GC3 (20%, 29% and 40% respectively) in their ftsZ CDS compared to their genomic GC3 content. In terms of GC3, the percentage of genes within the genome of an organism with GC3 below that of ftsZ (S5 Table), depicted in Fig 7, was found to relatively differ in contrast to Nc. Similar trend was also visible in case of rpoB. About 10% of the total 143 organisms have more than 75% genes in their genome with GC3 greater than that of both ftsZ and rpoB. A graphical representation of the comparative z-score of ftsZ and rpoB GC3 is shown in Fig 8. The z-score data of GC3 is provided as supplementary information in S6 Table. Two Gram negative bacterial species Bacteroides cellulosilyticus and Salinibacter ruber DSM 13855 were found to demonstrate the maximal ftsZ GC3 z-score.
Fig 7. A scatter plot showing the distribution of the percentage of genes with GC3 less than that of ftsZ and rpoB residing within the genome of each of the organism segregated on the basis of the non-pathogenic and pathogenic nature of the organism.

Fig 8. A scatter plot depicting the distribution of z-score for the GC3 of ftsZ and rpoB CDS of all the bacterial species included in this study.

The name of the organisms with relatively higher and lower GC3 z-score have been labelled in the plot.
Relative study of genomic GC content and ftsZ GC content
The guanine-cytosine (GC) composition of bacterial genomes is a very important taxonomic marker from the genomics perspective. The GC content of a genome as well as that of a gene have been reported to be a significant genomic indicator for delineating covalently closed circular plasmid DNA from chromosomal DNA [99], and for distinguishing between vertically and horizontally transferred genes [100]. In our study we found that, out of the 143 organisms, the ftsZ CDS of 49 organisms depicted greater than 10% GC content variation in comparison to their genomic GC content. Among these organisms, Coxiella burnetii RSA 493, Rickettsia conorii str. Malish 7, Staphylococcus aureus, Bacteroides cellulosilyticus, Fusobacterium nucleatum, Lactococcus lactis subsp. lactis Il1403, Anaerostipes hadrus DSM 3319 and Acinetobacter johnsonii XBB1 demonstrates 15% greater usage of GC residues in their ftsZ CDS in comparison to the whole genome GC content. In comparison to the genomic GC content, a relatively greater usage of GC residues (more than 20% to 68%) was observed in the ftsZ CDS of Francisella philomiragia subsp. philomiragia ATCC 25017, Staphylococcus capitis subsp. capitis, Clostridium butyricum, Prochlorococcus marinus str. AS9601, Buchnera aphidicola str. APS, Borrelia burgdorferi B31 and Flavobacterium hydatis. The GC content of ftsZ CDS in comparison to the genomic GC of Flavobacterium hydatis was an extraordinarily 68% greater. On the other hand, the GC content of ftsZ CDS in comparison to the genomic GC content of organisms like Bacillus mycoides, Capnocytophaga ochracea DSM 7271, Salinispora tropica CNB-440, Eikenella corrodens ATCC 23834 and Bordetella bronchiseptica 253 was found to be 2% to 6% lower. A comparative graphical depiction of the genomic GC and genic ftsZ GC content segregated on the basis of lifestyle is given in Fig 9. A Mann-Whitney U test was conducted to statistically validate the difference between the genomic GC content and the ftsZ genic GC content of the 143 species. The results suggest that the genomic GC content and ftsZ GC content differs significantly (U = 8536.50, p = 0.016). All these findings evidently suggest that the nucleotide composition of the gene coding for FtsZ protein in a large number of species deviates significantly from their genomic GC content. The deviation of GC content of a coding sequence or a patch of nucleotides from the genomic GC content is a possible pointer towards horizontal gene transfer or HGT [101], and our analysis using Mann-Whitney U test also points in that direction.
Fig 9.

A radial plot comparing the genomic GC content with genic ftsZ GC content in (a) non-pathogenic [NP], and (b) pathogenic bacterial species.
Relationship between GC1, GC2 and GC3
The relationship between GC1, GC2 and GC3 have been utilised in different studies to explore the mechanistic factor shaping coding usage pattern [78,102–104]. Utilizing Spearman’s Rank Order Correlation significant positive correlation was found to exist between the GC1 and GC2 of ftsZ (ρ = 0.754; p<0.01). Similar trend was also observed in the case of rpoB (ρ = 0.70; p<0.01). The linear relationship of GC3 with GC1 and GC3 with GC2 shown in Fig 10 for ftsZ CDS depicts a comparatively stronger positive association between GC3 and GC1 (R2 = 0.6525; p<0.01) with slope approaching 0. As seen in Fig 10, a similar trend is also reflected by the rpoB CDS. With respect to the relation between GC3 and GC2, both ftsZ (R2 = 0.335; p<0.01) and rpoB (R2 = 0.3883; p<0.01) demonstrated a comparatively weak association with a slope approaching relatively much closer to 0. The average of GC1 and GC2 was plotted against GC3 in a neutrality plot for the ftsZ CDS shown in Fig 11, and a significant positive correlation between the two is evident with a slope approaching 0 (y = 0.1404x + 0.4582; R2 = 0.5852, p<0.01) further suggesting that codon usage of ftsZ CDS is shaped by natural selection [104,105]. This trend was also found to be replicated by the rpoB CDS as depicted in Fig 11.
Fig 10.

A plot showing the linear relationship of GC3 values with GC1 values and GC3 with GC2 values of (a) ftsZ and (b) rpoB CDS. The green and blue dashed line represents the trend line of the association between GC3-GC1 and GC3-GC2 respectively. The equation for the line and R2 value for GC3-GC1 and GC3-GC2 association is depicted in green and blue colours.
Fig 11. A plot showing the relationship of the average of GC1 and GC2 values (GC1-GC2) plotted against GC3 values of ftsZ and rpoB CDS of all the bacterial species involved in this study.

The blue and green dashed line represents the trend of association between GC1-GC2 with GC3 of ftsZ and rpoB CDS respectively. The equation for the line and R2 value for ftsZ and rpoB CDS is depicted in blue and green colours respectively.
Analysis of the different codon usage parameters of the ftsZ CDS such as Nc, GC, GC1, GC2, GC3 and their interrelationships strongly suggest the fact that the codon usage pattern of ftsZ gene is relatively biased and is to a large extent shaped by forces of selection. Results obtained from the tandem analysis of a key housekeeping gene such as rpoB were found to be a mostly in line with that of ftsZ, emphasizing the significance of ftsZ as a key component in maintenance of bacterial cellular process such as cell division.
