

Abstract
Tissue-engineered skeletal muscle plays an important role not only in the field of regenerative medicine but also in emerging areas such as soft robotics, organ-on-a-chip disease models, and drug testing. However, further expansion of the applications of tissue-engineered skeletal muscle models requires a suitable method for their long-term storage and shipment. Cryopreservation has long been the standard for cell storage, but the freezing of three-dimensional tissues is accompanied by many complications due to heat and mass transfer limitations. In this study, we used a tissue-engineered skeletal muscle bioactuator as a model to characterize the effects of freezing on muscle viability, gene expression, myotube structure, and force generation. We optimized the protocol for cryopreservation by comparing outcomes when tissue was frozen undifferentiated and differentiated. Our optimized protocol, in which skeletal muscle was frozen undifferentiated, not only maintained cell viability but also led to a three-fold increase in force production compared to unfrozen muscle. Furthermore, we enhanced muscle lifetime through inhibition of cysteine proteases. The reported timeline for skeletal muscle tissue fabrication, freezing, revival, and long-term culture not only promotes a more streamlined fabrication process but also enables multisite collaborative research efforts through the shipment of preformed skeletal muscle constructs.
Impact Statement
The ability to freeze, revive, and prolong the lifetime of tissue-engineered skeletal muscle without incurring any loss of function represents a significant advancement in the field of tissue engineering. Cryopreservation enables the efficient fabrication, storage, and shipment of these tissues. This in turn facilitates multidisciplinary collaboration between research groups, enabling advances in skeletal muscle regenerative medicine, organ-on-a-chip models of disease, drug testing, and soft robotics. Furthermore, the observation that freezing undifferentiated skeletal muscle enhances functional performance may motivate future studies developing stronger and more clinically relevant engineered muscle.
Keywords: biobot, bioactuator, skeletal muscle, tissue engineering, cryopreservation, cysteine cathepsins
Introduction
Engineered skeletal muscle tissue has long been of interest to the tissue engineering community because of its potential to counteract loss of function resulting from volumetric muscle loss injuries or degenerative illness.1,2 More recently, efforts to engineer skeletal muscle have targeted applications beyond regenerative medicine, such as organ-on-a-chip models of disease,3–5 formation of in vitro neuromuscular junctions,6,7 and cell-based bioactuators for soft robotics.8–11 As these studies generate more functional tissues with different end-use applications, there is a growing need for a robust method of storing engineered skeletal muscle long term.
Cryopreservation is the gold standard for storing living cells that must be revived postpreservation.12 While cryopreservation of cell suspensions is a well-understood and characterized process, freezing and revival of engineered tissue have not been investigated or optimized with similar rigor, as the protocol must be customized to tissue type.13 Most cell freezing processes rely on the use of dimethyl sulfoxide (DMSO), a cryoprotectant that inhibits the formation of ice crystals that could lyse cells, in the freezing medium.14 However, it is unclear whether this medium composition is sufficient for preserving the morphology and function of myoblasts embedded within three-dimensional (3D) engineered skeletal muscle tissue. Since this has not been reported in the literature, to our knowledge, there is a need for the growing body of skeletal muscle tissue engineers to understand, optimize, and use a standardized protocol for long-term storage and revival of the tissue.
Engineering mature contractile skeletal muscle can take several days to weeks to form, depending on the protocol used.15–19 In addition to optimization of freezing medium composition, a comprehensive study of skeletal muscle cryopreservation also requires investigating the timeline for freezing and reviving engineered tissue. This protocol must take into account maturity of the tissue, undifferentiated myoblasts or differentiated myotubes, and yield an understanding of the advantages and disadvantages of freezing tissue in either state. Moreover, since all end-use applications of engineered skeletal muscle will require the tissue to be contractile in response to external stimulation, the effect of cryopreservation on muscle force production must also be well characterized.
This study aims to develop a protocol for freezing, revival, and long-term culture of engineered skeletal muscle tissue to keep the tissue intact, viable, metabolically active, and contractile. By providing a deeper understanding of the optimized timeline for muscle cryopreservation, this protocol generates guidelines for long-term storage and revival of skeletal muscle tissue, while introducing preliminary results of a method to enhance tissue lifetime by inhibition of cysteine proteases.
Materials and Methods
Stereolithographic 3D printing of ring molds and biobot skeletons
Molds for the formation of muscle “ring” structures were 3D printed using a modified form of the commercially available stereolithography apparatus (SLA 250/50; 3D Systems), as previously reported.15,16 These molds were fabricated using 20% w/v polyethylene glycol dimethacrylate (PEGDMA) of molecular weight 1000 g/mol (Polysciences) with 0.5% w/v Irgacure 2959 (Ciba). We used a biological micro robot (biobot) to characterize forces produced by the muscles.15,16 The skeleton of the biobots, onto which the muscle rings were transferred, was fabricated using 20% w/v polyethylene glycol diacrylate (PEGDA) of molecular weight 700 g/mol (Sigma-Aldrich) with 0.5% w/v Irgacure 2959. After fabrication, the parts were sterilized in 70% ethanol for 1 h, followed by storage in phosphate-buffered saline (PBS).
Seeding of C2C12 myoblast rings
C2C12 murine myoblasts transduced to express channelrhodopsin-2 (ChR2[H134R]) were cultured in growth medium (GM) consisting of Dulbecco's modified Eagle medium (DMEM) with l-glutamine and sodium pyruvate (Corning) supplemented with 10% v/v fetal bovine serum (FBS; Lonza), 1% v/v l-glutamine (Cellgro Mediatech), and 1% v/v penicillin–streptomycin (Cellgro Mediatech). Upon reaching ∼85% confluency, cells were trypsinized and resuspended at a concentration of 1E7 cells/mL within a matrix mixture of 30% v/v Matrigel (BD Biosciences), 4 mg/mL fibrinogen (Sigma-Aldrich), and 0.5 U mg/fibrinogen thrombin (Sigma-Aldrich) in GM supplemented with 1 mg/mL aminocaproic acid (ACA; Sigma-Aldrich) (GM+). The cell–matrix suspension (120 μL) was pipetted into the injection ring mold and incubated at 37°C for 1 h before being immersed in GM+.
Differentiation of skeletal muscle biobots
C2C12 myoblast tissues were cultured for 3 days postseeding in GM+ (refreshed daily). After 3 days, the myoblast rings were lifted from their molds and transferred to biobot skeletons, where they were immersed in differentiation medium (DM) consisting of DMEM supplemented with 10% v/v heat-inactivated horse serum (Lonza), 1% v/v l-glutamine, 1% v/v penicillin–streptomycin, 1 mg/mL ACA, and 50 ng/mL human insulin-like growth factor-1 (Sigma Aldrich) (DM++). The differentiation medium was refreshed daily for 7 days after ring transfer.
Freezing and thawing of undifferentiated myoblast rings
Twenty-four hours postseeding, undifferentiated C2C12 myoblast rings were removed from their molds and placed into cryogenic freezing tubes containing 1 mL warm GM supplemented with 5%, 10%, or 15% v/v DMSO (Fisher). Freezing tubes were placed in alcohol-free freezing containers (CoolCell; Corning) and stored in the −80°C freezer, where they were slowly frozen at a rate of 1°C/min. After 24 h in the freezer, the cryogenic freezing tubes were placed in a warm 37°C water bath for ∼3 min until completely thawed. The rings were then removed from the tubes, rinsed in PBS, and returned to their original ring molds to incubate for 48 h at 37°C. GM+ was refreshed daily. On day 4 postseeding, myoblast rings were transferred to biobot skeletons and differentiated for 7 days in DM++.
