

Summary
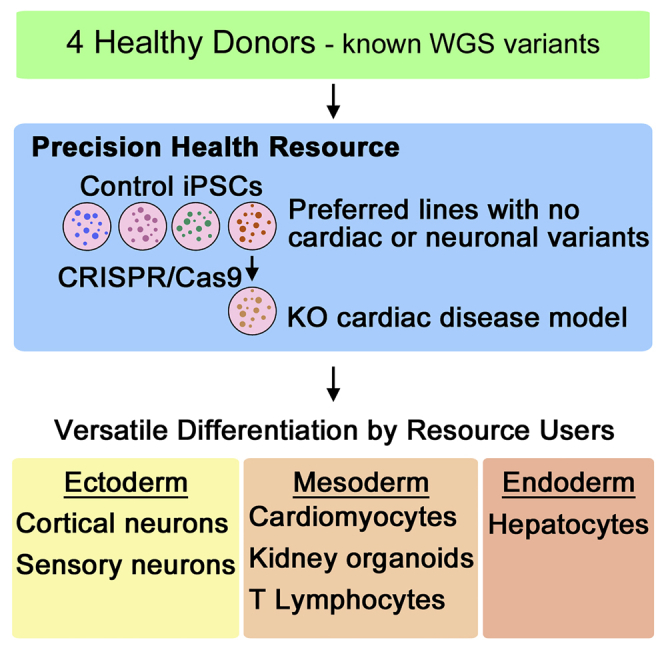
Induced pluripotent stem cells (iPSC) derived from healthy individuals are important controls for disease-modeling studies. Here we apply precision health to create a high-quality resource of control iPSCs. Footprint-free lines were reprogrammed from four volunteers of the Personal Genome Project Canada (PGPC). Multilineage-directed differentiation efficiently produced functional cortical neurons, cardiomyocytes and hepatocytes. Pilot users demonstrated versatility by generating kidney organoids, T lymphocytes, and sensory neurons. A frameshift knockout was introduced into MYBPC3 and these cardiomyocytes exhibited the expected hypertrophic phenotype. Whole-genome sequencing-based annotation of PGPC lines revealed on average 20 coding variants. Importantly, nearly all annotated PGPC and HipSci lines harbored at least one pre-existing or acquired variant with cardiac, neurological, or other disease associations. Overall, PGPC lines were efficiently differentiated by multiple users into cells from six tissues for disease modeling, and variant-preferred healthy control lines were identified for specific disease settings.
Keywords: Personal Genome Project Canada, control iPSCs, whole-genome sequencing, gene editing, cellular phenotyping, disease modeling
Graphical Abstract
Highlights
-
•
Precision health resource of high-quality control iPSC lines for disease modeling
-
•
Versatile differentiation into six functional cell types shown by resource users
-
•
CRISPR gene editing reveals expected phenotype of cardiomyopathy model
-
•
Variant annotation identified preferred lines for neurologic and cardiac disease
Ellis, Scherer, and colleagues apply precision health to upgrade iPSC quality for disease modeling. The resource provides control lines from four healthy individuals, clinical annotation of whole-genome variants, and identification of variant-preferred lines for neurologic and cardiac disease. Resource users demonstrated versatile differentiation into functional cells from six tissues, and CRISPR-edited cells phenocopied a cardiomyopathy model.
Introduction
The development of induced pluripotent stem cells (iPSC) led to rapid development of many stem cell-based models of disease (Takahashi and Yamanaka, 2016). Despite exponential growth in the application of iPSCs across multiple tissue- and organ-based systems, there remains no consistent consensus about which control lines should be used in disease-modeling studies. Over the past decade, choices for control cells have ranged from: (1) human embryonic stem cells (hESCs) that are considered healthy despite a medical history being unavailable, (2) iPSCs from healthy but unrelated individuals (Schwartzentruber et al., 2018), (3) iPSCs from unaffected family members who may have been phenotyped for the disease of interest, but with unknown broader health profile (Lan et al., 2013), and (4) isogenic pairs of iPSC lines derived through CRISPR-Cas9 gene editing (Deneault et al., 2018), or through non-random X chromosome inactivation status in female cells (Tchieu et al., 2010). Hundreds of sources of unrelated and related healthy iPSC lines exist and are widely available from individual labs, biobanks, and large iPSC-focused consortia, such as HipSci (Streeter et al., 2017).
Although there are genetically diverse lines to reflect heterogeneity found within the human population, all control lines are potentially compromised by genetic variants that may predispose to a phenotype or mask it (Hollingsworth et al., 2017). At present, disease modeling has focused on penetrant monogenic disorders that may be relatively unaffected by the presence of concurrent variants. However, we anticipate an emerging need for healthy controls with few disease variants as modeling of complex diseases builds toward assessing the impact of modifier genes or multigenic disorders that may involve multiple variants including noncoding variants in gene regulatory regions.
iPSCs carry additional variants compared with donor sequences (D’Antonio et al., 2018, Gore et al., 2011). This has made apparent the need for whole-genome sequencing (WGS) to identify the full set of potential disease-susceptibility variants present in such control lines (D’Antonio et al., 2018, Kilpinen et al., 2017, Popp et al., 2018). Although there are some common reprogramming-associated variants (Yoshihara et al., 2017), most variants appear to be present in the original mosaic source of cells reprogrammed (Abyzov et al., 2017). Some of these variants could affect downstream differentiations and baseline phenotypes of differentiated lineages (Hoekstra et al., 2017). Furthermore, most control lines are recruited for specific studies limited to a single tissue type or disease, and therefore their versatility for multilineage-directed differentiation into many functional cell types required for broad disease modeling research is not firmly established.
One way to limit the presence of potentially confounding variants is to reprogram cells from selected donors who have minimal variant load. In both the initial Personal Genome Project (PGP) and Personal Genome Project Canada (PGPC) publications, one aim was to generate iPSCs that would have extensive genomic characterization (Ball et al., 2012, Reuter et al., 2018). PGPC genotyped and clinically annotated the genomes of 56 apparently healthy individuals who consented to disclosure of their genome sequence and medical traits (Reuter et al., 2018). In addition to comprehensive annotation of all classes of constitutional genetic variants, these analyses also included their assessment of the mitochondrial genomes and their pharmacogenetic diplotypes. All healthy PGPC individuals harbor heterozygous variants of unknown significance in disease-relevant genes, but still had no overt disease phenotype at the time of initial assessment or at the start of this study. Here we report the iPSC resource generated from PGPC donors.
Our resource comprises multiple iPSC lines derived from two male and two female donors. One line each from both males and one female was subjected to multilineage-directed differentiation into cortical neurons, cardiomyocytes (CMs), and hepatocytes representative of the three germ layers. The morphology and function of the resulting cells were evaluated to assess the versatility of PGPC iPSC lines for in vitro studies of different tissues. To further evaluate the versatility of the resource, we shared the three best-characterized PGPC lines with pilot users for differentiation into kidney organoids, T lymphocytes, and sensory neurons. CRISPR gene editing of a known cardiomyopathy gene created an isogenic pair of lines for modeling a cardiac disorder. As variant annotation of the donors became available (Reuter et al., 2018), we performed WGS to search for iPSC line-specific variants that were distinct from donor PGPC blood variants, and surveyed off-target mutations in the gene edited line.