Detection of ‘core’ set of codons used in structuring of ftsZ CDS
The individual usage frequency of the 61 sense codons from the 143 organisms were calculated (S4 Table). Out of the 61 sense codons, the two non-degenerate codons coding for methionine and tryptophan were eliminated. For the remaining 18 amino acids, the 59 codons were grouped in to their degenerate classes of 2, 3, 4 and 6 codons. This analysis was performed to find out if there exists a preferred set of ‘core’ codons for each of the amino acids used in structuring of the ftsZ CDS. A Kruskal-Wallis one way analysis of variance on ranks was carried out for the amino acids coded by 3, 4 and 6 codons, whereas Mann-Whitney Rank Sum test was used to test the codon preference in the two codon family amino acids. The results established the fact that, out of the 18 amino acids, the codons of three amino acids namely aspartic acid, histidine and alanine are randomly utilised on a global scale for structuring the ftsZ CDS. On the other hand, the codons for the remaining 15 amino acids show a non-random utilization pattern. These amino acids include cysteine, glutamine, phenylalanine, glycine, isoleucine, lysine, leucine, asparagine, proline, glutamine, arginine, serine, threonine, valine and tyrosine. Table 1 contains the Mann-Whitney U statistic and the H-value with degrees of freedom for the Kruskal-Wallis one way analysis of variance on ranks with their corresponding p-value obtained from the tests. Our analysis using both the above mentioned robust inferential statistical tools suggest that for all the 18 amino acids (except aspartic acid, histidine and alanine) the differences in the median values among the codon groups are greater than would be expected by chance and hence there is a statistically significant difference at p = <0.001 level. This is a substantial finding suggesting the antiquity and conservation of a preferred set of codons in structuring of a vital gene such as ftsZ.
Table 1. Mann-Whitney U statistic and the H-value with degrees of freedom for the Kruskal-Wallis one way ANOVA on ranks on codon usage to detect ‘core’ set of codons used in structuring of ftsZ.
| Sl. No. | Amino acid | Degenerate codon family | Mann-Whitney U statistic | H-value with degrees of freedom (df) for the Kruskal-Wallis one way analysis of variance on ranks | p-value |
|---|---|---|---|---|---|
| 1 | Cys | 2 | 8029.50 | - | <0.001 |
| 2 | Glu | 2 | 5622.00 | - | <0.001 |
| 3 | Phe | 2 | 8375.50 | - | 0.008 |
| 4 | Lys | 2 | 7552.00 | - | <0.001 |
| 5 | Asn | 2 | 6426.00 | - | <0.001 |
| 6 | Gln | 2 | 6384.00 | - | <0.001 |
| 7 | His | 2 | 9211.00 | - | 0.145 |
| 8 | Asp | 2 | 9565.50 | - | 0.346 |
| 9 | Tyr | 2 | 8696.50 | - | 0.001 |
| 10 | Ile | 3 | - | 229.853, df = 2 | <0.001 |
| 11 | Gly | 4 | - | 273.266, df = 3 | <0.001 |
| 12 | Thr | 4 | - | 70.997, df = 3 | <0.001 |
| 13 | Val | 4 | - | 49.583, df = 3 | <0.001 |
| 14 | Ala | 4 | - | 3.777, df = 3 | 0.287 |
| 15 | Pro | 4 | - | 69.438, df = 3 | <0.001 |
| 16 | Leu | 6 | - | 144.051, df = 5 | <0.001 |
| 17 | Arg | 6 | - | 406.291, df = 5 | <0.001 |
| 18 | Ser | 6 | - | 76.847, df = 5 | <0.001 |
Interrelation of ftsZ codon deployment with lifestyle and Gram nature of bacteria
Sixty one separate variance analysis tests called two factor (or two way) ANOVA was performed to find out how the two major factors namely lifestyle (pathogenic or non-pathogenic), Gram nature and interaction of these two factors influence the coding composition of the ftsZ CDS in the selected organisms at p<0.01 level of significance. A critical analysis of the results show that the compositional bias of eight codons coding for six amino acids is influenced mostly by the Gram nature of the organisms and in some instances by the interaction of lifestyle and Gram nature. In our study, we find that the compositional bias of the codons AUG (methionine), UCA (serine), UAU (tyrosine) and UAC (tyrosine) is influenced solely by the Gram nature of the organism. On the other hand, the compositional frequency of the codons GGA (glycine), CUU (leucine), CUG (leucine) and ACA (threonine) is influenced by the interaction between the Gram nature of the organism and their lifestyle preference of being either pathogenic or non-pathogenic. The two way ANOVA results suggest that the codon organization of the ftsZ CDS is determined largely by the Gram nature and pathogenic/non-pathogenic nature of the organisms, and it is a non-randomly constituted sequence in terms of codon deployment.
Interrelation of amino acid deployment of ftsZ with lifestyle and Gram nature
To further comprehend the codon deployment pattern of the ftsZ CDS, a two way ANOVA was carried out by grouping the different codons according to the amino acids they code (for example alanine is coded by four codons and these four codons are clubbed into a single category to estimate the total frequency of alanine residues present in the CDS). Twenty discrete two way ANOVA analysis was carried out to find if the two factors namely lifestyle, Gram nature and interaction of these two factors influence the amino acid composition of the ftsZ CDS in the selected organisms (at p<0.01 level of significance) or, is the amino acid composition random in nature. All the post-hoc pairwise multiple comparison in the analysis was performed using the Holm-Sidak method of pairwise multiple comparison [106,107]. The results show that the compositional frequency of the amino acids glutamic acid, phenylalanine, leucine, valine, glutamine, threonine and tryptophan is influenced neither by the lifestyle nor the Gram nature of the organism. But, the frequency of the amino acids like aspartic acid, histidine, glycine, methionine, cysteine and tyrosine is influenced by the Gram nature of the organism (p<0.01 level). This shows that the compositional frequency of at least one amino acid from the four different chemical classes of amino acids is directly associated with the Gram nature of the bacteria. Another interesting observation is that the two sulphur containing amino acids methionine and cysteine are both involved in inducing compositional variability based on the wall nature of the bacterium. The hydroxymethyl side chain containing polar amino acid serine was found to be unique in the sense that a two factor ANOVA on composition frequency of serine detected that it is influenced both by the Gram nature and lifestyle of the organism. No amino acid other than serine was found to be influenced by the lifestyle of the organism. Thus, serine appears to be the only amino acid in the FtsZ protein which acts as a pointer to the lifestyle of the bacterial species considered in this study. In case of the compositional frequency of the remaining amino acids like alanine, isoleucine, proline, lysine, arginine and asparagine the effect of lifestyle was found to rest on the Gram nature of the organisms at p<0.01 level.