Freezing and thawing of differentiated muscle rings
For this set of experiments, the medium was changed to DM++ on day 3 postseeding of the cell matrix solution on the ring molds. However, muscle rings were left within the ring molds rather than being transferred to biobot skeletons. On day 9 postseeding, the central portion of the ring mold was detached from the rest of the mold, so that the structure could be lifted away with a spatula. The central portion of the mold and surrounding muscle ring were transferred to a cryogenic freezing tube containing 1 mL warm GM with 5%, 10%, or 15% v/v DMSO and frozen at −1°C/min to −80°C. After 24 h in the freezer, the tubes were thawed in a warm 37°C water bath and the mold-ring structures were rinsed in PBS. Muscle rings were removed from molds, transferred to biobot skeletons, and returned to culture in DM++ at 37°C.
MTS viability assay
MTS solution (CellTiter 96 AQueous One Solution Cell Proliferation Assay; Promega) was thawed in a 37°C water bath and combined with warm DMEM (without phenol red) in a 1:5 volume ratio to form a working reagent. Muscle rings were rinsed in PBS, placed in individual wells of a 24-well plate, and immersed in MTS working reagent. The reaction was allowed to proceed in the dark for 4 h at 37°C. A microplate reader (Synergy HT; BioTek) was used to measure absorbance at 490 nm, which was then used to calculate relative viability between control and experimental samples.
Live/dead staining and confocal imaging
Muscle rings were stained with 2 μM calcein AM and 4 μM ethidium homodimer-1 (Thermofisher) in PBS and incubated at 37°C for 1 h. To image, tissue samples were sliced length-wise along each side of the muscle ring. Confocal imaging was performed using the Zeiss LSM 710. To compare cell viability between tissue samples, the mean fluorescence intensity of calcein AM (green) and ethidium homodimer-1 (red) was measured in each image. A ratio of red to green fluorescence was measured to quantify the ratio of dead to live cells in tissue samples.
Passive tension measurement
Side-view images of the biobots were used to calculate passive tension by applying the Euler-Bernoulli beam bending theory (Equation 1), as previously described.15
 |
Fp is passive tension force, E is the Young's modulus of the PEGDA skeleton (319.4 kPa), I is the moment of inertia of the beam (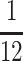 bh3
= 2.77E−14 m4), δmax is the maximum deflection of the beam, l is the distance between the muscle and the beam, and L is the length of the beam (6 mm).15
bh3
= 2.77E−14 m4), δmax is the maximum deflection of the beam, l is the distance between the muscle and the beam, and L is the length of the beam (6 mm).15
Electrical stimulation and active tension measurement
Biobots were detached from glass coverslips and placed in a dish of warm, serum-free DMEM. Two platinum electrodes were aligned parallel to the biobot to stimulate the muscle with 20 V, 50 ms pulses at 1 Hz. Videos of biobot muscle contraction were used to calculate active tension through motion tracking and application of a Kelvin–Voigt viscoelasticity model [Eq. (2)].15 The distance between the two pillars of the biobot was tracked over the course of 150 frames (∼25 s depending on frame rate) to calculate strain and determine the time-varying force, calculated by Equation (2). The peak values of active tension force were averaged, and the minimum value of active tension force was subtracted from this average peak value to determine the active tension value for each biobot.
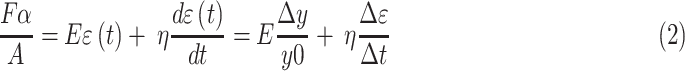 |
Fα is the active tension force, A is the contact area between the muscle ring and the biobot skeleton, E is the Young's modulus of the PEGDA skeleton (319.4 kPa), Δy is the change in length of the skeleton during a contraction, y0 is the original length of the skeleton in the passive state, η is the viscosity of the PEGDA skeleton (5.1 E−3 mPa · s), Δɛ is the change in strain between successive frames, and Δt is the time between frames.15
Quantitative reverse transcription PCR analysis of myogenic marker expression
Muscle rings were removed from biobot skeletons, rinsed in PBS, and flash-frozen for 5 min in liquid nitrogen to be stored at −80°C. Thawed rings were homogenized by vortexing in RNeasy lysis buffer (Qiagen) with 10 μL/mL B-mercaptoethanol and centrifuged using the QIAshredder (Qiagen) column. RNA was extracted using the RNeasy Plus RNA isolation kit (Qiagen) according to manufacturer's instructions. cDNA synthesis was done using qScript cDNA SuperMix (Quanta Biosciences) from 100 ng of RNA and reactions were performed as directed by the supplier. For quantitative PCR (qPCR), SsoFast EvaGreen Supermix (Bio-Rad) was added to cDNA and primers targeting the genes of interest and GAPDH (Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/tea). The cycling parameters were 30 s at 95°C, and 40 cycles of 5 s at 95°C and 20 s at 55°C, followed by melt-curve analysis of the product using a CFX Connect Real-Time System (Bio-Rad). Cycle threshold (Ct) values were used to calculate changes in expression level, relative to GAPDH and control samples by the 2−ΔΔCt method.20 Reaction efficiencies over the appropriate dynamic range were calculated to ensure linearity of the standard curve.
Immunohistochemical staining and lightsheet imaging of muscle rings
Muscle rings to be imaged were rinsed with PBS. To preserve the internal structure, the tissue was fixed on the skeleton structures using 4% v/v of paraformaldehyde for 20 min. Samples were later permeabilized using Triton-X, which was diluted to 0.25% v/v and used to permeabilize the tissue membrane for 20 min. After washing with PBS, samples were blocked and stored in 1% w/v bovine serum albumin (Sigma Aldrich) diluted in PBS with 0.1% v/v Tween 20 at 4°C. Mouse anti-myosin heavy chain (MF-20) and rabbit anti-α-actinin were used to stain for myosin and sarcomeres, respectively. AlexaFluor-647 anti-mouse (ThermoFisher) and AlexaFluor-633 anti-rabbit (ThermoFisher) were used to stain MF-20 and α-actinin primary antibodies, respectively, and incubated for 2 h at room temperature. To be able to image 3D tissue, stained samples were embedded in 1% agarose inside a 1 mL syringe. For imaging, the embedded sample was suspended in a chamber to be imaged by a Zeiss Z1 Lightsheet microscope.
Quantifying myotube dispersion
The directionality plugin for ImageJ (NIH) was used to determine the alignment of myotubes through Fourier component analysis, as previously demonstrated.21 A histogram was formed by determining the number of myotubes in each direction. A Gaussian fit was calculated from the highest peak, which represents the preferred orientation. The standard deviation of the Gaussian fit is referred to as the dispersion. The α-actinin fluorescent images were used with the directionality plugin to determine alignment.
Scanning electron microscopy imaging of muscle rings
Muscle rings were rinsed in PBS and fixed in 4% paraformaldehyde for 20 min. Samples were then rinsed in PBS for 5 min, followed by a series of 10-min immersions in 37%, 67%, and 95% ( × 3) ethanol to dehydrate the tissue. Afterward, samples were washed with hexamethyldisilazane and left to dry overnight at room temperature. Samples were then coated with gold palladium before adhering to a chuck covered with carbon tape. Mounted samples were imaged in a FEI Quanta FEG 450 ESEM.
