

Abstract
Bone marrow-derived mesenchymal cells (BM-MSCs) are able to differentiate into adipocytes, which can secrete adipokines to affect BM-MSC proliferation and differentiation. Recent evidences indicated that adipocytes can secrete fatty acid metabolites, such as palmitic acid methyl ester (PAME), which is able to cause vasorelaxation and exerts anti-inflammatory effects. However, effects of PAME on BM-MSC proliferation remain unclear. The aim of this study was to investigate the effect of PAME on human BM-MSC (hBM-MSC) proliferation and its underlying molecular mechanisms. hBM-MSCs were treated with PAME for 48 h and then subjected to various analyses. The results from the present study show that PAME significantly reduced the levels of G2/M phase regulatory proteins, cyclin-dependent kinase 1 (Cdk1), and cyclin B1 and inhibited proliferation in hBM-MSCs. Moreover, the level of Mdm2 protein decreased, while the levels of p21 and p53 protein increased in the PAME-treated hBM-MSCs. However, PAME treatment did not significantly affect apoptosis/necrosis, ROS generation, and the level of Cdc25C protein. PAME also induced intracellular acidosis and increased intracellular Ca2+ levels. Cotreatment with PAME and Na+/H+ exchanger inhibitors together further reduced the intracellular pH but did not affect the PAME-induced decreases of cell proliferation and increases of the cell population at the G2/M phase. Cotreatment with PAME and a calcium chelator together inhibited the PAME-increased intracellular Ca2+ levels but did not affect the PAME-induced cell proliferation inhibition and G2/M cell cycle arrest. Moreover, the half-life of p53 protein was prolonged in the PAME-treated hBM-MSCs. Taken together, these results suggest that PAME induced p53 stabilization, which in turn increased the levels of p53/p21 proteins and decreased the levels of Cdk1/cyclin B1 proteins, thereby preventing the activation of Cdk1, and eventually caused cell cycle arrest at the G2/M phase. The findings from the present study might help get insight into the physiological roles of PAME in regulating hBM-MSC proliferation.
1. Introduction
Mesenchymal stem cells (MSCs), found in bone marrow stroma, adipose, and many other tissues, are candidates for tissue regeneration due to their high proliferation rate and potential for multilineage differentiation [1]. Recent studies have suggested that MSCs may not only replace diseased tissues but also exert several trophic, regenerative, and anti-inflammatory effects [2]. However, the number of MSCs that can be obtained from a donor remains insufficient for cell therapy purpose [3]. Therefore, it is imperative to obtain the maximum number and expand the population in vitro in order to be practicable for use in clinical application.
Human bone marrow-derived MSCs (hBM-MSCs) have been studied extensively for many years and used in multiple clinical studies and trials. They are self-renewable and retain the potential to differentiate into pericytes, myofibroblasts, bone marrow stromal cells, osteocytes, osteoblasts, and endothelial cells, all of which support hematopoiesis and stable bone mass [4, 5]. In recent studies, gender and age show significant effect on the number of hBM-MSCs and their proliferative capacity [6, 7]. The decrease in the number of resident MSCs may be one of the most important factors responsible for reduction in bone formation and the subsequent increase in bone fragility [8].
Bone marrow-derived MSCs reside within specialized microenvironments. These stem cell niches are essential for preservation of their self-renewal and differentiation capacity [9, 10]. Bone marrow is composed of multiple cell types including adipocytes, which are one of the most abundant cell types in adult bone marrow and constitute approximately 15% of the bone marrow volume in young adults, rising up to 60% by the age of 65 years old [11]. It has been reported that the number of adipocytes correlates inversely with the hematopoietic activity of the bone marrow. Adipocyte-rich bone marrow has a decreased number of hematopoietic stem cells compared to the adipocyte-poor bone marrow [12]. These findings implicate that adipocytes are predominantly negative regulators in the bone marrow microenvironment.
It has been shown that the adipose tissue produces and secretes various adipokines and free fatty acids (FFA), which could potentially influence the bone marrow niche for tissue homeostasis and repair [13]. A recent study showed that perivascular adipose tissue can release palmitic acid methyl ester (PAME), causing vasorelaxation [14]. PAME is an endogenous fatty acid methyl ester (FAME), which has been reported to possess potent anti-inflammatory and antifibrotic activities [15–17]. However, the effects of PAME on hBM-MSC proliferation remain unclear.
p53 protein can induce both cell cycle arrest and cell death. The regulation of cell fate decision has been the focus of numerous studies. Cell cycle arrest driven by p53 requires the transcription of p21, which is a cyclin-dependent kinase inhibitor. The p53/p21 pathway has been shown to play a role in the modulation of the differentiation and proliferation of stem cells [18, 19]. In normal cells, p53 is kept at low levels by murine double minute 2 (Mdm2), which can bind to p53 and act as p53 ubiquitin ligase that negatively regulates p53 function in several cellular pathways, such as cell cycle and apoptosis. Previous studies have shown that the Mdm2 inhibitor can decrease cell proliferation in rat BM-MSCs (rBM-MSCs) and hBM-MSCs [20, 21]. However, the mechanism underlying PAME-induced p53 stabilization and consequent cell cycle arrest in hBM-MSCs has not been elucidated. In the present investigation, we delineated the signaling pathway involved in the PAME-inhibited hBM-MSC proliferation.
2. Materials and Methods
2.1. Chemicals
PAME and stearic acid methyl ester (SAME) were purchased from Sigma-Aldrich (St. Louis, MO, USA) and dissolved in 100% methanol. Antimycin A (AMA), cariporide (HOE 642), 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetra (acetoxymethyl ester) (BAPTA-AM), cycloheximide (CHX), ionomycin, and cantharidin (CTD) were purchased from Sigma-Aldrich. SC79 was purchased from Cayman Chemical Company (Ann Arbor, MI, USA); LB-100 was purchased from BioVision (Milpitas, CA, USA); ethyl isopropyl amiloride (EIPA) was purchased from Research Biochemical Incorporated (Natick, MA, USA).
2.2. Preparation and Culture of rBM-MSCs
Adult male Sprague-Dawley rats (270-350 g) were used as bone marrow cell donors. The rBM-MSCs were obtained from the tibias and the femurs of rats that were anesthetized using urethane (1.5 g/kg, i.p.) and sacrificed. The protocol was approved by the Institution of Animal Care and Use Committee of the Tzu Chi University (IACUC Approval No. 107003). Bone marrow cells were flushed out using phosphate-buffered saline (PBS) containing 1% penicillin/streptomycin (Gibco; Grand Island, NY, USA) and passed through a 100 μm nylon gauze. The mononuclear rBM-MSCs were isolated by Ficoll density gradient centrifugation on Ficoll-Paque™ PLUS (GE Healthcare; Uppsala, Sweden). The mononuclear rBM-MSCs were pipetted, gently washed with PBS, and then centrifuged twice for 5 min at 1,200 rpm. The final pellet was resuspended in α-MEM (Gibco) supplemented with 15% fetal bovine serum (FBS) (Gibco) and 1% penicillin/streptomycin. The rBM-MSCs were incubated at 37°C in a fully humidified atmosphere with 5% CO2, and the medium was changed every 72 h. After growing to 70-80% confluence, the cells were detached using 0.25% trypsin-EDTA (Gibco) and then neutralized by adding fresh medium. For experiments, rBM-MSCs were cultured in α-MEM supplemented with 1% FBS, 1% penicillin/streptomycin, and various concentrations (10, 30, 50, or 100 μM) of PAME for 48 h and incubated at 37°C in a fully humidified atmosphere with 5% CO2.
2.3. hBM-MSC Culture
The hBM-MSCs obtained from Sigma-Aldrich were cultured in α-MEM supplemented with 15% FBS and 1% penicillin/streptomycin. For experiments, hBM-MSCs were cultured in α-MEM supplemented with 1% FBS, 1% penicillin/streptomycin, and various concentrations (10, 30, 50, or 100 μM) of PAME for 48 h and incubated at 37°C in a fully humidified atmosphere with 5% CO2.
2.4. MTT Assay
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (Sigma-Aldrich) solution (5 mg/mL in PBS) was added to each 3.5 cm petri dish and incubated for 2 h at 37°C. Then, 1 mL of dimethyl sulfoxide (DMSO) (Sigma-Aldrich) was added to dissolve formazan crystals. The absorbance of each well was measured at 570 nm using an ELISA plate reader (Multiskan EX; Thermo, Waltham, MA, USA).