Phylogenetic and cluster based analysis of ftsZ CDS
The level of identity existing within the 143 FtsZ protein coding genes determined using Clustal Omega [83] demonstrated tremendous variation, with identity ranging from 20% to 98% among the species considered in this study. This suggests a tremendous degree of heterogeneity existing within the ftsZ CDS across the different types of bacterial species residing within the eubacterial domain that have arisen in course of evolution over a long period of time. To infer the phylogenetic relationship of such a gene sequence that demonstrates substantial heterogeneity, a phylogenetic analysis was performed. A phylogenetic tree depicting the evolutionary relationship between the 143 ftsZ CDS given in Fig 12 show the ftsZ gene of Gardnerella vaginalis 409–05 to reside with the Gram positive bacterial species. This is quite understandable since many Gram positive bacteria under different conditions have been found to behave as Gram variable [108]. The ftsZ CDS of the Gram positive bacterium Deinococcus radiodurans R1 was found to peculiarly reside with the Gram negative bacteria. Another such obscure behaviour was displayed by the ftsZ of the Gram positive bacterium Bacillus mycoides which clustered with the Gram negative bacteria Capnocytophaga ochracea DSM 7271 and Flavobacterium hydatis. The ftsZ CDS of Selenomonas noxia ATCC 43541 was found to display the maximum number of substitutions followed by Delftia acidovorans SPH-1, Bacillus mycoides, Chromobacterium subtsugae, Burkholderia gladioli, Ralstonia solanacearum GMI1000 and Chlamydophila pneumoniae CWL029. Apart from Bacillus mycoides, all these species are Gram negative. The ftsZ CDS of many pathogenic Gram negative species were found to display least amount of evolutionary change measured in terms of substitutions. These include Salmonella enterica subsp. enterica serovar Typhi str. CT18, Enterobacter, Shigella dysenteriae Sd197, Klebsiella oxytoca, Citrobacter amalonaticus, Yersinia pestis CO92, etc.
Fig 12. A phylogenetic tree depicting the evolutionary relationship between the 143 ftsZ CDS considered in this study constructed using MEGA X and annotated using iTOL.

The evolutionary history was inferred using the Neighbor-Joining method and the optimal tree was obtained using 1000 bootstrap replicates. The tree is drawn to scale, with branch lengths representing evolutionary distances in the units of number of base substitutions per site. The evolutionary distances were computed using the Tajima-Nei method and are the rate variation among sites was modeled with a gamma distribution (shape parameter = 1). The name of the organisms have been given in colours to match with their Gram nature with blue representing ftsZ CDS of Gram positive whereas red represents ftsZ CDS of Gram negative bacterial species. The Gram variable bacterium has been depicted using orange. The green circles at the node represents the relative bootstrap support for each branch with large circle representing higher confidence level. The purple dashed line branches represents ftsZ CDS of those organisms that have been utilized for analysing the relationship between codons constituting ftsZ CDS and SSE of FtsZ proteins.
The 143 ftsZ CDS were further subjected to clustering using CD-HIT with a 50% similarity threshold. A tabular account of the 17 clusters generated using CD-HIT along with the number of representative sequences for each cluster is given in Table 2. From the data given in Table 2, it is quite evident that the majority of the sequences are grouped together in the first two clusters which contains 43% of the total ftsZ CDS (41 sequences in Cluster 0 and 21 sequences in Cluster 1). The amino acid sequence of the corresponding ftsZ CDS representing the first four cluster i.e., Pseudoalteromonas luteoviolacea (Cluster 0), Cutibacterium avidum 44067 (Cluster 1), Streptococcus agalactiae 2603V/R (Cluster 2) and Burkholderia ubonensis MSMB22 (Cluster 3) were subjected to secondary structure prediction using SSpro 5.2 module of SCRATCH Protein Predictor (http://scratch.proteomics.ics.uci.edu/)[109]. SSpro catalogues three classes of secondary structures and based on that, the amino acid residues constituting the four FtsZ proteins have been identified as H (alpha helix), E (strand) and C (all the rest SSE). We have meticulously aligned the FtsZ amino acid sequences with the SSE mark-up sequence (S1 Fig) generated using Sspro 5.2. Using this alignment for each of the four representative sequence, we have identified the individual codons coding for the different amino acids within the three classes of SSE detected by Sspro (S2 Fig). We have analysed the RSCU values of the ftsZ CDS of Pseudoalteromonas luteoviolacea, Cutibacterium avidum 44067, Streptococcus agalactiae 2603V/R and Burkholderia ubonensis MSMB22 by splitting the sequences into the three different SSE classes (S7 Table).
Table 2. Clusters of ftsZ gene sequences generated using CD-HIT with a similarity threshold of 50 percent.
| Cluster at 50% identity | No. of sequences in the cluster | Representative sequence | Length of the representative sequence |
|---|---|---|---|
| Cluster 0 | 41 | Pseudoalteromonas luteoviolacea | 418 |
| Cluster 1 | 21 | Cutibacterium avidum 44067 | 417 |
| Cluster 2 | 9 | Streptococcus agalactiae 2603V/R | 419 |
| Cluster 3 | 6 | Burkholderia ubonensis MSMB22 | 399 |
| Cluster 4 | 4 | Butyrivibrio proteoclasticus B316 | 412 |
| Cluster 5 | 3 | Acinetobacter johnsonii XBB1 | 398 |
| Cluster 6 | 2 | Neisseria gonorrhoeae FA 1090 | 392 |
| Cluster 7 | 2 | Geobacter sulfurreducens PCA | 383 |
| Cluster 8 | 2 | Anaplasma marginale str. Florida | 412 |
| Cluster 9 | 1 | Fusobacterium nucleatum | 360 |
| Cluster 10 | 1 | Deinococcus radiodurans R1 | 371 |
| Cluster 11 | 1 | Helicobacter pylori 26695 | 385 |
| Cluster 12 | 1 | Arcobacter butzleri RM4018 | 377 |
| Cluster 13 | 1 | Chromobacterium subtsugae | 400 |
| Cluster 14 | 1 | Ralstonia solanacearum GMI1000 | 408 |
| Cluster 15 | 1 | Borrelia burgdorferi B31 | 399 |
| Cluster 16 | 1 | Selenomonas noxia ATCC 43541 | 412 |
Comparative RSCU analysis of the helix, strand and other structural elements constituting residues of ftsZ
A RSCU analysis of the sense codons used for coding the amino acids of the FtsZ protein of Pseudoalteromonas luteoviolacea, Cutibacterium avidum 44067, Streptococcus agalactiae 2603V/R and Burkholderia ubonensis MSMB22, based on the type of SSE was performed. An amino acid wise description of the RSCU of the sixty one sense codons used in structuring of the ftsZ CDS is described below.
Non Polar amino acids
Glycine: In case of glycine, the residues constituting the helix in proteins are encoded by the codons GGU, GGA, GGG and GGC of which GGC is used by all the four species. In the strand region, Cutibacterium utilizes all the four codons whereas Burkholderia and Pseudoalteromonas use only two codons GGU and GGC. Likewise, Streptococcus also prefers the codons GGU and GGG only. This suggests that in these three bacterial species there is a preference towards a certain subset of codons in coding the glycine residues positioned in the strand regions. In all the remaining secondary structural elements, all the organisms are found to use GGU, GGG and GGC.
Alanine: The amino acid alanine is near universally encoded by GCU, GCC, GCA and GCG. In the helix region, GCG and GCC codons are used by all the four organisms. All the four codons are found to be employed by Streptococcus. But in the strand region, GCU and GCA are used only by Streptococcus. In the remaining regions, all the four codons are used randomly by all the organisms.