E-64 culture and multiplex zymography
E-64 broad-spectrum cysteine protease inhibitor (Sigma-Aldrich) was added to DM++ at 0, 5, or 10 μM, beginning on day 3. Protocols for multiplex zymography to detect cathepsins have been previously optimized.22,23 Briefly, this modified sodium dodecyl sulfate–polyacrylamide gel electrophoresis used a gel impregnated with either soluble gelatin or soluble fibrinogen substrate. Samples (muscle tissue collected on days 3 and 12) were prepared in a nonreducing loading dye and separated by electrophoresis at 4°C. Gels were washed in a renaturing buffer to refold enzymes and incubated in an assay buffer overnight at 37°C for optimal protease activity, and then stained with Coomassie blue and destained with white bands indicative of protease activity. Gels were imaged with an ImageQuant LAS 4000 (GE Health care). Densitometry, calculated using ImageJ, was used to quantify the intensity of the white bands (indicating protease activity, normalized to day 3). Lifetime was defined as the number of days before rupture or breakdown of the tissue. Kaplan-Meier plots represent survival for muscle tissues that ruptured naturally (endpoints represented by drops in survival curves; treated as subjects that did not survive the study), as well as muscle tissues that remained intact at the time of publication (represented by upticks in survival curves; treated as subjects that “dropped out” of the study). Previously published data (Fig. 7d, black curves) have been included for comparison purposes.24 Average lifetime values were calculated using the time of publication as an endpoint for muscle tissues that did not spontaneously break as of that point.
FIG. 7.

Engineering biological machine lifespan through control of proteolytic degradation. (a) Fibrinogen zymography confirmed fibrinogenolysis by biobot catL on days 8 and 12, with 1 mg/mL ACA, both with (+) and without (−) daily electrical stimulation. (b) Cathepsin zymography identified a significant dose-dependent decrease in amount of CatL bound to engineered muscle tissue (75 kDa, CatL + tissue) for day 12 conditions (5 or 10 μM E-64 cysteine cathepsin inhibitor) compared to control samples cultured without E-64. (c) E-64 did not impact (i) cell viability, (ii) MCK activity, or (iii) short-term passive tension in E-64-cultured muscle strips on day 12. (iv) Long-term analysis demonstrated that the addition of E-64 increased tissue passive tension on days 30 and 90 (mean ± standard error of the mean; n = 4, day 12; n = 3–4, day 30; and n = 1–4, day 90). (d) Kaplan-Meier survival curve and heat map of E-64-cultured muscle strips (5 or 10 μM) cultured with ACA (n = 5–8). Previously, we reported that engineered muscle tissue cultured in DM with 0 or 1 mg/mL ACA resulted in average lifetimes of 8.2 ± 0.5 and 17 ± 3.9 days until rupture, respectively (black and red lines; data reproduced from Cvetkovic and Ferrall-Fairbanks24). Both in the absence and presence of ACA, E-64 significantly increased the lifetime of myotube rings. With the addition of 5 μM E-64, average lifetime extended significantly to 79.6 ± 9.4 days in the absence of ACA (p < 0.001; gray line) and 138.5 ± 16 days in the presence of ACA (p < 0.01; blue line). Some tissues were still intact as of day 200+, thus demonstrating a more potent effect of E-64 on long-term muscle tissue culture than ACA alone. Kaplan-Meier plots represent survival for muscle tissues that ruptured naturally (endpoints represented by drops in survival curves; treated as subjects that did not survive the study), as well as muscle tissues that remained intact at the time of publication (represented by upticks in survival curves; treated as subjects that “dropped out” of the study). Previously published data have been included for comparison purposes. Average lifetime values were calculated using the time of publication as an endpoint for muscle tissues, which did not spontaneously break as of that point. catL, cathepsin L; ACA, aminocaproic acid; MCK, muscle creatine kinase.
Muscle creatine kinase assay
Muscle rings were digested using RIPA buffer (Thermo Scientific). After sonication and centrifugation, protein extract supernatant was collected and used to determine muscle creatine kinase (MCK) production using a Liquid Creatine Kinase Reagent Set (Pointe Scientific). Absorbance was measured at 340 nm using a microplate reader.
Statistical analysis
In all bar graphs for freezing studies, data are represented as the mean ± standard deviation. For E-64, lifetime, and zymography studies, data are represented as the mean ± standard error of the mean. The students T test was used to determine significance, with *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
Freezing of tissue-engineered skeletal muscle
To study the effects of freezing on skeletal muscle, we engineered a versatile ring-shaped skeletal muscle bioactuator, originally developed by Raman et al.15,16 Skeletal muscle rings were formed from a cell–gel mixture of C2C12 myoblasts, fibrin, and Matrigel (Fig. 1a), which was added to a 3D printed PEGDMA mold (Fig. 1b) to achieve a ring-like shape. The cell–gel solution compacted within the mold over time to form a dense 3D ring structure. After 3 days within the mold, the myoblast ring was removed from the mold and transferred to a 3D printed PEGDA skeleton structure (Fig. 1c). The GM was changed to a differentiation medium, driving muscle ring differentiation over the course of the next 7 days (Fig. 1d). After differentiation of myoblasts to myotubes, the “biobot” was electrically stimulated to observe contraction of the tissue-engineered muscle ring. The 3D printed skeleton was designed such that contraction of the muscle ring causes deflection of the beam and the inward bending of the two pillars (Fig. 1c). The bending of the skeleton can be measured to quantify the force production of the muscle ring, enabling analysis of the functional performance of tissue-engineered skeletal muscle.
FIG. 1.

Skeletal muscle bioactuator fabrication. (a) Composition of cell–matrix solution used to form the muscle ring. (b) CAD image of two ring molds (top) and the skeleton structure (bottom) that are 3D printed using stereolithography. The skeleton is printed with its beam tethered to the bottom of a glass slide so that the muscle ring remains in tension throughout the differentiation process. (c) Side-view image of the mature bioactuator once it has been released from the glass slide. (d) On day 0, C2C12 myoblasts and matrix gel components are pipetted into a 3D printed polyethylene glycol dimethacrylate ring mold. The ring is allowed to solidify and compact in growth media for the next 3 days. On day 3, the compacted ring is removed from the mold and transferred to a 3D printed, asymmetric skeleton structure and the media are changed to differentiation media. On day 10, the skeletal muscle can be placed between two platinum electrodes and electrically stimulated to analyze force production of the mature muscle ring. 3D, three dimensional.
The engineered muscle was frozen at two different time points, before and after differentiation, to determine which method would be optimal for preservation of skeletal muscle's health and function. To cryopreserve tissue while undifferentiated, the myoblast ring was frozen after 24 h postcell seeding (Fig. 2a). DMSO was used as a cryoprotective agent due to its well-understood ability to prevent large ice crystal formation and permeate the cell membrane to reduce osmotic stress on cells.25 The myoblast ring was removed from the ring mold and submerged in freezing medium within a cryogenic freezing tube. The ring was found to be mechanically robust enough to withstand removal from the mold without rupture. Similarly, the tissue maintained its ring-like shape throughout the freezing process and could be easily returned to the ring mold postthaw, where it was allowed to remain in culture for an additional 2 days before being transferred to the skeleton structure (Fig. 2a). The total amount of time in GM within the ring mold, and in differentiation medium, while attached to the skeleton structure, was identical to that of an unfrozen muscle.
FIG. 2.

