

Abstract
For thousands of years opioids have been the first-line treatment option for pain management. However, the tolerance and addiction potential of opioids limit their applications in clinic. NFP, a MOR/KOR dual-selective opioid antagonist, was identified as a ligand that significantly antagonized the antinociceptive effects of morphine with lesser withdrawal effects than naloxone at similar doses. To validate the potential application of NFP in opioid addiction treatment, a series of in vitro and in vivo assays were conducted to further characterize its pharmacological profile. In calcium mobilization assays and MOR internalization studies, NFP showed the apparent capacity to antagonize DAMGO-induced calcium flux and etorphine-induced MOR internalization. In contrast to the opioid agonists DAMGO and morphine, cells pretreated with NFP did not show apparent desensitization and down regulation of the MOR. Though in vitro bidirectional transport studies showed that NFP might be a P-gp substrate, in warm-water tail-withdrawal assays it was able to antagonize the antinociceptive effects of morphine indicating its potential central nervous system activity. Overall these results suggest that NFP is a promising dual selective opioid antagonist that may have the potential to be used therapeutically in opioid use disorder treatment.
Keywords: NFP, dual-selective opioid antagonist, opioid use disorder, drug abuse and addiction, desensitization, down regulation
Graphical Abstract
NFP was able to antagonize the effect of morphine indicative of tolerance developed by Day 7.
1. Introduction
Opioids have been used for many years to manage pain (Manchikanti et al., 2008; Svendsen et al., 2005; Cohen et al., 2003). Opioid drugs, such as morphine, oxycodone, and hydrocodone, are still widely used clinically to treat pain. However, their clinical use is often accompanied by diverse side effects, such as abuse (acute and prolonged use), respiratory depression (acute and prolonged use), and constipation (acute use/prolonged use) (McNicol et al., 2003). In addition, prolonged use of opioid agonists can lead to differential of tolerance to analgesic versus side effects as well as the development of dependence. Of those adverse effects, opioid abuse and addiction have become a serious world-wide concern. It is reported that among 250 million people illegally using drugs all over the world, 13.2 percent of them preferentially abusing opioids (United Nations Office on Drugs and Crime, 2015). As of 2015, approximately 4.4 million people in the U.S. alone were reported to abuse opioids (Center for Behavioral Health Statistics and Quality, 2015). Moreover, the number of deaths resulting from opioid overdose has increased four-fold since 1999 (Muhuri et al., 2013). Therefore, therapeutics to address the addiction/abuse effects of opioids is highly desirable.
Among three major subtypes of opioid receptors (Pande et al., 1996; Petrillo et al., 2003; Xu et al., 1996), the mu opioid receptor (MOR) is the primary pharmacological target for most known opioids (Remesic et al., 2017). Detoxification and maintenance therapy are two of the most commonly used approaches to treat opioid addiction. However, about 40-60% of patients relapse after treating with medications for opioid addiction, such as methadone and buprenorphine (Fig. 1) (National Institute on Drug Abuse, 2012). It has been reported that opioid antagonists, such as naltrexone and naloxone (Fig. 1), displayed the ability to manage opioid addiction and reduce relapse (Chen et al., 2010; George et al., 2010; Minozzi et al., 2011) while there are some side effects seen in patients using these drugs, such as dysphoria, depression, even suicide (Miotto et al., 2002; Ritter et al., 2002). Additionally, patients taking high doses of these drugs have a higher risk of hepatotoxicity, and cardiovascular and pulmonary problems (van Dorp et al., 2007; Dhopesh et al., 2000; Hallinan et al., 2005). Some of the side effects of these opioid antagonists may be related with their low selectivity to the MOR over the other two opioid receptors (Valtchanova-Matchouganska et al., 2004; Filliol et al., 2000). For example, it has been found that the release of dynorphin, resulting in activation of the kappa receptor (KOR), may contribute to mood with opioid withdrawal (Butelman et al., 2012). Interestingly, De Marco et al. recently reported a new KOR partial agonist with less harmful side effects (De Marco et al., 2018). Therefore, it would be intriguing to further examine whether partial agonism or antagonism of KOR may provide a new avenue for opioid use disorder therapeutics.
Fig. 1.

The chemical structures of opioid receptor agonists and antagonists.
As a continuous effort to develop opioid receptor selective ligands (Li et al., 2009; Yuan et al., 2012; Williams et al., 2016; Zheng et al., 2018; Obeng et al., 2018; Obeng et al., 2019), NFP (17-cyclopropylmethyl-3,14-dihydroxy-4,5-epoxy-6-[(3’-fluoro-4’-pyridyl)acetamido]mo rphinan, Fig. 1), a MOR/KOR dual-selective antagonist of NAP derivatives (Li et al.,2009, Fig. 1), was recently identified in our laboratory (Zheng et al., 2019). NFP showed a high binding affinity to the MOR (Ki = 0.36 nM) with 436-fold selectivity to the MOR over the DOR with a single-digit nanomolar binding affinity to the KOR (Ki = 4.80 ± 0.26 nM). Further studies demonstrated that NFP effectively antagonized the antinociceptive effects of morphine while it failed to precipitate withdrawal symptoms in morphine-dependent mice as compared to naloxone. Herein we report further exploration of the pharmacological properties of NFP using functional in vitro and in vivo assays for its potential applications in opioid use disorder treatment.
2. Materials and methods
2.1. Calcium mobilization assay
A Chinese hamster ovary cell line stably expressing the mouse μ opioid receptor (mMOR-CHO) was used for this assay (Obeng, et al., 2018, 2019). The cells were transfected with Gαqi5 for 4 h and then plated (3,000,000 cells/well) to black 96-well plates with clear bottoms (Greiner Bio-One). After 24 h of incubation, the culture media was removed and the cells were washed with assay buffer (50 ml HBSS, 1 ml HEPES, 250 μl probenecid, 50 μ 1mM CaCl2, 50 μl 1mM MgCl2). The hydrochloride salt of NFP was dissolved in water as a stock solution for assay (1 M).
For agonist assays, cells were then incubated with 50 μl/well loading buffer (6 ml assay buffer, 24 μl Fluo4-AM solution (Invitrogen), 12 μl probenecid solution) for 45 min. Following incubation, different concentrations of the test compounds were added by FlexStation3 microplate reader (Molecular Devices) and read at ex494/em516. Each concentration was run in triplicate.
For antagonism studies, the cells were incubated with the same loading buffer as the agonist assay for 45 min. Then, different concentrations of the test compounds (20 μL/well) were manually added to each well followed by another 15 min incubation. After that, the solution of DAMGO in assay buffer (500 nM) or just assay buffer (blank) was added by FlexStation3 microplate reader (Molecular Devices) and read at ex 494/em 516. Each concentration was run in triplicate.
The corresponding Emax and EC50 or IC50 value of each compound was calculated by non-linear regression using GraphPad Prism 6.0 (GraphPad Software, San Diego, CA).