Valine: It is encoded by four codons GUU, GUC, GUA and GUG of which in the helix region, GUG is used by all the four organisms. In contrast to the helix region, in the strand region Pseudoalteromonas uses all the valine triplets whereas Cutibacterium and Streptococcus was found to use only three. In all the remaining regions, all the four organisms use the codons GUG and GUC.
Methionine: Since methionine is coded by a single codon AUG, we observed that for all the three regions, the codon AUG is preferred by all the four species.
Leucine: It is one of the three amino acids which is encoded by six different codons UUA, UUG, CUU, CUC, CUG and CUA. In the helix elements, the codon UUG was used by all the organisms whereas the codon CUA is totally absent in the helix region. In the strand region, Burkholderia, Cutibacterium, Streptococcus and Pseudoalteromonas was found to use a specific subset of leucine codons. No organism was found to use all the 6 codons. In the remaining regions, it was observed that the codon CUA is not used by any of the species. The codon CUG is used by three organisms except Streptococcus.
Isoleucine: In the helix regions, the codon AUC is used by all the organisms whereas AUA remains totally absent. But in the strand regions, codon AUA is used only by Pseudoalteromonas which also uses the other two codons AUU and AUC. Burkholderia uses only AUC but Streptococcus uses AUU and AUC. In the rest of the remaining regions, AUC is used by three of the organisms except Streptococcus. AUA is not used by any of the organisms.
Proline: In the helix regions, codon CCA is used by only two organisms–Pseudoalteromonas and Streptococcus whereas codon CCC is used by a single organism, Cutibacterium. Three organisms use the codon CCU except Burkholderia. In the strand regions, out of the four codons of proline, CCC is used by Cutibacterium and CCU by Pseudoalteromonas. The rest two codons aren’t used. In case of the remaining secondary structural elements, Cutibacterium is found to use all the four codons.
Phenylalanine: It is encoded by two codons–UUU and UUC. Considering the codon usage of the phenylalanine residues in the helix regions, the codon UUU is used by Pseudoalteromonas and Streptococcus whereas UUC is used by all the organisms except Pseudoalteromonas. But in strand elements, codon UUU is only used by Streptococcus and Pseudoalteromonas. The use of UUC is totally avoided here. In case of the remaining secondary structural elements, UUU codon is used by all the four species.
Tryptophan: The amino acid tryptophan is encoded by a single codon UGG in a near universal manner. In case of helix elements of ftsZ CDS, this amino acid is totally absent. In the strand elements, UGG is used only by Streptococcus whereas in the remaining elements, tryptophan is found to be used by Burkholderia and Streptococcus.
Tyrosine: In the helix regions, we found that the codon UAC is used by Burkholderia alone. Similarly Pseudoalteromonas use the codon UAU. In strand regions, UAU remains totally absent whereas UAC is used by Burkholderia alone. UAC is not used by Pseudoalteromonas. In the remaining regions, UAU is found to be used by the organisms Streptococcus and Pseudoalteromonas.
Polar basic amino acids
Histidine: In the helix regions, histidine is coded by CAC in three of the organisms except Streptococcus. Similarly, another codon CAU is preferred in the helix regions buy all the three organisms except Cutibacterium. In the extended strand regions, our analysis shows that the amino acid histidine is not used by any of the organisms. For the rest of the remaining secondary structural elements CAU is preferred by all the four organisms except Streptococcus which uses CAC.
Lysine: This amino acid is encoded by two codons—AAA and AAG. In the helix regions lysine is found to be coded by the homo triplet AAA in the studied organisms except Burkholderia. AAG was found to be employed by all the four organisms. In the strand regions, the triplet AAA is used by the organisms Pseudoalteromonas and Streptococcus whereas AAG is preferred by Burkholderia and Cutibacterium. For the remaining secondary structural elements, the preference for AAA is restricted to Pseudoalteromonas and Streptococcus, a scenario exactly similar to the strand region.
Arginine: This is one of the three amino acid which is encoded by six degenerate codons–CGU, CGC, CGA, CGG, AGA and AGG. In the helix regions, none of the four organisms were found to use the codon CGA. The remaining organisms display preference towards the use of specific subset of codons. In the strand regions only three codons AGA, CGC and CGU are used out of the six. This suggests the preference of the organism towards specific codons for encoding the amino acids that have the propensity to be included in the strand regions of FtsZ protein. In the remaining structural elements, out of the six codons two are totally absent and this are CGG and AGA. The codon CGU is found to be used by Cutibacterium, Pseudoalteromonas, Streptococcus and comparatively in lesser frequency by Burkholderia.
Polar acidic amino acids
Aspartic acid: In the helix region, all the four organisms use the codons GAU and GAC, but the frequency of GAU usage by Cutibacterium is very low. In contrast to the helix regions, in strand regions we found that Pseudoalteromonas does not use aspartic acid. Streptococcus use GAU alone whereas GAC is used by Burkholderia and Cutibacterium.
Glutamic acid: This amino acid is represented by the codons GAA and GAG. In the helix, GAG is used by all the four organisms whereas GAA is used by all except Cutibacterium. In the strand regions, GAA is used by Burkholderia and Streptococcus whereas GAG is used by Cutibacterium alone. Both the codons are found to remain absent in Pseudoalteromonas. In the rest of the structural elements, GAG is preferred by all the organisms.
Polar neutral amino acids
Serine: It is encoded by six codons—UCU, UCC, UCA, UCG, AGU, AGC. A preferential usage of certain codons encoding the different amino residues constituting the different structural elements was also observed in case of this amino acid. In the helix regions, AGC is used by all the organisms except Cutibacterium. The codon AGU UCA and UCC is found to be preferred by Streptococcus and Cutibacterium. Burkholderia and Cutibacterium was found to prefer UCG, whereas Pseudoalteromonas and Streptococcus use UCU. The use of the codon UCU by Pseudoalteromonas was found to be comparatively higher than the rest of the organisms. In the strand regions, the codon AGU and UCU were found to be avoided by all the four organisms. In the rest of the structural elements, AGC was found to be preferred by all the four organisms. All the four organisms use the codon UCG, but in Burkholderia the frequency of usage is relatively greater.
Threonine: In the helix elements, ACC and ACA are preferred by three organisms. ACC remains absent in Pseudoalteromonas whereas ACA is absent in Burkholderia. All the four organisms preferentially use the codon ACG. In the extended strand elements, ACG is used by all the organisms whereas ACA and ACU codons are used by Streptococcus and Pseudoalteromonas and ACC is preferred by Burkholderia and Cutibacterium.
Asparagine: This amino acid is encoded in general by two codons–AAU and AAC. In the helix regions, AAC is preferred by all the organisms. Likewise codon AAU is also used by all the organisms. But in the strand region, AAU is used by two organisms Pseudoalteromonas and Streptococcus. The codon AAC is employed by all the organisms except Pseudoalteromonas. In the remaining structural elements, we did not observe any fixed preference for a particular codon in the organisms considered in this analysis.