(a) Timeline for freezing of bioactuators before differentiation. Twenty-four hours postseeding, the ring is removed from culture and placed in a cryogenic freezing vial with 1 mL of medium. The ring is slowly frozen at −1°C/min and left at −80°C for 24 h. The ring is rapidly thawed for 3 min in a water bath, transferred back to the ring mold, and maintained in culture for 24 h. On day 4, the ring is transferred to the skeleton and differentiation begins. Evaluation of the ring occurs on day 11 (rather than day 10) to account for the “null time” while frozen. (b) Timeline for freezing of bioactuators after differentiation. Similar to the standard protocol for bioactuator fabrication, differentiation begins on day 3. However, the muscle ring is differentiated within the ring mold. The entire central region of the ring mold is frozen with the muscle to prevent the muscle from losing its shape. After being thawed, the muscle ring is transferred directly from the mold to the skeleton and left in culture to recover at least 24 h before evaluation. (c) Images of differentiated muscle rings within the ring mold, demonstrating how the central portion of the ring mold is detached from the rest of the mold to be frozen with the muscle ring.
The second cryopreservation method involved freezing the muscle after differentiation, 9 days postcell seeding (Fig. 2b). This time point of freezing occurs 2 days after the skeletal muscle rings first exhibit macroscale contraction, indicating presence of differentiated myotubes.15 The timeline for muscle differentiation before freezing was identical to that of the unfrozen muscle. However, rather than transferring the myoblast ring to the skeleton structure on day 3, the tissue was left to differentiate within the mold. This was done to avoid freezing the 3D printed skeleton structure. When the differentiated muscle was frozen while attached to the skeleton, the entire structure collapsed inward such that the pillars of the skeleton touched and no contraction of the muscle could be visualized (Supplementary Fig. S1). The process of freezing likely contributed to microstructural changes within the PEGDA, which allowed the muscle to easily compact the weakened structure once it was thawed. To avoid this issue, we attempted to freeze the muscle ring by itself, but found that it could not retain its shape throughout the freezing process. The passive tension of the differentiated myotubes caused the ring to compact inward such that the ring was too small to be transferred to the skeleton. To avoid these complications, we removed the elevated central portion of the ring mold along with the muscle ring to preserve the ring shape throughout the freezing process (Fig. 2c). After thawing, the muscle was simply removed from the mold and transferred to the 3D printed skeleton, where it was cultured for another 24 h.
Effect of freezing on cell viability
To effectively freeze and revive biological tissues without incurring a loss of function, it is imperative to prevent significant cell death. In the case of skeletal muscle constructs, a reduction in cell number would lead to a decrease in net force production. Furthermore, the presence of dead cells may impair the process of myoblast fusion and differentiation, thus decreasing maturity and function of single myotubes.
To examine the effects of freezing on engineered skeletal muscle tissue, we began by first freezing muscle in the undifferentiated state (1 day postcell seeding). MTS viability assays were performed to optimize freezing medium composition and quantify the effect of the freezing process on cell viability. Muscle rings were frozen in the same GM in which they were cultured. DMSO serves as a cryoprotectant25 and was supplemented to the freezing medium at concentrations of 5%, 10%, and 15% v/v. Muscle rings were slowly frozen at −1°C/min to −80°C and were thawed rapidly after being frozen for 24 h. MTS assays showed that undifferentiated muscle frozen in GM with 5% DMSO had a viability of 87% ± 6.5%, rings frozen with 10% DMSO had a viability of 93% ± 8.4%, and rings frozen with 15% DMSO had a viability of 84% ± 8.0% (Fig. 3a). Only rings frozen in 10% DMSO did not exhibit significantly lowered viability compared to unfrozen controls. For this reason, all muscle tissues in future studies were frozen in GM supplemented with 10% DMSO.
FIG. 3.

(a) MTS viability of undifferentiated and differentiated muscle rings after being frozen for 24 h in growth medium with varying concentrations of DMSO (mean ± SD, n = 3). Viability is normalized to that of the unfrozen control. (b) MTS viability of undifferentiated muscle rings after being frozen for varying time periods in growth media with 10% DMSO (mean ± SD, n = 3). (c) MTS viability of undifferentiated and differentiated muscle rings frozen for 48 h at −80°C or 24 h in −80°C followed by 24 h in liquid nitrogen (mean ± SD, n = 3). (d, e) Confocal images of differentiated muscle treated with calcein AM (green) to visualize live cells and ethidium homodimer-1 (red) to visualize dead cells. Scale bar = 200 μm. (f) Ratio of mean fluorescence intensity of ethidium homodimer-1 (red) to calcein AM (green) in unfrozen and frozen differentiated muscle rings (mean ± SD, n = 4,5) (*p < 0.05, **p < 0.01). DMSO, dimethyl sulfoxide; SD, standard deviation.
To examine the effect of total time frozen on cell viability, undifferentiated muscle rings were frozen at −80°C for periods of 1 day, 1 week, 1 month, and 2 months. MTS viability assays showed that for all time periods, the viability of frozen undifferentiated muscle was never significantly lowered, compared to the unfrozen controls (Fig. 3b). While these results are promising, it is important to note that storage in liquid nitrogen is standard for long-term cryopreservation. To determine whether the process of transferring tissues to liquid nitrogen may affect cell viability, undifferentiated muscle rings were stored for 24 h at −80°C and transferred to liquid nitrogen for an additional 24 h before thawing. Muscle rings that were frozen for 48 h at −80°C maintained a viability of 82% ± 10%, while those that were transferred to liquid nitrogen had a viability of 103% ± 24%. These results show that GM supplemented with 10% DMSO serves as an adequate freezing medium for undifferentiated muscle, which can be frozen for long periods of time at −80°C or in liquid nitrogen without a significant reduction in cell viability.
The freezing medium was also optimized for the freezing of differentiated muscle rings. Like undifferentiated muscle, differentiated muscle frozen with 10% DMSO maintained the highest viability (90% ± 10%), compared to muscle frozen in 5% DMSO (83% ± 8.7% viability) or 15% DMSO (79% ± 16% viability) (Fig. 3a). Furthermore, differentiated muscle frozen for 48 h at −80°C maintained a viability of 87% ± 16%, while muscle that was transferred to liquid nitrogen for 24 h maintained a similar viability of 88% ± 17%. Outcomes were also compared when differentiated tissues were frozen in differentiation medium compared to GM (Supplementary Fig. S2). Differentiated muscle frozen in differentiation medium had a viability of 77% ± 7.7%, while muscle frozen in GM had a viability of 81% ± 6.9%. The viability of differentiated muscle was significantly reduced when frozen in either medium, and no significant difference between viability of muscle frozen in the two mediums was observed.
It is important to note that while the measured viability of frozen differentiated muscle does not vary significantly between batches, there is some discrepancy between batches as to whether viability significantly varies from that of the unfrozen control. This may be due to small variance in the density of cells seeded or the mixture of cells within the extracellular matrix (ECM). From these conflicting MTS studies, we cannot confirm whether or not the freezing of differentiated muscle significantly affects cell viability, as this appears to vary by batch.
Live/dead staining of unfrozen and frozen differentiated muscle with calcein AM to label live cells green and ethidium homodimer-1 to label dead cells red (Fig. 3d, e) was used to visualize the viability of cells throughout the thickness of the muscle ring. Imaging revealed no significant difference in the ratio of red to green fluorescence between unfrozen muscle (0.26 ± 0.09) and frozen differentiated muscle (0.3 ± 0.08), indicating no significant change in viability (Fig. 3f).