2.2. Downregulation and desensitization study
2.2.1. Incubation of mMOR-CHO cells with opioid ligands
mMOR-CHO cells were grown in culture media (DMEM/F12 media, 10% FBS, 1% penicillin/streptomycin, 0.5% G418) for 5 days in an incubator set at 37 °C with 5% CO2 and 95% humidity. On the fifth day when the cells were confluent, the culture media was removed and the cells were rinsed with 5 ml PBS. The cells were then treated with DAMGO (5 μM), morphine (5 μM), nalbuphine (1 μM), NFP (1 μM), naltrexone (1 μM) and vehicle (0.02% DMSO) dissolved in DMEM/F12 media and incubated for 24 h. After incubation, the treatment media was removed and the cells were washed three times with 10 ml phosphate-buffered saline (PBS). 5 ml PBS was added to each dish and the cells were then scraped off the dishes using a scraper. The cells were then centrifuged at 1,000 x g for 10 min at 4 °C. After centrifugation, the supernatant was decanted and membrane buffer (50 mM Tris, 3 mM MgCl2, and 1 mM EGTA, pH 7.4) was added to each sample. The cells were then homogenized and centrifuged again at 50,000 x g at 4 °C for 10 min. The supernatant was decanted and the cells were homogenized again in membrane buffer. A Bradford assay was conducted to determine the concentration of the membrane protein. The membrane protein preparations were then stored at −80 °C.
2.2.2. mMOR receptor saturation assay
Membranes were homogenized in membrane buffer and centrifuged at 50,000 x g for 10 min. This step was repeated to ensure that the drugs were completely removed from the receptor. The supernatant was then decanted and membranes were re-suspended in binding buffer (50 mM Tris-HCl, 3 mM MgCl2, and 0.2 mM EGTA (pH 7.4). A Bradford assay was conducted to determine the protein concentration. The MOR-CHO membranes (30 μg protein/sample) were then incubated in binding buffer containing varying concentrations of [3H]naloxone (specific activity = 66.58 Ci/mmol) in a 0.5 ml total volume for 90 min at 30 °C. Nonspecific binding was determined using 5 μM naltrexone. The incubation was terminated by rapid filtration and bound radioactivity was determined as described previously (Selley et al., 1998). KD and Bmax values were determined by non-linear regression using GraphPad Prism 6.0 (GraphPad Software, San Diego, CA).
2.2.3. [35S]GTPγS binding assay
Ligand-stimulated [35S]GTPγS binding was performed as described previously (Selley et al., 1998). Briefly, membranes from the treated mMOR-CHO cells (10 μg of protein) were incubated with 0.1 nM [35S]GTPγS (specific radioactivity was 1250 Ci/mmol) and 20 μM GDP for 90 min at 30 °C with varying concentrations of DAMGO in assay buffer (50 mM Tris-HCl, 3 mM MgCl2, 100 mM NaCl, 0.2 mM EGTA, pH 7.4). Nonspecific binding was determined with 20 μM unlabeled GTPγS and basal binding was determined in the absence of MOR ligand. The incubation was terminated by rapid filtration through GF/B glass fiber filters and rinsed three times with ice-cold wash buffer (50 mM Tris-HCl, pH 7.2). Bound radioactivity was determined by liquid scintillation spectrophotometry at 95% efficiency for 35S. Net-stimulated [35S]GTPγS binding was defined as ligand-stimulated minus basal binding. All assays were determined in duplicate and repeated at least 4 times. Concentration-effect and saturation binding data were analyzed by non-linear regression using GraphPad Prism 6.0 (GraphPad Software, San Diego, CA).
2.2.4. Statistical Analysis
One-way ANOVA followed by the posthoc Dunnett test were performed to assess significance using Prism 6.0 software (GraphPad Software, San Diego, CA).
2.3. MOR internalization
2.3.1. Cell culture
Neuro2A murine neuroblastoma cells (N2A cells) stably transfected with the rat MOR cDNA epitope-tagged with HA at N-terminus (N2A-HA-rMOR) were established previously (Obeng, et al., 2019) and clones H38 and H16 expressing MOR at 1-2 pmole/mg protein were used in the study. Cells were cultured in 10-cm dishes at 37°C with 5% CO2 in humidified air in MEM (Minimum Essential Medium, ref 41500, Gibco, NY) supplemented with 10% FBS and penicillin, streptomycin and amphotericin (A5955, Sigma, MO) and grew to 80% confluence.
2.3.2. MOR internalization
MOR internalization experiments were conducted as described previously (Obeng, et al., 2019) Briefly, N2A-HA-rMOR cells were sub-cultured onto coverslips placed in 6-well plates at 300,000 cells per well. Forty-eight h later, mouse anti-HA.11 antibodies (Clone 16B12, BioLegend, CA) were added at 1:1000 to cell medium and incubated for 1 h. NFP (final 10 μM) or vehicle was added and incubated for 15 min followed by addition of etorphine (final 10 μM) or vehicle and incubated for another 15 min. Cells were cooled on ice, washed, and fixed with 4% paraformaldehyde (PFA) in PB for 15 min, and washed. Cells were then incubated with Alexa Fluor594 goat anti-mouse IgG (1:1000) (A11005, Invitrogen, OR) on a shaker with light protection for 2 h at room temperature. After washing, the cells on cover slips were mounted on slides using mounting medium (H-1200, Vector Laboratories, CA) and cured overnight. The images were acquired by a Nikon Eclipse TE300 fluoresce microscope coupled to a digital camera (MagnaFire) using 20X objective and processed with ImageJ.
2.4. Caco-2 bidirectional transport assay
2.4.1. Transport of NFP in Caco-2 cell line
Caco-2 cells (passage 48; ATCC, Manassas, VA) were plated on 12 mm, 0.4 μm, #3460 - Clear Transwell® inserts (Corning Incorporated, Corning, NY) at an initial seeding density of 90,000 cells/well. The cells were cultured in DMEM supplemented with 10 % FBS and non-essential amino acids for 23 days. On day 24, the medium was removed and the transwells were rinsed with PBS. HBSS buffer (pH 7.4) was added to both apical (0.5 ml) and basolateral (1.5 ml) chambers and the initial transepithelial electrical resistance (TEER) values were recorded. The blank HBSS buffer in apical or basolateral chambers was replaced by NFP solutions in HBSS (20 μM) with or without P-glycoprotein (P-gp) inhibitor (elacridar, 1 μM) (Matsson et al., 2009). 200 μl aliquots were collected from the receiver chamber up to 2 h. Acetonitrile (50 μl was added to each sample (50 μl, which was vortexed then centrifuged at 10,000g for 5 min at 20°C. The supernatant was then used for analysis by LC-MS as described below. At the end of 2 h, the solution in both chambers was replaced with HBSS and post-experiment TEER values were measured. The integrity of the monolayers was confirmed by studying the transport of lucifer yellow, a low permeability marker (Maharao et al. 2017) The permeability directional ratio (PDR) was calculated as the ratio of basolateral-to-apical permeability to apical-to-basolateral permeability (Papp, B-A / Papp, A-B). Two-way ANOVA followed by Bonferroni’s multiple comparisons test was performed for determining statistical significance for the permeability values (F (dFn, dFd): interaction F (1, 8) = 184, P < 0.0001; treatment F (1, 8) = 267, P < 0.0001), while Student’s t-test was performed to compare the PDR values. All statistical analyses were performed using GraphPad Prism 7.0 (GraphPad Software, Inc., San Diego, CA). Results were considered to be statistically significant if P values were lower than 0.05.
