

Abstract
Context
Reduced β-cell mass, impaired islet function, and dedifferentiation are considered causal to development of hyperglycemia and type 2 diabetes. In human cohort studies, changes of islet cell–specific expression patterns have been associated with diabetes but not directly with in vivo insulin secretion.
Objective
This study investigates alterations of islet gene expression and corresponding gene variants in the context of in vivo glycemic traits from the same patients.
Methods
Fasting blood was collected before surgery, and pancreatic tissue was frozen after resection from 18 patients undergoing pancreatectomy. Islet tissue was isolated by laser capture microdissection. Islet transcriptome was analyzed using microarray and quantitative RT-PCR. Proteins were examined by immunohistochemistry and western blotting. The association of gene variants with insulin secretion was investigated with oral glucose tolerance test (OGTT)-derived insulin secretion measured in a large cohort of subjects at increased risk of type 2 diabetes and with hyperglycemic clamp in a subset.
Results
Differential gene expression between islets from normoglycemic and hyperglycemic patients was prominent for the glycolytic enzyme ALDOB and the obesity-associated gene FAIM2. The mRNA levels of both genes correlated negatively with insulin secretion and positively with HbA1c. Islets of hyperglycemic patients displayed increased ALDOB immunoreactivity in insulin-positive cells, whereas α- and δ-cells were negative. Exposure of isolated islets to hyperglycemia augmented ALDOB expression. The minor allele of the ALDOB variant rs550915 associated with significantly higher levels of C-peptide and insulin during OGTT and hyperglycemic clamp, respectively.
Conclusion
Our analyses suggest that increased ALDOB expression in human islets is associated with lower insulin secretion.
Transcriptomic analysis of hyper- vs normoglycemic human islet tissue points to a differential expression of the Aldolase B (ALDOB) gene. An ALDOB variant is associated with altered insulin secretion.
Appropriate insulin secretion and β-cell mass are prerequisites of the maintenance of glucose homeostasis. Insufficient insulin secretion is considered the ultimate cause for development of type 2 diabetes mellitus (1, 2). To date, multiple changes have been implicated in the development of insulin deficiency. The most prominent concepts involve defective insulin secretion, insufficient adaptation of insulin synthesis, reduced β-cell mass, and increased α-cell mass with inappropriate high glucagon secretion. These changes are triggered by metabolic stress factors (i.e., hyperglycemia, hyperlipidemia, and other factors from the diabetogenic milieu) (3). Obesity accentuates metabolic stress and, when accompanied by a genetic predisposition, accelerates the development of type 2 diabetes mellitus. Several of the diabetes-associated single nucleotide polymorphisms (SNPs) are located within genes essential for islet function (4, 5).
Considerable efforts have been undertaken to decipher expressional changes in whole islets and individual islet cells (β-, α-, and δ-cells) in association with hyperglycemia and type 2 diabetes (6–10). Some of the genes differentially altered in type 2 diabetic islets are involved in glucose metabolism (SLC2A2), Ca2+ trafficking (TMEM37) mitochondrial metabolism (GPD2 and FXYD2), cell cycling (P21/CIP), and fatty acid (FFAR4), insulin (IR), and IGF-1 (IGF1R) receptor signaling (6–8, 11).
Most human islets used for scientific purposes are obtained from deceased organ donors. In these studies, a potential bias could arise from post mortem alteration of mRNA levels. Furthermore, human islets and consequently the RNA preparations are usually analyzed after enzymatic digestion of pancreatic tissue. Although the enzymatic isolation of cells allows the separation of islets from exocrine tissue and their dissociation into single cells, such procedures are known to alter unstable mRNAs. Indeed, considerable differences between the expression profile of human type 2 diabetic islets isolated enzymatically or by laser capture microdissection (LCM) have been discussed recently (11). Specifically, LCM-collected islet tissue from patients with type 2 diabetes displayed lower mRNA levels of inflammatory markers and increased levels of ALDOB and FAIM2 mRNA compared with enzyme-digested, isolated type 2 diabetic islets. The current study conducted a direct comparison between expressional changes in human islet tissue and in vivo insulin secretion. This goal was achieved by assessing the in vivo secretory capacity of insulin via a hyperglycemic clamp, a procedure that circumvents potential changes in blood glucose triggered by other mechanisms (i.e., peripheral glucose handling). Glucagon-like peptide 1 (GLP-1) and arginine infusion on top of the hyperglycemic clamp further allows the evaluation of maximal secretory capacity.
Materials and Methods
Human pancreas biobank
Patients (n = 18) undergoing elective surgery for removal of operable pancreatic tumors participated in this ongoing study after providing written informed consent. For each patient, a brief clinical history was obtained. Fasting blood samples were collected prior to anesthesia and pancreatic surgery to assess metabolic traits. Tumor-free pancreatic tissue was dissected from surgical resections by a trained pathologist. The collection of human material and the study were approved by the Ethics Commission of the Medical Faculty of the University of Tuebingen (#697/2011BO1 and #355/2012BO2). Classification of normal glucose tolerance, prediabetes, or diabetes was performed according to clinical history and fasting glucose and HbA1c levels using criteria of the American Diabetes Association (12). Clinical characteristics of the patients are summarized in Supplemental Table 1.
Isolation of pancreatic islet tissue by LCM
Islet tissue was isolated using a protocol previously described (11). Briefly, the freshly resected pancreatic tissue was embedded in cryomolds containing Tissue-Tek O.C.T. compound (Sakura Finetek GmbH, Staufen, Germany). The cryomolds were frozen in isopropyl alcohol precooled in a dry ice–ethanol mixture and stored at −80°C. For the LCM procedure, pancreatic slices (10-μm thick) were dehydrated in ethanol, incubated in xylene, and air dried for 5 minutes. The laser microdissection of islet tissue was performed with a PALM MicroBeam (Zeiss, Oberkochen, Germany). Islets of Langerhans were identified by means of autofluorescence. Islet area was selected with the “freehand” selection tool and microdissected using the laser beam. Islet tissue from 40 dehydrated cryosections/pancreatic samples was lysed in 100 μL extraction buffer (Arcturus Pico pure RNA isolation kit; Applied Biosystems, Foster City, CA) and the supernatant stored at −80°C.
RNA extraction and amplification
Total RNA of islet tissue from 18 patients was isolated using the same protocol as previously described using a commercial kit (Arcturus Pico pure RNA isolation kit) (7, 11). RNA integrity numbers were determined using a bioanalyzer (Agilent Technologies, Santa Clara, CA). RNA samples with RNA integrity numbers >6.0 were processed in transcriptome analyses.
Transcriptome analysis
Affymetrix microarray
For this analysis, 10 to 25 ng of total RNA was amplified using the Ovation RNA Amplification System V2 and subsequently labeled with Biotin using the Encore™ Biotin Module (NuGEN Technologies, San Carlos, CA). The length distribution of the amplified cDNA products was determined using an Agilent 2100 Bioanalyzer (Agilent Technologies). Hybridization of biotin-labeled cDNA to Affymetrix HG_U133Plus2.0 GeneChip microarrays (Affymetrix, Santa Clara, CA) was performed at ATLAS Biolabs GmbH (Berlin, Germany) as described in Solimena et al. (11).