Force production of frozen muscle tissue
To understand how freezing 3D tissues of undifferentiated myoblasts impacts their eventual development into mature myotubes, we began by measuring their force production postdifferentiation. Differentiated skeletal muscle that had previously been frozen for 24 h in the undifferentiated state surprisingly produced significantly higher active contractile force (174 ± 82 μN) than the unfrozen control (45 ± 17 μN) (Fig. 4a; Supplementary Movies S1 and S2). Passive tension, however, was not affected by the freezing process (Fig. 4b). Active and passive tension of a single frozen undifferentiated muscle ring were plotted with time to visualize the change in active force production as the muscle contracts under 1 Hz stimulation (Fig. 4c). Muscle that had been frozen undifferentiated also contracted spontaneously, while unfrozen muscle did not undergo any spontaneous contraction (Supplementary Fig. S3 and Supplementary Movie S3).
FIG. 4.

(a) Active tension produced by differentiated skeletal muscle rings stimulated electrically at 1 Hz (n = 7, 6, and 5). (b) Passive tension of differentiated skeletal muscle rings (n = 7, 6, and 5). (c) Active tension (solid line) and passive tension (dotted line) plotted with time for a single frozen undifferentiated biobot stimulated at 1 Hz. (d) Active tension of four frozen differentiated biobots recovered measured 24 and 48 h postthaw. (e) RT-qPCR fold change in mRNA expression of various myogenic markers in differentiated muscle, which had previously been frozen while undifferentiated. Expression is normalized to the unfrozen control (mean ± SD, n = 3). (f) RT-qPCR fold change in mRNA expression in muscle frozen differentiated (mean ± SD, n = 3). (*p < 0.05, **p < 0.01). RT-qPCR, quantitative reverse transcription PCR.
Skeletal muscle rings that were frozen after differentiation were found to produce significantly lower active tension compared to unfrozen and frozen differentiated muscle (Supplementary Movie S4). When frozen differentiated muscle rings were allowed to recover for 24 h in culture postthawing, only one out of four frozen muscle rings demonstrated macroscale contraction in response to stimulation (5.3 μN) (Fig. 4d). When these same four muscle rings were allowed to recover in culture for another 24 h (for a total of 48 h postthaw), all muscle rings increased in active tension, by an average of 10 μN (Fig. 4d). However, even after 2 days of recovery, average active tension of the muscle (11.7 ± 8.5 μN) was still significantly lower than unfrozen controls (Fig. 4a). Passive tension, however, was not affected by the freezing process (Fig. 4b). Freezing medium composition was altered in an attempt to maintain higher functional performance of the muscle. FBS was increased from 10% to 90% v/v, and DMSO was increased from 10% to 20% v/v, but no improvement in force production was achieved (Supplementary Fig. S4).
Effect of freezing on myogenic marker expression
To further understand the effects that freezing in the undifferentiated state has on muscle development, and to shed light on the molecular mechanisms, which may be responsible for the observed increase in force production, we used RT-qPCR to analyze the changes in gene expression associated with myoblast freezing. The following myogenic markers were measured: desmin, myostatin, myogenic factor 5, myogenic factor 6, myosin heavy chain 1, MyoD, myogenin, and troponin 1. Differentiated muscle that had been frozen for 24 h in the undifferentiated state showed significantly increased expression of myogenic factor 6 (1.9 ± 0.4-fold) and myosin heavy chain 1 (1.7 ± 0.3-fold), compared to unfrozen controls (Fig. 4e). Myogenic factor 6 (MYF6) was highly expressed in terminally differentiated myotubes or myofibers, and upregulation may indicate that the frozen undifferentiated muscle had achieved a more advanced state of differentiation.26 Myosin heavy chain 1 (MYH1) is also a late-stage myogenic marker, essential for force-generating muscle contraction.27
RT-qPCR was also used to evaluate the changes in myogenic marker expression associated with freezing of differentiated muscle. Muscle rings were frozen for 24 h in GM with 10% DMSO and were recovered in culture for 48 h postthawing. Increased expression of desmin (1.7 ± 0.3-fold), myogenic factor 5 (1.5 ± 0.1-fold), MyoD (2.5 ± 0.7-fold), and myogenin (2.0 ± 0.1-fold) was observed, while MYF6 showed decreased expression (0.5 ± 0.1-fold), compared to the unfrozen control (Fig. 4f). Increased expression of early-stage myogenic markers (MYOD, MYF5, and DES) and mid-stage myogenic marker MYOG, together with decreased expression of terminal differentiation marker MYF6, could indicate a relative increase in the presence of myoblasts compared to mature myotubes.27 Compared to frozen undifferentiated muscle, frozen differentiated muscle exhibits significantly higher expression of MYF5, MYOD, and MYOG and lower expression of MYF6 (Supplementary Fig. S5).
Effect of freezing on myotube structure
To visualize any change to myotube structure caused by the freezing process, skeletal muscle samples were labeled for α-actinin, myosin, and cell nucleus (4′,6-diamidino-2-phenylindole) (Fig. 5a–c). Lightsheet microscopy was used to analyze myotube alignment, thickness, and striation, which all play a critical role in force generation of 3D skeletal muscle tissue.1 Mature muscle tissue that had been frozen while undifferentiated showed a dispersion angle of 10.1 ± 2.8° while the unfrozen control had a dispersion of 9.3 ± 2.1°, indicating no effect of freezing in the undifferentiated state on development of aligned myotubes (Fig. 5d). Frozen undifferentiated muscle also exhibited highly striated, mature myotubes (Fig. 5e). The average myotube width of frozen undifferentiated muscle rings (10.1 ± 0.4 μm) was similar to that of unfrozen tissue (8.6 ± 1.2 μm) (Fig. 5f). Both the unfrozen and frozen undifferentiated tissues contained visibly striated myotubes as indicated by the α-actinin stain, with sarcomere intervals of 1.2 ± 0.02 and 1.2 ± 0.03 μm, respectively (Supplementary Fig. S6).
FIG. 5.

(a–c) Lightsheet images of differentiated skeletal muscle rings stained with a-actinin (red), myosin (green), and 4′,6-diamidino-2-phenylindole (blue). Scale bar = 100 μm. (d) Dispersion angle of myotube directionality shows loss of myotube alignment in frozen differentiated muscle (n = 12) compared to unfrozen muscle (n = 10) and frozen undifferentiated muscle (n = 11). (e) Magnified image of a bundle of myotubes within a frozen undifferentiated muscle ring, showing clearly distinguished striation indicated by a-actinin bands. (f) Average myotube width obtained by measuring the widest diameter of 10 random myotubes within each muscle ring image (mean ± SD, n = 3). (*p < 0.05, **p < 0.01).