2.4.2. LC-MS conditions
Chromatographic separation was achieved using a Waters HPLC system (Waters Corporation, Milford, MA). The analyte NFP was separated on a Thermo Scientific Hypersil BDS C18 column (50 × 4.6 mm, 3 μm; Waltham, MA) with isocratic elution (aq. 0.1% formic acid and acetonitrile, 85:15). The flow rate was 1.0 ml/min and the temperature of the column was maintained at 35 °C. The injection volume was 40 μl. The MS system consisted of an Acquity QDa mass spectrometer (Waters Corporation, Milford, MA) equipped with electrospray ionization in the positive ion detection mode. The run time for each sample was 5.20 min. Data acquisition and processing were performed using Empower 3 software (Waters Corporation, Milford, Massachusetts, USA). The retention time for NFP was found to be 2.99 min and had an m/z value of 466.20 ([NFP + H]+).
2.5. Warm-Water Tail-Withdrawal Test
2.5.1. Animals
Adult male C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) aged approximately 8 weeks at the start of studies were housed in a temperature-controlled (20-22°C) AAALAC-accredited facility in which they had ad libitum access to food and water. The mice were maintained on a 12 h/12 h light-dark cycle (0600-1800 lights on) for the duration of the experiment and were tested during the light segment of this cycle. Mice arrived at the vivarium housed 4/cage, and following one-week habituation were separated into individual cages. Mice were allowed to acclimate to individual caging for at least 24 h and then were randomly assigned to the various treatment conditions before the start of studies. Experimenters were blinded to these treatment conditions during the duration of the experiment and data analysis. No adverse events occurred during the experiment and no mice were excluded from data analysis. Protocols and procedures (Animal Welfare Assurance Number D16-00180) were approved by the Institutional Animal Care and Use Committee (IACUC) at Virginia Commonwealth University Medical Center and complied with the recommendations of the IASP (International Association for the Study of Pain).
2.5.2. Apparatus and drugs
A commercial warm water bath (Model # JBN5 US; Grant Instruments Ltd., Cambridge, UK) maintained at 52.5 ± 0.5°C was used to assess nociception. Tail withdrawal latencies were measured with a digital stopwatch (Model # 14-649-7; Fisher Scientific, Pittsburgh, PA). Morphine sulfate was obtained from the National Institute on Drug Abuse Drug Supply Program. All compounds were dissolved in sterile saline (Fisher Scientific, Pittsburgh, PA; Cat. # 125EZ-7002) and were administered s.c. in a volume equivalent to 10 ml/kg body weight.
2.5.3. Warm-water tail-withdrawal tests
Mice were randomly assigned to either the Vehicle Group or the 0.3 mg/kg NFP Group (N=6/group). Each group was tested twice; once on Day 1 prior to chronic dosing with morphine, and once on Day 7 following chronic dosing.
Day 1 Tests:
At the start of testing of Day 1, each mouse was placed in a restraint cloth fashioned from a surgical drape, and the distal 3 cm of its tail was submerged in the warm water bath to determine its baseline withdrawal latency. A digital stopwatch was used to record the amount of time that elapsed between tail immersion and tail withdrawal (i.e., tail-withdrawal latency). Immediately after that, mice received consecutive injections of saline (i.e., morphine's vehicle) and their scheduled NFP condition (i.e., vehicle or 0.3 mg/kg NFP), and were returned to their home cage.
After a 30-min pretreatment period had elapsed, tail-withdrawal latencies were re-determined, and mice were immediately injected with the lowest dose of morphine (1 mg/kg). Following the 30-min pretreatment period, tail-withdrawal latencies were re-determined, and the next highest dose of morphine was administered (i.e., acutely 2.2 mg/kg resulting in a cumulative dose of 3.2 mg/kg). This process was repeated with cumulative doses of 10 and 32 mg/kg morphine. A 10-s cutoff time was imposed across all assessments to minimize potential tissue damage.
Days 2-6:
On non-test days (i.e., Days 2-6) at a similar time each afternoon (between 1400 and 1500 h), mice received two injections one right after the other, either 10 mg/kg morphine + NFP vehicle (saline; Vehicle Group) or 10 mg/kg morphine + 0.3 mg/kg NFP (NFP Group).
Day 7 Tests:
Approximately 24 h after receiving the last set of injections on Day 6, mice were re-assessed in the warm-water tail-withdrawal procedure with cumulative morphine dosese identical to that described for Day 1.
2.5.4. Data Analysis
Tail-withdrawal latencies were recorded for each mouse and data were expressed as percent maximum possible effect (%MPE) as follows: [(test-baseline)/(10-baseline)*100]. Control data represented %MPE values obtained after administration of saline (morphine vehicle) and either 0.3 mg/kg NFP or saline (NFP's vehicle) on the respective test day. Within-group analyses were conducted separately for %MPE data generated during Day 1 and Day 7 for each group using a repeated measure ANOVA with morphine dose as the within-subjects factor. Fisher’s LSD post-hoc tests were used to identify significant differences of morphine dose vs control. Separate two-way ANOVAs comparing %MPE scores between the Vehicle and NFP groups during Day 1 and Day 7 were additionally conducted. Data were subsequently analyzed using Fisher’s LSD post-hoc tests comparing Vehicle and NFP groups at each morphine dose on each day. All statistical tests were conducted using microcomputer software (Prism 8 for Mac OSX, GraphPad Software, Inc., San Diego, CA), and all types of comparisons were considered statistically significant if P < 0.05.
2.6. Statistical analysis
The data of each assay were repeated at least three times and presented as mean values ± S.E.M. GraphPad Prism 6.0 or 7.0 (GraphPad Software, San Diego, CA) was used to statistical analyze the data of each assay. The statistical significance was determined using GraphPad Prism 6.0 or 7.0 by Student's t-test or two-way ANOVAs with the indicated post-hoc test. Results were considered to be statistically significant if P ≤ 0.05.
3. Results
3.1. Calcium mobilization assay
The calcium mobilization assay in mMOR-CHO cells transfected with chimeric Gaqi5 proteins was used to measure functional receptor activation at the effector level, downstream of G-protein activation (Selly et al., 1998). After stimulated by a ligand, the conformation of the GPCR changes, thus activating its corresponding intracellular downstream messenger pathway. As a result of these variations, cytosolic calcium is released which can be measured using a fluorescent calcium indicator (Zhu et al., 2008). DAMGO and naltrexone were used as control compounds. The assay was carried out as described previously (Yuan et al., 2013). As seen in Fig. 2A, the full MOR agonist DAMGO displayed a concentration-dependent, robust stimulation of Ca2+ signal (EC50 = 36.32 ± 1.85 nM). However, NFP did not induce any apparent Ca2+ mobilization as compared with DAMGO, even at the highest concentration tested (3 μM), indicating a lack of agonist efficacy. Therefore, the ability of NFP to antagonize the DAMGO-stimulated Ca2+ signal was determined (Fig. 2B). Both NFP and naltrexone inhibited DAMGO (500 nM)-induced calcium flux in a concentration-dependent manner, which confirmed that NFP acted as an antagonist similar to naltrexone. Compared to naltrexone, however, NFP inhibited the DAMGO stimulated calcium flux with a 12-fold lower potency.