Laboratory measurements and assessment of β-cell function in vivo
Fasting venous blood samples were obtained prior to pancreatic surgery. Glucose levels were measured using the hexokinase method. Plasma insulin and C-peptide were determined by an immunoassay with the ADVIA Centaur XP Immunoassay System (Siemens Healthineers, Eschborn, Germany). HbA1c measurements were performed using the Tosoh glycohemoglobin analyzer HLC-723G8 (Tosoh Bioscience, Tokyo, Japan). β-cell function was computed from fasting glucose and C-peptide levels with the updated homeostasis model assessment (HOMA2) (13). Insulin secretion was expressed with the %B index adjusted for insulin resistance, also calculated as HOMA2 index (HOMA2-IR).
Immunohistochemistry
Paraffin-embedded, pancreatic serial sections from 12 to 17 patients were incubated with primary antibodies against insulin (1:1000; A0564; Dako, Carpinteria, CA) and glucagon (1:600; #13091; Santa Cruz Biotechnology, Santa Cruz, CA). The specific antibody binding was visualized using the Opti-View amplification system (Multimer Technology; Roche-Ventana, Tucson, AZ). Hematoxylin-eosin was used as counterstaining. Insulin- and glucagon-positive stained areas were used for quantification of α- and β-cell areas within the islets using the ImageJ software (National Institutes of Health, Bethesda, MD).
For immunofluorescence and confocal microscopy, the pancreatic sections and cultured human islet cells from organ donors were incubated with primary antibodies against insulin (1:200; mouse, #8304; Abcam, Cambridge, MA), glucagon (1:500; mouse, #10988; Abcam), somatostatin (1:1000; mouse, 14-9751-80; Invitrogen, Carlsbad, CA), and aldolase B (1:100; rabbit, #18065-1-AP; Proteintech, Rosemont, IL). The primary antibodies were visualized with goat anti-rabbit IgG-Alexa Fluor 488 (for aldolase B) and goat anti-mouse IgG-Alexa Fluor 546 (for insulin, glucagon, and somatostatin). Nuclei were stained with TO-PRO3.
Human subjects of the cross-sectional validation cohort (Tuebingen Family cohort) and hyperglycemic clamp study
The Tuebingen Family (TUEF) study currently includes >3000 healthy subjects from Southern Germany with an increased risk for type 2 diabetes as determined by a family history of diabetes, an impaired fasting glycemia, a body mass index (BMI) ≥27 kg/m2, and/or previous gestational diabetes (14). From this study, 2645 participants with complete data sets of anthropometric assessment (sex, age, and BMI) and five-point oral glucose tolerance test (OGTT) including glucose, insulin, C-peptide measurements, and genotypes were selected for association analysis. In the hyperglycemic clamp study, participants underwent hyperglycemic clamps at 10 mM glucose with a stepwise GLP-1 infusion and an arginine bolus, as described earlier (15, 16). Genotyped participants were evaluated in the current analyses (n = 70). Both studies adhered to the Declaration of Helsinki and were approved by the Ethics Committee of the Eberhard Karls University of Tuebingen. Written informed consent was obtained from all participants.
Genotyping, association analysis, and data from open-access repositories
The TUEF participants were genotyped on a genome-wide scale using Illumina’s Infinium® Global Screening Array-24 v1.0 BeadChip (Illumina, San Diego, CA) which was developed based on phase 3 data of the 1,000 Genomes Project and depicts 700,078 SNPs. Within the genomic region of ALDOB including 2 kb of 5′-flanking sequence, 21 SNPs were identified on the array. Among these SNPs, only two, rs550915 and rs533017, revealed minor allele frequencies >0.05 and were selected for association testing due to statistical power limitations of the study population. Both SNPs were successfully genotyped (call rates >99%), in the Hardy-Weinberg equilibrium (P ≥ 0.4), and nonlinked (R2 = 0.34).
Publicly available genotype-phenotype association data were accessed via the Type 2 Diabetes Knowledge Portal (accessed 16 March 2018; http://www.type2diabetesgenetics.org/variantInfo/variantInfo/rs550915#). We interrogated the Genotype-Tissue Expression (GTEx) project database for information on the association of a SNP of interest with gene transcription (17). Single-cell RNA-sequencing data in islet cells were analyzed from data shared from the work of Enge et al. (18).
Culture of isolated human islets and western blotting
Isolated human islets obtained from the European Consortium for Islet Transplantation (Geneva, Switzerland and Milan, Italy) and dispersed human islet cells (19) were cultured for 72 hours with medium change every 24 hours in CMRL1066 medium supplemented with 5, 10, 15, or 20 mM glucose, 2 mM l-glutamine, 10 mM HEPES, and 10% fetal calf serum. At the end of the culture period, the islets were lysed for western blotting, and the islet cells were used for immunostaining (19). Protein lysates were subjected to SDS-PAGE, proteins transferred on nitrocellulose membranes (Hahnemühle, Dassel, Germany). Membranes were incubated for 1 hour in Tris-buffered saline–Tween supplemented with 5% milk, overnight with primary antibodies against aldolase B (Proteintech) or tubulin (Cell Signaling Technology, Danvers, MA), followed by 1-hour incubation with horseradish peroxidase–coupled secondary anti-rabbit IgG (GE Healthcare, München, Germany). Proteins were detected using ChemiDoc Touch Imaging System (Bio-Rad Laboratories, Hercules, CA). Analyses were performed using Bio-Rad Image laboratory software.
Statistics
The microarray data from the Affymetrix Human Genome Array HG_U133Plus2.0 were analyzed after quality control and normalization. Differential gene expression was tested with the limma package in R. Linear models were fitted using robust fit, and the log-fold change was determined by empirical Bayes moderation of the SEs toward a common value. For the visualization of the correlation of gene expression with clinical traits, the relationship between two variables was plotted with GraphPad Prism (GraphPad Software Inc, La Jolla, CA) and Pearson correlation coefficients are provided. All other analyses were performed in R. The evaluation of SNP association with insulin secretion indices from the OGTT [area under the curve (AUC)0–30min C-peptide/AUC0–30min glucose and AUC0–120min C-peptide/AUC0–120min glucose] was carried out by multiple linear regression analysis (least squares method) adjusted for potential confounders (sex, age, BMI and insulin sensitivity). The series of insulin levels during the hyperglycemic clamp study was analyzed with a repeated-measures ANOVA approach using sex, age, and insulin sensitivity as covariates. The genotype was modeled using additive inheritance in all tests. Values (P < 0.05) were considered statistically significant.