Glutamine: In case of helix regions, codons CAA and CAG are used by three organisms. The use of the amino acid glutamine in the helix region was absent in Cutibacterium. CAG is used by Burkholderia, Pseudoalteromonas and Streptococcus. The codon CAA was not used by any of the organisms in the strand regions. In the other secondary structural elements, the codon CAG is used by all the organisms whereas the codon CAA is used by all the organisms except Burkholderia.
Cysteine: In the helix elements, the codon UGU was avoided by all the organisms. The use of this sulphur containing amino acid in the helix regions of Burkholderia and Cutibacterium are found to be fulfilled by the codon UGC. Apart from the helix structural elements, cysteine was found to be totally absent in the other secondary structural elements in all the four organisms.
Our study clearly shows that a differential RSCU pattern is evident in the coding nature of the various SSE of the FtsZ proteins from different bacteria. It may be suggested that this variation could be attributed to the differential folding pattern of the different domain region of the FtsZ protein. The FtsZ protein has two major domains—one is the GTPase domain and the other is the C-terminal domain. Our findings suggest that the use of specific codons coding for the amino acids in the different SSE of the FtsZ protein is less organism specific but more codon specific. The helix regions demonstrates a comparative higher bias towards use of specific codons in coding the amino acids than the strand or the other SSE regions.
Conclusions
The FtsZ protein is ubiquitous in bacteria and plays a vital role in bacterial cell division. From the evolutionary stand point it might be regarded as the counterpart of the eukaryotic tubulin protein. Our study of the gene sequences coding for FtsZ from 142 bacterial species demonstrated that about one third of the selected organisms depicted more than ten percent GC variation in their ftsZ CDS compared to their genomic GC content. Thus, our study clearly suggest that the nucleotide composition of the gene coding for FtsZ protein in a large number of species deviates significantly from their genomic GC content. The codon usage pattern demonstrated that the ftsZ gene of about 93% of the organism show relatively biased codon usage profile and the same is largely influenced by natural selection. Parallel analysis of the housekeeping gene rpoB also demonstrated the resemblance of ftsZ codon usage pattern with a housekeeping gene. In this study, we have also captured the existence of a ‘core’ set of codons in the structuring of the ftsZ gene despite the presence of a varying degree of identity among the ftsZ sequences. This is probably due to the constraint exerted by nature to maintain form and function in an important physiological protein like FtsZ that plays a major role in a critical bacterial cellular process like cell division. By the utilization of inferential statistical methods such as a two way ANOVA, we were able to capture the influence of Gram nature of the bacteria and their lifestyle pattern on the amino acid compositional frequency of the FtsZ protein. Finally, a phylogenetic and cluster based analysis followed by an amino acid wise comparative RSCU analysis of the different secondary structural elements of the FtsZ protein tied with the ftsZ CDS, demonstrated the presence of bias towards specific triplet codons coding the amino acids of the different secondary structural elements of a multi domain protein like FtsZ. In conclusion, it may be stated that the ftsZ gene coding for an indispensable cell division protein called FtsZ in a large number of bacteria, differing in terms of cellular morphology, physiology, biochemistry and a host of other features displays a significantly biased codon usage pattern with an extremely inflated GC content. Along with the existence of a preferred ‘core’ set of codons, the different SSE of the multi-domain FtsZ protein was also found to display bias towards specific synonymous codons particularly in the helix and strand regions. All these suggest that in an indispensable and vital protein such as FtsZ, there is an inherent tendency to maintain form for optimized performance in spite of the extrinsic variability in coding features.
Supporting information
(TXT)
(TXT)
(DOC)
(TXT)
(XLS)
(XLS)
(XLS)
(TXT)
(TXT)
(JPG)
{kind=link}
(JPG)
{kind=link}
Acknowledgments
The authors are grateful to Prof. Subhasis Mukhopadhyay and Late Prof. A. K. Bothra for their support and encouragement. The authors are also thankful to all the reviewers for their constructive suggestions which has helped a great deal to improve the quality of the study.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
The authors received no specific funding for this work.
References
- 1.Powell JR, Dion K (2015) Effects of codon usage on gene expression: empirical studies on Drosophila. Journal of molecular evolution 80: 219–226. 10.1007/s00239-015-9675-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang L, Xing H, Yuan Y, Wang X, Saeed M, et al. (2018) Genome-wide analysis of codon usage bias in four sequenced cotton species. PLOS ONE 13: e0194372 10.1371/journal.pone.0194372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou Z, Dang Y, Zhou M, Li L, Yu C-h, et al. (2016) Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proceedings of the National Academy of Sciences 113: E6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belalov IS, Lukashev AN (2013) Causes and Implications of Codon Usage Bias in RNA Viruses. PLOS ONE 8: e56642 10.1371/journal.pone.0056642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prat Y, Fromer M, Linial N, Linial M (2009) Codon usage is associated with the evolutionary age of genes in metazoan genomes. BMC Evolutionary Biology 9: 285 10.1186/1471-2148-9-285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.LaBella AL, Opulente DA, Steenwyk JL, Hittinger CT, Rokas A (2019) Variation and selection on codon usage bias across an entire subphylum. PLOS Genetics 15: e1008304 10.1371/journal.pgen.1008304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shields DC, Sharp PM (1987) Synonymous codon usage in Bacillus subtilis reflects both translational selection and mutational biases. Nucleic Acids Res 15: 8023–8040. 10.1093/nar/15.19.8023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharp PM, Stenico M, Peden JF, Lloyd AT (1993) Codon usage: mutational bias, translational selection, or both? Biochem Soc Trans 21: 835–841. 10.1042/bst0210835 [DOI] [PubMed] [Google Scholar]
- 9.Ikemura T (1985) Codon usage and tRNA content in unicellular and multicellular organisms. Mol Biol Evol 2: 13–34. 10.1093/oxfordjournals.molbev.a040335 [DOI] [PubMed] [Google Scholar]