Results
Isolation and Pluripotency Characterization of PGPC iPSC Lines
We invited PGPC donors to participate in this iPSC study, and selected two male (PGPC3 and PGPC17) and two female donors (PGPC14 and PGPC1) (Reuter et al., 2018). We collected peripheral blood to isolate and reprogram CD34+ cells using non-integrating Sendai viruses. Approximately 120 clones from each donor were picked and qualitative metrics (colony morphology and low levels of spontaneously differentiated cells) were used to select lines for characterization. iPSC lines were maintained in feeder-free conditions and tested for Sendai virus clearance at passage (P)8 to 10. Sendai virus-negative lines were sent for karyotyping between P13 and P15. At least four karyotypically normal cell lines were found from each donor, with standard characterization results summarized in Table S1 and representative data shown in Figure S1. All cell lines stained positive for both cell surface (SSEA4 and TRA-1-60) and nuclear (OCT4 and NANOG) undifferentiated markers (Figure S1). We tested functional pluripotency by spontaneously differentiating embryoid bodies followed by staining for markers of all three germ layers—ectoderm (TUBB3), mesoderm (SMA), and endoderm (AFP) (Figure S1). All female lines had skewed X chromosome inactivation as revealed by androgen receptor assays consistent with preservation of an inactive X chromosome observed in isogenic female lines (Figure S1). These data confirm basic pluripotency status of our resource and cells were expanded and banked at passages ranging from P14 to P16.
We chose to focus on one cell line from the first three donors for deeper characterization as PGPC1 was recruited much later. PGPC3_75, PGPC14_26, and PGPC17_11 were selected for further phenotyping based on qualitative metrics regarding their growth rate, morphology, and relative low rate of spontaneous differentiation. RNA sequencing was analyzed online using Pluritest, and all lines cluster to the pluripotency quadrant (Figure S1). As explained in detail below, we validated the pluripotency and explored the versatility of all three lines for multilineage-directed differentiation to excitatory cortical neurons, CMs, and hepatocytes as representatives of cells derived from ectoderm, mesoderm, and endoderm respectively.
At this point the WGS data of all the PGPC participants became available and were annotated for coding variants defined by the American College of Medical Genetics (Richards et al., 2015). Two heterozygous variants of uncertain clinical significance (VUS) associated with electrophysiological alterations in cardiac disease (Table S2) were identified in PGPC3 (TRPM4) and PGPC14 (KCNE2), respectively. VUS that could affect neurologic function were found in cells derived from PGPC14 and PGPC17 (Table S3). We therefore prioritized PGPC3 as a preferred line for neuronal models and PGPC17 as a variant-preferred line for cardiac models based on their pre-existing variants. The newest PGPC1 female lines are available only with variant annotation (Table S2) and pluripotency characterization as part of the resource.
Ectodermal Differentiation into Active Cortical Neurons
To evaluate PGPC iPSC-derived neurons, we infected PGPC lines and a previously published control iPSC line (WT37) (Cheung et al., 2011) with lentivirus bearing doxycycline-inducible Ngn2 to generate homogeneous populations of excitatory cortical neurons (Zhang et al., 2013). Neurons were induced with doxycycline for 1 week and selected with puromycin and cytarabine (Ara-C) (Deneault et al., 2018) then re-seeded for morphological analysis in co-cultures with mouse astrocytes after an additional 5 weeks (Figure 1A). To measure single neurons, we sparsely labeled 6-week cultured neurons by transfection with ubiquitous expressing GFP plasmid in two batches. Neurons were identified by staining with pan-neuronal marker MAP2 (Figure 1B). Soma area, dendritic length, and neuronal complexity of the PGPC neurons determined by Sholl analysis were similar to the wild-type control (Figures 1C–1E).
Figure 1.

Active Neurons Generated from PGPC iPSCs Display Similar Dendrite Morphology and Network Circuitry
(A) Differentiation scheme to generate excitatory cortical neurons by induction of Ngn2. Transduced iPSCs were dissociated to single cells and plated in the presence of ROCK inhibitor. At D1, medium was changed to CM1 and Ngn2 was induced by incubating with doxycycline until D7. Puromycin was added from D2 to D4 to remove any non-transduced cells. Culture medium was changed to CM2 on D3. Ara-C was added from D6-8 to remove any remaining dividing cells. On D8, neurons were re-seeded for downstream assays in BrainPhys medium.
(B) Representative immunocytochemistry image of iPSC-derived neurons after 6 weeks in culture labeled with DAPI and MAP2 and sparsely labeled with GFP (independent experiments = 2; technical replicates = 20). Scale bar represents 100 μm. Color channels were independently altered to adjust contrast for publication.
(C–E) Plots of (C) soma area, (D) total dendrite length, and (E) number of intersections (Sholl analysis) from 6-week-old neurons (independent experiments n = 2; technical replicates per batch = 20). (C and D) Boxplots indicate median values. (E) Mean values were plotted with error bars indicating SE. Statistically significant pairwise comparisons indicated by ∗ are inset.
(F and G) (F) Representative raster plots of PGPC3 neurons from a single-well of recordings collected by micro-electrode arrays at different time points (two independent experiments each with eight technical replicates). Each spike is indicated by a black line, blue lines represent bursts defined by at least five spikes each separated by an inter-spike interval of no more than 100 ms. (G) Bursts are absent after treatment of week 7 neurons with CNQX (left). Bursts begin to return after compound removal and replacement with fresh basal medium (right).
To investigate the activity of the variant-preferred PGPC3 neurons, we collected weekly micro-electrode array (MEA) recordings (Axion BioSystems) for extracellular electrophysiology measurements (Deneault et al., 2018) over 6 weeks (weeks 2–7). Representative raster plots of PGPC3_75 showed progression of spontaneous activity at 3 weeks compared with development of network bursts at 5 and 7 weeks (Figure 1F). At the 7-week time point, we observed synchronous firing across multiple electrodes (minimum 8/16 electrodes) within wells, indicative of neural circuit formation as measured by network burst frequencies. Neurons displayed weighted mean firing rates ranging from 5 to 7.5 Hz and network burst frequencies ranging from 0.1 to 0.35 Hz, which were comparable with or more active (∼5 and ∼0.1 Hz, respectively) than our previously published MEA results from Ngn2-derived neurons (Deneault et al., 2018). To confirm that recorded activity was due to synaptic transmission from glutamatergic excitatory neurons, we treated cells with an α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor inhibitor—6-cyano-7-nitroquinoxaline-2,3-dione (CNQX)—which abolished network bursting (Figure 1G). Network bursting began to recover 2 h after washing out CNQX. These findings demonstrate differentiation of three PGPC lines into neurons and that the variant-preferred PGPC3 line was spontaneously active in network circuits.