Results
Alterations of islet gene expression associate with HbA1c and in vivo insulin secretion
In the mRNA microarray data, several genes showed differential expression in patients with diabetes or increased HbA1c >6% (Supplemental Fig. 1). The most highly differentially expressed genes showed good correlation with previously published data (Supplemental Fig. 2) (11). Among the top upregulated genes (−1 ≤ log fold change ≥ 1; Supplemental Fig. 1), only aldolase B (ALDOB) and Fas apoptotic inhibitory molecule 2 (FAIM2) displayed a substantial, negative correlation with in vivo insulin secretion (expressed as HOMA2%B; Fig. 1A and 1B). Moreover, ALDOB and FAIM2 displayed a considerable positive correlation with HbA1c (Fig. 1C and 1D). However, the expression level of FAIM2 (mean log2 intensity 6.5) is much lower than that of ALDOB (mean log2 intensity 9.7) (Fig. 1).
Figure 1.

Correlation of mRNA levels with HbA1c and in vivo insulin secretion (HOMA2%B*) in LCM-cut islet tissue. The mRNA levels determined by Affymetrix array of LCM-isolated islet tissue (n = 18), HbA1c, fasting blood glucose, and in vivo insulin secretion of the same patients were assessed as described in Materials and Methods. Correlations of islet mRNAs with (A and B) insulin secretion expressed as HOMA2%B* or with (C–H) plasma HbA1c. *Adjusted for insulin resistance (HOMA2-IR).
Several other mRNAs, namely the glucose transporter SLC2A2 (GLUT2), the fatty acid receptor 4 (FFAR4), and two transmembrane proteins TMEM37 (Ca-channel inhibitory γ subunit) and TMEM27, associated negatively with HbA1c (Fig. 1E–1H). These proteins are specifically expressed in β-cells and were reported to be decreased in type 2 diabetic islets (7, 11). However, the mRNA levels of SLC2A2, FFAR4, TMEM37, and TMEM27 as well as the β-cell–specific isoform ALDOA displayed no correlation with HOMA2%B (data not shown).
ALDOB gene variation associates with insulin secretion in humans
If ALDOB plays a causal role in the impairment of insulin secretion during development of type 2 diabetes, genetic variation of ALDOB could associate with changes in insulin secretion. Therefore, we investigated the association of two ALDOB SNPs in 2645 genotyped individuals of our TUEF cohort with insulin secretion assessed from 5-point OGTT. The minor allele of rs550915 in the ALDOB gene region associated with a significantly higher insulin secretion (Fig. 2A). Similarly, when insulin secretion was assessed from hyperglycemic clamp to circumvent the effects of glucose handling, insulin secretion was higher in minor allele carriers of rs550915 (n = 70; Fig. 2B). The effect was most prominent during the last phase of the clamp involving GLP-1 and arginine administration. From seven SNPs of the FAIM2 gene identified in our TUEF cohort, none was associated with alterations in insulin secretion (data not shown). These results endorse the prior assumption that ALDOB directly interferes with insulin secretion.
Figure 2.

Effect of ALDOB SNP rs550915 A>C on insulin secretion derived from five-point OGTT and hyperglycemic clamp. (A) Insulin secretion is given as AUC0–30min C-peptide/AUC0–30min glucose adjusted for sex, age, BMI, and OGTT-derived insulin sensitivity by multiple linear regression modeling. The SNP was tested in the additive inheritance model. The red boxes indicate the interquartile range cut into two pieces by the median; the whiskers extend to the first quartile − 1.5 × (interquartile range) and to the third quartile + 1.5 × (interquartile range). The green diamonds indicate the mean and 95% CI of the mean. (B) The hyperglycemic clamp was started using a body weight–adapted intravenous bolus of 20% glucose solution at min 0 and a continuous infusion adjusted to a target glucose level of 10 mM. GLP-1 infusion (bolus 4.5 pmol/kg, followed by a continuous administration with 1.5 pmol kg−1 min−1) was started at min 120. An arginine bolus (5 g, injected over 45 s) was administered at 180 min. Insulin was assessed from venous blood samples at the respective time points. Shown are geometric means with 95% CIs for n = 70 (nAA= 42, nAC = 25, and nCC = 3) individuals.
The expression of aldolase B in β-cells is augmented by glucose
To elucidate the cell type expressing ALDOB, pancreatic sections were immunostained for ALDOB, insulin, somatostatin, and glucagon (Fig. 3A–3C). In pancreatic islets from patients with HbA1c <6%, ALDOB immunostaining was undetectable. In tissue slices of patients with HbA1c >6%, ALDOB was detected in some cells within the islets. The ALDOB-positive cells displayed insulin immunoreactivity and were glucagon and somatostatin negative (Fig. 3A–3C). Of note, there was a poor overlapping between ALDOB and insulin signals, which suggests no colocalization of ALDOB and insulin within these cells (Fig. 3C and Supplemental Fig. 3A). The same distribution pattern of ALDOB was observed in isolated human islet β-cells from organ donors upon exposure to 20 mM glucose for 48 hours (Supplemental Fig. 3B). These results suggest that hyperglycemia induces expression of ALDOB in islet β-cells. In agreement, previous studies reported an elevation of AldoB mRNA in rodent islets exposed to high glucose concentrations (20). To confirm these findings, we examined whether high glucose alters the expression of ALDOB in human islets isolated from organ donors. ALDOB protein was already detectable in the islets cultured at 5 mM glucose, and its level was significantly increased upon islets exposure to 15 mM or 20 mM glucose (Fig. 3D and 3E).
Figure 3.