Fig. 2.

Calcium mobilization assay of NFP as an agonist (A) or an antagonist (B). The EC50 of DAMGO is 36.32 ± 1.85 nM, IC50 values of naltrexone and NFP are 6.62 ± 45 nM and 76.09 ± 2.50 nM, respectively. Data are presented as mean values ± S.E.M (n = 3).
3.2. Downregulation and desensitization study
To validate the potential application of NFP in opioid addiction treatments, it is important to study its capacity to desensitize and/or downregulate opioid receptors. Here, the effect of NFP on MOR-mediated G-protein activation and MOR Bmax levels was examined after prolonged exposure of MOR-CHO cells to the drugs. DAMGO (opioid agonist), morphine (opioid agonist), nalbuphine (opioid partial agonist), and naltrexone (opioid antagonist) were used as control compounds for comparison. Concentration-effect and saturation binding data were analyzed by non-linear regression using GraphPad Prism 6.0 (GraphPad Software, San Diego, CA). One-way ANOVA followed by the post-hoc Dunnett test were performed to assess significance using Prism 6.0 software (GraphPad Software, San Diego, CA).
As shown in Fig. 3, compared with the vehicle treated group, the concentration-effect curves of the opioid agonists, morphine and DAMGO, showed an apparent right-shift. This was confirmed by curve-fitting analysis: the DAMGO EC50 values in cells treated with morphine and DAMGO increased about 6- and 4-fold, respectively (Table 1), indicating that morphine and DAMGO desensitized the MOR. Conversely, the DAMGO EC50 values in nalbuphine, naltrexone and NFP treated cells were similar to the DAMGO EC50 value for the vehicle treated cells (Table 1). There was no significant effect of prior treatment on the DAMGO Emax values compared to vehicle-treated cells. These results demonstrated that prior treatment of cells with the opioid agonists morphine or DAMGO desensitized MOR mediated G-protein activation compared with the vehicle pretreated cells. However, the cells pretreated with opioid partial agonists (nalbuphine and NFP) or the opioid antagonist (naltrexone) did not desensitize the activation of G-proteins mediated by the MOR. This conclusion was further confirmed by the efficiency of receptors determined as Emax/EC50 (Table 1). These results showed that pretreatment of cells with morphine and DAMGO significantly reduced the efficiency of MOR activation by DAMGO, while the cells pretreated with nalbuphine, naltrexone, and NFP did not show noticeable desensitization compared to the vehicle group.
Fig. 3.

The concentration-effect curves of DAMGO in vehicle (0.02% DMSO), DAMGO (5 μM), Morphine (5 μM), NFP (1 μM), naltrexone(1 μM), and nalbuphine (1 μM) pretreated mMOR-CHO cells. Data presented means ± S.E.M. and repeated at least 4 times in independent experiments (Obeng et al., 2019).
Table 1.
Bmax, KD values and EC50 and Emax values of [3H]naloxone saturation binding and GTPγS functional assay, respectively in opioid ligand-pretreated MOR-CHO cell membranes.
| Pretreatment | Bmax (pmol/mg) |
KD (nM) | EC50 (nM) | Emax (net fmol/mg) | Efficiency (Emax/EC50) |
|---|---|---|---|---|---|
| Vehicleb | 2.90 ± 0.24 | 1.16 ± 0.23 | 27.54 ± 3.78 | 123.04 ± 11.93 | 4.47 |
| DAMGOb | 1.49 ± 0.12a | 1.96 ± 0.07 | 120.83 ± 6.97 | 121.62 ± 13.78 | 1.01 |
| Morphineb | 1.58 ± 0.10a | 1.84 ± 0.28 | 168.43 ± 6.25 | 140.63 ± 6.59 | 0.83 |
| NFP | 2.10 ± 0.15 | 3.22 ±0.32a | 53.95 ± 9.87 | 156.20 ± 13.83 | 2.90 |
| Nalbuphineb | 3.66 ± 0.26 | 1.70 ± 0.10 | 48.40 ± 5.65 | 165.63 ± 6.52 | 3.42 |
| Naltrexoneb | 3.86 ± 0.68 | 3.84 ± 0.44a | 21.85 ± 0.78 | 126.73 ± 3.97 | 5.80 |
Data are mean ± S.E.M. of Bmax, KD, EC50 and Emax values derived from non-linear regression analysis of [3H]naloxone saturation binding and DAMGO-stimulated [35S]GTPγS binding curves in memberanes from mMOR-CHO cells pretreated with the indicated ligand or vehicle (n = 4).
: P < 0.05, different from corresponding value in vehicle-pretreated cells.
: Values previously published (Obeng et al., 2018).
As described previously, prolonged treatment with agonists may also induce desensitization in the number of opioid receptors. Therefore, it is important to discern whether the compound induces MOR downregulation in addition to desensitization. To address this question, a mMOR-CHO saturation binding assay using [3H]-naloxone was applied to determine KD and Bmax values (Kostenis et al., 2005; Allouche et al., 2008; Christie et al., 2008; Obeng et al., 2018; Obeng et al., 2019). As the results demonstrated (Table 1), compared with vehicle pretreated group, there was significant MOR downregulation seen as a decrease in Bmax in cells pretreated with morphine and DAMGO [F (5, 5.195) = 9.708, P = 0.0117], whereas nalbuphine, naltrexone and NFP did not induce significant downregulation of the MOR. Cells pretreated with naltrexone and NFP had almost three times greater [3H]-naloxone KD values compared to the vehicle group [F (5, 7.525) = 22.56, P = 0.0002], which was not seen in cells pretreated with morphine, DAMGO and nalbuphine.
3.3. MOR internalization
As described previously, desensitization and downregulation of opioid receptors may occur following prolonged binding of opioid agonists to the receptors. Desensitization and downregulation of receptors may co-occur with internalization via different mechanisms (Kaplan et al., 2011). Following internalization, receptors may be recycled or downregulated. NFP did not cause significant desensitization or downregulation of the MOR. Here we examined if NFP promoted MOR internalization. N2A-HA-rMOR-N2A cell line was used for this assay because CHO cells have small cytosolic volume and it is difficult to visualize receptor internalization. The opioid agonist etorphine was used as a positive control. Compared with the control group (Fig. 4A), etorphine (10 μM, Fig. 4B) distinctly induced internalization of the MOR, whereas NFP did not. (10 μM, Fig. 4C). Interestingly, when etorphine and NFP were incubated together (10 μM respectively, Fig. 4D), internalization of the MOR induced by etorphine was reduced. These results indicated that NFP can antagonize the MOR internalization induced by etorphine. This result was consistent with those of the downregulation and desensitization studies.
Fig. 4.

The MOR internalization assay result of NFP in N2A-HA-rMOR-N2A cell line. (A) Control panel; (B) Etorphine panel; (C) NFP panel; (D) NFP+Etorphine panel. Etorphine was used as positive control. Two different HA-rMOR-N2A cell lines were used for this assay and both of them showed similar results. One of those results (H38 cells) was presented here. Each experiment was repeated twice.