Mesodermal Differentiation into Contractile Cardiomyocytes
PGPC iPSCs were differentiated into CMs using a STEMdiff Cardiomyocyte Differentiation Kit (Figure 2A). We observed beating cells at D8 with all lines. Contractile cultures were dissociated to single CMs at D16 for flow cytometry. The proportion of cardiac troponin T (cTNT)-positive cells was routinely between 75% and 85% (Figure 2B). The D16 CMs were re-seeded into 24-well plates and matured for an additional 17 days to D33. Immunostaining showed that D33 CMs were a mixture of round and cylindrical-shaped CMs and most cells positively stained for cTNT, myosin light chain variant 2 (MLC2V—a ventricular marker), and the sarcomere marker α-actinin (Figure 2C).
Figure 2.

PGPC iPSCs Differentiate to Beating Contractile Cardiomyocytes
(A) Differentiation scheme to generate CMs using STEMdiff Cardiomyocyte Differentiation Kit. iPSCs were dissociated to single cells, plated in 12-well plates, and allowed to reach 85%–90% confluency before beginning differentiation.
(B) D16 CMs were dissociated to single cells for reseeding and a proportion was labeled with anti-cTNT-fluorescein isothiocyanate and subjected to flow cytometry (independent experiments ≥3).
(C) Representative images of immunocytochemistry staining of D30 PGPC17 CMs labeled with DAPI, anti-MLC2V (both), and anti-cTNT (left) or anti-α-actinin (right) (independent experiments = 2). Scale bars represent 100 μm. Color channels were independently altered to adjust contrast for publication.
(D) Representative traces of spontaneous Ca2+ transients of PGPC CMs at D31 measured by relative fluorescence intensity (independent experiments n ≥ 3; technical replicates per batch ≥2).
(E and F) Plots of (E) beat rate and (F) Ca2+ transient amplitudes.
(G) Representative xCELLigence data of D40 PGPC17 CMs showing impedance changes (BAmp: defined as the cell index value between lowest and highest points within a beat waveform) reflecting CM beat waveform and absolute extracellular voltage tracings over a 20-s recording (independent experiments = 3; technical replicates ≥3).
Intracellular Ca2+ transients were measured by loading D31 and D34 CMs with Fluo-4 AM dye. Fluorescence intensity ratios were plotted against time to calculate the Ca2+ transient amplitude and rate (Figures 2D–2F). All three PGPC CMs had similar average beat rates and amplitudes. To measure contractility of PGPC17_11 in a complementary method and to determine extracellular electrophysiology, an xCELLigence Real-Time Cell Analysis (RTCA) Cardio ExtraCellular Recording (ECR) system was used. In brief, contracting CMs were recorded every 3 h for ∼25 days after reseeding (Figure 2G). Contractility of CMs was evaluated via impedance readouts as beats per minute (bpm) and beating amplitude (BAmp) defined as the cell index value between lowest and highest points within a beat waveform. Beat rate averaged 36 bpm (range 32–49 bpm) with average amplitude 0.04 a.u. (range 0.027–0.05 a.u.). Extracellular field potential spike amplitudes defined as the difference between the lowest and highest recorded voltages ranged from 0.12 to 0.55 mV. These experiments demonstrate differentiation of three PGPC lines into beating CMs and highlight the potential value of using PGPC17 for CRISPR gene editing for cardiac disease modeling.
Endodermal Differentiation into Enzymatically Active Hepatocytes
For endodermal differentiation we generated hepatocyte-like cells (HLCs) (Figure 3A). Differentiated cells were characterized at multiple stages to monitor quality and efficiency. At D4, over 95% of cells co-expressed definitive endoderm (DE) markers CXCR4 and cKIT (data not shown). DE cells were induced to generate foregut (FG) progenitors as indicated with the increase in FG markers FOXA2 and GATA6 (normalized to iPSCs) compared with DE (Figure 3B). FG progenitors were further specified to hepatoblasts (HBs) followed by maturation to HLCs by D25 where clear upregulation of respective mRNAs was assessed by qPCR (normalized to fetal liver) (Figure 3B). Over 95% of HLCs tested positive via flow cytometry for hepatocyte markers including albumin (ALB), alpha fetoprotein (AFP), alpha-1-antitrypsin (A1AT), and CYP3A7 (Figure 3C), and further supported by immunostaining for AFP, ALB, CYP3A7, and HNF4A (Figure 3D). Measuring functional activity of HBs (D14) and HLCs (D25) was performed using a p450-glo assay (Promega). As expected, HLCs had significantly more enzymatic activity of CYP3A7 as measured by luminescence as compared with HBs. Treatment with 1 μM ketoconazole inhibited enzymatic activity of CYP3A7 to levels observed in HBs (Figure 3E). These results demonstrate differentiation of the three PGPC lines into hepatocytes that produce active enzymes.
Figure 3.

Enzymatically Active HLCs Are Generated from PGPC iPSCs
(A) Hepatocyte-like cell differentiation scheme. iPSCs were dissociated to single cells and maintained in ROCK inhibitor for 24 h to support survival. From D1 to D4 cells are transferred to STEMdiff definitive endoderm (DE) differentiation kit. From D5 to D9 cells were switched to serum-free differentiation (SFD)-based medium with activin A for 4 days and basic fibroblast growth factor (bFGF) for 2 days adding B27 without retinoic acid (RA). Medium was changed every other day, then on D9 cells were switched to low glucose DMEM supplemented with SB-431542 and bone morphogenic protein (BMP4) from day 9 to day 15. bFGF was added back to the medium during D9 to D15 while B27 with RA was supplemented from D9 to D25 with medium changed every other day. From D15 to D25 hepatocyte growth factor (HGF), dexamethasone, and oncostatin M (OSM) were added to culture medium. From D15 to D21, cells were cultured in a mixture of low glucose DMEM/Ham's F12 (3:1) medium, and at D22 cells were cultured in a mixture of high glucose DMEM/Ham's F12 (3:1) medium. Bright-field images highlight morphology changes during differentiation. Scale bars represent 100 μm.
(B) Heatmaps indicating log2 fold change of marker gene expression normalized to iPSCs (top) or to fetal liver (bottom) (independent experiments = 3).
(C) Dissociated D25 PGPC14 HLCs were labeled with anti-ALB and anti-AFP (left) or anti-alpha-1-AT and anti-CYP3A7 (right) and subjected to flow cytometry (independent experiments = 3).
(D) Representative immunocytochemistry images of D25 PGPC14 HLCs labeled with DAPI and anti-AFP, anti-albumin, anti-CYP3A7, or anti-HNF4A (independent experiments = 3). Scale bars represent 100 μm. Color channels were independently altered to adjust contrast for publication.
(E) Log scale plot of luminescence of each cell line measuring P450 enzymatic activity assayed at D14 and D25 with or without ketoconazole as an inhibitor (independent experiments n = 3; technical replicates for untreated samples = 2, treated samples = 1). Statistical significance was determined by Dunn's test between all samples and ∗ indicate pairs where p < 0.05.