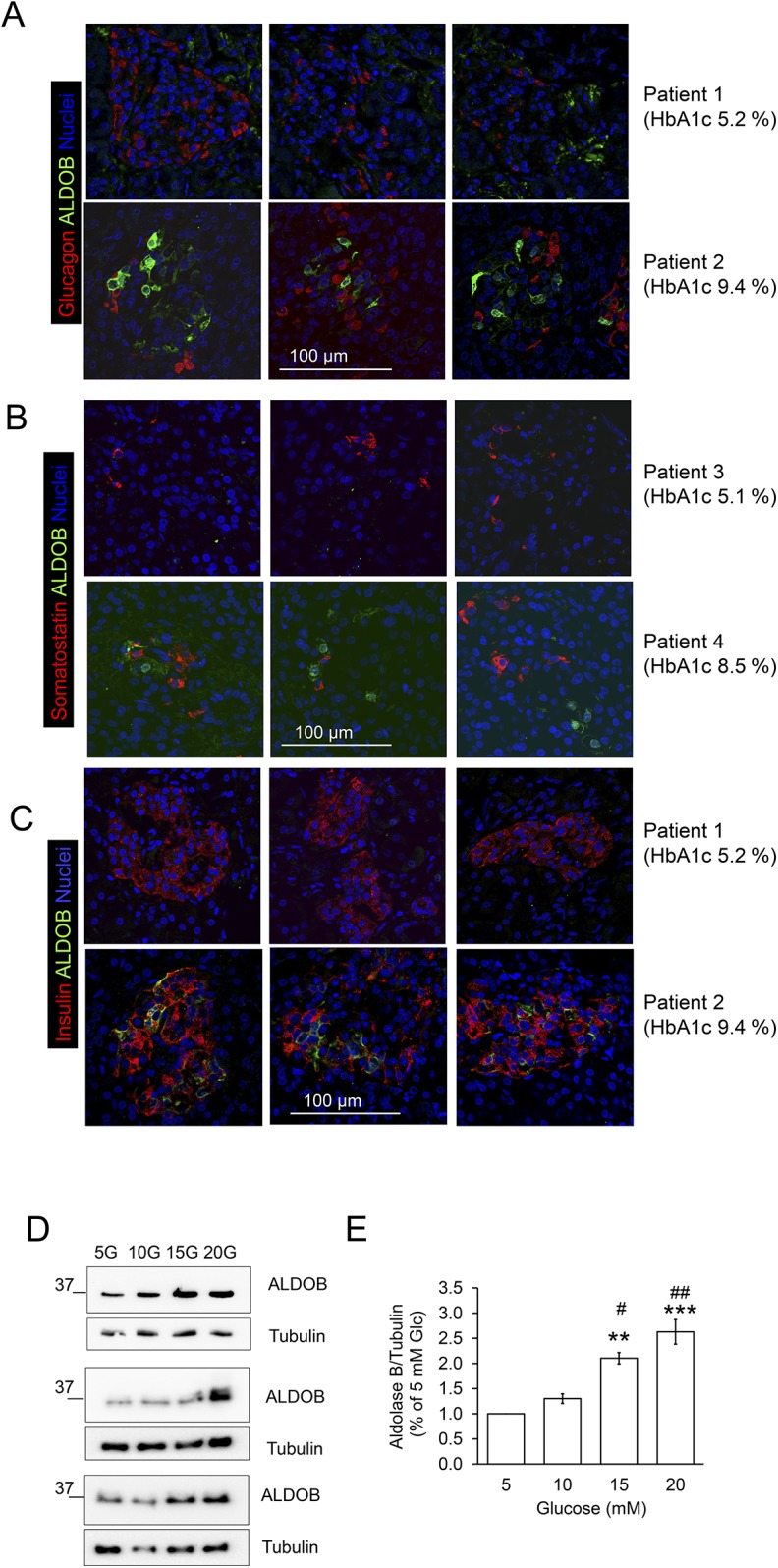
Expression of aldolase B protein is increased by hyperglycemia in human islets. (A–C) Human pancreatic slices from the samples analyzed by Affymetrix array were immunostained for ALDOB, insulin, glucagon, and somatostatin as described in Materials and Methods. Representative confocal pictures of (A) ALDOB (green), glucagon (red), and nuclei (blue), (B) ALDOB (green), somatostatin (red), and nuclei (blue), and (C) ALDOB (green), insulin (red), and nuclei (blue) in two patients with HbA1c <5.5% and HbA1c >8%. (D and E) Isolated human islets were cultured for 72 h in the presence of different glucose concentration as indicated and described in Materials and Methods. (D) Western blots of ALDOB and (E) quantitative analysis presented as mean ± SEM of n = 3 independent experiments. **P < 0.005; ***P < 0.001 vs 5 mM glucose; #P < 0.05; ##P < 0.005 vs 10 mM glucose.
LCM-cut islet tissue is enriched in β-cells
The composition of LCM-cut tissue samples with respect to endocrine cell ratio was evaluated by analyzing insulin, glucagon, and somatostatin mRNA levels using quantitative RT-PCR (qRT-PCR) (Table 1). The relative insulin mRNA level was 2500-fold higher than that of the housekeeping gene RPS13 and ∼100-fold higher than glucagon and somatostatin mRNA levels. This proportion differs from the one observed in cultured human islets isolated from organ donors (19). The insulin mRNA level of LCM-cut islet tissue, determined by the same qRT-PCR method, was 10-fold higher than that of isolated islets from organ donors, whereas the mRNA level of glucagon was 10-fold lower. This observation implies an enrichment of β-cells over α-cells when using the LCM as method for islet isolation.
Table 1.
Relative mRNA Levels Assessed by Quantitative PCR of Insulin (INS), Glucagon (GCG), and Somatostatin (SST) in 12 RNA Samples From LCM-Isolated Islet Tissue
|
Relative mRNA Level (ΔCt)
|
|||
|---|---|---|---|
| Gene Symbol | Mean | SEM | n |
| INS | 2466.15 | 349.86 | 12 |
| GCG | 35.31 | 5.76 | 12 |
| SST | 18.43 | 2.62 | 12 |
| RPS13 | 1.00 | — | 12 |
The housekeeping gene RPS13 was set to 1.
Abbreviation: Ct, threshold cycle.
Endocrine cell differentiation markers and α/β-cell areas do not correlate with HbA1c
β-cell dedifferentiation is a mechanism proposed to be, at least partly, responsible for reduced β-cell mass and impaired β-cell function (21). The mRNA level of the β-cell differentiation marker PDX1 as well as other β- and α-cell–specific markers (MAFB, NEUROD1, and FOXO1) was remarkably similar between individuals and did not correlate with HbA1c (Supplemental Fig. 4).
To examine a possible association of β- and α-cell mass with glycemia, we assessed the area of insulin- and glucagon-immunostained islet cells in the pancreatic resections. Insulin immunostaining varied between 39% and 76% of total islet area, but there was no correlation with either HbA1c or BMI (61.06 ± 3.4% of total islet area in n = 10 subjects with HbA1c <6% and 60.56 ± 2.7% of total islet area in n = 7 subjects with HbA1c >6%) (Fig. 4A and 4B). Similarly, glucagon immunostaining varied between 13% and 39% of islet area, but there was no correlation between α-cell area and HbA1c or BMI (Fig. 4C and 4D). The average α-cell area was 23.8 ± 3.1% of total islet area in n = 9 subjects with HbA1c <6% and 28.7 ± 1.7% of total islet area in n = 9 subjects with HbA1c > 6%. Furthermore, the highly variable mRNA levels of insulin, glucagon, and somatostatin did also not correlate to HbA1c, in our subjects at least (Fig. 4E–4G). These results suggest that dedifferentiation and loss of β-cell mass are not major causes of the development of frequent hyperglycemic episodes (i.e., increased HbA1c).
Figure 4.

β-Cell and α-cell areas and the expression level of the islet hormones did not correlate with either HbA1c or BMI. (A and B) β-cell area (expressed as percentage of total islet area) determined by insulin immunostaining in n = 17 pancreatic resections. (C and D) α-Cell area (expressed as percentage of total islet area) determined by glucagon immunostaining in n = 18 pancreatic resections as described in Materials and Methods. HbA1c and BMI were from the same patients. The relative mRNA levels of (E) insulin (INS), (F) glucagon (GCG), and (G) somatostatin (SST) were assessed by qRT-PCR as described in Materials and Methods in n = 12 LCM-isolated islet tissue samples.
Furthermore, no mRNA levels examined correlated with BMI, suggesting that obesity had no major impact on β-cell–specific gene expression in our cohort (Supplemental Fig. 5). Because vitamin D may modulate insulin secretion (22), we also assessed plasma concentrations of 25-hydroxy-vitamin D. No correlation of vitamin D with any of the examined parameters was found (data not shown).