3.4. Caco-2 bidirectional transport assay
As reported previously (Zheng et al., 2019) NFP showed evident MOR antagonism in in vivo studies with an AD50 value of 2.82 (1.34-5.94) mg/kg with a 95% confidence level (CL). Additionally, NFP precipitated dramatically fewer withdrawal symptoms compared with naloxone at the same doses. These characteristics were maintained even at the highest dose (100 mg/kg) tested. However, the potency of NFP was lower than that of naloxone (AD50 = 0.05mg/kg) (Li et al., 2009). To address this discrepancy, a bidirectional transport (apical-to-basolateral and basolateral-to-apical) assay was conducted to simulate and assess the bidirectional permeability of NFP in the human gastrointestinal tract and test whether NFP is a P-gp substrate (Mitra et al., 2011).
The net TEER values for the transwells greater than 200 Ω.cm2, and a Papp of <1 × 106 cm/s for lucifer yellow confirmed the integrity of the Caco-2 monolayers (Maharao et al., 2017). As seen in Fig. 5, the basolateral-to-apical permeability (Papp, B-A, efflux) of NFP (20 μM) is much higher than the apical-to-basolateral permeability (Papp, A-B, absorption). However, when the P-gp inhibitor elacridar (Matsson et al., 2009) (10 μM) was added, it reduced the B-A transport significantly more than A-B transport. Elacridar reduced the PDR value of NFP from 7.0 ± 0.5 to 1.3 ± 0.3, consistent with P-gp transport activity. A similar phenomenon is also seen with a well-known P-gp substrate, digoxin. In a related study, the PDR of digoxin decreased from 5.1 to 1.2 after elacridar was added (Mitra, P., 2011). These results suggest that NFP may be a P-gp substrate.
Fig. 5.

The result of bidirectional transport assay for NFP. Papp values of tested compounds in apical-to-basolatoral (A-B) and basolateral-to-apical (B-A) directions were determined by incubating Caco-2 monolayers at 37 °C for 2 h with NFP (20 μM). Data represented means ± S.E.M. (n =3); two-way ANOVA results: F (dFn, dFd): interaction F (1, 8) = 184, P < 0.0001; treatment F (1, 8) = 267, P < 0.0001; Bonferroni post-test results: *** indicates P < 0.0001, ns indicates no significant difference.
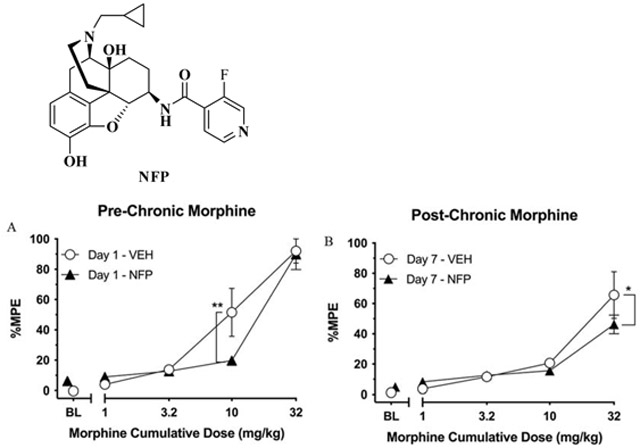
3.5. Warm-water tail-withdrawal assay
As reported previously, NFP was shown to act as a centrally acting antagonist (Zheng et al., 2019) without apparent desensitization and downregulation effects. Moreover, it did not precipitate significant withdrawal symptoms as compared with naloxone, even at high doses (Zheng et al., 2019). To further evaluate its centrally acting pharmacological features, a warm-water tail-withdrawal test was used in a morphine cumulative-dosing mouse model.
Day 1 results of morphine cumulative dosing tests for both groups are presented in Fig. 6A. Two-way ANOVA indicated a significant main effect of morphine [F (4, 40) = 62.05, P < 0.0001], no effect of pretreatment condition (i.e., saline or 0.3 mg/kg NFP) [F (1, 10) = 1.164, P = 0.3060], and a significant interaction [F (4, 40) = 2.851, P = 0.0360]. Within-subjects post-hoc analysis showed that morphine produced significant increases in %MPE relative to the respective control condition at doses of 10 (P < 0.0001) and 32 (P < 0.0001) mg/kg in the vehicle-pretreated group, and of 32 (p < 0.0001) mg/kg in the NFP-pretreated group. Further, between-subjects analysis revealed that 10 mg/kg morphine produced significantly less antinociception in the NFP group relative to the vehicle-treated group (P = 0.0013) demonstrating that NFP reduces the antinociceptive effects of morphine at 10 mg/kg. However, this antagonism was surmounted by increasing the dose to 32 mg/kg morphine where there was no difference in antinociception between groups (P = 0.8201).
Fig. 6.

The result of the warm-water tail-withdrawal test. (A) Effects of vehicle (“VEH”, i.e., saline) or NFP (0.3 mg/kg, s.c.) pretreatment on warm-water tail-withdrawal latencies following cumulative doses of morphine (1-32 mg/kg, s.c.) expressed as percent maximum possible effect (%MPE). Values represent the mean (values ± S.E.M.) %MPE of 6 mice. Unfilled circles represent %MPE values following saline pretreatment. Filled triangles represent results following pretreatment with 0.3 mg/kg NFP. ** indicates NFP pretreatment resulted in a lower %MPE score than the vehicle treated group; P = 0.0013. (B) Results of cumulative doses of morphine on warm-water tail-withdrawal latencies in the Vehicle and NFP Groups following five days of dosing with 10 mg/kg morphine + either saline or 0.3 mg/kg NFP. * indicates NFP pretreatment resulted in a lower %MPE score than the vehicle treated group; P = 0.0138.
Six days after the initial morphine cumulative-dosing experiment, the effects of chronic 0.3 mg/kg NFP or saline co-treatment with morphine were assessed in an identical tail-withdrawal procedure (Fig. 6B). Two-way ANOVA indicated that there was a significant main effect of morphine [F (4, 40) = 36.55, P < 0.0001) and a significant interaction [F (4, 40) = 2.737, P = 0.0420), but no effect of pretreatment condition [F (1, 10) = 0.01645, P = 0.9005]. Within-subjects post-hoc analysis indicated that morphine only produced a significant increase in %MPE relative to control conditions at a dose of 32 mg/kg for both the vehicle-pretreated (P < 0.0001) and NFP-pretreated (P < 0.0001) groups. Morphine produced significantly less antinociception in the NFP group relative to the Vehicle Group at the dose of 32 mg/kg (P = 0.0138).