2.5. Hoechst 33342 Staining
The cells were fixed in 10% formaldehyde for 1 h at room temperature (RT) and then permeabilized for 15 min at RT with 0.15% Triton X-100. After blocking with 1% bovine serum albumin (BSA) in PBS for 15 min at RT, nuclei were stained with 2 μM Hoechst 33342 (Sigma-Aldrich) and then incubated for 15 min at RT. Images were taken at 40x magnifications using a confocal microscope (Nikon C2 Si+; Melville, NY, USA) and analyzed using NIS-Elements imaging software.
2.6. BrdU Assay
Cell proliferation was evaluated by measuring 5-bromo-2′-deoxyuridine (BrdU) incorporation using a cell proliferation assay kit (Millipore; Burlington, MA, USA) following the manufacturer's instructions. Briefly, BrdU was added to each well of the 96-well plate and incubated for 24 h. The cells were washed with PBS for several times and then fixed with fixing solution for 30 min at RT, washed intensively, and then incubated with anti-BrdU monoclonal antibody for 1 h at RT. After washing, the cells were incubated with peroxidase-conjugated goat anti-mouse IgG for 30 min at RT, washed with PBS, and then incubated in TMB peroxidase substrate for 30 min at RT. The plate was read at 450 nm by ELISA plate reader (Multiskan EX; Thermo).
2.7. Western Blot Analysis
Proteins were extracted from hBM-MSCs and lysed in RIPA lysis buffer (Millipore, USA) containing 1% protease inhibitor (Calbiochem; San Diego, CA, USA) and 0.5% phosphatase inhibitor (Calbiochem). Protein concentration was determined using a BSA Protein Assay kit (Bio-Rad; Hercules, CA, USA). Electrophoresis sample buffer (1 M Tris-HCl, pH 6.8, 5% 2-mercaptoethanol, 20% glycerol, 10% SDS, and 0.5% bromophenol blue) was added to the cell lysates and boiled for 10 min at 110°C. Then, 30 μg of the protein sample was loaded into a 10% SDS-polyacrylamide gel, subjected to electrophoresis, and then transferred to PVDF membrane (Millipore). The membrane was treated with 5% BSA in tween-tris-buffered saline (T-TBS) buffer (0.05% Tween 20, 200 mM Tris-HCl, pH 7.4, and 1.5 M NaCl) for 1 h at RT to block the nonspecific IgGs and then incubated with primary antibodies diluted in T-TBS buffer overnight at 4°C. The information of primary and secondary antibodies used in this study is shown as follows: primary antibodies including anti-Cdk1, anti-p21, anti-cyclin B1, anti-Cdc25C, anti-p53, and anti-Mdm2 were in 1,000 dilution; anti-β-actin purchased from GeneTex (Irvine, CA, USA) was in 10,000 dilution. Secondary antibodies including peroxidase-conjugated goat anti-rabbit IgG and anti-mouse IgG were diluted in T-TBS buffer (1 : 5,000). The resulting bands were analyzed using Image-Pro Plus software and normalized to β-actin.
2.8. Cytosolic Reactive Oxygen Species Analysis
hBM-MSCs were incubated with 2.5 μM CM-H2DCFDA (Molecular Probes; Eugene, OR, USA) for 30 min at 37°C. Images were objective at 20x magnifications using a confocal microscope (Nikon C2 Si+) and analyzed using NIS-Elements imaging software.
2.9. Quantitative Real-Time PCR
Total RNAs were extracted from hBM-MSCs using TRIzol reagent (Ambion; Carlsbad, CA, USA) according to the manufacturer's instructions. Total cDNAs were synthesized with Verso™ cDNA Kit (Thermo) using 3 μg of total RNAs. Quantitative real-time PCR was performed by mixing the cDNA with the Maxima SYBR Green qPCR Master Mix (2X) and ROX Solution (Thermo) and then detected using the ABI PRISM 7300 Real-Time PCR System (Applied Biosystems; Waltham, MA, USA). The following PCR primers were used: Cdk1, forward: 5′-TCA GGA TTT TCA GAG CTT TGG GCA CTC-3′, reverse: 5′-GCC ATT TTG CCA GAA ATT CGT TTG G-3′; cyclin B1, forward: 5′-TGC CCC TGC AGA AGA AGA CCT GTG T-3′, reverse: 5′-TGT TTC CAG CTT CCC GAC CCA GT-3′; p53, forward: 5′-CCC CTC CTG GCC CCT GTC ATC TTC-3′, reverse: 5′-GCA GCG CCT CAC AAC CTC CGT CAT-3′; p21, forward: 5′-GAG GCC GGG ATG AGT TGG GAG GAG-3′, reverse: 5′-CAG CCG GCG TTT GGA GTG GTA GAA-3′; and GAPDH, forward: 5′-TGC ACC ACC AAC TGC TTA GC-3′, reverse: 5′-GGC ATG GAC TGT GGT CAT GAG-3′. All gene expression was analyzed using the comparative Ct method (2-ΔΔCt), where ΔΔCt = ΔCt (sample) − ΔCt (reference) relative to GAPDH levels.
2.10. Flow Cytometry
Cell death, ROS, and intracellular pH analyses were carried out using FACSCalibur Flow Cytometer (Becton Dickinson Biosciences; San Jose, CA, USA). Cell cycle and intracellular Ca2+ analyses were carried out using Gallios™ Flow Cytometer (Beckman Coulter; Brea, CA, USA).
2.10.1. Cell Death Analysis
Apoptosis/necrosis was examined using Annexin V-FITC Apoptosis Kit Plus (BioVision). The excitation/emission was detected at 488/530 nm wavelength.
2.10.2. Cell Cycle Analysis
hBM-MSCs were dissociated into single cells with 0.25% trypsin, fixed in 1 mL 70% ethanol for 1 h at -20°C, and then centrifuged for 5 min at 2,000 rpm. The supernatant was removed, and Triton X-100 (0.1%) was added to permeabilize the hBM-MSCs, which were then treated with DNase-free RNase A (0.2 mg/mL) (Sigma-Aldrich) and stained with propidium iodide (PI) (20 μg/mL) for 30 min at RT. The excitation/emission was detected at 488/585 nm wavelength.
2.10.3. Reactive Oxygen Species Analysis
hBM-MSCs were incubated with 2.5 μM MitoSOX™ Red reagent (Molecular Probes). The excitation/emission was detected at 510/580 nm wavelength.
2.10.4. Intracellular Ca2+ Analysis
hBM-MSCs were incubated with 2.5 μM Fluo-3-AM (Invitrogen; Carlsbad, CA, USA). The excitation/emission was detected at 488/525 nm wavelength.
2.10.5. Intracellular pH Analysis
hBM-MSCs were incubated with 200 nM 2′,7′-bis(carboxyethyl)-5,6-carboxyfluorescein acetoxymethyl ester (BCECF-AM; Molecular Probes). The calibration buffer (140 mM KCl, 2 mM CaCl2, 1.2 mM MgSO4, 10 mM HEPES, 11 mM glucose, and 10 μM nigericin) was adjusted to pH 6.5 with KOH. Nigericin, a K+/H+ ionophore, was used to set the internal pH to the external pH in the absence of a K+ gradient across the cell membrane so that [K+]i/[K+]o equals to [H+]i/[H+]o. The excitation/emission was detected at 488/530 nm wavelength.
2.11. GC-MS Analysis
The rat bone marrow flush solutions were flushed out using 0.025 mL/g body weight PBS supplemented with 1% penicillin/streptomycin and 0.025 mL/g body weight α-MEM supplemented with 1% FBS and 1% penicillin/streptomycin. The samples were extracted with methanol to solubilize the organic compounds, vortexed, sonicated, and collected by centrifuge according to a previous report [14]. The supernatant was transferred to screw cap tubes with polytetrafluoroethylene/silicone septa in the caps. Samples were analyzed using a Hewlett-Packard (HP, Palo Alto, CA, USA) 6890 series II chromatograph coupled to a HP 5973 mass detector equipped with a G1512A automatic injector with BPX5 5% phenyl polysilphenylene-siloxane capillary column (25 m × I.D.0.22 mm; film thickness 0.25 μm). The ionization energy was 70 eV. The carrier gas was He (flow 0.6 mL/min). The temperature of the injection block was 250-300°C. The GC oven temperature was programmed as follows: initial temperature 90°C followed by a temperature increase of 15°C/min up to 240°C and second rate of 10°C/min to the final temperature of 300°C. The mass spectrum was obtained by scanning from m/z 50 to 550. Data were acquired and analyzed using Hewlett-Packard G1701AA version 0.300 ChemStation Software.