In summary, ALDOB expression is increased in islets from hyperglycemic subjects, an event that is associated with reduced insulin secretion.
Discussion
The current study shows that islet ALDOB mRNA levels negatively correlate with in vivo insulin secretion in humans. A direct effect of ALDOB on insulin secretion is suggested by the association of a common variant in ALDOB with insulin secretion evaluated using OGTT and hyperglycemic clamp studies in humans. Hyperglycemic clamp is the gold standard for the assessment of insulin secretion, allowing the estimation of insulin secretion independently from endogenous changes of blood glucose levels.
An upregulation of ALDOB and FAIM2 expression with hyperglycemia has been documented previously (11). Despite the limited sample size in our study, the expressional alterations mirror recent data obtained from a larger set (n = 103) of LCM-cut islet tissue (Supplemental Fig. 2) (11).
In agreement with the transcriptome analysis of LCM-cut islet tissue, chronic exposure to high glucose augmented ALDOB protein in human islets isolated from organ donors. Similarly, in mature, functional rodent β-cells, AldoB expression is repressed, but strongly upregulated by hyperglycemia (7, 20, 23). The confocal laser scanning imaging of immunostained pancreatic tissue and isolated human islet cells suggests that ALDOB-expressing islet cells are β-cells, as ALDOB and insulin signals were detected in the same cells. Single islet cell transcriptomics data suggest that only a minor fraction of β-cell population expresses ALDOB (Supplemental Fig. 6) (7, 18). However, an increased expression of ALDOB was also detected in β-, δ-, and γ- cells from subjects with type 2 diabetes (Supplemental Fig. 7) (7). That dedifferentiation of β-cells underlies this process is unlikely, because the mRNA levels of β-cell–specific genes (PDX1, MAFB, INS, and NEUROD1), as well as β- and α-cell mass, were not different between patients with high and low HbA1c. The transcripts of NANOG and POU5F1B were only modestly elevated in the hyperglycemic islets (data not shown), which, similarly to previous studies, suggests no considerable dedifferentiation of type 2 diabetic β-cells (9, 11).
Our observations suggest that increased expression of ALDOB in β-cells affects in vivo insulin secretion. A recent study with a β-cell–specific Raptor knockout mouse showed increased expression of AldoB in the islets and impaired glucose-induced insulin secretion (GIIS) and ATP production (24). Similarly, β-cell–specific NeuroD1 knockout mice display impaired GIIS and upregulated AldoB in their β-cells (25). Noteworthy, the juvenile, glucose-unresponsive β-cells express high levels of ALDOB and have high glycolytic flux and low oxidative metabolism (23, 26). Thus, a chronic upregulation of ALDOB in adult β-cells might shift the glucose metabolism away from ATP production, an effect that could impair GIIS. ALDOB catalyzes the cleavage of fructose 1,6-bisphosphate and fructose 1-phosphate. The glycolytic intermediates DHAP and GA3P are primary sources for methylglyoxal production (27). ALDOB seems to be involved in glucose-dependent overproduction of methylglyoxal (28). Methylglyoxal accumulation predicts endothelial dysfunction in type 2 diabetes (29). In β-cells, methylglyoxal-induced protein glycation and reactive oxygen species accumulation impair mitochondrial function and insulin secretion (30). Further experiments are required to elucidate whether ALDOB mediates these events in human β-cells of subjects with type 2 diabetes.
The association of rs550915 in ALDOB with altered insulin secretion both in OGTT and hyperglycemic clamp studies indicates that ALDOB modulates insulin secretion (Fig. 2A and 2B). Data from the GTEx project indicate that this SNP is an expression quantitative trait locus. The minor allele of rs550915 is significantly associated with lower ALDOB expression in the tibial nerve (P = 0.000015; normalized effect size = −0.39). Islets are not specifically sampled in the GTEx project, but an association with expression in pancreas was not seen. However, rs550915 is a proposed multitissue expression quantitative trait locus in GTEx data, suggesting that it regulates gene expression of ALDOB in several tissues. In genome-wide association studies (with results accessed from www.type2diabetesgenetics.org), only one GWAS showed a nominal association of rs550915 with type 2 diabetes (31). The discrepancy between effects on insulin secretion and type 2 diabetes prevalence could derive from compensatory glycemic effects of the variant in other tissues. ALDOB is prominently expressed in liver and kidney, two organs involved in glucose disposal and reabsorption. Effects of ALDOB in islets could be potentially offset by effects in these organs (32, 33). In our study, the endogenous glucose handling was circumvented using a hyperglycemic clamp, which enabled us to investigate alterations of in vivo insulin secretion (Fig. 2B).
The other prominently upregulated gene in hyperglycemic patients was the obesity-related gene FAIM2 (34). FAIM2 expression in healthy islet cells is low (7). Recent publications reported the upregulation and increased methylation of FAIM2 in type 2 diabetic islets, but its function in β-cells is currently unknown (11, 34, 35). We also found no association between SNPs of FAIM2 and insulin secretion in our cohorts. As corroborated by the low expression level, this observation prompted us to attribute a rather minor role to FAIM2 in insulin secretion.
In contrast to the upregulation of ALDOB and FAIM2, expression of the β-cell proteins SLC2A2, FFAR4, TMEM37, and TMEM27 correlated negatively with hyperglycemia, confirming recent observations in type 2 diabetic islets (6, 11). The reduced SLC2A2 mRNA level points to a functional defect of β-cells (36). Nonetheless, we found no correlation of SLC2A2 with in vivo insulin secretion. FFAR4, a long-chain fatty acids receptor, is expressed in β-, α-, and δ-cells and augments glucagon and somatostatin secretion (7, 37, 38). Again, we found no correlation with insulin secretion. Although a reduced expression of TMEM37, the regulatory γ subunit of the voltage-gated Ca2+ channel, was found in type 2 diabetic islets, TMEM37 downregulation in INS-1E cells augmented insulin secretion (11). The negative correlation between TMEM27 and HbA1c confirms previous reports (11, 39). TMEM27 supports GIIS and was proposed as a marker of β-cell mass (7, 40). This study did not uncover a correlation of TMEM27 with insulin secretion, nor was β-cell area reduced in patients with high HbA1c.
In conclusion, our data suggest that an upregulation of ALDOB in human β-cells occurs upon the development of hyperglycemia and may contribute to impairment of insulin secretion in humans.
Acknowledgments
We thank Dr. Louise Fritsche, Andreas Vosseler, and Anja Dessecker [German Research Center for Environmental Health (HMGU)/Institute for Diabetes Research and Metabolic Diseases, Eberhard Karls University of Tuebingen] for patient recruitment and study management, Ulrike Schmidt, Kubrom Bekure (HMGU/Institute for Diabetes Research and Metabolic Diseases), Birgit Schreiner (Department of Internal Medicine IV, University Hospital of Tuebingen), Harald Grallert, and Jennifer Kriebel (Research Unit of Molecular Epidemiology/HMGU) for work in the genotyping of the TUEF study.