4. Discussion
As previously reported, NFP is a MOR/KOR dual-selective partial agonist (Zheng et al., 2019), showing 34.96% efficacy relative to DAMGO and 26.71% efficacy relative to U50,488H with single-digit nanomolar potency to the MOR and double-digit nanomolar potency to the KOR. NFP showed much lower binding affinity to the DOR (Zheng et al., 2019). In the studies presented herein, it was shown that NFP did not induce apparent calcium flux as compared with the opioid agonist DAMGO. Conversely, NFP was able to inhibit the mobilization of calcium induced by DAMGO, thus confirming its ability to act as an MOR antagonist. Interestingly, the results of calcium assay for NFP were contrary to the [35S]GTPγS assay results in which NFP was shown to act as a partial agonist with 34.97% efficacy relative to DAMGO (Zheng et al., 2019). One possible explanation is that the chimeric G-protein Gαqi5 was used for calcium mobilization assay. Kostenis et al. (2005) reported that the use of a Gα-protein can significantly affect the pharmacological profiles of compounds when they interact with receptors. For example, ipsapirone, a known agonist at 5-HT1A receptors, did not show any calcium flux signal using Gα-protein in a calcium mobilization assay. The inhibition of Ca2+ flux might be induced by the direct binding of NFP with Gβγ subunits of G proteins and the voltage activation of Ca2+ channel pore opening decrease (Zamponi et al., 1998, 2002) though more experimental evidence would be needed to support this hypothesis. On the other hand, the results of the calcium flux assay were consistent with the in vivo studies of NFP, substantiating its ability to effectively block the antinociceptive effects of morphine (Zheng et al., 2019).
The phenomenon of desensitization and tolerance in chronic opioid use was elaborated during the 1980’s (Allouche et al., 2014). As a result of chronic exposure to opioid agonists such as morphine, the ligand-mediated opioid receptor response may decrease due to impaired signal transduction inside the cell. This reduced signal transduction is then followed by a downregulation in the number of opioid receptors, which can be measured as Bmax. Contrary to opioid agonists, opioid antagonists and/or opioid partial agonists can induce upregulation and sensitization of opioid receptors. This method provides an appropriate way to study the tolerance and desensitization of receptors by opioid ligands. To carry out this assay, mMOR-CHO cells were pretreated with DAMGO, morphine, nalbuphine, NFP and naltrexone for 24 h. The results demonstrated that cells pretreated with opioid agonists significantly increased the EC50 of DAMGO and morphine (Table 1). On the other hand, groups pretreated with opioid partial agonists (nalbuphine and NFP) or an opioid antagonist (naltrexone) did not significantly change the DAMGO EC50 values (Table 1) or receptor efficiency. Therefore, pretreating the cells with opioid agonists induced the desensitization of the MOR, whereby pretreatment of an opioid partial agonist or antagonist did not result in the desensitization of the MOR. Due to the possible feedback mechanism between desensitization and downregulation, the possible contribution of MOR downregulation to its desensitization was discerned by the saturation assay using [3H] naloxone.
Compared with the vehicle group, the densities of the MOR (Bmax) decreased significantly in DAMGO and morphine groups, while the binding affinity of [3H] naloxone remained unchanged (Table 1). There was no apparent change in MOR density in nalbuphine, naltrexone and NFP pretreated groups; however, there was a significant decrease in binding affinity of [3H] naloxone in groups pretreated with naltrexone and NFP. The groups pretreated with DAMGO and morphine induced desensitization and downregulation of the MOR, but did not affect the binding affinity of [3H] naloxone to the MOR. One possible explanation is internalization of the receptor (Koch and Höllt, 2008). It is possible that some of the MOR receptors are internalized, thus affecting the binding of [3H]-naloxone. The downregulation assay is a method to measure the density of MOR on the membrane. However, as a result of opioid agonist pretreatment, it is possible that some of these binding sites are internalized, which may affect the binding of [3H] naloxone in the saturation binding assay. The MOR internalization assay (Fig. 4) confirmed that opioid agonists produce MOR internalization. Furthermore, NFP reduced %MPE induced by 32 mg/kg morphine from Day 1 to Day 7 from 89.93% to 46.24%, respectively, which suggested that NFP was able to antagonize the effect of morphine indicative of tolerance that developed by Day 7 (Fig. 6). The observation that %MPE induced by 32 mg/kg morphine was significantly lower in the NFP group relative to the vehicle group further documents its potent antagonistic properties.
Though NFP may be an in vitro P-gp substrate (Fig. 5), in vivo studies showed that it can behave as a centrally acting agent (the LogP value of NFP is 1.44, predicted by ChemDraw 18.2). Our previous study showed a PDR for NAP of 131 (Mitra et al., 2011), nearly twice that of NFP. Comparing the chemical structures of NAP and NFP, the fluorine atom on the pyridyl ring may play a key role in improving the CNS penetration capability of NFP. Hagmann reported that fluorination can optimize the penetration of non-CNS drugs crossing BBB (Hagmann et al., 2008). The improved PDR value of NFP over NAP may enhance its ability to cross the BBB and block the antinociceptive effects of morphine. Furthermore, many drugs on the market (5-15%) are fluorinated compounds and more than 100 fluorinated drug candidates are under their clinical trials (Hagmann et al., 2008). Fluorination not only improves these compounds’ metabolic stability but also optimizes their physicochemical properties (Christie et al., 2008; Hagmann et al., 2008; Mahar Doan et al., 2002). Fluorination may improve the physicochemical properties of NFP, such as solubility and log D value, and optimize its metabolic stability. Those changes may also enhance NFP’s ability to cross BBB. This may help explain why NFP is an in vitro P-gp subtract but works as a centrally acting agent in in vivo studies.
5. Conclusion
Taken together, the pharmacological profile of NFP was further evaluated to determine its potential applications in opioid tolerance and opioid use disorder treatment. NFP displayed apparent opioid antagonism in the calcium flux assay. Additionally, in desensitization studies, it was shown that NFP did not produce desensitization and downregulation of the MOR. In fact, NFP showed the potential ability to antagonize internalization of the MOR following desensitization by opioid agonists. Though NFP may be a P-gp substrate at in vitro level, the in vivo studies demonstrated its ability to block morphine’s antinociceptive effects in a 7 day warm-water tail-withdrawal assay, which indicate its potential application for opioid use disorder. Overall, current in vitro and in vivo studies suggest that NFP may serve as a promising therapeutic candidate for opioid use disorder treatment without producing tolerance.
Acknowledgements
The authors are grateful to NIDA Drug Supply Program for providing the free base of naltrexone and morphine sulfate. The work is partially supported by NIH/NIDA Grant DA024022, DA044855 and DA050311 (Y.Z.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Drug Abuse or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest
The authors declare no conflict of interest.