Muscle tissue that had been frozen after differentiation, however, had a significantly higher dispersion angle of 15.7 ± 6.2°. Compared to both frozen undifferentiated and unfrozen muscle, the increase in dispersion was significantly higher, indicating a disruption to myotube alignment caused by the process of freezing differentiated muscle (Fig. 5d). This disruption to myotube alignment is consistent with that observed from live/dead staining (Fig. 3d, e). The average myotube width within the frozen tissue was 8.6 ± 0.8 μm, similar to that of the unfrozen tissue, but significantly lower compared with the frozen undifferentiated muscle (Fig. 5f). Frozen differentiated myotubes also showed striation, with an average sarcomere interval of 1.1 ± 0.12 μm, similar to that of the unfrozen and frozen undifferentiated tissues, indicating no decrease in the force generation capability of individual myotubes (Supplementary Fig. S6).28,29
Effect of freezing on ECM microstructure
To further understand the mechanism by which the process of freezing undifferentiated muscle promotes development of mature muscle with increased force production, we investigated the structure of the ECM. We used scanning electron microscopy (SEM) to image the surface structure of both unfrozen muscle and muscle frozen in the undifferentiated state. Unfrozen, undifferentiated samples appeared to have a thick, smooth matrix coating along the surface of the ring (Fig. 6a), while undifferentiated muscle that had been frozen seemed to show an apparent thinning of matrix (Fig. 6b). By the time muscle rings that had been frozen in the undifferentiated state were allowed to differentiate, however, their surfaces seemed to be smoother. There was no distinguishable difference between matrices of the frozen versus unfrozen differentiated muscle rings after differentiation (Fig. 6c, d).
FIG. 6.

Scanning electron microscopy images of muscle ring surface. (a) Unfrozen, undifferentiated muscle ring, showing appearance of thick ECM coating. (b) Frozen, undifferentiated muscle ring, showing apparent thinning of ECM coating compared to the unfrozen, undifferentiated muscle. (c) Unfrozen, differentiated muscle ring. (d) Differentiated muscle ring, which had been previously frozen while undifferentiated, showing ECM coating similar in appearance to that of the unfrozen, differentiated muscle. Scale bar = 50 μm. ECM, extracellular matrix.
E-64 treatment extends biobot lifetime and functionality
Beyond cryopreservation, we sought to examine an in vitro treatment to further extend lifetime and thus functionality of engineered skeletal muscle tissue. Our previous work assessed the effect of ACA, a serine protease inhibitor, on engineered muscle tissue lifetime and structural integrity.24 We extended this study to examine whether a broad-spectrum cysteine protease inhibitor could be more effective in preserving engineered muscle tissue matrix and thus lifetime and functionality. As we previously demonstrated that engineered muscle tissues secrete cathepsin L (CatL), we adapted a multiplex zymography protocol utilizing a fibrinogen substrate (in place of gelatin) to verify that the cathepsins secreted by myotubes within the muscle strips can degrade the fibrin(ogen) matrix in the presence of 1 mg/mL ACA (Fig. 7a). Then, we explored the effects of adding E-64, a broad-spectrum, small molecule cysteine cathepsin inhibitor,30–32 to DM++ beginning on day 3. Proteolytic activity was assayed by zymography on day 12 after treatment with low (5 μM) and high (10 μM) concentrations of E-64. The CatL bound to engineered muscle tissue (75 kDa, CatL + tissue) with E-64 treatment at both low and high concentrations was significantly reduced on day 12 in a dose-dependent manner compared to control samples cultured in DM++ alone (Fig. 7b).
With limited reporting of E-64 concentrations in cell culture media in prior literature,33–35 especially for engineered skeletal muscle, we next tested whether E-64 adversely affected cell health or maturation within the skeletal muscle tissue. On day 12, there was no significant difference in cell viability (measured by MTS assay), passive tension, or the production of MCK (which can serve as a quantitative gauge of myogenesis in mature skeletal muscle36) of tissue with either low or high concentrations of E-64 compared to the control (Fig. 7c, i–iii). Furthermore, long-term analysis demonstrated that the addition of E-64 increased tissue passive tension at days 30 and 90, with a low E-64 dose in DM++ treatment providing the biggest increase in tension force (Fig. 7c, iv). There was no observed significant difference, however, when increasing E-64 from low to high doses.
Finally, we examined whether E-64 could significantly affect lifetime of muscle rings before rupture. Previously, we reported that engineered muscle tissue cultured in DM with 0 or 1 mg/mL ACA resulted in average lifetimes of 8.2 ± 0.5 and 17 ± 3.9 days until rupture, respectively (Fig. 7d, black lines).24 Both in the absence (0 mg/mL) and presence (1 mg/mL) of ACA, E-64 significantly increased the lifetime of these muscle rings from the time of fabrication (day 0). With the addition of 5 μM E-64, average lifetime extended significantly to 79.6 ± 9.4 days in the absence of ACA (p < 0.001) and 138.5 ± 16 days in the presence of ACA (p < 0.01) (Fig. 7d, red lines). No significant difference was observed with 5 μM E-64 with the addition of ACA alone.
Discussion
Cryopreservation serves as an effective method for the long-term storage of living cells for many applications. However, the process of freezing is known to have many detrimental effects on living cells. Osmotic intolerance, toxicity of cryoprotectants, chilling or cold shock, and intracellular ice formation can all contribute to cell death or loss of function as cells undergo the freeze-thaw process.37 These negative effects are especially apparent in freezing of bulk tissues, which possess differing heat and mass transfer effects that prevent even temperature and cryoprotectant distribution. Our results were consistent in showing the process of freezing to have no negative effects on 3D constructs of undifferentiated myoblasts, but much more detrimental effects when freezing occurred after the 3D constructs had been differentiated into a dense skeletal muscle tissue.
The process of cryopreserving differentiated skeletal muscle resulted in a small reduction of cell viability, which varied by batch and was not always statistically significant. The most detrimental effect of freezing on differentiated muscle appeared to be the loss of myotube alignment, which likely contributed to the resulting loss of function as demonstrated by significantly decreased force production.21 Skeletal muscle that had been frozen while differentiated showed increased expression of MYOD, which is known to activate in response to cell damage and can induce cell apoptosis. Desmin is a protein that helps maintain sarcomere alignment and regulate muscle contraction, and its increased expression could indicate a defense mechanism as frozen myotubes attempt to maintain alignment disrupted by the freezing process. Even after efforts to alter freezing medium composition and allow muscle rings to recover for up to 48 h in medium postthawing, functional performance of the muscle could not be recovered after freezing. For this reason, we do not recommend cryopreservation of tissue-engineered skeletal muscle postdifferentiation.
After freezing skeletal muscle while undifferentiated, we were surprised to observe an increase in functional performance of the resulting differentiated skeletal muscle. Active tension was increased, as was expression of myosin heavy chain 1, which is integral in the process of skeletal muscle contraction. Myogenic factor 6 expression was also increased, indicating late-stage differentiation as myotubes begin to form myofibers. Myotube alignment was maintained, and myotube width was significantly higher compared with frozen differentiated muscle. Overall, the process of freezing skeletal muscle before differentiation increased muscle performance, and thus, we recommend the protocol indicated in Figure 2a for optimal freezing of tissue-engineered skeletal muscle.