Financial Support: This study was supported by Grant 01GI0925 from the German Federal Ministry of Education and Research to the German Center for Diabetes Research. The project was supported by the Innovative Medicines Initiative Joint Undertaking (Grant 155005) for IMIDIA, which received financial contributions from the European Union’s Seventh Framework Program (FP7/2007–2013). This project has received funding from the Innovative Medicines Initiative 2 Joint Undertaking under Grant 115881 (RHAPSODY). This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme and European Federation of Pharmaceutical Industries and Associations. This work is supported by the Swiss State Secretariat for Education, Research and Innovation under Contract Number 16.0097. The opinions expressed and arguments used in this study do not necessarily reflect the official views of these funding bodies. The GTEx Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health and by the National Cancer Institute, National Human Genome Research Institute, National Heart, Lung, and Blood Institute, National Institute on Drug Abuse, National Institute of Mental Health, and National Institute of Neurological Disorders and Stroke. The data used for the analyses described in this study were obtained from the GTEx portal at https://gtexportal.org/home/eqtls/bySnp?snpId=rs550915&tissueName=, all on 16 March 2018.
Author Contributions: F.G., B.A.J., H.S., G.K., M.P., M.H., B.S., F.F., A.K., S.N., A.P., A.F., and R.W. recruited patients and obtained tissue and blood samples. F.G., H.S., A.M.S., E.L.-G., G.K., M.P., S.H., M. Schütz., M. Stadion, A.S., F.M., M.I., T.F., P.P.N., S.U., and R.W. performed experiments and analyzed data. F.G., H.S., S.U., and R.W. prepared figures and wrote the manuscript. F.G., B.A.J., H.S., A.M.S., E.L.-G., G.K., M.P., S.H., M.H., M. Schütz, M. Stadion, A.S., F.M., M.I., B.S., F.F., T.F., P.P.N., A.K., S.N., S.W., A.P., A.F., D.R., M. Solimena, H.-U.H., S.U., and R.W. approved the final version of the manuscript. M. Solimena, H.-U.H., S.U., and R.W. conceived and designed the study.
Disclosure Summary: The authors have nothing to disclose.
Glossary
Abbreviations:
- AUC
area under the curve
- BMI
body mass index
- GIIS
glucose-induced insulin secretion
- GLP-1
glucagon-like peptide 1
- GTEx
Genotype-Tissue Expression
- HOMA2
updated homeostasis model assessment
- LCM
laser capture microdissection
- OGTT
oral glucose tolerance test
- qRT-PCR
quantitative RT-PCR
- SNP
single nucleotide polymorphism
- TUEF
Tuebingen Family
References
- 1. Meier JJ, Bonadonna RC. Role of reduced β-cell mass versus impaired β-cell function in the pathogenesis of type 2 diabetes. Diabetes Care. 2013;36(Suppl 2):S113–S119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gerber PA, Rutter GA. The role of oxidative stress and hypoxia in pancreatic beta-cell dysfunction in diabetes mellitus. Antioxid Redox Signal. 2017;26(10):501–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Poitout V, Amyot J, Semache M, Zarrouki B, Hagman D, Fontes G. Glucolipotoxicity of the pancreatic beta cell. Biochim Biophys Acta. 2010;1801(3):289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lyssenko V, Jonsson A, Almgren P, Pulizzi N, Isomaa B, Tuomi T, Berglund G, Altshuler D, Nilsson P, Groop L. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med. 2008;359(21):2220–2232. [DOI] [PubMed] [Google Scholar]
- 5. Staiger H, Machicao F, Fritsche A, Häring HU. Pathomechanisms of type 2 diabetes genes. Endocr Rev. 2009;30(6):557–585. [DOI] [PubMed] [Google Scholar]
- 6. Marselli L, Thorne J, Dahiya S, Sgroi DC, Sharma A, Bonner-Weir S, Marchetti P, Weir GC. Gene expression profiles of Beta-cell enriched tissue obtained by laser capture microdissection from subjects with type 2 diabetes. PLoS One. 2010;5(7):e11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Segerstolpe Å, Palasantza A, Eliasson P, Andersson EM, Andréasson AC, Sun X, Picelli S, Sabirsh A, Clausen M, Bjursell MK, Smith DM, Kasper M, Ämmälä C, Sandberg R. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab. 2016;24(4):593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ottosson-Laakso E, Krus U, Storm P, Prasad RB, Oskolkov N, Ahlqvist E, Fadista J, Hansson O, Groop L, Vikman P. Glucose-induced changes in gtene expression in human pancreatic islets: causes or consequences of chronic hyperglycemia. Diabetes. 2017;66(12):3013–3028. [DOI] [PubMed] [Google Scholar]
- 9. Lawlor N, George J, Bolisetty M, Kursawe R, Sun L, Sivakamasundari V, Kycia I, Robson P, Stitzel ML. Single-cell transcriptomes identify human islet cell signatures and reveal cell-type-specific expression changes in type 2 diabetes. Genome Res. 2017;27(2):208–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ndiaye FK, Ortalli A, Canouil M, Huyvaert M, Salazar-Cardozo C, Lecoeur C, Verbanck M, Pawlowski V, Boutry R, Durand E, Rabearivelo I, Sand O, Marselli L, Kerr-Conte J, Chandra V, Scharfmann R, Poulain-Godefroy O, Marchetti P, Pattou F, Abderrahmani A, Froguel P, Bonnefond A. Expression and functional assessment of candidate type 2 diabetes susceptibility genes identify four new genes contributing to human insulin secretion. Mol Metab. 2017;6(6):459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Solimena M, Schulte AM, Marselli L, Ehehalt F, Richter D, Kleeberg M, Mziaut H, Knoch KP, Parnis J, Bugliani M, Siddiq A, Jörns A, Burdet F, Liechti R, Suleiman M, Margerie D, Syed F, Distler M, Grützmann R, Petretto E, Moreno-Moral A, Wegbrod C, Sönmez A, Pfriem K, Friedrich A, Meinel J, Wollheim CB, Baretton GB, Scharfmann R, Nogoceke E, Bonifacio E, Sturm D, Meyer-Puttlitz B, Boggi U, Saeger HD, Filipponi F, Lesche M, Meda P, Dahl A, Wigger L, Xenarios I, Falchi M, Thorens B, Weitz J, Bokvist K, Lenzen S, Rutter GA, Froguel P, von Bülow M, Ibberson M, Marchetti P. Systems biology of the IMIDIA biobank from organ donors and pancreatectomised patients defines a novel transcriptomic signature of islets from individuals with type 2 diabetes. Diabetologia. 2018;61(3):641–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. American Diabetes Association (2) Classification and diagnosis of diabetes. Diabetes Care. 2015;38(Suppl):S8–S16. [DOI] [PubMed] [Google Scholar]