2.12. Statistical Analyses
Experimental data was presented as means ± SEM and compared with unpaired t-tests. Data obtained from three or more groups were subjected to one-way ANOVA followed by Fisher's least significant difference test, and p values < 0.05 were considered significant.
3. Results
3.1. Detection of PAME and SAME in Rat Bone Marrow
Previous studies indicated that FAMEs including PAME and SAME were released from the superior cervical ganglion and retina [22, 23]. Initially, we used GC-MS to generate a five-point calibration curve obtained from analysis of methanol-extracted solutions containing a standard PAME and SAME in five different concentrations (1 μM, 5 μM, 10 μM, 50 μM, and 100 μM), respectively (Figures 1(a) and 1(b)). This calibration curve was used for the quantitative analysis of PAME and SAME concentrations in the bone marrow flushing fluid. The rat femurs and tibias were excised, and the rat bone marrow (rBM) was flushed out with PBS. PAME and SAME in the bone marrow flushing PBS were detected and quantified by GC-MS. The PAME and SAME concentrations in the flushing PBS are 51.59 μM and 32.66 μM, respectively (Figures 1(c) and 1(d)). Previous studies have shown that the release of PAME from superior cervical ganglion, retinal, and perivascular adipose tissue is calcium-dependent [14, 22, 23]. We also used the medium to harvest the rBM, to determine whether the concentration of PAME and SAME in the flushing medium is different from those flushed with PBS. Our results show that the PAME and SAME concentrations in the flushing medium are 44.44 μM and 30.87 μM, respectively (Figures 1(c) and 1(d)), and they are not statistically significantly different from the concentrations obtained from the flushing PBS. These data indicate that the bone marrow contains FAMEs, including PAME and SAME.
Figure 1.

Detection of FAMEs in rBM and effects of FAMEs on cell proliferation in rBM-MSCs and hBM-MSCs. Calibration curves were generated for quantification of PAME (a) and SAME (b) using methanol solutions containing PAME and SAME in five different concentrations (1 μM, 5 μM, 10 μM, 50 μM, and 100 μM). Both flushing PBS and flushing medium contain PAME (c) and SAME (d) from rBM (n = 3). PAME (10-100 μM), but not SAME, concentration-dependently inhibited proliferation of rBM-MSCs (n = 3-9) (e) and hBM-MSCs (n = 5-15) (f). All data represent mean ± SEM. ∗p < 0.05, versus the control group. Con: control; rBM: rat bone marrow; Veh: vehicle.
3.2. Effects of PAME on rBM-MSC Proliferation
The effect of PAME on rBM-MSC proliferation was evaluated using the MTT assay. As shown in Figure 1(e), treatment with PAME (10-100 μM) in medium containing 1% FBS for 48 h significantly reduced rBM-MSC proliferation in a concentration-dependent manner. To further examine the specificity of PAME on rBM-MSC proliferation inhibition, the proliferation effect of SAME, a structural analog of FAME, was tested. Treatment with SAME at a range of concentrations (10-100 μM) for 48 h did not significantly affect the proliferation of rBM-MSCs (Figure 1(e), right panel). Methanol (1 : 1,000), the vehicle used in the incubation medium, did not significantly affect the viability of rBM-MSCs (Figure 1(e), left panel).
3.3. Effects of PAME on hBM-MSC Proliferation, Apoptosis, and Necrosis
The effect of PAME on the proliferation of hBM-MSCs was evaluated using MTT, Hoechst 33342 staining, and BrdU assay. In order to examine the proper FBS concentrations in the culture medium required for the PAME-induced proliferation inhibition, hBM-MSCs were incubated in the culture medium supplemented with PAME (50 μM) and various concentrations of FBS (0%, 1%, 3%, 5%, 10%, and 15%) for 48 h. Our results show that PAME in the culture medium containing 1% or 3% FBS significantly inhibited hBM-MSC proliferation, and PAME had the strongest inhibition in the medium containing 1% FBS (). Furthermore, treatment with PAME (10-100 μM) in the medium containing 1% FBS for 48 h significantly reduced the proliferation of hBM-MSCs in a concentration-dependent manner (Figure 1(f)). On the other hand, treatment with SAME (10-100 μM) in the medium containing 1% FBS for 48 h did not significantly affect hBM-MSC proliferation (Figure 1(f), right panel). Methanol (1 : 1,000), the vehicle used in the incubation medium, did not significantly affect the viability of hBM-MSCs (Figure 1(f), left panel). We also evaluated the effect of the PAME on the growth of hBM-MSCs by Hoechst 33342 staining assay. As shown in Figure 2(a), treatment with 50 or 100 μM PAME for 48 h significantly reduced the number of hBM-MSCs. The inhibitory effect of PAME on the hBM-MSC proliferation was further confirmed by immunocytochemical detection of BrdU incorporation. As illustrated in Figure 2(b), BrdU incorporation, a DNA synthesis indicator, was significantly reduced in the hBM-MSCs exposed for 48 h to 50 μM PAME, but not vehicle. To confirm that the PAME-induced reduction in the number of hBM-MSCs is not due to cell apoptosis and necrosis were examined by flow cytometry using Annexin V-FITC Apoptosis Kit Plus, which includes Annexin V-FITC for detecting apoptosis and SYTOX green dye for detecting necrosis. Figure 2(c) shows the representative histograms of the PAME-treated hBM-MSCs stained with Annexin V-FITC Apoptosis Kit Plus. Treatment of hBM-MSCs with 50 or 100 μM PAME for 48 h did not significantly affect the number of viable cells (Figure 2(d)), apoptotic cells (Figure 2(e)), and necrotic cells (Figure 2(f)). In contrast, treatment for 3 h with 20 μM AMA, which served as a positive control for cell death induction, reduced the number of viable cells and increased the number of apoptotic and necrotic cells. These results indicate that the PAME-reduced hBM-MSC cell number was due to proliferation inhibition, but not induction of cell death.
Figure 2.

Effects of PAME on cell growth and death in hBM-MSCs. (a) Treatment with PAME (50 and 100 μM) for 48 h inhibited hBM-MSC proliferation. This inhibition was blocked by 0.5% BSA (n = 3). (b) PAME (50 μM)-induced proliferation inhibition in hBM-MSCs was confirmed by BrdU assay (n = 7). (c) Histogram plots of viable (M1), apoptotic (M2), and necrotic cells (M3) distinguished by flow cytometric analysis using Annexin V-FITC and SYTOX green dye staining. The (d) viable cells, (e) apoptotic cells, and (f) necrotic cells were not significantly different between the PAME-treated group and the control group (n = 6). AMA served as a positive control for cell death induction (n = 3). All data represent mean ± SEM. ∗p < 0.05, versus the control group; #p < 0.05, versus the PAME group. Con: control; AMA: antimycin A; BSA: bovine serum albumin; Veh: vehicle.
3.4. Effects of PAME on hBM-MSC Cell Cycle Progression
The effect of hBM-MSCs on cell cycle was investigated using PI for nucleic acid staining followed by flow cytometric analysis. PAME (50 μM) significantly increased the cell population at the G2/M phase and decreased in the G0/G1 phase but did not significantly affect the S phase and the sub-G1 phase (Figures 3(a) and 3(b)). These results suggest that PAME induced cell cycle arrest at the G2/M phase. To confirm that PAME induced the G2/M phase arrest, the mRNA and protein levels of the G2/M regulatory proteins, Cdk1 and cyclin B1, were examined in hBM-MSCs. Quantitative RT-PCR assay demonstrated that the mRNA levels of both Cdk1 and cyclin B1 were significantly decreased in the PAME-treated group as compared with the control group (Figure 3(c)). The protein levels of Cdk1 (Figure 3(d)) and cyclin B1 (Figure 3(e)) detected by Western blot analyses also decreased in the PAME-treated group. Methanol (1 : 1,000), the vehicle used in the incubation medium, did not significantly affect the protein levels of Cdk1 (Figure 3(d)) and cyclin B1 (Figure 3(e)). Moreover, treatment with PAME significantly increased the protein levels of p53 (Figure 4(a)) and p21 (Figure 4(b)), but not Cdc25C (Figure 4(c)) in hBM-MSCs.
Figure 3.