Utility of the Resource––Mesodermal Differentiation into Kidney Organoids and T Cells
To test the utility of PGPC lines as a resource, we made them available to pilot users. Unlike monolayer differentiations described above, human kidney organoids are 3D structures generated from iPSCs consisting of multiple cell types and resembling early embryonic human kidney tissue (Takasato et al., 2015) (Figure S2A).
This protocol entailed a 7-day monolayer culture with directed differentiation toward posterior streak mesoderm (PSM) and subsequently to anterior and posterior intermediate mesoderm (AIM and PIM, respectively). This was accomplished by applying the canonical WNT-signaling activator, CHIR99021 (CHIR), followed by a switch to fibroblast growth factor 9 (FGF9) and heparin. Timing of the FGF9/heparin switch (between D3 and D5 of differentiation) determined the relative proportion of AIM versus PIM and thus fewer or more nephrons. For all experiments, we made this factor switch on D5. During this course, the PSM marker T (brachyury) was transiently induced followed by AIM marker GATA3 and PIM marker HOXD11 as measured by qPCR and expression was normalized to iPSCs at D0 (Figure 4A). After 7 days of monolayer differentiation cells were aggregated, transferred to Transwell membranes, pulsed with CHIR, and then treated with FGF9/heparin for an additional 5 days. Aggregates began reorganization and formed nephron-like structures. mRNA level analysis of D25 organoids (normalized to D7 pre-aggregated cells) showed induction of markers of different nephron segments, endothelial, and stromal cells (Figure 4A). Immunofluorescence imaging of D18 cross-sections of organoids showed glomerular structures—positive for podocyte marker Wilms tumor 1—as well as tubular structures, both proximal—labeled with lectin (LTL)—and distal—positive for E-cadherin (Figure 4B). These results show that 3D kidney organoid structures are produced by the PGPC lines.
Figure 4.

Generation of Kidney Organoids and T Lymphocytes
(A) Heatmaps indicating log2 fold change of marker gene expression normalized to iPSCs (left) or D7 (right) (independent experiments [PGPC3/17 = 1, PGPC14 ≥ 2]; technical replicates ≥3).
(B) Representative images of immunohistochemistry of kidney organoid sections labeled with anti-WT1 (Wilms tumor 1), anti-LTL (labeled with lectin), and anti-ECAD (E-cadherin) antibodies (independent experiments ≥3 for each line). Scale bars represent 100 μm. Color channels were independently altered to adjust contrast for publication.
(C) Average differentiation efficiency of iPSC lines after magnetic-activated cell sorting for CD34 (independent experiments = 4; SEM).
(D) T lymphocyte marker expression during T cell maturation from HSPCs.
(E) D8+14 cells were labeled with anti-CD34, anti-CD7, and anti-CD5 antibodies and measured with flow cytometry (independent experiments = 3).
(F) D8+42 cells were labeled with anti-CD34, anti-CD5, anti-CD7, anti-CD4, anti-CD8b, anti-CD3, anti-T cell receptor alpha/beta, and anti-T cell receptor gamma/delta then measured by flow cytometry (independent experiments = 3).
To evaluate the potential to generate hematopoietic stem/progenitor cells (HSPCs) and mature T cells, we compared the PGPC lines with iPSC11 (Alstem Cell Advancements) in an embryoid body differentiation protocol with feeder-free adaptation (Figure S2B). All three PGPC lines gave rise to CD34+ HSPCs with a similar proficiency as iPSC11 cells (Figure 4C) and were magnetic-activated cell sorted at D8 (Figure S2C). PGPC14 and 17 were most enriched for CD34+ HSPCs and cocultured on OP9-DL4 cells. Next, multi-color flow cytometry was used to simultaneously measure different cell populations at D8+14 and D8+42 to assess the ability of HSPCs to differentiate to T lymphocytes, a hallmark of definitive hematopoietic potential of HSPCs (Figure 4D). At D8+14 (shown in Figure 4E), early T-lineage progenitor cells (proT cells marked as CD34+ CD7+) could be observed transitioning to more developmentally matured T-lineage cells (CD34– CD7+), with a subpopulation co-expressing a pan-T cell marker (CD7+ CD5+). PGPC14 exhibited a prolonged proT stage (70%), while PGPC17 showed a more rapid transition (17%) compared with iPSC11 (53%). Simultaneous assessment of mature T-lineage markers on culture D8+42 detected the presence of double-positive (DP) CD4+ CD8+ T-lineage cells, T cell receptors (TCR+) and a T cell-specific marker (CD3+). At this time point, the PGPC lines showed similar propensity as iPSC11 to generate TCR α/β (39%–45%) cells, but only PGPC17 produced rare TCR γ/δ bearing (0.1%) DP cells. We conclude that PGPC lines differentiate into HSPC that mature into T cells but with different maturation dynamics that may require line-specific protocol optimization.
Utility of the Resource––Sensory Neuron Protocol Optimization and Subtype Identification
PGPC17_11 was selected to optimize differentiation into peripheral sensory neurons (PSNs) using a small-molecule inhibitor protocol adapted from (Chambers et al., 2012) (Figure 5A). Whole-cell patch-clamp recordings were used to assess excitability. At 2 weeks post-induction, all PSNs responded to sustained current injection with transient spiking but, by 4 weeks, half the neurons had switched to repetitive spiking (Figure 5B). The action potential waveform also experienced significant changes, which included an increase in amplitude (Figure 5C) and a decrease in width (Figure 5D) among both transient and repetitive spiking 4-week-old neurons compared with 2-week-old neurons. Among 4-week-old neurons, repetitive spiking neurons had a significantly lower rheobase (current threshold) than transient spiking neurons (Figure 5E). Additional membrane properties are described in Figures S3A–S3F.
Figure 5.

PGPC17-Derived Sensory Neurons Are Predominately Non-peptidergic
(A) Sensory neuron differentiation scheme. The LDN and SB drug combination was applied between D0 and D5 with the CHIR, DAPT, SU, and NGF combination starting on D2 through D11. Starting on D4, N2 medium was added in increasing 25% increments replacing mTeSR1. Dividing cells were eliminated using Ara-C on D10. N2 medium was changed twice weekly thereafter.
(B–E) Representative spiking patterns to sustained somatic current injection (B). At 2 weeks, all cells spike transiently (independent experiments = 3; technical replicates ≥5), whereas at 4 weeks there is a significant increase in the proportion of repetitively spiking neurons (independent experiments = 4; technical replicates ≥2) compared with transiently spiking neurons (independent experiments = 4; technical replicates ≥2) (p < 0.0001, chi-squared test). The action potential waveform experienced a significant increase in amplitude (C) and a significant decrease in width (D) between 2 and 4 weeks post-induction. Rheobase was significantly lower in repetitive spiking neurons than in transient spiking neurons at 2 or 4 weeks post-induction (E). ∗p < 0.05 based on Mann-Whitney U tests.
(F) Ca2+ revealed a significant decrease in the proportion of neurons responsive to capsaicin between 2 and 4 weeks (p < 0.00001, chi-squared test), whereas the proportion of neurons responsive to GABA (p = 0.0001) or ATP (p = 0.042) significantly increased.