- 10.Bulmer M (1991) The selection-mutation-drift theory of synonymous codon usage. Genetics 129: 897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tuller T, Waldman YY, Kupiec M, Ruppin E (2010) Translation efficiency is determined by both codon bias and folding energy. Proceedings of the National Academy of Sciences 107: 3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu C, Cai X, Chen Q, Zhou H, Cai Y, et al. (2011) Factors Affecting Synonymous Codon Usage Bias in Chloroplast Genome of Oncidium Gower Ramsey. Evolutionary bioinformatics online 7: 271–278. 10.4137/EBO.S8092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu C, Dong J, Tong C, Gong X, Wen Q, et al. (2013) Analysis of Synonymous Codon Usage Patterns in Seven Different Citrus Species. Evolutionary Bioinformatics 9: EBO.S11930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chithambaram S, Prabhakaran R, Xia X (2014) The Effect of Mutation and Selection on Codon Adaptation in <em>Escherichia coli</em> Bacteriophage. Genetics 197: 301 10.1534/genetics.114.162842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plotkin JB, Kudla G (2011) Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet 12: 32–42. 10.1038/nrg2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharp PM, Matassi G (1994) Codon usage and genome evolution. Curr Opin Genet Dev 4: 851–860. 10.1016/0959-437x(94)90070-1 [DOI] [PubMed] [Google Scholar]
- 17.Quax TE, Claassens NJ, Soll D, van der Oost J (2015) Codon Bias as a Means to Fine-Tune Gene Expression. Mol Cell 59: 149–161. 10.1016/j.molcel.2015.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song H, Gao H, Liu J, Tian P, Nan Z (2017) Comprehensive analysis of correlations among codon usage bias, gene expression, and substitution rate in Arachis duranensis and Arachis ipaënsis orthologs. Scientific Reports 7: 14853 10.1038/s41598-017-13981-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang R, Zhang L, Wang W, Zhang Z, Du H, et al. (2018) Differences in Codon Usage Bias between Photosynthesis-Related Genes and Genetic System-Related Genes of Chloroplast Genomes in Cultivated and Wild Solanum Species. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sahoo S, Das SS, Rakshit R (2019) Codon usage pattern and predicted gene expression in Arabidopsis thaliana. Gene: X 2: 100012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hershberg R, Petrov DA (2008) Selection on codon bias. Annu Rev Genet 42: 287–299. 10.1146/annurev.genet.42.110807.091442 [DOI] [PubMed] [Google Scholar]
- 22.Bennetzen JL, Hall BD (1982) Codon selection in yeast. J Biol Chem 257: 3026–3031. [PubMed] [Google Scholar]
- 23.Gouy M, Gautier C (1982) Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res 10: 7055–7074. 10.1093/nar/10.22.7055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frumkin I, Lajoie MJ, Gregg CJ, Hornung G, Church GM, et al. (2018) Codon usage of highly expressed genes affects proteome-wide translation efficiency. Proceedings of the National Academy of Sciences of the United States of America 115: E4940–E4949. 10.1073/pnas.1719375115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salim HMW, Cavalcanti ARO (2008) Factors influencing codon usage bias in genomes. Journal of the Brazilian Chemical Society 19: 257–262. [Google Scholar]
- 26.Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, et al. (2011) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 39: D38–51. 10.1093/nar/gkq1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hasegawa M, Yasunaga T, Miyata T (1979) Secondary structure of MS2 phage RNA and bias in code word usage. Nucleic Acids Res 7: 2073–2079. 10.1093/nar/7.7.2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arjes HA, Lai B, Emelue E, Steinbach A, Levin PA (2015) Mutations in the bacterial cell division protein FtsZ highlight the role of GTP binding and longitudinal subunit interactions in assembly and function. BMC Microbiology 15: 209 10.1186/s12866-015-0544-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bi E, Lutkenhaus J (1991) FtsZ ring structure associated with division in Escherichia coli. Nature 354: 161–164. 10.1038/354161a0 [DOI] [PubMed] [Google Scholar]
- 30.Sun Q, Margolin W (1998) FtsZ dynamics during the division cycle of live Escherichia coli cells. Journal of bacteriology 180: 2050–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stricker J, Maddox P, Salmon ED, Erickson HP (2002) Rapid assembly dynamics of the <em>Escherichia coli</em> FtsZ-ring demonstrated by fluorescence recovery after photobleaching. Proceedings of the National Academy of Sciences 99: 3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson DE, Gueiros-Filho FJ, Erickson HP (2004) Assembly Dynamics of FtsZ Rings in <em>Bacillus subtilis</em> and <em>Escherichia coli</em> and Effects of FtsZ-Regulating Proteins. Journal of Bacteriology 186: 5775 10.1128/JB.186.17.5775-5781.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Y, Erickson HP (2005) Rapid in vitro assembly dynamics and subunit turnover of FtsZ demonstrated by fluorescence resonance energy transfer. The Journal of biological chemistry 280: 22549–22554. 10.1074/jbc.M500895200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pogliano J, Pogliano K, Weiss DS, Losick R, Beckwith J (1997) Inactivation of FtsI inhibits constriction of the FtsZ cytokinetic ring and delays the assembly of FtsZ rings at potential division sites. Proceedings of the National Academy of Sciences of the United States of America 94: 559–564. 10.1073/pnas.94.2.559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michie KA, Löwe J (2006) Dynamic Filaments of the Bacterial Cytoskeleton. Annual Review of Biochemistry 75: 467–492. 10.1146/annurev.biochem.75.103004.142452 [DOI] [PubMed] [Google Scholar]
- 36.Romberg L, Levin PA (2003) Assembly Dynamics of the Bacterial Cell Division Protein FtsZ: Poised at the Edge of Stability. Annual Review of Microbiology 57: 125–154. 10.1146/annurev.micro.57.012903.074300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Margolin W (2005) FTSZ AND THE DIVISION OF PROKARYOTIC CELLS AND ORGANELLES. Nature reviews Molecular cell biology 6: 862–871. 10.1038/nrm1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.OLIVEIRA JR AF, FOLADOR EL, GOMIDE ACP, GOES-NETO A, AZEVEDO VAC, et al. (2018) Cell Division in genus Corynebacterium: protein-protein interaction and molecular docking of SepF and FtsZ in the understanding of cytokinesis in pathogenic species. Anais da Academia Brasileira de Ciências 90: 2179–2188. 10.1590/0001-3765201820170385 [DOI] [PubMed] [Google Scholar]
- 39.Erickson HP (1997) FtsZ, a tubulin homologue in prokaryote cell division. Trends Cell Biol 7: 362–367. 10.1016/S0962-8924(97)01108-2 [DOI] [PubMed] [Google Scholar]
- 40.Stokes KD, Osteryoung KW (2003) Early divergence of the FtsZ1 and FtsZ2 plastid division gene families in photosynthetic eukaryotes. Gene 320: 97–108. 10.1016/s0378-1119(03)00814-x [DOI] [PubMed] [Google Scholar]
- 41.Ma S, Ma S (2012) The development of FtsZ inhibitors as potential antibacterial agents. ChemMedChem 7: 1161–1172. 10.1002/cmdc.201200156 [DOI] [PubMed] [Google Scholar]