References
- Allouche S, Noble F, Marie N, 2014. Opioid receptor desensitization: mechanisms and its link to tolerance. Front. Pharmacol 5, 280. doi: 10.3389/fphar.2014.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butelman ER, Yuferov V, Kreek MJ, 2012. κ-Opioid receptor/dynorphin system: genetic and pharmacotherapeutic implications for addiction. Trends Neurosci. 35, 587–596. doi: 10.1016/j.tins.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Center for Behavioral Health Statistics and Quality, 2015. Behavioral health trends in the United States: results from the 2014 national survey on drug use and health. HHS Pulication No. SMA 15-4927, NSDUH Ser. H-50. [Google Scholar]
- Chen H, Wu J, Zhang J, Hashimoto K, 2010. Recent topics on pharmacotherapy for amphetamine-type stimulants abuse and dependence. Curr. Drug Abuse Rev 3, 222–238. doi: 10.2174/1874473711003040222. [DOI] [PubMed] [Google Scholar]
- Christie MJ, 2008. Cellular neuroadaptations to chronic opioids: tolerance, withdrawal and addiction. Br. J. Pharmacol 154, 384–396. doi: 10.1038/bjp.2008.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MZ, Easley MK, Ellis C, Hughes B, Ownby K, Rashad BG, Rude M, Taft E, Westbrooks JB, JCAHO., 2003. Cancer pain management and the JCAHO’s pain standards: an institutional challenge. J. Pain Symptom Manage 25, 519–527. doi: 10.1016/S0885-3924(03)00068-X. [DOI] [PubMed] [Google Scholar]
- De Marco R, Bedini A, Spampinato S, Comellini L, Zhao J, Artali R, Gentilucci L, 2018. Constraining endomorphin-1 by β,α-hybrid dipeptide/heterocycle scaffolds: identification of a novel κ-opioid receptor selective partial agonist. J. Med. Chem 61, 5751–5757. doi: 10.1021/acs.jmedchem.8b00296. [DOI] [PubMed] [Google Scholar]
- Dhopesh VP, Taylor KR, Burke WM, 2000. Survey of hepatitis B and C in addiction treatment unit. Am. J. Drug Alcohol Abuse 26, 703–707. doi: 10.1081/ADA-100101903. [DOI] [PubMed] [Google Scholar]
- Filliol D, Ghozland S, Chluba J, Martin M, Matthes HW, Simonin F, Befort K, Gavériaux-Ruff C, Dierich A, LeMeur M, Valverde O, Maldonado R, Kieffer BL, 2000. Mice deficient for delta- and mu opioid receptors exhibit opposing alterations of emotional responses. Nat. Genet 25, 195–200. doi: 10.1038/76061. [DOI] [PubMed] [Google Scholar]
- George S, Ekhtiari H, 2010. Naltrexone in the treatment of opioid dependence. Br. J.Hosp. Med (Lond) 71, 568–570. doi: 10.12968/hmed.2010.71.10.78943. [DOI] [PubMed] [Google Scholar]
- Hagmann WK, 2008. The many roles for fluorine in medicinal chemistry. J. Med. Chem 51, 4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- Hallinan R, Byrne A, Amin J, Dore GJ, 2005. Hepatitis C virus prevalence and outcomes among injecting drug users on opioid replacement therapy. J. Gastroenterol. Hepatol 20, 1082–1086. doi: 10.1111/j.1440-1746.2005.03882.x [DOI] [PubMed] [Google Scholar]
- Handal KA, Schauben JL, Salamone FR, 1983. Naloxone. Ann. Emerg. Med 12, 438–445. doi: 10.1016/S0196-0644(83)80343-6. [DOI] [PubMed] [Google Scholar]
- Kaplan E, Binyaminy B, Gafni M, Keren O, Sarne Y, 2011. Membrane-delimited proteolytic regulation of opioid receptors. Brain Res. 1386, 25–34. doi: 10.1016/j.brainres.2011.02.040. [DOI] [PubMed] [Google Scholar]
- Koch T, Höllt V, 2008. Role of receptor internalization in opioid tolerance and dependence. Pharmacol. Ther 117, 199–206. doi: 10.1016/j.pharmthera.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Kostenis E, Waelbroeck M, Milligan G, 2005. Techniques: promiscuous Galpha proteins in basic research and drug discovery. Trends Pharmacol. Sci 26, 595–602. doi: 10.1016/j.tips.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Li G, Aschenbach LC, Chen J-Y, Cassidy MP, Stevens DL, Gabra BH, Selley DE, Dewey WL, Westkaemper RB, Zhang Y, 2009. Design, synthesis, and biological evaluation of 6α- and 6β-N-heterocyclic substituted naltrexamine derivatives as μ opioid receptor selective antagonists. J. Med. Chem 52, 1416–1427. doi: 10.1021/jm801272c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharao NV, Joshi AA, Gerk PM, 2017. Inhibition of glucuronidation and oxidative metabolism of buprenorphine using GRAS compounds or dietary constituents/supplements: in vitro proof of concept. Biopharm. Drug Dispos 38, 139–154. doi: 10.1002/bdd.2050. [DOI] [PubMed] [Google Scholar]
- Mahar Doan KM, Humphreys JE, Webster LO, Wring SA, Shampine LJ, Serabjit-Singh CJ, Adkison KK, Polli JW, 2002. Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J. Pharmacol. Exp. Ther 303, 1029–1037. doi: 10.1124/jpet.102.039255. [DOI] [PubMed] [Google Scholar]
- Manchikanti L, Singh A, 2008. Therapeutic opioids: a ten-year perspective on the complexities and complications of the escalating use, abuse, and nonmedical use of opioids. Pain Physician 11, S63–88. [PubMed] [Google Scholar]
- Matsson P, Pedersen JM, Norinder U, Bergström CA, Artursson P, 2009. Identification of novel specific and general inhibitors of the three major human ATP-binding cassette transporters P-gp, BCRP and MRP2 among registered drugs. Pharm. Res 26, 1816–31. doi: 10.1007/s11095-009-9896-0. [DOI] [PubMed] [Google Scholar]
- McNicol E, Horowicz-Mehler N, Fisk RA, Bennett K, Gialeli-Goudas M, Chew PW, Lau J, Carr D, 2003. Management of opioid side effects in cancer-related and chronic noncancer pain: a systematic review. J. Pain 4, 231–256. doi: 10.1016/S1526-5900(03)00556-X. [DOI] [PubMed] [Google Scholar]
- Minozzi S, Amato L, Vecchi S, Davoli M, Kirchmayer U, Verster A, 2011. Oral naltrexone maintenance treatment for opioid dependence. Cochrane Database Syst. Rev 4, CD001333. doi: 10.1002/14651858.CD001333.pub4. [DOI] [PubMed] [Google Scholar]
- Miotto K, McCann M, Basch J, Rawson R, Ling W, 2002. Naltrexone and dysphoria: fact or myth? Am. J. Addict 11, 151–160. doi: 10.1080/10550490290087929. [DOI] [PubMed] [Google Scholar]
- Mitra P, Venitz J, Yuan Y, Zhang Y, Gerk PM, 2011. Preclinical disposition (in vitro) of novel μ-opioid receptor selective antagonists. Drug Metab. Dispos 39, 1589–1596. doi: 10.1124/dmd.111.038588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhuri PK, Gfroerer JC and Davies MC, 2013. CBHSQ data review. Center for Behavioral Health Statistics and Quality, SAMHSA 1–17. [Google Scholar]
- National Institute on Drug Abuse, 2012. Principles of drug addiction treatment: a research-based guide, third ed. U.S. Government Printing Office, Washington, DC. [Google Scholar]