PAME induces G2/M cell cycle arrest in hBM-MSCs. The cells were treated with PAME (50 μM) for 48 h prior to the flow cytometric and Western blot analyses. (a) Representative histograms of cell distribution at the sub-G1 phase (M1), G0/G1 phase (M2), S phase (M3), and G2/M phase (M4) detected by flow cytometry using PI staining. (b) PAME (50 μM) significantly increased the cell population at the G2/M phase (M4) and decreased at the G0/G1 phase (M2) (n = 8). PAME significantly decreased the levels of (c) Cdk1 and cyclin B1 mRNA (n = 8) and (d, e) protein (n = 5). The levels of mRNA and protein were examined by qRT-PCR and Western blot analyses, respectively. All data represent mean ± SEM. ∗p < 0.05, versus the control group. Con: control; Veh: vehicle; PI: propidium iodide.
Figure 4.

Involvement of the p53-p21 pathway and ROS in the PAME-induced G2/M cell cycle arrest in hBM-MSCs. Treatment with PAME (50 μM) for 48 h increased the protein levels of (a) p53 (n = 9) and (b) p21 (n = 11), but not (c) Cdc25C (n = 8). (d) PAME did not significantly affect the level of mitochondrial ROS. The top panel shows that the mitochondrial ROS was detected by flow cytometric analysis using MitoSOX™ Red reagent; the bottom panel shows a graph of quantitation of these data. AMA-treated hBM-MSCs were used as a positive control for mitochondrial ROS production (n = 5). (e) PAME did not significantly affect the level of cytosolic ROS. H2O2-treated hBM-MSCs served as a positive control for cytosolic ROS production. The top panel shows ROS image detected by confocal microscopy using CM-H2DCFDA; the bottom panel shows a graph of quantitation of these data. At least 20 cells from 2 randomly selected fields were scored in each experiment to determine the DCFDA intensity (n = 5). All data represent mean ± SEM. ∗p < 0.05, versus the control group. AMA: antimycin A.
3.5. Effects of PAME on ROS Production
Cellular generation of ROS is central to redox signaling. Since ROS have been demonstrated to act as upstream signals triggering the p53 activation, we next examined the mitochondrial ROS production in the PAME-treated hBM-MSCs by flow cytometric analysis using MitoSOX™ Red reagent. As shown in Figure 4(d), treatment with PAME (50 μM) in 1% FBS-containing medium for 48 h did not significantly affect the mitochondrial ROS level in hBM-MSCs. We also examined the cytosolic ROS production in hBM-MSCs treated with PAME (50 μM) by confocal microscopy using CM-H2DCFDA. As demonstrated in Figure 4(e), PAME did not significantly affect the cytosolic ROS level. In these studies, the productions of ROS in the hBM-MSCs treated with 100 nM AMA for 30 min and 100 μM H2O2 for 1 h were used as a positive control for ROS production, respectively.
3.6. Effects of Intracellular Acidosis on the PAME-Inhibited hBM-MSC Cell Proliferation
Since arachidonic acid methyl ester (AAME) has been reported to be able to induce intracellular acidosis in rat cardiac myocytes and cerebellar granule cells [24, 25], we examined whether PAME could induce intracellular acidosis, hence inhibiting hBM-MSC proliferation. Treatment of hBM-MSCs with PAME (50 μM) for 48 h significantly reduced the level of BCECF, an intracellular ratiometric pH indicator. Moreover, cotreatment with PAME and a Na+/H+ exchanger blocker, cariporide (HOE 642) or ethyl isopropyl amiloride (EIPA), further reduced the level of BCECF (Figure 5(a)). Methanol (1 : 1,000), the vehicle used in the incubation medium, did not significantly affect the intracellular acidosis of hBM-MSCs (Figure 5(a)). However, cotreatment with PAME and HOE 642 or EIPA did not further reduce the PAME-induced reduction in the hBM-MSC cell number (Figure 5(c)). Moreover, cotreatment with PAME and HOE 642 together did not further increase the PAME-increased percentage of the cell number at the G2/M phase (Figure 5(d)). Taken together, these data suggest that the PAME-induced hBM-MSC proliferation inhibition was not due to intracellular acidosis.
Figure 5.

Involvement of intracellular acidosis and Ca2+ in the PAME-inhibited hBM-MSC proliferation. (a) Treatment with PAME (50 μM) for 48 h induced the intracellular acidosis, and this effect was enhanced by cotreatment with a Na+/H+ exchanger blocker, HOE 642 or EIPA. The top panel shows the BCECF intensity, an indicator of cytosolic pH, detected using flow cytometric analysis; the bottom panel shows a graph of quantitation of these data (n = 6). (b) Treatment with PAME (50 μM) for 48 h increased the intracellular Ca2+, and this effect was abolished by cotreatment with BAPTA-AM, a Ca2+ chelator. The top panel shows the Fluo-3 intensity, a fluorescence indicator of intracellular Ca2+; the bottom panel shows a graph of quantitation of these data (n = 6). (c) The PAME-decreased cell population was not significantly affected by cotreatment with HOC 642 or EIPA or BAPTA-AM. The cell population was detected by MTT assay (n = 8-20). (d) The PAME-increased cell accumulation at the G2/M phase was not significantly affected by cotreatment with HOE 642 or BAPTA-AM. The cell population at the G2/M phase was detected by flow cytometry using PI staining (n = 4). All data represent mean ± SEM. ∗p < 0.05, versus the control group; #p < 0.05, versus the PAME group. Con: control; Veh: vehicle; EIPA: ethyl isopropyl amiloride; HOE 642: cariporide; PI: propidium iodide.
3.7. Effects of Intracellular Ca2+ on the PAME-Inhibited hBM-MSC Cell Proliferation
Cytosolic Ca2+ concentrations fluctuate in an ordered manner along the cell cycle, suggesting that Ca2+ might be involved in regulating cell proliferation. Since p53 can modulate intracellular Ca2+ homeostasis [26], we examined whether PAME could affect the intracellular Ca2+ concentrations, hence inhibiting hBM-MSC proliferation. Treatment of hBM-MSCs with PAME (50 μM) for 48 h significantly increased the level of Fluo-3, an intracellular Ca2+ indicator. Methanol (1 : 1,000), the vehicle used in the incubation medium, did not significantly affect the intracellular Ca2+ of hBM-MSCs (Figure 5(b)). Moreover, cotreatment with PAME and BAPTA-AM (0.5 μM), a Ca2+ chelator, abolished the PAME-increased level of Fluo-3-AM (Figure 5(b)) but did not significantly affect the PAME-induced proliferation inhibition in hBM-MSCs (Figure 5(c)) and the PAME-increased percentage of the cell number at the G2/M phase (Figure 5(d)), suggesting that the PAME-induced hBM-MSC proliferation inhibition was not due to an increase of intracellular Ca2+ concentration.
3.8. Effects of PAME on p53 Protein Stabilization
We further investigated whether PAME-increased levels of p53 and p21 proteins were due to increases of transcription and/or translation. The quantitative RT-PCR assay demonstrated that PAME significantly increased the mRNA level of p21, but not p53, in hBM-MSCs (Figure 6(a)). These findings suggest that PAME might increase the p53 protein stability. To address this issue, the levels of Mdm2 protein, a negative regulator of p53, and p53 protein degradation rate were examined. Western blot analyses showed that PAME (50 μM) treatment reduced the protein levels of Mdm2 in hBM-MSCs (Figure 6(b)). Moreover, treatment with PAME for 6 h followed by cycloheximide (CHX), a protein synthesis inhibitor, for 0.5 to 6 h significantly prolonged the half-life of p53 protein (Figure 6(c)). Thus, PAME can indeed reduce the rate of p53 degradation, hence causing p53 protein stabilization.
Figure 6.