- 42.Hurley KA, Santos TMA, Nepomuceno GM, Huynh V, Shaw JT, et al. (2016) Targeting the Bacterial Division Protein FtsZ. Journal of Medicinal Chemistry 59: 6975–6998. 10.1021/acs.jmedchem.5b01098 [DOI] [PubMed] [Google Scholar]
- 43.Ojima I, Kumar K, Awasthi D, Vineberg JG (2014) Drug discovery targeting cell division proteins, microtubules and FtsZ. Bioorg Med Chem 22: 5060–5077. 10.1016/j.bmc.2014.02.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dai K, Lutkenhaus J (1991) ftsZ is an essential cell division gene in Escherichia coli. Journal of bacteriology 173: 3500–3506. 10.1128/jb.173.11.3500-3506.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Erickson HP (1995) FtsZ, a prokaryotic homolog of tubulin? Cell 80: 367–370. 10.1016/0092-8674(95)90486-7 [DOI] [PubMed] [Google Scholar]
- 46.Erickson HP, Taylor DW, Taylor KA, Bramhill D (1996) Bacterial cell division protein FtsZ assembles into protofilament sheets and minirings, structural homologs of tubulin polymers. Proceedings of the National Academy of Sciences 93: 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Popp D, Iwasa M, Narita A, Erickson HP, Maéda Y (2009) FtsZ condensates: an in vitro electron microscopy study. Biopolymers 91: 340–350. 10.1002/bip.21136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Romberg L, Simon M, Erickson H (2001) Polymerization of FtsZ, a Bacterial Homolog of Tubulin. The Journal of biological chemistry 276: 11743–11753. 10.1074/jbc.M009033200 [DOI] [PubMed] [Google Scholar]
- 49.Eisenberg E, Levanon EY (2013) Human housekeeping genes, revisited. Trends Genet 29: 569–574. 10.1016/j.tig.2013.05.010 [DOI] [PubMed] [Google Scholar]
- 50.Lai Q, Liu Y, Yuan J, Du J, Wang L, et al. (2014) Multilocus Sequence Analysis for Assessment of Phylogenetic Diversity and Biogeography in Thalassospira Bacteria from Diverse Marine Environments. PLOS ONE 9: e106353 10.1371/journal.pone.0106353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Supek F, Skunca N, Repar J, Vlahovicek K, Smuc T (2010) Translational selection is ubiquitous in prokaryotes. PLoS Genet 6: e1001004 10.1371/journal.pgen.1001004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roller M, Lucić V, Nagy I, Perica T, Vlahovicek K (2013) Environmental shaping of codon usage and functional adaptation across microbial communities. Nucleic acids research 41: 8842–8852. 10.1093/nar/gkt673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Botzman M, Margalit H (2011) Variation in global codon usage bias among prokaryotic organisms is associated with their lifestyles. Genome biology 12: R109–R109. 10.1186/gb-2011-12-10-r109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hart A, Cortés MP, Latorre M, Martinez S (2018) Codon usage bias reveals genomic adaptations to environmental conditions in an acidophilic consortium. PLOS ONE 13: e0195869 10.1371/journal.pone.0195869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carbone A, Képès F, Zinovyev A (2004) Codon Bias Signatures, Organization of Microorganisms in Codon Space, and Lifestyle. Molecular Biology and Evolution 22: 547–561. 10.1093/molbev/msi040 [DOI] [PubMed] [Google Scholar]
- 56.Yang DC, Blair KM, Salama NR (2016) Staying in Shape: the Impact of Cell Shape on Bacterial Survival in Diverse Environments. Microbiology and Molecular Biology Reviews 80: 187 10.1128/MMBR.00031-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller SI, Salama NR (2018) The gram-negative bacterial periplasm: Size matters. PLOS Biology 16: e2004935 10.1371/journal.pbio.2004935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Navarro Llorens JM, Tormo A, Martínez-García E (2010) Stationary phase in gram-negative bacteria. FEMS Microbiology Reviews 34: 476–495. 10.1111/j.1574-6976.2010.00213.x [DOI] [PubMed] [Google Scholar]
- 59.Gontang EA, Fenical W, Jensen PR (2007) Phylogenetic Diversity of Gram-Positive Bacteria Cultured from Marine Sediments. Applied and Environmental Microbiology 73: 3272 10.1128/AEM.02811-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Muto A, Osawa S (1987) The guanine and cytosine content of genomic DNA and bacterial evolution. Proc Natl Acad Sci U S A 84: 166–169. 10.1073/pnas.84.1.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lightfield J, Fram NR, Ely B (2011) Across Bacterial Phyla, Distantly-Related Genomes with Similar Genomic GC Content Have Similar Patterns of Amino Acid Usage. PLOS ONE 6: e17677 10.1371/journal.pone.0017677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marin M (2008) Folding at the rhythm of the rare codon beat. Biotechnol J 3: 1047–1057. 10.1002/biot.200800089 [DOI] [PubMed] [Google Scholar]
- 63.Saunders R, Deane CM (2010) Synonymous codon usage influences the local protein structure observed. Nucleic Acids Research 38: 6719–6728. 10.1093/nar/gkq495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, et al. (2013) GenBank. Nucleic acids research 41: D36–D42. 10.1093/nar/gks1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garrity GM, Boone DR, Castenholz RW (2001) Bergey's Manual of Systematic Bacteriology. New York: Springer-Verlag. [Google Scholar]
- 66.Brenner DJ, Krieg NR, Staley JT, Garrity GM (2005) Bergey's Manual of Systematic Bacteriology. New York: Springer-Verlag. [Google Scholar]
- 67.Vos P, Garrity G, Jones D, Krieg NR, Ludwig W, et al. (2009) Bergey's Manual of Systematic Bacteriology. New York: Springer-Verlag. [Google Scholar]
- 68.Krieg NR, Ludwig W, Whitman WB, Hedlund BP, Paster BJ, et al. (2010) Bergey's Manual of Systematic Bacteriology. New York: Springer-Verlag. [Google Scholar]
- 69.Whitman WB, Goodfellow M, Kämpfer P, Busse H-J, Trujillo ME, et al. (2012) Bergey's Manual of Systematic Bacteriology. New York: Springer-Verlag. [Google Scholar]
- 70.Wright F (1990) The 'effective number of codons' used in a gene. Gene 87: 23–29. 10.1016/0378-1119(90)90491-9 [DOI] [PubMed] [Google Scholar]
- 71.Peden JF (1999) Analysis of Codon Usage [Doctoral Thesis]: University of Nottingham. [Google Scholar]
- 72.Supek F, Vlahovicek K (2004) INCA: synonymous codon usage analysis and clustering by means of self-organizing map. Bioinformatics 20: 2329–2330. 10.1093/bioinformatics/bth238 [DOI] [PubMed] [Google Scholar]
- 73.Fuglsang A (2006) Estimating the "effective number of codons": the Wright way of determining codon homozygosity leads to superior estimates. Genetics 172: 1301–1307. 10.1534/genetics.105.049643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu X (2013) A more accurate relationship between 'effective number of codons' and GC3s under assumptions of no selection. Comput Biol Chem 42: 35–39. 10.1016/j.compbiolchem.2012.11.003 [DOI] [PubMed] [Google Scholar]