- 13. Levy JC, Matthews DR, Hermans MP. Correct homeostasis model assessment (HOMA) evaluation uses the computer program. Diabetes Care. 1998;21(12):2191–2192. [DOI] [PubMed] [Google Scholar]
- 14. Stefan N, Machicao F, Staiger H, Machann J, Schick F, Tschritter O, Spieth C, Weigert C, Fritsche A, Stumvoll M, Häring HU. Polymorphisms in the gene encoding adiponectin receptor 1 are associated with insulin resistance and high liver fat. Diabetologia. 2005;48(11):2282–2291. [DOI] [PubMed] [Google Scholar]
- 15. Schäfer SA, Tschritter O, Machicao F, Thamer C, Stefan N, Gallwitz B, Holst JJ, Dekker JM, ’t Hart LM, Nijpels G, van Haeften TW, Häring HU, Fritsche A. Impaired glucagon-like peptide-1-induced insulin secretion in carriers of transcription factor 7-like 2 (TCF7L2) gene polymorphisms [published corrections appear in Diabetologia 2008;51(1):208 and 2009;52(3):557]. Diabetologia. 2007;50(12):2443–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fritsche A, Stefan N, Hardt E, Schützenauer S, Häring H, Stumvoll M. A novel hyperglycaemic clamp for characterization of islet function in humans: assessment of three different secretagogues, maximal insulin response and reproducibility. Eur J Clin Invest. 2000;30(5):411–418. [DOI] [PubMed] [Google Scholar]
- 17. GTEx Consortium Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348(6235):648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Enge M, Arda HE, Mignardi M, Beausang J, Bottino R, Kim SK, Quake SR. Single-cell analysis of human pancreas reveals transcriptional signatures of aging and somatic mutation patterns. Cell. 2017;171(2):321–330.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gerst F, Wagner R, Kaiser G, Panse M, Heni M, Machann J, Bongers MN, Sartorius T, Sipos B, Fend F, Thiel C, Nadalin S, Königsrainer A, Stefan N, Fritsche A, Häring HU, Ullrich S, Siegel-Axel D. Metabolic crosstalk between fatty pancreas and fatty liver: effects on local inflammation and insulin secretion. Diabetologia. 2017;60(11):2240–2251. [DOI] [PubMed] [Google Scholar]
- 20. Wang H, Maechler P, Antinozzi PA, Hagenfeldt KA, Wollheim CB. Hepatocyte nuclear factor 4alpha regulates the expression of pancreatic beta -cell genes implicated in glucose metabolism and nutrient-induced insulin secretion. J Biol Chem. 2000;275(46):35953–35959. [DOI] [PubMed] [Google Scholar]
- 21. Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell. 2012;150(6):1223–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kadowaki S, Norman AW. Dietary vitamin D is essential for normal insulin secretion from the perfused rat pancreas. J Clin Invest. 1984;73(3):759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dhawan S, Tschen SI, Zeng C, Guo T, Hebrok M, Matveyenko A, Bhushan A. DNA methylation directs functional maturation of pancreatic β cells. J Clin Invest. 2015;125(7):2851–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ni Q, Gu Y, Xie Y, Yin Q, Zhang H, Nie A, Li W, Wang Y, Ning G, Wang W, Wang Q. Raptor regulates functional maturation of murine beta cells. Nat Commun. 2017;8:15755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gu C, Stein GH, Pan N, Goebbels S, Hörnberg H, Nave KA, Herrera P, White P, Kaestner KH, Sussel L, Lee JE. Pancreatic beta cells require NeuroD to achieve and maintain functional maturity. Cell Metab. 2010;11(4):298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Henquin JC, Nenquin M. Immaturity of insulin secretion by pancreatic islets isolated from one human neonate. J Diabetes Investig. 2018;9(2):270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Phillips SA, Thornalley PJ. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur J Biochem. 1993;212(1):101–105. [DOI] [PubMed] [Google Scholar]
- 28. Liu J, Mak TC, Banigesh A, Desai K, Wang R, Wu L. Aldolase B knockdown prevents high glucose-induced methylglyoxal overproduction and cellular dysfunction in endothelial cells. PLoS One. 2012;7(7):e41495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Groener JB, Oikonomou D, Cheko R, Kender Z, Zemva J, Kihm L, Muckenthaler M, Peters V, Fleming T, Kopf S, Nawroth PP. Methylglyoxal and advanced glycation end products in patients with diabetes - what we know so far and the missing links [published online ahead of print 13 April 2017]. Exp Clin Endocrinol Diabetes. doi: 10.1055/s-0043-106443. [DOI] [PubMed]
- 30. Fiory F, Lombardi A, Miele C, Giudicelli J, Beguinot F, Van Obberghen E. Methylglyoxal impairs insulin signalling and insulin action on glucose-induced insulin secretion in the pancreatic beta cell line INS-1E [published correction appears in Diabetologia 2012;55(1):272.] Diabetologia. 2011;54(11):2941–2952. [DOI] [PubMed] [Google Scholar]
- 31. Williams AL, Jacobs SB, Moreno-Macías H, Huerta-Chagoya A, Churchhouse C, Márquez-Luna C, García-Ortíz H, Gómez-Vázquez MJ, Burtt NP, Aguilar-Salinas CA, González-Villalpando C, Florez JC, Orozco L, Haiman CA, Tusié-Luna T, Altshuler D; SIGMA Type 2 Diabetes Consortium . Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature. 2014;506(7486):97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Agius L. Substrate modulation of aldolase B binding in hepatocytes. Biochem J. 1996;315(Pt 2):651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yañez AJ, Ludwig HC, Bertinat R, Spichiger C, Gatica R, Berlien G, Leon O, Brito M, Concha II, Slebe JC. Different involvement for aldolase isoenzymes in kidney glucose metabolism: aldolase B but not aldolase A colocalizes and forms a complex with FBPase. J Cell Physiol. 2005;202(3):743–753. [DOI] [PubMed] [Google Scholar]