To further characterize phenotype, we imaged the Ca2+ responses evoked by brief application of various agonists. Neurons exhibiting a robust Ca2+ response to KCl application were considered healthy and their responses to capsaicin, GABA, and ATP were tested (Figure 5F). At 2 weeks post-induction, 44.6% of neurons responded to the TrpV1 agonist capsaicin, but that number fell to 10% by week 4 (p < 0.00001). TrpV1 is a marker of peptidergic nociceptors, but is broadly expressed among immature PSNs and is developmentally downregulated (Cavanaugh et al., 2011). Our data suggest that iPSC-derived PSNs follow a similar developmental program. Low TrpV1 expression at 4 weeks suggests that repetitive spiking PSNs represent predominantly non-peptidergic nociceptors (Zeisel et al., 2018), whereas the transient spiking neurons are most likely mechanoreceptors. The proportion of neurons responsive to GABA increased over time (p = 0.0001), as did the proportion responsive to the purinergic receptor agonist ATP (p = 0.042). These results demonstrate that PGPC17 was successfully differentiated into active neurons with a non-peptidergic nociceptor or mechanoreceptor phenotype.
Utility of the Resource––WGS Analysis
To identify iPSC-specific variants we obtained whole-genome sequences of each PGPC line to compare with their respective donor blood sequences (Table 1). On average, we identified 1,502 novel nucleotide variants (range: 1,169–1,981) and 0.5 novel copy-number variants (range: 0–1) per clone. Twenty variants (range: 18–24) affected exonic gene regions: 14 non-synonymous (range: 12–16) and 1.75 loss of function (range: 1–3). PGPC1-73 had a likely pathogenic stopgain variant in the chromatin remodeler BPTF, which may disrupt normal gene expression, and particularly neuronal differentiations. We did not identify any other known pathogenic sequence variants in reprogrammed cell lines. Three loss-of-function variants, although not associated with human disease, were in genes with high haploinsufficiency scores and known function in embryonic development (PGPC14_26: TRIM71 and FRMD4A: PGPC17_11: ROBO2, Table S3). For PGPC14_26, we also identified an intronic 16-kb deletion of uncertain significance in IL1RAPL1, a gene associated with impaired synaptogenesis and neurodevelopmental deficits. Re-annotation of the PGP donor and cell-line derivative sequences will be important as variant databases mature (Costain et al., 2018).
Table 1.
Reprogrammed PGPC iPSC Lines: Novel Genomic Variants (Overview)
| SNVs/Indels |
CNVs |
|||||
|---|---|---|---|---|---|---|
| All | Exonic | Non-synonymous | Loss of Function | All | Exonic | |
| PGPC_1 | ||||||
| Genome-wide | 1,622 | 1 | ||||
| All genes | 684 | 24 | 16 | 3 | 1 | 1 |
| OMIM genes | 133 | 5 | 4 | 1 | 0 | 0 |
| Constrained genesa | 204 | 7 | 7 | 3 | 0 | 0 |
| PGPC_3 | ||||||
| Genome-wide | 1,981 | 0 | ||||
| All genes | 847 | 19 | 12 | 1 | 0 | 0 |
| OMIM genes | 172 | 3 | 3 | 0 | 0 | 0 |
| Constrained genesa | 216 | 6 | 5 | 0 | 0 | 0 |
| PGPC_14 | ||||||
| Genome-wide | 1,235 | 1 | ||||
| All genes | 499 | 18 | 13 | 2 | 1 | 0 |
| OMIM genes | 104 | 3 | 2 | 0 | 1 | 0 |
| Constrained genesa | 150 | 8 | 8 | 2 | 1 | 0 |
| PGPC_17 | ||||||
| Genome-wide | 1,169 | 0 | ||||
| All genes | 466 | 23 | 14 | 1 | 0 | 0 |
| OMIM genes | 95 | 6 | 6 | 1 | 0 | 0 |
| Constrained genesa | 113 | 5 | 5 | 1 | 0 | 0 |
| PGPC17_11 MYBPC3_KO | ||||||
| Genome-wide | 917 | 1 | ||||
| All genes | 382 | 17 | 9 | 1 | 0 | 0 |
| OMIM genes | 85 | 4 | 2 | 0 | 0 | 0 |
| Constrained genesa | 35 | 7 | 4 | 0 | 0 | 0 |
PGPC1_73, PGPC3_75, PGPC14_26, and PGPC17_11 were compared with the sequence data obtained from whole blood. PGPC17_11 MYBPC3_KO was compared with the PGPC17_11 reprogrammed line. CNV, copy-number variant; indel, insertion/deletion; SNV, single-nucleotide variant.
pLI>0.9 (http://exac.broadinstitute.org/).
Utility of the Resource––CRISPR/Cas9 Gene Editing and Phenotyping
To edit a gene for cardiac phenotyping, we targeted a region of MYBPC3 where frameshifts are associated with hypertrophic cardiomyopathy by using gRNAs for CRISPR-Cas9-directed non-homologous end-joining (Skarnes et al., 2019). We nucleofected PGPC17_11 iPSCs with a pSpCas9(BB)-2A-Puro vector containing guide RNA (gRNA) sequences targeting MYPBC3 (Figure 6A). Transfected cells were selected with puromycin treatment and resistant colonies were isolated and expanded. A karyotypically normal sub-clone bearing an apparent homozygous frameshift mutation was identified in the MYBPC3_KO line by Sanger sequencing (Figures 6A and 6B). To characterize genetic changes, we performed WGS. On-target compound heterozygote MYBPC3 frameshifts were shown to be an 8-bp insertion at chr11:47,359,282insGTGCAGGA, and a large >260-bp insertion at the same position in the other allele. This insertion did not map to the human genome and was not detected using our PCR-based sequencing due to the size of the insertion. To characterize potential off-target effects in the MYBPC3_KO cells, we first used benchling.com's prediction tool to identify the top 49 off-target sites. We searched 100 base pairs up- and downstream of each predicted site and found zero novel variants within these regions. When we looked for overall novel genomic variation, 917 new single-nucleotide variants and one intergenic 32-kb deletion (chr18:12,137,685–12,169,689) were found (Table 1). None of these variants were likely pathogenic, similar to our other reported gene edited lines (Deneault et al., 2018).
Figure 6.

Derivation of MYBPC3-knockout iPSC that Display a Cardiomyopathy Phenotype
(A) Exon 24 of MYBPC3 was targeted by CRISPR/Cas9 and Sanger sequencing of one clone PGPC17_11-MYBPC3#4 (hereby identified as MYBPC3_KO) identified an out-of-frame insertion resulting in an early stop codon.
(B) Normal karyotype was confirmed before differentiation and characterization.
(C) Western blot probing for MYBPC3 using iPSC lysate as a negative control and parental CM lysate as a positive control. No full-length or truncated forms of MYBPC3 were detected on the blot using near infrared detection. Revert total protein stain was used to show similar amounts of protein were added to each lane.