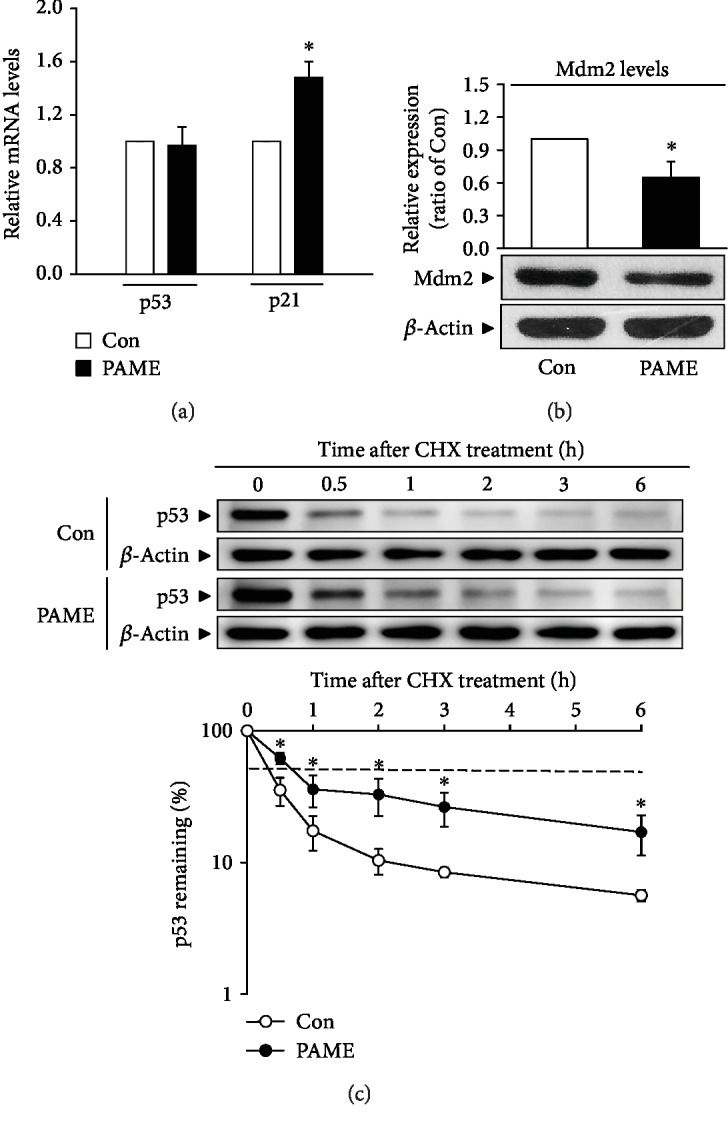
Effects of PAME on p53 protein stability. PAME significantly increased the (a) mRNA level of p21 but not p53 (n = 5) and reduced the (b) level of Mdm2 protein (n = 10). The mRNA levels were detected by qRT-PCR, and the protein level was determined by Western blot analysis. (c) PAME prolonged the degradation of p53 protein in hBM-MSCs. The dotted line indicates 50% abundance of p53, and the half-life (t1/2) of p53 protein in each group is shown in the bottom panel. hBM-MSCs were treated with PAME (50 μM) for 6 h followed by CHX (5 μM) treatment and then subjected to protein level detection by Western blot analysis (n = 4-6). All data represent mean ± SEM. ∗p < 0.05, versus the control group. Con: control; CHX: cycloheximide.
4. Discussion
hBM-MSCs have been widely investigated for their potential therapeutic applications. However, the number of hBM-MSCs in bone marrow is not high enough for therapeutic purpose. Therefore, experimental and clinical investigators continue to search for new methods to expand the isolated hBM-MSCs in vitro for obtaining a sufficient number. In the present study, we demonstrated that PAME significantly reduced the proliferation in rBM-MSCs and hBM-MSC (Figures 1(e) and 1(f)). These findings are different from the results of two other groups [27, 28]. The discrepancy between our results and their results might be due to the difference in the PAME concentrations used. In our study, the concentration of PAME used is 50 μM, which is close to the concentration detected in the flushing medium of rat bone marrow and did not cause cell death but significantly reduced the proliferation in both rBM-MSCs and hBM-MSCs (Figures 1 and 2). On the other hand, the concentration of PAME used in their study was 333 μM or above, which is much higher than the concentration used in our study.
PAME, one of the most abundant fatty acids in mammalian cells [29], represents an endogenous naturally occurring FAME [30]. It has been reported that endogenous PAME is able to inhibit phagocytosis in primary rat Kupffer cells [15, 31]; to inhibit fibrotic effects [17, 32, 33], inflammation [16, 34], and oxidative stress [35]; to induce vasodilation [14, 22, 23, 36, 37]; and to prevent nonalcoholic steatohepatitis [38]. However, the molecular mechanisms underlying PAME-induced biologic effects are still unclear. In the present study, we demonstrated that treatment with PAME induced cell cycle arrest at the G2/M phase through upregulation of p53 protein, which has been indicated in regulating the stress-induced G2/M transition of the mitotic cell cycle. The mechanism by which p53 regulates the G2/M transition involves the regulation of Cdk1, which is essential for the cell cycle entry into mitosis [39]. Repression of cyclin B1 and Cdk1 by p53 enforces the arrest. However, p53-induced repression of cyclin B1 and Cdk1 might involve p21 upregulation. p21, a transcriptional target of p53, can inhibit the activity of Cdks and participate in the G2 checkpoint. p21 inhibits the Cdk activity by direct binding to the Cdk/cyclin complexes. Although p53 has other targets that do not affect the Cdk1 activity but also contribute to the G2 arrest, our data suggest that PAME induced G2/M cell cycle arrest through the p53/p21-mediated reduction of the protein levels of Cdk1 and cyclin B1 in hBM-MSCs (Figures 3, 4(a), and 4(b)). Previous studies have shown that PAME can enhance cerebral blood flow (CBF), thereby alleviating neuronal cell death in the CA1 region of the hippocampus after cerebral ischemia. Furthermore, PAME also exerts other nonvasodilation-dependent neuroprotective effects (i.e., antiapoptosis) [40, 41]. In the present study, our data revealed that PAME did not cause apoptosis and necrosis in hBM-MSCs (Figures 2(e) and 2(f)) but did increase the expression of p21 (Figure 4(b)), which has been indicated to act as an inhibitor of apoptosis in a number of systems [42]. The antiapoptosis/necrosis caused by accumulation of p21 might contribute to the PAME-induced neuronal protection after ischemia.
Previous studies have shown that PAME reduces CCl4-induced or lipopolysaccharide- (LPS-) induced phosphorylation of inhibitory kappa B (IκBα), nuclear translocation of nuclear factor-κB (NF-κB), and subsequent proinflammatory cytokine production in vitro and in vivo [15, 16, 33, 34]. However, the mechanism underlying the PAME-regulated NF-κB level has not been clarified. It has been shown that ectopic expression of p53 enhanced NF-κB DNA binding but blocked its transactivation function [43, 44]. In the present study, we showed that PAME significantly increased the p53 protein level (Figure 4(a)), which may be involved in the PAME-reduced nuclear NF-κB level and inflammatory response. On the other hand, NF-κB can reduce the p53 levels by upregulating the Mdm2 expression [44, 45]. Therefore, PAME may upregulate the p53 protein level through downregulating the Mdm2 expression caused by reducing the nuclear translocation of NF-κB.
Acidosis has been demonstrated to increase the p53 protein levels and the mRNA levels of p21 and Mdm2 [46]. In the present study, we demonstrated that PAME significantly induced intracellular acidosis in hBM-MSCs (Figure 5(a)). Maintenance of acid-base homeostasis in extracellular fluids and cytoplasm is essential for cell proliferation and differentiation. It has been shown that acidity reduces the proliferation and viability of rBM-MSCs and human adipose-derived MSCs [47, 48]. hBM-MSCs are highly sensitive to small shifts in external pH. It has been reported that incubation of PMC-22 cells at pH 6.5 medium for 48 h results in marked decreases of cell population at the S phase and accumulation of cells at the G1 phase [49]. Application of sublethal acidic stress (pH 3.3, 37°C) for 25 min induces G1 arrest in Jurkat T-lymphocytes and G2 arrest in A301 cells [50]. However, cotreatment with PAME and HOE 642 or EIPA together did not affect the PAME-induced G2/M arrest in hBM-MSCs (Figure 5(d)), suggesting that the PAME-induced G2/M arrest is not caused by intracellular acidosis induction.
Although PI staining can be used to assess cell cycle states, it cannot determine that the cells in the S phase are actually cycling. In Figure 2(b), we used BrdU to detect the progress of S phase; the results showed that PAME can significantly inhibit the DNA synthesis. Although using BrdU pulse/chase experiment can more easily identify cells in the G1, S, and G2 phases and define the cell cycle kinetics [51], this technique might not be suitable for examining the cell cycle kinetics in stem cells due to a very low ratio of stem cells in the S phase. We did try to detect the cell cycle kinetics of hBM-MSCs using the FITC BrdU Flow Kit (BrdU and 7-AAD), but the results showed a very low proportion of BrdU-positive cells. Therefore, prepulse with BrdU cannot chase the cell cycle kinetics effectively.