- Obeng S, Yuan Y, Jali A, Selley DE, Zhang Y, 2018. In vitro and in vivo functional profile characterization of 17-cyclopropylmethyl-3, 14β-dihydroxy-4, 5α-epoxy-6α-(isoquinoline-3-carboxamido) morphinan (NAQ) as a low efficacy mu opioid receptor modulator. Eur. J. Pharmacol 827, 32–40. doi: 10.1016/j.ejphar. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeng S, Jali A, Zheng Y, Wang H, Schwienteck K, Chen C, Stevens DL, Akbarali H, Dewey WL, Banks ML, Liu-Chen LY, 2019. Characterization of 17-cyclopropylmethyl-3, 14β-dihydroxy-4, 5α-epoxy-6α-(indole-7-carboxamido) morphinan (NAN) as a novel opioid receptor modulator for opioid use disorder treatment. ACS chemical neuroscience, doi: 10.1021/acschemneuro.9b00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande AC, Pyke RE, Greiner M, Cooper SA, Benjamin R, Pierce MW, 1996. analgesic efficacy of the kappa-receptor agonist, enadoline, in dental surgery pain. Clin. Neuropharmacol 19, 92–97. [DOI] [PubMed] [Google Scholar]
- Peng X, Knapp BI, Bidlack JM, Neumeyer JL 2007. Pharmacological properties of bivalent ligands containing butorphan linked to nalbuphine, naltrexone, and naloxone at μ, δ, and κ opioid receptors. J. Med. Chem 50, 2254–2258. doi: 10.1021/jm061327z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrillo P, Angelici O, Bingham S, Ficalora G, Gamier M, Zaratin PF, Petrone G, Pozzi O, Sbacchi M, Stean TO, Upton N, Dondio GM, Scheideler MA, 2003. Evidence for a selective role of the delta-opioid agonist [8R-(4bS*,8aalpha,8abeta,12bbeta)]7,10-dimethyl-1-methoxy-11-(2-methylprop yl)oxycarbonyl 5,6,7,8,12,12b-hexahydro-(9H)-4,8-methanobenzofuro[3,2-e]pyrrolo[2,3-g]isoqu inoli ne hydrochloride (SB-235863) in blocking hyperalgesia associated with inflammatory and neuropathic pain responses. J. Pharmacol. Exp. Ther 307, 1079–1089. doi: 10.1124/jpet.103.055590. [DOI] [PubMed] [Google Scholar]
- Remesic M, Hruby VJ, Porreca F, Lee YS, 2017. Recent advances in the realm of allosteric modulators for opioid receptors for future therapeutics. ACS. Chem. Neurosci 8, 1147–1158, doi: 10.1021/acschemneuro.7b00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter AJ, 2002. Naltrexone in the treatment of heroin dependence: relationship with depression and risk of overdose. Aust. N. Z. J. Psychiatry 36, 224–228. doi: 10.1046/j.1440-1614.2002.01012.x。 [DOI] [PubMed] [Google Scholar]
- Selley DE, Liu Q, Childers SR, 1998. Signal transduction correlates of mu opioid agonist intrinsic efficacy: receptor-stimulated [35S]GTPγS binding in mMOR-CHO cells and rat thalamus. J. Pharmacol. Exp. Ther 285, 496–505. [PubMed] [Google Scholar]
- Svendsen KB, Andersen S, Arnason S, Arnér S, Breivik H, Heiskanen T, Kalso E, Kongsgaard UE, Sjogren P, Strang P, Bach FW, Jensen TS, 2005. Breakthrough pain in malignant and nonmalignant diseases: a review of prevalence, characteristics and mechanisms. Eur. J. Pain 9, 195–206. doi: 10.1016/j.ejpain.2004.06.001. [DOI] [PubMed] [Google Scholar]
- United Nations Office on Drugs and Crime, 2015. World Drug Report 2015. United Nations, New York. [Google Scholar]
- Valtchanova-Matchouganska A, Missankov A, Ojewole JA, 2004. Evaluation of the antidysrhythmic effects of delta- and kappa-opioid receptor agonists and antagonists on calcium chloride-, adrenaline- and ischemia/reperfusion-induced arrhythmias in rats. Methods Find Exp. Clin. Pharmacol 26, 31–38. doi: 10.1358/mf.2004.26.1.793470. [DOI] [PubMed] [Google Scholar]
- van Dorp E, Yassen A, Dahan A, 2007. Naloxone treatment in opioid addiction: the risks and benefits. Expert. Opin. Drug Saf 6, 125–132. doi: 10.1517/14740338.6.2.125. [DOI] [PubMed] [Google Scholar]
- Williams DA, Zheng Y, David BG, Yuan Y, Zaidi SA, Stevens DL, Scoggins KL, Selley DE, Dewey WL, Akbarali HI, Zhang Y, 2016. 6β-N-heterocyclic substituted naltrexamine derivative BNAP: a peripherally selective mixed MOR/KOR ligand. ACS. Chem. Neurosci 7,1120–1129. doi: 10.1021/acschemneuro.6b00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XJ, Hao JX, Wiesenfeld-Hallin Z, 1996. Nociceptin or antinociceptin: potent spinal antinociceptive effect of orphanin FQ/nociceptin in the rat. Neuroreport 7, 2092–2094. [PubMed] [Google Scholar]
- Yuan Y, Arnatt CK, El-Hage N, Dever SM, Jacob JC, Selley DE, Hauser KF, Zhang Y, 2013. A bivalent ligand targeting the putative mu opioid receptor and chemokine receptor CCR5 heterodimers: binding affinity versus functional activities. Medchemcomm. 4, 847–851. doi: 10.1039/C3MD00080J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Elbegdorj O, Chen J, Akubathini SK, Zhang F, Stevens DL, Beletskaya IO, Scoggins KL, Zhang Z, Gerk PM, Selley DE, Akbarali I, Dewey WL, Zhang Y, 2012. Design, synthesis, and biological evaluation of 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(4'-pyridyl)carboxamido]morphinan derivatives as peripheral selective μ opioid receptor Agents. J. Med. Chem 55, 10118–10129. doi: 10.1021/jm301247n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Snutch TP, 1998. Modulation of voltage-dependent calcium channels by G proteins. Curr. Opin. Neurobiol 8, 351–356. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Snutch TP, 2002. Modulating modulation: crosstalk between regulatory pathways of presynaptic calcium channels. Mol. Interv 2, 476–478. doi: 10.1124/mi.2.8.476. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Obeng S, Wang H, Jali AM; Peddibhotla B, Williams D, Zou C, Stevens DL, Dewey WL, Akbarali HI, Selley DE, Zhang Y, 2019. Design, synthesis, and biological evaluation of the third generation 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(4'-pyridyl)carboxamido]morphinan (NAP) derivatives as mu/kappa opioid receptor dual selective ligands. J. Med. Chem 62, 561–574. doi: 10.1021/acs.jmedchem.8b01158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Obeng S, Wang H, Stevens DL, Komla E, Selley DE, Dewey WL, Akbarali HI, Zhang Y, 2018. Methylation Products of 6β- N-Heterocyclic Substituted Naltrexamine Derivatives as Potential Peripheral Opioid Receptor Modulators. ACS. Chem. Neurosci 9, 3028–3037. doi: 10.1021/acschemneuro.8b00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu T, Fang LY, Xie X, 2008. Development of a universal high-throughput calcium assay for G-protein coupled receptors with promiscuous G-protein Galpha15/16. Acta. Pharmacol. Sin, 2008, 29(4), 507–516. doi: 10.1111/j.1745-7254.2008.00775.x. [DOI] [PubMed] [Google Scholar]