(D) Representative xCELLigence traces of PGPC17 and MYBPC3-KO CMs showing beat amplitude (cell index) and extracellular voltage recordings over a 20-s sweep at D40 (independent experiments = 2; technical replicates ≥8). MYBPC3-KO CMs showed a higher beat amplitude compared with isogenic control CMs suggestive of hypercontractility as seen in hypertrophic cardiomyopathy.
To examine the consequences of the frameshifts on MYBPC3 protein, we generated CMs as described in Figure 3 and collected protein lysates from PGPC17 parental and MYBPC3_KO iPSC-CMs. Western blots were unable to detect MYBPC3 protein in the KO clone (Figure 6C). We matured CMs until D36–D44 to look for phenotypic evidence of hypertrophic cardiomyopathy as predicted by loss of MYBPC3. Indeed, xCELLigence assays detected increased BAmp in the MYBPC3_KO-CMs compared with the parental line at D42 (0.08 and 0.04 a.u., respectively) while having similar beat rates (41 to 36 bpm, respectively). Recently, Cohn et al. (2019) generated a frameshift in MYBPC3 using cells from the American PGP and observed similar phenotypes. Our findings demonstrate the utility of PGPC17_11 for gene editing to produce isogenic cell lines for cardiac phenotyping.
WGS Analysis of Publicly Available HipSci Lines
Since we found that all our iPSC lines have pre-existing and/or novel variants of potential concern when considering experiments for different lineages, we analyzed downloaded genome sequencing data of five publicly available HipSci lines suggested as healthy controls (HPSI0114i.kolf_2, HPSI0214i.kucg_2, HPSI0214i.wibj_2, HPSI0314i.hoik_1, and HPSI0314i.sojd_3). Across all five samples, 89%–96% of the genome was covered at least 20× (quality metrics in Table S4). We interpreted likely pathogenic variants, loss-of-function constraint gene variants, and VUS as described previously (Reuter et al., 2018) (Supplemental Information).
Two likely pathogenic variants were found in kolf_2 and one in sojd_3 that were predicted to have clinical relevance if identified in humans and could also affect experimental assays. kolf_2 had a substitution of two adjacent nucleotides, disrupting exon-intron boundaries of one COL3A1 allele. One variant was within canonical splice site c.3526-1G>A, and likely to cause out-of-frame exon skipping. If splicing was preserved, the second nucleotide change would result in a likely pathogenic missense alteration p.(Gly1176Ser). COL3A1 haploinsufficiency is associated with dysfunctional connective tissue, such as in the vascular system, skin, intestine, lung, and uterus, and causes vascular type (IV) Ehlers-Danlos syndrome. The same kolf_2 line also harbored a heterozygous 19-bp deletion p.(Pro197Hisfs∗12) in ARID2. The variant was likely pathogenic for Coffin-Siris syndrome, a neurodevelopmental disorder with variable skeletal and organ manifestations. These likely pathogenic variants were also confirmed in the kolf2-C1 subline (Skarnes et al., 2019). Finally, sojd_3 harbored a likely pathogenic heterozygous nonsense variant p.(Gln348∗) in BCOR. This X-linked gene encodes a transcriptional corepressor with important functions in early embryonic development of various tissues. Females with heterozygous BCOR defects may exhibit oculofaciocardiodental syndrome. None of these likely pathogenic variants had been previously reported, and we cannot determine if they were present in the donor genomes, or arose during reprogramming, and could therefore be mosaic. We also identified several loss-of-function variants of uncertain significance in constrained genes in hoik_1 and kucg_2, mostly with known functions in early development (as in PTK2, ZNF398, UBE3C, CDC37, and TNS3; Table S3) and many VUS (Supplemental Information). We did not identify pathogenic or likely pathogenic variants in wibj_2, which suggests it is the variant-preferred line among this subset.
Discussion
Here we generated a high-quality resource of versatile iPSC control lines for use in disease modeling studies. These cells have the benefit of both annotated genomic variants and demonstrated multilineage-directed differentiation into functional cortical neurons, CMs, and hepatocytes. Pilot users showed that the lines can be used to generate kidney organoids, T lymphocytes, or to identify specific subtypes of active sensory neurons. We also performed gene editing, which revealed a preliminary phenotype in isogenic MYBPC3 KO CMs similar to another isogenic pair (Cohn et al., 2019).
Apart from their versatility, the main advantage of these blood-derived footprint-free lines is the clinical annotation of potentially disease-associated variants that may affect cellular phenotypes. Variant analysis in the PGPC participants' blood had revealed heterozygous variants of unknown significance in all individuals (Reuter et al., 2018). This observation suggests that it may not be possible to isolate universal control lines and reinforces the importance for WGS in characterizing control lines, especially as clinical annotation gains precision with ongoing variant discoveries. WGS has the advantage of allowing detection of coding variants, CNVs, and noncoding variants, although the latter have not yet been fully explored in these lines. Knowledge of the donors' genomes allowed predictions on how to prioritize control lines for use as tissue specific controls. For example, as PGPC3 and PGPC14 had variants that could predispose to altered cardiac channel function, PGPC17 was deemed to be the preferred line for the study of cardiac disease. PGPC3, however, were variant preferred for neurological disorders. Consistent with a precision health approach, this strategy would allow matching of genotyped iPSC controls to the disease being modeled.
WGS has previously determined that iPSC lines have variants that differ from those in the donor. Our WGS data reveals that the reprogrammed lines have more than a thousand new SNVs each, whereas only two new CNVs were detected likely due to previous selection for normal karyotype. Most variants were of uncertain significance, with new variants of potential concern found in two of four of the blood (CD34+ cell)-derived PGPC lines (PGPC1_73 and PGPC14_26 in Table S3). Genome sequencing of the MYBPC3 KO line showed more than 900 additional SNVs compared with the unedited iPSC line. None of the new variants were near potential gRNA cut sites, suggesting that they were not off-target and were indeed novel mutations. These analyses highlight that iPSC lines harbor variants of potential concern that are not found in the donor blood. Moreover, our annotation of five healthy control lines from the HipSci consortium that were generated from fibroblasts discovered likely pathogenic variants in two lines and additional loss-of-function variants in constrained genes in two other lines, leaving only wibj_2 as a preferred healthy control line. Since donor WGS is not available for the HipSci lines, it is not possible to determine whether these potentially damaging variants were pre-existing or were captured during fibroblast reprogramming. In contrast, our precision health resource identified >1,000 new variants in each iPSC line, consistent with numbers reported for fibroblast reprogramming (Abyzov et al., 2017). We propose that clinical annotation of WGS data is an important quality control measure of iPSC lines, and its expanded use will identify the best source of healthy control cells to reprogram to find additional variant-preferred lines for disease modeling.