Intracellular Ca2+ rise is involved in controlling numerous cellular processes, such as cell proliferation, differentiation, and apoptosis. It has been reported that an increase of intracellular Ca2+ results in activation of p53/p21 and induces cell cycle arrest in melanoma cells [52]. Moreover, growth inhibition of human keratinocytes caused by high Ca2+ has been shown to be mediated by p21 upregulation [53, 54]. In the present study, we showed that PAME increased intracellular Ca2+ level (Figure 5(b)). However, cotreatment with PAME and BAPTA-AM, a Ca2+ chelator, did not affect the PAME-induced G2/M arrest in hBM-MSCs (Figure 5(d)), suggesting that the PAME-induced G2/M arrest is not caused by increases of the intracellular Ca2+. In addition, it has been indicated that p53 can directly bind to the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pump at the sarco/endoplasmic reticulum (SR/ER), changing its oxidative state and thus leading to an increased Ca2+ load, followed by an enhanced transfer to mitochondria [55]. This signaling has also been recently proved to be important for M phase progression [56]. Unfortunately, the SR/ER to mitochondrial Ca2+ transmission is poorly sensitive to BAPTA-AM. In order to further rule out the changes of Ca2+ in SR/ER and mitochondria, which might affect M phase progression, we used Mag-Fluo4 and Rhod-2 to measure the SR/ER and mitochondrial Ca2+ concentration, respectively. The data show that PAME caused a significant increase in the SR/ER Ca2+ but did not significantly affect the mitochondrial Ca2+ (). These data suggest that the PAME-induced G2/M arrest is not caused by increases of mitochondrial Ca2+. The physiological significance of PAME-increased intracellular acidosis and intracellular Ca2+ deserves further investigation.
Mdm2 has been demonstrated to play a major role in regulating hematopoietic stem cell survival [57]. Moreover, previous studies have shown that Mdm2 inhibitors can inhibit cell proliferation in rBM-MSCs and hBM-MSCs [20, 21]. Dysfunction of the Mdm2/p53 axis has been linked to cell proliferation and apoptosis. Cdc25C is a dual-specificity phosphatase that promotes cell cycle entry into mitosis by removing the inhibitory phosphates on Cdks. Mdm2-promoted Cdc25C protein degradation has been shown to delay cell cycle progression through the G2/M phase [58]. However, treatment with PAME did not significantly affect the levels of Cdc25C protein in hBM-MSCs (Figure 4(c)). In the present study, we demonstrated that treatment with PAME induced hBM-MSC cell cycle arrest at the G2/M phase via a p53-dependent pathway.
It has been shown that Akt, a protein serine/threonine kinase involved in regulating cell growth, proliferation, and apoptosis processes, can enhance the Mdm2-mediated ubiquitination and degradation of p53 [59]. Akt activation is controlled by protein phosphatase 2A (PP2A), a serine/threonine phosphatase involved in mitotic progression and cellular responses to DNA damage. A recent study demonstrated that PP2A leads to cell cycle arrest through the p53/p21 pathway in hepatocytes [60]. Another study provide evidence showing that PP2A blocks HA22T cell proliferation through suppressing the PI3K/Akt/Mdm2-mediated p53 activation [61]. In the present study, we showed that treatment with PAME significantly increased the protein level of PP2A and decreased the protein level of p-Akt. However, cotreatment with PAME and a PP2A inhibitor, cantharidin (CTD) or LB-100, or an Akt activator, SC79, did not affect the PAME-induced proliferation inhibition in hBM-MSCs (). These findings suggest that activation of Akt and upregulation of PP2A might not participate in the PAME-induced hBM-MSC proliferation inhibition.
Although excessive proliferation of hBM-MSCs has not been reported to be associated with clinical disease, previous studies have shown that hBM-MSCs can be recruited to the tumor microenvironment, subsequently promoting proliferation, invasion, survival, tumorigenicity, and migration in a variety of cancers [62, 63]. Moreover, BM-MSCs enhance the chemoresistance of ovarian cancer by releasing miR-1180 and upregulating the glycolytic level [64]. Our data showed that PAME induced cell cycle arrest and reduced cell numbers in BM-MSCs (Figures 1(e), 1(f), 2(a), 2(b), and 3). We also found that PAME can increase the p53 expression in A549 cancer cell line (). Therefore, use of PAME to reduce hBM-MSCs might be a potential application for treating cancer diseases.
5. Conclusions
Our results suggest that PAME downregulated Mdm2, which in turn caused p53 stabilization, subsequently increasing the protein levels of p53 as well as p21 and decreasing the levels of Cdk1/cyclin B1 protein, and eventually caused cell cycle arrest at the G2/M phase. We propose a model of the molecular mechanism underlying PAME-induced cell cycle arrest in hBM-MSCs as shown in Figure 7.
Figure 7.
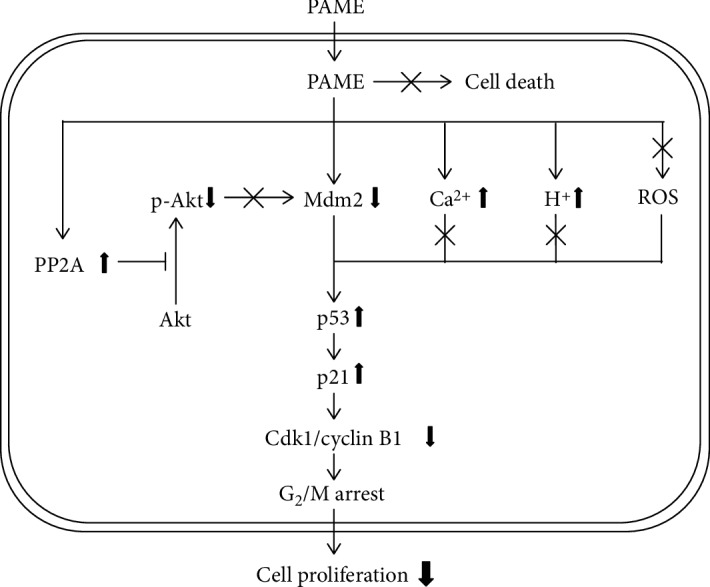
Schematic representation of the PAME-induced G2/M arrest in hBM-MSCs. PAME decreased the expression of Mdm2, which in turn reduced the degradation of p53, subsequently upregulating the expression of Cdk1 and cyclin B1 through suppression of the p21 protein level, and eventually caused cell cycle arrest at the G2/M phase.
Acknowledgments
This study was supported by Ministry of Science and Technology, Taiwan (Grant MOST 105-2320-B-320-015), and by Tzu Chi University, Taiwan (Grant TCMRC-P-103009).
Data Availability
All materials are available from the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
Supplementary Materials
Supplement 1: effects of FBS concentrations on the PAME-inhibited hBM-MSC proliferation. Supplement 2: effects of PAME on SR/ER and mitochondrial [Ca2+] in hBM-MSCs. Supplement 3: involvement of the Akt and PP2A in the PAME-inhibited hBM-MSC proliferation. Supplement 4: effects of PAME on the p53 protein level in A549 cells.