Using this protocol, tissue-engineered skeletal muscle rings can be efficiently fabricated in large batches and stored for future use, eliminating the time-consuming process of preparing 3D printed molds, expanding C2C12 cell cultures, and seeding the cell–gel solution each time a skeletal muscle tissue is needed for experimentation. This fabrication efficiency could facilitate the integration of bioactuators into laboratory courses to train the next generation of engineers to build systems using biological materials.38 Similarly, skeletal muscle tissue rings could be fabricated by one laboratory, and then frozen and shipped to various locations for further applications. Once received, the frozen muscle ring would simply need to be thawed and cultured in differentiation medium, making this a simple method for laboratories without tissue engineering expertise to integrate skeletal muscle tissues into their research. The ability to ship premade bioactuators would be instrumental for advancement of skeletal muscle-powered robotics, as many robotics-focused research groups may not have the experience or resources required for tissue engineering. Similarly, the efficient fabrication and shipment of large batches of muscle tissues may prove useful in preclinical drug testing applications, or in future clinical studies in which tissue-engineered skeletal muscle is grafted in vivo for the repair of functional muscle.39
The goal of this study was to optimize a protocol for cryopreservation of tissue-engineered skeletal muscle and characterize the effects of freezing on the muscle. However, the surprising discovery that freezing of undifferentiated muscle tissue leads to enhanced functional performance of differentiated muscle may prove to be equally impactful. Currently, tissue engineering efforts have not been able to attain the morphology and level of differentiation found in native skeletal muscle, resulting in low force generation and little clinical utility.2 The mechanisms by which this freezing-induced enhancement of force generation occurs may lead to breakthroughs in the engineering of more highly differentiated and clinically relevant skeletal muscle. However, further studies are necessary to determine and control the exact mechanisms by which freezing-induced changes occur.
It is known that tissue engineering scaffolds can be tuned in many ways to optimize growth and development of seeded cells. Pore size of scaffold material, for instance, is known to play a critical role in cell attachment and outgrowth, oxygen transfer, and perfusion of nutrients and waste.40 In the case of myoblast differentiation, it has been shown that matrices with elongated pores induce the formation of more aligned myotubes capable of higher net force production, while also increasing the efficiency of myotube formation.41 We believe that ice crystal formation associated with the freezing process may have caused changes to the microstructure of the tissue's ECM. The process of freezing ECM has been previously demonstrated to increase collagen network pore size, in addition to increasing fibril diameter.42 An increase in porosity of ECM of frozen undifferentiated muscle may have enabled more efficient cell migration and fusion, leading to the formation of more mature myotubes capable of higher force production.
The SEM images in Figure 5 indicate a change to the ECM structure caused by freezing of undifferentiated muscle tissue. The observation of a thinned ECM in the frozen undifferentiated tissue, which appears to recover in thickness by the time the tissue has differentiated, may suggest that frozen undifferentiated muscle was able to secrete its own matrix. This increase in ECM secretion may have been stimulated by the freezing-induced increase in pore size, which has previously been shown to stimulate the increased ECM secretion of chondrocytes.43 Furthermore, ECM composition is known to affect skeletal muscle force generation, and the naturally secreted matrix was likely optimal for development of stronger muscle tissues.17 However, further studies are needed to confirm this hypothesis and analyze the effects of pore size and ECM fibril thickness on muscle differentiation. Similarly, imaging of the microstructure throughout the 3D tissue and analysis of ECM composition differences in frozen and unfrozen muscle will be necessary to examine the effects of freezing on ECM.
Fibrin-based and fibrinogen-based engineered tissues are playing an increasingly prominent role in biohybrid living systems,15,21,44,45 with fibrin-based constructs experiencing further exposure to nontraditional proteolytic environments. Our previous characterization of engineered skeletal muscle tissues demonstrated the activity of proteolytic enzymes (such as myotube-secreted catL) that can degrade the fibrin-based ECM and thus lead to diminished mechanical integrity and functionality over time.24 Along with recently described modeling,46 we underscore catL as an alternative fibrin(ogen)lytic protease. In this study, we show preliminary evidence that secreted cathepsin had fibrinogenolytic activity (Fig. 7a) as detected by a fibrinogen zymography protocol adapted from the well-established multiplex cathepsin zymography. Furthermore, culturing the engineered muscle tissues with E-64—a low-cost, potent, and irreversible broad-spectrum cathepsin inhibitor—resulted in a significant and dose-dependent reduction in the amount of active CatL bound to muscle strip matrix proteins (Fig. 7b). Furthermore, E-64 significantly increased the average lifespan—with some tissues still morphologically intact as of day 200+ (Fig. 7d)—thus demonstrating a more potent effect on long-term muscle tissue culture than ACA alone. These results further support previous evidence24 that CatL proteolysis destabilizes engineered muscle tissues. In summary, inhibiting cysteine cathepsin proteolytic activity can lengthen life expectancy of biological machines without a concomitant negative effect on cell health or functionality in muscle tissues.
Conclusion
In this study, we optimized a protocol for the cryopreservation of tissue-engineered skeletal muscle constructs before differentiation. This protocol not only maintains cell viability, myotube structure, and alignment but also leads to increased force generation, as well as the increased expression of late-stage myogenic markers. Further studies are needed to determine the mechanisms by which the freezing of undifferentiated muscle tissue leads to the differentiation of stronger skeletal muscle. Specifically, studies that examine the effects of freezing on ECM and the role of these ECM changes in myogenesis may advance the engineering of stronger, more efficient skeletal muscle. We also demonstrated, in this study, that catL produced by these muscle strips has fibrinogenolytic activity, which further supported the catL-mediated destabilization of muscle strip integrity. The ability to freeze, ship, and control both the lifetime and functional contractility of engineered skeletal muscle will help drive advances in clinical and robotic applications of these tissues.
Supplementary Material
Acknowledgments
This work was funded by National Science Foundation (NSF) Science and Technology Center Emergent Behavior of Integrated Cellular Systems (EBICS) Grant CBET0939511. L.G. was funded by NSF Research Traineeship in Understanding the Brain (NRT-UtB): Training the Next Generation of Researchers in Engineering and Deciphering of Miniature Brain Machinery, Grant 1735252.
Disclosure Statement
No competing financial interests exist.