- 75.Šmarda P, Bureš P, Horová L, Leitch IJ, Mucina L, et al. (2014) Ecological and evolutionary significance of genomic GC content diversity in monocots. Proceedings of the National Academy of Sciences 111: E4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McCutcheon JP, Moran NA (2010) Functional convergence in reduced genomes of bacterial symbionts spanning 200 My of evolution. Genome Biol Evol 2: 708–718. 10.1093/gbe/evq055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lassalle F, Périan S, Bataillon T, Nesme X, Duret L, et al. (2015) GC-Content evolution in bacterial genomes: the biased gene conversion hypothesis expands. PLoS genetics 11: e1004941–e1004941. 10.1371/journal.pgen.1004941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sharp PM, Bailes E, Grocock RJ, Peden JF, Sockett RE (2005) Variation in the strength of selected codon usage bias among bacteria. Nucleic acids research 33: 1141–1153. 10.1093/nar/gki242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aho KA (2013) Foundational and Applied Statistics for Biologists Using R. New York: Chapman and Hall/CRC. [Google Scholar]
- 80.Spiegel MR (1961) Schaum's outline of theory and problems of statistics: Schaum Pub. Co. [Google Scholar]
- 81.Shang M LF, Hua J, Wang K, (2011) Analysis on codon usage of chloroplast genome of Gossypium hirsutum. Scientia Agricultura Sinica 44: 245–253. [Google Scholar]
- 82.Zhou Y, Skidmore ST (2018) A Reassessment of ANOVA Reporting Practices: A Review of Three APA Journals. Journal of Methods and Measurement in the Social Sciences; Vol 8, No 1 (2017). [Google Scholar]
- 83.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, et al. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7: 539 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li W, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22: 1658–1659. 10.1093/bioinformatics/btl158 [DOI] [PubMed] [Google Scholar]
- 85.Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol 35: 1547–1549. 10.1093/molbev/msy096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4: 406–425. 10.1093/oxfordjournals.molbev.a040454 [DOI] [PubMed] [Google Scholar]
- 87.Felsenstein J (1985) CONFIDENCE LIMITS ON PHYLOGENIES: AN APPROACH USING THE BOOTSTRAP. Evolution 39: 783–791. 10.1111/j.1558-5646.1985.tb00420.x [DOI] [PubMed] [Google Scholar]
- 88.Tajima F, Nei M (1984) Estimation of evolutionary distance between nucleotide sequences. Mol Biol Evol 1: 269–285. 10.1093/oxfordjournals.molbev.a040317 [DOI] [PubMed] [Google Scholar]
- 89.Letunic I, Bork P (2019) Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Research 47: W256–W259. 10.1093/nar/gkz239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Letunic I, Bork P (2007) Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23: 127–128. 10.1093/bioinformatics/btl529 [DOI] [PubMed] [Google Scholar]
- 91.Magnan CN, Baldi P (2014) SSpro/ACCpro 5: almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, machine learning and structural similarity. Bioinformatics (Oxford, England) 30: 2592–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang S-F, Su M-W, Tseng S-P, Li M-C, Tsao C-H, et al. (2016) Analysis of codon usage preference in hemagglutinin genes of the swine-origin influenza A (H1N1) virus. Journal of Microbiology, Immunology and Infection 49: 477–486. 10.1016/j.jmii.2014.08.011 [DOI] [PubMed] [Google Scholar]
- 93.Sharp PM, Tuohy TM, Mosurski KR (1986) Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res 14: 5125–5143. 10.1093/nar/14.13.5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pal A, Banerjee R, Mondal UK, Mukhopadhyay S, Bothra AK (2015) Deconstruction of Archaeal Genome Depict Strategic Consensus in Core Pathways Coding Sequence Assembly. PLOS ONE 10: e0118245 10.1371/journal.pone.0118245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Quiroz-Castañeda RE, Amaro-Estrada I, Rodríguez-Camarillo SD (2016) Anaplasma marginale: Diversity, Virulence, and Vaccine Landscape through a Genomics Approach. BioMed Research International 2016: 9032085 10.1155/2016/9032085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wallach JC, Samartino LE, Efron A, Baldi PC (1997) Human infection by Brucella melitensis: an outbreak attributed to contact with infected goats. FEMS Immunology & Medical Microbiology 19: 315–321. [DOI] [PubMed] [Google Scholar]
- 97.Contini C, Seraceni S, Cultrera R, Castellazzi M, Granieri E, et al. (2010) Chlamydophila pneumoniae Infection and Its Role in Neurological Disorders. Interdisciplinary Perspectives on Infectious Diseases 2010: 273573 10.1155/2010/273573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.White P, Murphy M, Moss J, Armstrong G, Spencer SP (2007) Chronic fatigue syndrome or myalgic encephalomyelitis. BMJ 335: 411 10.1136/bmj.39316.472361.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nishida H (2012) Evolution of genome base composition and genome size in bacteria. Frontiers in microbiology 3: 420–420. 10.3389/fmicb.2012.00420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Garcia-Vallve S, Romeu A, Palau J (2000) Horizontal gene transfer in bacterial and archaeal complete genomes. Genome Res 10: 1719–1725. 10.1101/gr.130000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lawrence JG, Ochman H (2002) Reconciling the many faces of lateral gene transfer. Trends Microbiol 10: 1–4. 10.1016/s0966-842x(01)02282-x [DOI] [PubMed] [Google Scholar]
- 102.Liu Q (2012) Mutational Bias and Translational Selection Shaping the Codon Usage Pattern of Tissue-Specific Genes in Rice. PLOS ONE 7: e48295 10.1371/journal.pone.0048295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.He B, Dong H, Jiang C, Cao F, Tao S, et al. (2016) Analysis of codon usage patterns in Ginkgo biloba reveals codon usage tendency from A/U-ending to G/C-ending. Scientific Reports 6: 35927 10.1038/srep35927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Song H, Liu J, Song Q, Zhang Q, Tian P, et al. (2017) Comprehensive Analysis of Codon Usage Bias in Seven Epichloë Species and Their Peramine-Coding Genes. Frontiers in microbiology 8: 1419–1419. 10.3389/fmicb.2017.01419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sueoka N (1988) Directional mutation pressure and neutral molecular evolution. Proc Natl Acad Sci U S A 85: 2653–2657. 10.1073/pnas.85.8.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Holm S (1979) A Simple Sequentially Rejective Multiple Test Procedure. Scandinavian Journal of Statistics 6: 65–70. [Google Scholar]
- 107.Aickin M, Gensler H (1996) Adjusting for multiple testing when reporting research results: the Bonferroni vs Holm methods. American Journal of Public Health 86: 726–728. 10.2105/ajph.86.5.726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Beveridge TJ (1990) Mechanism of gram variability in select bacteria. Journal of bacteriology 172: 1609–1620. 10.1128/jb.172.3.1609-1620.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cheng J, Randall AZ, Sweredoski MJ, Baldi P (2005) SCRATCH: a protein structure and structural feature prediction server. Nucleic acids research 33: W72–W76. 10.1093/nar/gki396 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TXT)
(TXT)
(DOC)
(TXT)
(XLS)
(XLS)
(XLS)
(TXT)
(TXT)
(JPG)
(JPG)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.