References
- 1.Zhang J., Huang X., Wang H., et al. The challenges and promises of allogeneic mesenchymal stem cells for use as a cell-based therapy. Stem Cell Research & Therapy. 2015;6(1):p. 234. doi: 10.1186/s13287-015-0240-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Griffin M. D., Elliman S. J., Cahill E., English K., Ceredig R., Ritter T. Concise review: adult mesenchymal stromal cell therapy for inflammatory diseases: how well are we joining the dots? Stem Cells. 2013;31(10):2033–2041. doi: 10.1002/stem.1452. [DOI] [PubMed] [Google Scholar]
- 3.Ikebe C., Suzuki K. Mesenchymal stem cells for regenerative therapy: optimization of cell preparation protocols. BioMed Research International. 2014;2014:11. doi: 10.1155/2014/951512.951512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bielby R., Jones E., McGonagle D. The role of mesenchymal stem cells in maintenance and repair of bone. Injury. 2007;38(1):S26–S32. doi: 10.1016/j.injury.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 5.Uccelli A., Moretta L., Pistoia V. Mesenchymal stem cells in health and disease. Nature Reviews Immunology. 2008;8(9):726–736. doi: 10.1038/nri2395. [DOI] [PubMed] [Google Scholar]
- 6.Phinney D. G., Kopen G., Righter W., Webster S., Tremain N., Prockop D. J. Donor variation in the growth properties and osteogenic potential of human marrow stromal cells. Journal of Cellular Biochemistry. 1999;75(3):424–436. doi: 10.1002/(SICI)1097-4644(19991201)75:3<424::AID-JCB8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 7.Fafian-Labora J., Fernandez-Pernas P., Fuentes I., et al. Influence of age on rat bone-marrow mesenchymal stem cells potential. Scientific Reports. 2015;5(1, article 16765) doi: 10.1038/srep16765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yao W., Guan M., Jia J., et al. Reversing bone loss by directing mesenchymal stem cells to bone. Stem Cells. 2013;31(9):2003–2014. doi: 10.1002/stem.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gattazzo F., Urciuolo A., Bonaldo P. Extracellular matrix: a dynamic microenvironment for stem cell niche. Biochimica et Biophysica Acta (BBA) - General Subjects. 2014;1840(8):2506–2519. doi: 10.1016/j.bbagen.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moore K. A., Lemischka I. R. Stem cells and their niches. Science. 2006;311(5769):1880–1885. doi: 10.1126/science.1110542. [DOI] [PubMed] [Google Scholar]
- 11.Morris E. V., Edwards C. M. The role of bone marrow adipocytes in bone metastasis. Journal of Bone Oncology. 2016;5(3):121–123. doi: 10.1016/j.jbo.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naveiras O., Nardi V., Wenzel P. L., Hauschka P. V., Fahey F., Daley G. Q. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature. 2009;460(7252):259–263. doi: 10.1038/nature08099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J., Liu X., Zuo B., Zhang L. The role of bone marrow microenvironment in governing the balance between osteoblastogenesis and adipogenesis. Aging and Disease. 2016;7(4):514–525. doi: 10.14336/AD.2015.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee Y. C., Chang H. H., Chiang C. L., et al. Role of perivascular adipose tissue-derived methyl palmitate in vascular tone regulation and pathogenesis of hypertension. Circulation. 2011;124(10):1160–1171. doi: 10.1161/CIRCULATIONAHA.111.027375. [DOI] [PubMed] [Google Scholar]
- 15.Cai P., Kaphalia B. S., Ansari G. A. S. Methyl palmitate: inhibitor of phagocytosis in primary rat Kupffer cells. Toxicology. 2005;210(2-3):197–204. doi: 10.1016/j.tox.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 16.El-Demerdash E. Anti-inflammatory and antifibrotic effects of methyl palmitate. Toxicology and Applied Pharmacology. 2011;254(3):238–244. doi: 10.1016/j.taap.2011.04.016. [DOI] [PubMed] [Google Scholar]
- 17.Sharawy M. H., el-Agamy D. S., Shalaby A. A., Ammar E. S. M. Protective effects of methyl palmitate against silica-induced pulmonary fibrosis in rats. International Immunopharmacology. 2013;16(2):191–198. doi: 10.1016/j.intimp.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 18.Hong H., Takahashi K., Ichisaka T., et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460(7259):1132–1135. doi: 10.1038/nature08235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruan Z. B., Zhu L., Yin Y. G., Chen G. C. Inhibitor of p53-p21 pathway induces the differentiation of human umbilical cord derived mesenchymal stem cells into cardiomyogenic cells. Cytotechnology. 2016;68(4):1257–1265. doi: 10.1007/s10616-015-9886-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bajelan B., Zaki-Dizaji M., Darabi S., Rajaei F. The effects of Nutlin-3 on morphology, cellular proliferation, and apoptosis in rat primary mesenchymal stem cells. Journal of Cellular Physiology. 2019;234(7):11424–11430. doi: 10.1002/jcp.27798. [DOI] [PubMed] [Google Scholar]
- 21.Daniele S., Giacomelli C., Pietrobono D., et al. Long lasting inhibition of Mdm2-p53 interaction potentiates mesenchymal stem cell differentiation into osteoblasts. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2019;1866(5):737–749. doi: 10.1016/j.bbamcr.2019.01.012. [DOI] [PubMed] [Google Scholar]
- 22.Lin H. W., Liu C. Z., Cao D., et al. Endogenous methyl palmitate modulates nicotinic receptor-mediated transmission in the superior cervical ganglion. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(49):19526–19531. doi: 10.1073/pnas.0810262105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee Y.-C., Chang H.-H., Liu C.-H., et al. Methyl palmitate: a potent vasodilator released in the retina. Investigative Ophthalmology & Visual Science. 2010;51(9):4746–4753. doi: 10.1167/iovs.09-5132. [DOI] [PubMed] [Google Scholar]
- 24.Wu M.-L., Chan C.-C., Su M.-J. Possible mechanism(s) of arachidonic acid–induced intracellular acidosis in rat cardiac myocytes. Circulation Research. 2000;86(3):e55–e62. doi: 10.1161/01.res.86.3.e55. [DOI] [PubMed] [Google Scholar]
- 25.Chen W.-H., Chen C.-R., Yang K.-T., et al. Arachidonic acid-induced H+ and Ca2+ increases in both the cytoplasm and nucleoplasm of rat cerebellar granule cells. The Journal of Physiology. 2001;537(2):497–510. doi: 10.1111/j.1469-7793.2001.00497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giorgi C., Bonora M., Pinton P. Inside the tumor: p53 modulates calcium homeostasis. Cell Cycle. 2015;14(7):933–934. doi: 10.1080/15384101.2015.1010973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng H. P., Wang T. T., Chen W., Wang C. Y., Chen D. F., Shen J. G. Characterization of chemical components in extracts from _Si-wu_ decoction with proliferation-promoting effects on rat mesenchymal stem cells. Bioorganic & Medicinal Chemistry. 2008;16(9):5109–5114. doi: 10.1016/j.bmc.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 28.Wang T. T., Chen W., Zeng H. P., Chen D. F. Chemical components in extracts from Plastrum testudinis with proliferation-promoting effects on rat mesenchymal stem cells. Chemical Biology & Drug Design. 2012;79(6):1049–1055. doi: 10.1111/j.1747-0285.2012.01361.x. [DOI] [PubMed] [Google Scholar]
- 29.Lau C. H. E., Tredwell G. D., Ellis J. K., Lam E. W. F., Keun H. C. Metabolomic characterisation of the effects of oncogenic PIK3CA transformation in a breast epithelial cell line. Scientific Reports. 2017;7(1, article 46079) doi: 10.1038/srep46079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fukuda J.-I., Mizukami E., Imaichi K. Production of methyl esters of fatty acids as artifacts during the concentration of methanolic extracts of serum or plasma lipids. Journal of Biochemistry. 1967;61(5):657–658. doi: 10.1093/oxfordjournals.jbchem.a128597. [DOI] [PubMed] [Google Scholar]
- 31.Sarkar S., Khan M. F., Kaphalia B. S., Ansari G. A. S. Methyl palmitate inhibits lipopolysaccharide-stimulated phagocytic activity of rat peritoneal macrophages. Journal of Biochemical and Molecular Toxicology. 2006;20(6):302–308. doi: 10.1002/jbt.20150. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez-Rivera A., Galicia-Moreno M., Reyes-Gordillo K., et al. Methyl palmitate prevents CCl4-induced liver fibrosis. Journal of Applied Toxicology. 2008;28(8):1021–1026. doi: 10.1002/jat.1368. [DOI] [PubMed] [Google Scholar]
- 33.Mantawy E. M., Tadros M. G., Awad A. S., Hassan D. A. A., El-Demerdash E. Insights antifibrotic mechanism of methyl palmitate: impact on nuclear factor kappa B and proinflammatory cytokines. Toxicology and Applied Pharmacology. 2012;258(1):134–144. doi: 10.1016/j.taap.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 34.Saeed N. M., El-Demerdash E., Abdel-Rahman H. M., Algandaby M. M., Al-Abbasi F. A., Abdel-Naim A. B. Anti-inflammatory activity of methyl palmitate and ethyl palmitate in different experimental rat models. Toxicology and Applied Pharmacology. 2012;264(1):84–93. doi: 10.1016/j.taap.2012.07.020. [DOI] [PubMed] [Google Scholar]