- 34. Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, Lango Allen H, Lindgren CM, Luan J, Mägi R, Randall JC, Vedantam S, Winkler TW, Qi L, Workalemahu T, Heid IM, Steinthorsdottir V, Stringham HM, Weedon MN, Wheeler E, Wood AR, Ferreira T, Weyant RJ, Segrè AV, Estrada K, Liang L, Nemesh J, Park JH, Gustafsson S, Kilpeläinen TO, Yang J, Bouatia-Naji N, Esko T, Feitosa MF, Kutalik Z, Mangino M, Raychaudhuri S, Scherag A, Smith AV, Welch R, Zhao JH, Aben KK, Absher DM, Amin N, Dixon AL, Fisher E, Glazer NL, Goddard ME, Heard-Costa NL, Hoesel V, Hottenga JJ, Johansson A, Johnson T, Ketkar S, Lamina C, Li S, Moffatt MF, Myers RH, Narisu N, Perry JR, Peters MJ, Preuss M, Ripatti S, Rivadeneira F, Sandholt C, Scott LJ, Timpson NJ, Tyrer JP, van Wingerden S, Watanabe RM, White CC, Wiklund F, Barlassina C, Chasman DI, Cooper MN, Jansson JO, Lawrence RW, Pellikka N, Prokopenko I, Shi J, Thiering E, Alavere H, Alibrandi MT, Almgren P, Arnold AM, Aspelund T, Atwood LD, Balkau B, Balmforth AJ, Bennett AJ, Ben-Shlomo Y, Bergman RN, Bergmann S, Biebermann H, Blakemore AI, Boes T, Bonnycastle LL, Bornstein SR, Brown MJ, Buchanan TA, Busonero F, Campbell H, Cappuccio FP, Cavalcanti-Proença C, Chen YD, Chen CM, Chines PS, Clarke R, Coin L, Connell J, Day IN, den Heijer M, Duan J, Ebrahim S, Elliott P, Elosua R, Eiriksdottir G, Erdos MR, Eriksson JG, Facheris MF, Felix SB, Fischer-Posovszky P, Folsom AR, Friedrich N, Freimer NB, Fu M, Gaget S, Gejman PV, Geus EJ, Gieger C, Gjesing AP, Goel A, Goyette P, Grallert H, Grässler J, Greenawalt DM, Groves CJ, Gudnason V, Guiducci C, Hartikainen AL, Hassanali N, Hall AS, Havulinna AS, Hayward C, Heath AC, Hengstenberg C, Hicks AA, Hinney A, Hofman A, Homuth G, Hui J, Igl W, Iribarren C, Isomaa B, Jacobs KB, Jarick I, Jewell E, John U, Jørgensen T, Jousilahti P, Jula A, Kaakinen M, Kajantie E, Kaplan LM, Kathiresan S, Kettunen J, Kinnunen L, Knowles JW, Kolcic I, König IR, Koskinen S, Kovacs P, Kuusisto J, Kraft P, Kvaløy K, Laitinen J, Lantieri O, Lanzani C, Launer LJ, Lecoeur C, Lehtimäki T, Lettre G, Liu J, Lokki ML, Lorentzon M, Luben RN, Ludwig B, Manunta P, Marek D, Marre M, Martin NG, McArdle WL, McCarthy A, McKnight B, Meitinger T, Melander O, Meyre D, Midthjell K, Montgomery GW, Morken MA, Morris AP, Mulic R, Ngwa JS, Nelis M, Neville MJ, Nyholt DR, O’Donnell CJ, O’Rahilly S, Ong KK, Oostra B, Paré G, Parker AN, Perola M, Pichler I, Pietiläinen KH, Platou CG, Polasek O, Pouta A, Rafelt S, Raitakari O, Rayner NW, Ridderstråle M, Rief W, Ruokonen A, Robertson NR, Rzehak P, Salomaa V, Sanders AR, Sandhu MS, Sanna S, Saramies J, Savolainen MJ, Scherag S, Schipf S, Schreiber S, Schunkert H, Silander K, Sinisalo J, Siscovick DS, Smit JH, Soranzo N, Sovio U, Stephens J, Surakka I, Swift AJ, Tammesoo ML, Tardif JC, Teder-Laving M, Teslovich TM, Thompson JR, Thomson B, Tönjes A, Tuomi T, van Meurs JB, van Ommen GJ, Vatin V, Viikari J, Visvikis-Siest S, Vitart V, Vogel CI, Voight BF, Waite LL, Wallaschofski H, Walters GB, Widen E, Wiegand S, Wild SH, Willemsen G, Witte DR, Witteman JC, Xu J, Zhang Q, Zgaga L, Ziegler A, Zitting P, Beilby JP, Farooqi IS, Hebebrand J, Huikuri HV, James AL, Kähönen M, Levinson DF, Macciardi F, Nieminen MS, Ohlsson C, Palmer LJ, Ridker PM, Stumvoll M, Beckmann JS, Boeing H, Boerwinkle E, Boomsma DI, Caulfield MJ, Chanock SJ, Collins FS, Cupples LA, Smith GD, Erdmann J, Froguel P, Grönberg H, Gyllensten U, Hall P, Hansen T, Harris TB, Hattersley AT, Hayes RB, Heinrich J, Hu FB, Hveem K, Illig T, Jarvelin MR, Kaprio J, Karpe F, Khaw KT, Kiemeney LA, Krude H, Laakso M, Lawlor DA, Metspalu A, Munroe PB, Ouwehand WH, Pedersen O, Penninx BW, Peters A, Pramstaller PP, Quertermous T, Reinehr T, Rissanen A, Rudan I, Samani NJ, Schwarz PE, Shuldiner AR, Spector TD, Tuomilehto J, Uda M, Uitterlinden A, Valle TT, Wabitsch M, Waeber G, Wareham NJ, Watkins H, Wilson JF, Wright AF, Zillikens MC, Chatterjee N, McCarroll SA, Purcell S, Schadt EE, Visscher PM, Assimes TL, Borecki IB, Deloukas P, Fox CS, Groop LC, Haritunians T, Hunter DJ, Kaplan RC, Mohlke KL, O’Connell JR, Peltonen L, Schlessinger D, Strachan DP, van Duijn CM, Wichmann HE, Frayling TM, Thorsteinsdottir U, Abecasis GR, Barroso I, Boehnke M, Stefansson K, North KE, McCarthy MI, Hirschhorn JN, Ingelsson E, Loos RJ, MAGICProcardis Consortium . Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42(11):937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dayeh T, Volkov P, Salö S, Hall E, Nilsson E, Olsson AH, Kirkpatrick CL, Wollheim CB, Eliasson L, Rönn T, Bacos K, Ling C. Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 2014;10(3):e1004160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McCulloch LJ, van de Bunt M, Braun M, Frayn KN, Clark A, Gloyn AL. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: implications for understanding genetic association signals at this locus. Mol Genet Metab. 2011;104(4):648–653. [DOI] [PubMed] [Google Scholar]
- 37. Stone VM, Dhayal S, Brocklehurst KJ, Lenaghan C, Sörhede Winzell M, Hammar M, Xu X, Smith DM, Morgan NG. GPR120 (FFAR4) is preferentially expressed in pancreatic delta cells and regulates somatostatin secretion from murine islets of Langerhans. Diabetologia. 2014;57(6):1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Suckow AT, Polidori D, Yan W, Chon S, Ma JY, Leonard J, Briscoe CP. Alteration of the glucagon axis in GPR120 (FFAR4) knockout mice: a role for GPR120 in glucagon secretion. J Biol Chem. 2014;289(22):15751–15763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Altirriba J, Gasa R, Casas S, Ramírez-Bajo MJ, Ros S, Gutierrez-Dalmau A, Ruiz de Villa MC, Barbera A, Gomis R. The role of transmembrane protein 27 (TMEM27) in islet physiology and its potential use as a beta cell mass biomarker. Diabetologia. 2010;53(7):1406–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Akpinar P, Kuwajima S, Krützfeldt J, Stoffel M. Tmem27: a cleaved and shed plasma membrane protein that stimulates pancreatic beta cell proliferation. Cell Metab. 2005;2(6):385–397. [DOI] [PubMed] [Google Scholar]