Disease modeling has generally used two to three lines from each individual to account for variability in reprogramming. To account for 1,000–2,000 novel variants in each line compared with the parental genome, this study provides another rationale for studying multiple lines from each individual. With this in mind, we generated a resource of four to five iPSC lines each from two males and two females, all with standard pluripotency characterization available. We also performed multilineage-directed differentiation on a single line from three individuals, assuming that single lines from three to four individuals can account for inter-individual variability. One highly characterized line is therefore available from three PGPC participants, and preferred lines are likely to be of high utility for gene editing studies that compare the phenotype of isogenic cells. Ultimately, users of the resource will select one or more lines from each PGPC participant depending on their research strategy. Future efforts to apply our precision health approach to the characterization of additional control lines in cell repositories should increase the numbers of variant-preferred iPSC banked for gene editing and disease modeling studies.
Overall, our resource upgrades the quality of existing healthy iPSC lines in two ways. First our identification of novel variants of potential concern after reprogramming suggest that a subset of lines may not accurately reflect the phenotype of the original donor in some tissues. To address this concern, our precision health resource provides variant-preferred lines as controls for cardiac or neurological disease modeling and for use in gene editing strategies to create isogenic pairs of mutant and control cells. Second, our exhaustive characterization of multilineage-directed differentiation by pilot users provides strong evidence that the lines can be broadly applied by the disease modeling community.
Experimental Procedures
Reprogramming of PGPC iPSCs was performed under the approval of the Canadian Institutes of Health Research Stem Cell Oversight Committee, and the Research Ethics Board of The Hospital for Sick Children, Toronto. Blood cells were reprogrammed with Sendai virus to deliver reprogramming factors, and iPSCs were maintained in feeder-free conditions with mTeSR1 (STEMCELL Technologies); see Supplemental Information. WGS was performed on Illumina HighSeq X and analyzed as described previously (Reuter et al., 2018). A vector-based CRISPR/Cas9 approach was used to mutagenize MYBPC3, further described in Supplemental Information. Detailed descriptions of differentiations, characterizations, and functional assays are summarized in figures and Supplemental Information. Overexpression of Ngn2 induced iPSCs to differentiate to glutamatergic neurons. Extracellular electrophysiology recordings were collected with an Axion Maestro MEA reader (Axion Biosystems) micro-electrode array as described in the Supplemental Information. CMs were differentiated using STEMdiff Cardiomyocyte Differentiation Kits (STEMCELL Technologies). CM calcium imaging was captured by loading cells with Fluo-4 dye and taking images at 4 Hz for 30 s. Contractile and electrical activity was recorded with an xCELLigence RTCA CardioECR (ACEA Biosciences). CYP3A7 was measured using a p450-Glo assay kit (Promega) as per the manufacturer's protocol. Whole-cell electrophysiology recordings where made at room temperature with an Axopatch 200B (Molecular Devices) from borosilicate patch electrodes. Ca2+ imaging was performed on sensory neurons incubated in Ca2+ green-1 AM dye (Thermo Fisher Scientific) at room temperature. Images were acquired at 25 Hz using a NeuroCCD-SM256 imaging system (RedShirt Imaging).
Author Contributions
M.R.H., M.S.R., S.W.S., and J.E. designed the research project. M.R.H. and J.E. supervised the project. M.S.R. performed WGS clinical annotation and off-target analyses. M.R.H., N.T., and W.W. contributed to the CRISPR experiments. M.R.H., W.W., N.T., J.L., S.S., J.M., L.S.L., P.M.B., A.P., A.R., and G.M. contributed to iPSC isolation, characterization and differentiation. Cells studied by each lab group: iPSC by J.E. and S.W.S., cortical neurons by J.E., cardiomyocytes by S.M. and J.E., hepatocytes by B.M.K., kidney by N.D.R. and J.E., T cells by J.C.Z.-P. and M.K.A., sensory neurons by S.A.P. and J.E. C.K., D.d.C.R., J.H., P.P., M.R., E.C.M., and M.J.S. provided technical help. M.R.H., M.S.R., N.T., J.L., J.M., L.S.L., P.M.B., J.C.Z.-P., M.K.A., S.A.P., N.D.R., B.M.K., S.M., S.W.S., and J.E. wrote the manuscript with comments from all co-authors. Specific contributions of the co-corresponding authors: S.W.S. lab obtained donor blood for reprogramming and performed WGS analyses and annotation on iPSCs; J.E. lab generated iPSCs, cortical neurons, cardiomyocytes, kidney organoids, and sensory neurons.
Acknowledgments
The research was supported by grants from the University of Toronto McLaughlin Centre (MC-2014-06) and Ontario Brain Institute Province of Ontario Neurodevelopmental Disorders Network (to J.E. and S.W.S.; IDS-11-02), GlaxoSmithKline––Canadian Institutes of Health Research (CIHR) Chair in Genome Sciences (to S.W.S.), Ted Rogers Center for Heart Research Strategic Innovation grant (to J.E. and S.M.), Heart and Stroke Foundation Chair (to S.M.), CIHR Team Grant (to B.M.K.; THC-135232), Tier I Canada Research Chair and CIHR Foundation Grant (to N.D.R.; SOP-155609), CIHR Chronic Pain Network-SPOR (to S.A.P. and J.E.; 2017-007), Medicine by Design New Ideas grants (to J.E. and M.K.A., MBDNICL-2017-03; and J.C.Z.-P. and M.K.A., C1TPA-2016-20), NSERC RGPIN grant (to M.K.A., 05333-14), and Fellowship support from SickKids RestraComp award (to J.M and S.S.). We thank B. Thiruvahindrapuram, T. Nalpathamkalam, W.W. Sung, Z. Wang, and G. Kaur for bioinformatic support; The Center for Applied Genomics (TCAG; a CGEn node), the SickKids-UHN Flow and Mass Cytometry Facility and the SickKids Imaging Facility for technical support. The Ngn2/rtTA lentiviral constructs were gifts from T.C. Südhof, and we thank N.N. Kasri and K. Linda for technical advice. We thank the Personal Genome Project Canada, Cheryl Cytrynbaum, Ny Hoang, and Barbara Kellem for genomic data and collecting samples for reprogramming, and the PGPC blood donors for volunteering to participate in this research. M.R.H. became an employee of STEMCELL technologies Inc during peer review of this report.
Published: December 5, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2019.11.003.
Contributor Information
Stephen W. Scherer, Email: stephen.scherer@sickkids.ca.
James Ellis, Email: jellis@sickkids.ca.
Accession Numbers
WGS datasets are available from EGA: EGAS00001003684 and RNA sequencing datasets are available from the GEO: GSE132012. iPSC lines are available upon request.