References
- 1. Ostrovidov S., Hosseini V., Ahadian S., et al. Skeletal muscle tissue engineering: methods to form skeletal myotubes and their applications. Tissue Eng Part B Rev 20, 403, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bian W., and Bursac N. Tissue engineering of functional skeletal muscle: challenges and recent advances. IEEE Eng Med Biol Mag 27, 109, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Uzel S.G.M., Pavesi A., and Kamm R.D. Microfabrication and microfluidics for muscle tissue models. Prog Biophys Mol Biol 115, 279, 2014 [DOI] [PubMed] [Google Scholar]
- 4. Smith A.S.T., Davis J., Lee G., Mack D.L., and Kim D.H. Muscular dystrophy in a dish: engineered human skeletal muscle mimetics for disease modeling and drug discovery. Drug Discov Today 21, 1387, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Raman R., Grant L., Seo Y., et al. Damage, healing, and remodeling in optogenetic skeletal muscle bioactuators. Adv Healthc Mater 6, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Uzel S.G.M., Platt R.J., Subramanian V., et al. Microfluidic device for the formation of optically excitable, three-dimensional, compartmentalized motor units. Sci Adv 2, e1501429, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cvetkovic C., Rich M.H., Raman R., Kong H., and Bashir R. A 3D-printed platform for modular neuromuscular motor units. Microsyst Nanoeng 3, 17015, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Duffy R.M., and Feinberg A.W. Engineered skeletal muscle tissue for soft robotics: fabrication strategies, current applications, and future challenges. Wiley Interdiscip Rev Nanomater Nanobiotechnol 6, 178, 2014 [DOI] [PubMed] [Google Scholar]
- 9. Ricotti L., Trimmer B., Feinberg A.W., et al. Biohybrid actuators for robotics: a review of devices actuated by living cells. Sci Robot 2, eaaq0495, 2017 [DOI] [PubMed] [Google Scholar]
- 10. Raman R., and Bashir R. Biomimicry, biofabrication, and biohybrid systems: the emergence and evolution of biological design. Adv Healthc Mater 6, 2017 [DOI] [PubMed] [Google Scholar]
- 11. Cvetkovic C., Raman R., Chan V., et al. Three-dimensionally printed biological machines powered by skeletal muscle. Proc Natl Acad Sci U S A 111, 10125, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mazur P. Freezing of living cells: mechanisms and implications. Am J Physiol 247, C125, 1984 [DOI] [PubMed] [Google Scholar]
- 13. Day A.G.E., Bhangra K.S., Murray-Dunning C., Stevanato L., and Phillips J.B. The effect of hypothermic and cryogenic preservation on engineered neural tissue. Tissue Eng Part C Methods 23, 575, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Best B.P. Cryoprotectant toxicity: facts, issues, and questions. Rejuvenation Res 18, 422, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Raman R., Cvetkovic C., and Bashir R. A modular approach to the design, fabrication, and characterization of muscle-powered biological machines. Nat Protoc 12, 519, 2017 [DOI] [PubMed] [Google Scholar]
- 16. Raman R., Cvetkovic C., Uzel S.G.M., et al. Optogenetic skeletal muscle-powered adaptive biological machines. Proc Natl Acad Sci U S A 113, 3497, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hinds S., Bian W., Dennis R.G., and Bursac N. The role of extracellular matrix composition in structure and function of bioengineered skeletal muscle. Biomaterials 32, 3575, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dennis R.G., Kosnik P.E., Gilbert M.E., and Faulkner J.A. Excitability and contractility of skeletal muscle engineered from primary cultures and cell lines. Am J Physiol Cell Physiol 280, C288, 2001 [DOI] [PubMed] [Google Scholar]
- 19. Sakar M.S., Neal D., Boudou T., et al. Formation and optogenetic control of engineered 3D skeletal muscle bioactuators. Lab Chip 12, 4976, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Livak K.J., and Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods 25, 402, 2001 [DOI] [PubMed] [Google Scholar]
- 21. Pagan-Diaz G.J., Zhang X., Grant L., et al. Simulation and fabrication of stronger, larger, and faster walking biohybrid machines. Adv Funct Mater 23, 28, 2018 [Google Scholar]
- 22. Li W.A., Barry Z.T., Cohen J.D., et al. Detection of femtomole quantities of mature cathepsin K with zymography. Anal Biochem 401, 91, 2010 [DOI] [PubMed] [Google Scholar]
- 23. Wilder C.L., Park K.Y., Keegan P.M., and Platt M.O. Manipulating substrate and pH in zymography protocols selectively distinguishes cathepsins K, L, S, and v activity in cells and tissues. Arch Biochem Biophys 516, 52, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cvetkovic C., Ferrall-Fairbanks M.C., Ko E., et al. Investigating the life expectancy and proteolytic degradation of engineered skeletal muscle biological machines. Sci Rep 7, 3775, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McGann L.E., and Walterson M.L. Cryoprotection by dimethyl sulfoxide and dimethyl sulfone. Cryobiology 24, 11, 1987 [DOI] [PubMed] [Google Scholar]
- 26. Zammit P.S. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin Cell Dev Biol 72, 19, 2017 [DOI] [PubMed] [Google Scholar]
- 27. Dominov J.A., Dunn J.J., and Miller J.B. Bcl-2 expression identifies an early stage of myogenesis and promotes clonal expansion of muscle cells. J Cell Biol 142, 537, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. ter Keurs H.E., Iwazumi T., and Pollack G.H. The sarcomere length-tension relation in skeletal muscle. J Gen Physiol 72, 565, 1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lemke S.B., and Schnorrer F. Mechanical forces during muscle development. Mech Dev 144, 92, 2017 [DOI] [PubMed] [Google Scholar]
- 30. Hanada K., Tamai M., Yamagishi M., Ohmura S., Sawada J., and Tanaka I. Isolation and characterization of e-64, a new thiol protease inhibitor. Agric Biol Chem 42, 523, 1978 [Google Scholar]
- 31. Matsumoto K., Mizoue K., Kitamura K., Tse W.C., Huber C.P., and Ishida T. Structural basis of inhibition of cysteine proteases by E-64 and its derivatives. Biopolymers 51, 99, 1999 [DOI] [PubMed] [Google Scholar]
- 32. Hanada K., Tamai M., Ohmura S., Sawada J., Seki T., and Tanaka I. Structure and synthesis of E–64, a new thiol protease inhibitor. Agric Biol Chem 42, 529, 1978 [Google Scholar]
- 33. Kherif S., Lafuma C., Dehaupas M., et al. Expression of matrix metalloproteinases 2 and 9 in regenerating skeletal muscle: a study in experimentally injured and mdx muscles. Dev Biol 205, 158, 1999 [DOI] [PubMed] [Google Scholar]
- 34. Wilder C.L., Walton C., Watson V., et al. Differential cathepsin responses to inhibitor-induced feedback: E-64 and cystatin C elevate active cathepsin S and suppress active cathepsin L in breast cancer cells. Int J Biochem Cell Biol 79, 198, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Keegan P.M., Wilder C.L., and Platt M.O. Tumor necrosis factor alpha stimulates cathepsin K and V activity via juxtacrine monocyte-endothelial cell signaling and JNK activation. Mol Cell Biochem 367, 65, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chamberlain J.S., Jaynes J.B., and Hauschka S.D. Regulation of creatine kinase induction in differentiating mouse myoblasts. Mol Cell Biol 5, 484, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Woods E.J., Thirumala S., Badhe-Buchanan S.S., Clarke D., and Mathew A.J. Off the shelf cellular therapeutics: factors to consider during cryopreservation and storage of human cells for clinical use. Cytotherapy 18, 697, 2016 [DOI] [PubMed] [Google Scholar]
- 38. Raman R., Mitchell M., Perez-Pinera P., Bashir R., and DeStefano L. Design and integration of a problem-based biofabrication course into an undergraduate biomedical engineering curriculum. J Biol Eng 10, 10, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Juhas M., Ye J., and Bursac N. Design, evaluation, and application of engineered skeletal muscle. Methods 99, 81, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Loh Q.L., and Choong C. Three-dimensional scaffolds for tissue engineering applications: role of porosity and pore size. Tissue Eng Part B Rev 19, 485, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bian W., Juhas M., Pfeiler T.W., and Bursac N. Local tissue geometry determines contractile force generation of engineered muscle networks. Tissue Eng Part A 18, 957, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Han B., Miller J.D., and Jung J.K. Freezing-induced fluid-matrix interaction in poroelastic material. J Biomech Eng 131, 021002, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lien S.M., Ko L.Y., and Huang T.J. Effect of pore size on ECM secretion and cell growth in gelatin scaffold for articular cartilage tissue engineering. Acta Biomater 5, 670, 2009 [DOI] [PubMed] [Google Scholar]
- 44. Kamm R.D., and Bashir R. Creating living cellular machines. Ann Biomed Eng 42, 445, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Janmey P.A., Winer J.P., and Weisel J.W. Fibrin gels and their clinical and bioengineering applications. J R Soc Interface 6, 1, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ferrall-Fairbanks M.C., West D.M., Douglas S.A., Averett R.D., and Platt M.O. Computational predictions of cysteine cathepsin-mediated fibrinogen proteolysis. Protein Sci 27, 714, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.