- 35.El-Agamy D. S., Elkablawy M. A., Abo-Haded H. M. Modulation of cyclophosphamide-induced cardiotoxicity by methyl palmitate. Cancer Chemotherapy and Pharmacology. 2017;79(2):399–409. doi: 10.1007/s00280-016-3233-1. [DOI] [PubMed] [Google Scholar]
- 36.Crankshaw D. J., Walsh J. M., Morrison J. J. The effects of methyl palmitate, a putative regulator from perivascular fat, on the contractility of pregnant human myometrium. Life Sciences. 2014;116(1):25–30. doi: 10.1016/j.lfs.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 37.Wang N., Kuczmanski A., Dubrovska G., Gollasch M. Palmitic acid methyl ester and its relation to control of tone of human visceral arteries and rat aortas by perivascular adipose tissue. Frontiers in Physiology. 2018;9:p. 583. doi: 10.3389/fphys.2018.00583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chi Y., You X. Kinetics of hydrogen abstraction reactions of methyl palmitate and octadecane by hydrogen atoms. The Journal of Physical Chemistry A. 2019;123(14):3058–3067. doi: 10.1021/acs.jpca.8b08802. [DOI] [PubMed] [Google Scholar]
- 39.Nurse P. Universal control mechanism regulating onset of M-phase. Nature. 1990;344(6266):503–508. doi: 10.1038/344503a0. [DOI] [PubMed] [Google Scholar]
- 40.Lin H. W., Saul I., Gresia V. L., Neumann J. T., Dave K. R., Perez-Pinzon M. A. Fatty acid methyl esters and Solutol HS 15 confer neuroprotection after focal and global cerebral ischemia. Translational Stroke Research. 2014;5(1):109–117. doi: 10.1007/s12975-013-0276-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee R. H. C., Couto e Silva A., Possoit H. L. E., et al. Palmitic acid methyl ester is a novel neuroprotective agent against cardiac arrest. Prostaglandins, Leukotrienes, and Essential Fatty Acids. 2019;147:6–14. doi: 10.1016/j.plefa.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gartel A. L., Tyner A. L. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Molecular Cancer Therapeutics. 2002;1(8):639–649. [PubMed] [Google Scholar]
- 43.Kawauchi K., Araki K., Tobiume K., Tanaka N. Activated p53 induces NF-κB DNA binding but suppresses its transcriptional activation. Biochemical and Biophysical Research Communications. 2008;372(1):137–141. doi: 10.1016/j.bbrc.2008.05.021. [DOI] [PubMed] [Google Scholar]
- 44.Pal S., Bhattacharjee A., Ali A., Mandal N. C., Mandal S. C., Pal M. Chronic inflammation and cancer: potential chemoprevention through nuclear factor kappa B and p53 mutual antagonism. Journal of Inflammation. 2014;11(1):p. 23. doi: 10.1186/1476-9255-11-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ehsanian R., Van Waes C., Feller S. M. Beyond DNA binding - a review of the potential mechanisms mediating quinacrine’s therapeutic activities in parasitic infections, inflammation, and cancers. Cell Communication and Signaling. 2011;9(1):p. 13. doi: 10.1186/1478-811X-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.LaMonte G., Tang X., Chen J. L. Y., et al. Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer & Metabolism. 2013;1(1):p. 23. doi: 10.1186/2049-3002-1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H., Liang C., Tao Y., et al. Acidic pH conditions mimicking degenerative intervertebral discs impair the survival and biological behavior of human adipose-derived mesenchymal stem cells. Experimental Biology and Medicine (Maywood, N.J.) 2012;237(7):845–852. doi: 10.1258/ebm.2012.012009. [DOI] [PubMed] [Google Scholar]
- 48.Fliefel R., Popov C., Troltzsch M., Kuhnisch J., Ehrenfeld M., Otto S. Mesenchymal stem cell proliferation and mineralization but not osteogenic differentiation are strongly affected by extracellular pH. Journal of Cranio-Maxillo-Facial Surgery. 2016;44(6):715–724. doi: 10.1016/j.jcms.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 49.Taylor I. W., Hodson P. J. Cell cycle regulation by environmental pH. Journal of Cellular Physiology. 1984;121(3):517–525. doi: 10.1002/jcp.1041210310. [DOI] [PubMed] [Google Scholar]
- 50.Kim J. Y., Cheng X., Wolfl S. Acidic stress induced G1 cell cycle arrest and intrinsic apoptotic pathway in Jurkat T-lymphocytes. Experimental Cell Research. 2017;350(1):140–146. doi: 10.1016/j.yexcr.2016.11.015. [DOI] [PubMed] [Google Scholar]
- 51.Teng T., Mercer C. A., Hexley P., Thomas G., Fumagalli S. Loss of tumor suppressor RPL5/RPL11 does not induce cell cycle arrest but impedes proliferation due to reduced ribosome content and translation capacity. Molecular and Cellular Biology. 2013;33(23):4660–4671. doi: 10.1128/MCB.01174-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nicolau-Galmes F., Asumendi A., Alonso-Tejerina E., et al. Terfenadine induces apoptosis and autophagy in melanoma cells through ROS-dependent and -independent mechanisms. Apoptosis. 2011;16(12):1253–1267. doi: 10.1007/s10495-011-0640-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sakaguchi M., Miyazaki M., Takaishi M., et al. S100C/A11 is a key mediator of Ca2+-induced growth inhibition of human epidermal keratinocytes. The Journal of Cell Biology. 2003;163(4):825–835. doi: 10.1083/jcb.200304017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakaguchi M., Sonegawa H., Nukui T., et al. Bifurcated converging pathways for high Ca2+- and TGFβ-induced inhibition of growth of normal human keratinocytes. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(39):13921–13926. doi: 10.1073/pnas.0500630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Giorgi C., Bonora M., Sorrentino G., et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(6):1779–1784. doi: 10.1073/pnas.1410723112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao H., Li T., Wang K., et al. AMPK-mediated activation of MCU stimulates mitochondrial Ca2+ entry to promote mitotic progression. Nature Cell Biology. 2019;21(4):476–486. doi: 10.1038/s41556-019-0296-3. [DOI] [PubMed] [Google Scholar]
- 57.Abbas H. A., Maccio D. R., Coskun S., et al. Mdm2 is required for survival of hematopoietic stem cells/progenitors via dampening of ROS-induced p53 activity. Cell Stem Cell. 2010;7(5):606–617. doi: 10.1016/j.stem.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Giono L. E., Resnick-Silverman L., Carvajal L. A., St Clair S., Manfredi J. J. Mdm2 promotes Cdc25C protein degradation and delays cell cycle progression through the G2/M phase. Oncogene. 2017;36(49):6762–6773. doi: 10.1038/onc.2017.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ogawara Y., Kishishita S., Obata T., et al. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. The Journal of Biological Chemistry. 2002;277(24):21843–21850. doi: 10.1074/jbc.M109745200. [DOI] [PubMed] [Google Scholar]
- 60.He C., Qiu Y., Han P., et al. ER stress regulating protein phosphatase 2A-B56γ, targeted by hepatitis B virus X protein, induces cell cycle arrest and apoptosis of hepatocytes. Cell Death & Disease. 2018;9(7):p. 762. doi: 10.1038/s41419-018-0787-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dung T. D., Day C. H., Binh T. V., et al. PP2A mediates diosmin p53 activation to block HA22T cell proliferation and tumor growth in xenografted nude mice through PI3K-Akt-MDM2 signaling suppression. Food and Chemical Toxicology. 2012;50(5):1802–1810. doi: 10.1016/j.fct.2012.01.021. [DOI] [PubMed] [Google Scholar]
- 62.Liu C., Feng X., Wang B., et al. Bone marrow mesenchymal stem cells promote head and neck cancer progression through periostin-mediated phosphoinositide 3-kinase/Akt/mammalian target of rapamycin. Cancer Science. 2018;109(3):688–698. doi: 10.1111/cas.13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zheng Y., Wang G., Chen R., Hua Y., Cai Z. Mesenchymal stem cells in the osteosarcoma microenvironment: their biological properties, influence on tumor growth, and therapeutic implications. Stem Cell Research & Therapy. 2018;9(1):p. 22. doi: 10.1186/s13287-018-0780-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gu Z. W., He Y. F., Wang W. J., Tian Q., Di W. MiR-1180 from bone marrow-derived mesenchymal stem cells induces glycolysis and chemoresistance in ovarian cancer cells by upregulating the Wnt signaling pathway. Journal of Zhejiang University-Science B. 2019;20(3, article 334):219–237. doi: 10.1631/jzus.B1800190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement 1: effects of FBS concentrations on the PAME-inhibited hBM-MSC proliferation. Supplement 2: effects of PAME on SR/ER and mitochondrial [Ca2+] in hBM-MSCs. Supplement 3: involvement of the Akt and PP2A in the PAME-inhibited hBM-MSC proliferation. Supplement 4: effects of PAME on the p53 protein level in A549 cells.
Data Availability Statement
All materials are available from the corresponding author.