Supplemental Information
References
- Abyzov A., Tomasini L., Zhou B., Vasmatzis N., Coppola G., Amenduni M., Pattni R., Wilson M., Gerstein M., Weissman S. One thousand somatic SNVs per skin fibroblast cell set baseline of mosaic mutational load with patterns that suggest proliferative origin. Genome Res. 2017;27:512–523. doi: 10.1101/gr.215517.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball M.P., Thakuria J.V., Zaranek A.W., Clegg T., Rosenbaum A.M., Wu X., Angrist M., Bhak J., Bobe J., Callow M.J. A public resource facilitating clinical use of genomes. Proc. Natl. Acad. Sci. U S A. 2012;109:11920–11927. doi: 10.1073/pnas.1201904109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanaugh D.J., Chesler A.T., Braz J.M., Shah N.M., Julius D., Basbaum A.I. Restriction of transient receptor potential vanilloid-1 to the peptidergic subset of primary afferent neurons follows its developmental downregulation in nonpeptidergic neurons. J. Neurosci. 2011;31:10119–10127. doi: 10.1523/JNEUROSCI.1299-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers S.M., Qi Y., Mica Y., Lee G., Zhang X.J., Niu L., Bilsland J., Cao L., Stevens E., Whiting P. Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat. Biotechnol. 2012;30:715–720. doi: 10.1038/nbt.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung A.Y.L., Horvath L.M., Grafodatskaya D., Pasceri P., Weksberg R., Hotta A., Carrel L., Ellis J. Isolation of MECP2-null Rett syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum. Mol. Genet. 2011;20:2103–2115. doi: 10.1093/hmg/ddr093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn R., Thakar K., Lowe A., Ladha F.A., Pettinato A.M., Romano R., Meredith E., Chen Y.-S., Atamanuk K., Huey B.D. A contraction stress model of hypertrophic cardiomyopathy due to sarcomere mutations. Stem Cell Reports. 2019;12:71–83. doi: 10.1016/j.stemcr.2018.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costain G., Jobling R., Walker S., Reuter M.S., Snell M., Bowdin S., Cohn R.D., Dupuis L., Hewson S., Mercimek-Andrews S. Periodic reanalysis of whole-genome sequencing data enhances the diagnostic advantage over standard clinical genetic testing. Eur. J. Hum. Genet. 2018;26:740–744. doi: 10.1038/s41431-018-0114-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Antonio M., Benaglio P., Jakubosky D., Greenwald W.W., Matsui H., Donovan M.K.R., Li H., Smith E.N., D’Antonio-Chronowska A., Frazer K.A. Insights into the mutational burden of human induced pluripotent stem cells from an integrative multi-omics approach. Cell Rep. 2018;24:883–894. doi: 10.1016/j.celrep.2018.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deneault E., White S.H., Rodrigues D.C., Ross P.J., Faheem M., Zaslavsky K., Wang Z., Alexandrova R., Pellecchia G., Wei W. Complete disruption of autism-susceptibility genes by gene editing predominantly reduces functional connectivity of isogenic human neurons. Stem Cell Reports. 2018;11:1211–1225. doi: 10.1016/j.stemcr.2018.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore A., Li Z., Fung H.-L., Young J.E., Agarwal S., Antosiewicz-Bourget J., Canto I., Giorgetti A., Israel M.A., Kiskinis E. Somatic coding mutations in human induced pluripotent stem cells. Nature. 2011;471:63–67. doi: 10.1038/nature09805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra S.D., Stringer S., Heine V.M., Posthuma D. Genetically-informed patient selection for iPSC studies of complex diseases may aid in reducing cellular heterogeneity. Front. Cell. Neurosci. 2017;11:1–8. doi: 10.3389/fncel.2017.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth E.W., Vaughn J.E., Orack J.C., Skinner C., Khouri J., Lizarraga S.B., Hester M.E., Watanabe F., Kosik K.S., Imitola J. iPhemap: an atlas of phenotype to genotype relationships of human iPSC models of neurological diseases. EMBO Mol. Med. 2017;9:1742–1762. doi: 10.15252/emmm.201708191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpinen H., Goncalves A., Leha A., Afzal V., Alasoo K., Ashford S., Bala S., Bensaddek D., Casale F.P., Culley O.J. Common genetic variation drives molecular heterogeneity in human iPSCs. Nature. 2017;546:370–375. doi: 10.1038/nature22403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan F., Lee A.S., Liang P., Sanchez-Freire V., Nguyen P.K., Wang L., Han L., Yen M., Wang Y., Sun N. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013;12:101–113. doi: 10.1016/j.stem.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp B., Krumbiegel M., Grosch J., Sommer A., Uebe S., Kohl Z., Plötz S., Farrell M., Trautmann U., Kraus C. Need for high-resolution genetic analysis in iPSC: results and lessons from the ForIPS consortium. Sci. Rep. 2018;8:1–14. doi: 10.1038/s41598-018-35506-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter M.S., Walker S., Thiruvahindrapuram B., Whitney J., Cohn I., Sondheimer N., Yuen R.K.C., Trost B., Paton T.A., Pereira S.L. The Personal Genome Project Canada: findings from whole genome sequences of the inaugural 56 participants. Can. Med. Assoc. J. 2018;190:E126–E136. doi: 10.1503/cmaj.171151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–423. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber J., Foskolou S., Kilpinen H., Rodrigues J., Alasoo K., Knights A.J., Patel M., Goncalves A., Ferreira R., Benn C.L. Molecular and functional variation in iPSC-derived sensory neurons. Nat. Genet. 2018;50:54–61. doi: 10.1038/s41588-017-0005-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarnes W.C., Pellegrino E., McDonough J.A. Improving homology-directed repair efficiency in human stem cells. Methods. 2019;164–165:18–28. doi: 10.1016/j.ymeth.2019.06.016. [DOI] [PubMed] [Google Scholar]
- Streeter I., Harrison P.W., Faulconbridge A., Flicek P., Parkinson H., Clarke L. The human-induced pluripotent stem cell initiative—data resources for cellular genetics. Nucleic Acids Res. 2017;45:D691–D697. doi: 10.1093/nar/gkw928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 2016;17:183–193. doi: 10.1038/nrm.2016.8. [DOI] [PubMed] [Google Scholar]
- Takasato M., Er P.X., Chiu H.S., Maier B., Baillie G.J., Ferguson C., Parton R.G., Wolvetang E.J., Roost M.S., De Sousa Lopes S.M.C. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature. 2015;526:564–568. doi: 10.1038/nature15695. [DOI] [PubMed] [Google Scholar]
- Tchieu J., Kuoy E., Chin M.H., Trinh H., Patterson M., Sherman S.P., Aimiuwu O., Lindgren A., Hakimian S., Zack J.A. Female human iPS cells retain an inactive X-chromosome. Cell Stem Cell. 2010;7:329–342. doi: 10.1016/j.stem.2010.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara M., Araki R., Kasama Y., Sunayama M., Abe M., Nishida K., Kawaji H., Hayashizaki Y., Murakawa Y. Hotspots of de novo point mutations in induced pluripotent stem cells. Cell Rep. 2017;21:308–315. doi: 10.1016/j.celrep.2017.09.060. [DOI] [PubMed] [Google Scholar]
- Zeisel A., Hochgerner H., Lönnerberg P., Johnsson A., Memic F., van der Zwan J., Häring M., Braun E., Borm L.E., La Manno G. Molecular architecture of the mouse nervous system. Cell. 2018;174:999–1014. doi: 10.1016/j.cell.2018.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Pak C., Han Y., Ahlenius H., Zhang Z., Chanda S., Marro S., Patzke C., Acuna C., Covy J. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78:785–798. doi: 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.