

Abstract
Aims
To investigate the accuracy of phenotypic early‐onset ataxia (EOA) recognition among developmental conditions, including developmental coordination disorder (DCD) and hypotonia of central nervous system origin, and the effect of scientifically validated EOA features on changing phenotypic consensus.
Method
We included 32 children (4–17y) diagnosed with EOA (n=11), DCD (n=10), and central hypotonia (n=11). Three paediatric neurologists independently assessed videotaped motor behaviour phenotypically and quantitatively (using the Scale for Assessment and Rating of Ataxia [SARA]). We determined: (1) phenotypic interobserver agreement and phenotypic homogeneity (percentage of phenotypes with full consensus by all three observers according to the underlying diagnosis); (2) SARA (sub)score profiles; and (3) the effect of three scientifically validated EOA features on phenotypic consensus.
Results
Phenotypic homogeneity occurred in 8 out of 11, 2 out of 10, and 1 out of 11 patients with EOA, DCD, and central hypotonia respectively. Homogeneous phenotypic discrimination of EOA from DCD and central hypotonia occurred in 16 out of 21 and 22 out of 22 patients respectively. Inhomogeneously discriminated EOA and DCD phenotypes (5 out of 21) revealed overlapping SARA scores with different SARA subscore profiles. After phenotypic reassessment with scientifically validated EOA features, phenotypic homogeneity changed from 16 to 18 patients.
Interpretation
In contrast to complete distinction between EOA and central hypotonia, the paediatric motor phenotype did not reliably distinguish between EOA and DCD. Reassessment with scientifically validated EOA features could contribute to a higher phenotypic consensus.
Early‐onset ataxia (EOA) and central hypotonia motor phenotypes were reliably distinguished.
EOA and developmental coordination disorder (DCD) motor phenotypes were not reliably distinguished.
The EOA and DCD phenotypes have different profiles of the Scale for Assessment and Rating of Ataxia.
Short abstract
Early‐onset ataxia (EOA) and central hypotonia motor phenotypes were reliably distinguished.
EOA and developmental coordination disorder (DCD) motor phenotypes were not reliably distinguished.
The EOA and DCD phenotypes have different profiles of the Scale for Assessment and Rating of Ataxia.
This article's abstract has been translated into Spanish and Portuguese.
Follow the links from the http://onlinelibrary.wiley.com/doi/10.1111/dmcn.14355/abstract to view the translations.
This article is commented on by Baxter on page https://doi.org/10.1111/dmcn.14376 of this issue.
Resumen
Fenotipos pediátricos motores en ataxia de inicio temprano, trastorno del desarrollo de la coordinación e hipotonía de origen central
Objetivos
Investigar la precisión del reconocimiento fenotípico de ataxia de inicio temprano (EOA) con respecto a trastornos del desarrollo, incluido el trastorno del desarrollo de la coordinación (TDC) y la hipotonía de origen central. Investigar el efecto de las características científicamente validadas de EOA sobre el consenso fenotípico entre los evaluadores.
Método
Se incluyeron 32 niños (4‐17 años) diagnosticados con EOA (n = 11), TDC (n = 10) e hipotonía central (n = 11). Tres neurólogos pediátricos evaluaron de forma independiente el comportamiento motor grabado en video en cuanto a las características fenotípica y cuantitativa (utilizando la Escala de evaluación y calificación de la ataxia [SARA]). Determinamos: (1) coincidencia fenotípica entre los observadores y homogeneidad fenotípica (porcentaje de fenotipos con consenso total de los tres observadores según el diagnóstico subyacente), (2) perfiles de (sub)puntajes en el SARA y (3) el efecto sobre el consenso fenotípico de tres características de EOA validadas científicamente.
Resultados
La homogeneidad fenotípica ocurrió en 8 de 11, 2 de 10 y 1 de 11 pacientes con EOA, DCD e hipotonía central, respectivamente. La discriminación fenotípica homogénea de EOA con respecto a TDC e hipotonía central se produjo en 16 de 21 y 22 de 22 pacientes, respectivamente. Los fenotipos EOA y TDC que no fueron discriminados de manera homogénea por los observadores (5 de 21) revelaron superposición en los puntajes del SARA con diferentes perfiles en los subpuntajes del SARA. Después de una reevaluación fenotípica con características EOA científicamente validadas, la homogeneidad fenotípica cambió de 16 a 18 pacientes.
Interpretación
En contraste con la distinción completa entre EOA e hipotonía central, el fenotipo motor pediátrico no distinguió confiablemente entre EOA y TDC. La evaluación en base a características EOA científicamente validadas podría contribuir a un mayor consenso fenotípico.
Resumo
Fenótipos motores pediátricos na ataxia de início precoce, transtorno do desenvolvimento da coordenacão, e hipotonia central
Objetivos
Investigar a acurácia do reconhecimento fenotípico da ataxia de início precoce (AIP) entre condições desenvolvimentais, incluindo o transtorno do desenvolvimento da coordenação (TDC) e a hipotonia de origem no sistema nervoso central, e o efeito de aspectos cientificamente validados da AIP na modificação do consenso fenotípico.
Método
Incluímos 32 crianças (4–17a) diagnosticadas com AIP (n=11), TDC (n=10), e hipotonia central (n=11). Três neurologistas pediátricos avaliaram de maneira independente por meio de vídeo o comportamento motor tanto por meio do fenótiopo quanto quantitativamente (usando a Escala para Avaliação e Pontuação da Ataxia) [EAPA]). Determinamos: (1) a concordânica fenotípica inter‐observadores e a homogeneidade fenotípica (porcentagem de fenótipos com consenso completo pelos três observadores de acordo com o diagnóstico de base, (2) perfis segundo os (sub)escores da EAPA, e (3) o efeito de três aspectos cientificamente validados da AIP sobre o consenso fenotípico.
Resultados
A homogeneidade fenotípica ocorreu em 8 entre 12, 2 entre 10, e 1 entre 11 pacientes com AIP, TDC, e hipotonia central, respectivamente. A discriminação fenotípica homogênea da AIP com relação ao TDC e hipotonia central ocorreu em 16 entre 21 e 21 entre 22 pacientes, respectivamente. A discriminação não homogêna dos fenótipos AIP e TDC (5 em 21) revelou escores da EAPA que sobrepõem com diferentes perfis de subescores da EAPA. Após reavaliação fenotípica com aspectos cientificamente validados da AIP, a homogeneidade fenotípica mudou de 16 para 18 pacientes.
Interpretação
Em contraste com a completa distinção entre AIP e hipotonia central, o fenótipo motor pediátrico não distinguiu confiavelmente entre AIP e TDC. A reavaliação com aspectos cientificamente valiaddos da AIP pode contribuir para um maior consenso fenotípica. contrast to complete distinction between EOA and central hypotonia, the paediatric motor phenotype did not reliably distinguish between EOA and DCD. eassessment with scientifically validated EOA features could contribute to a higher phenotypic consensus.
Abbreviations
- DCD
Developmental coordination disorder
- EOA
Early‐onset ataxia
- PBS
Paediatric Balance Scale
- SARA
Scale for Assessment and Rating of Ataxia
Reliable phenotypic recognition of early‐onset ataxia (EOA) among other developmental disorders with coordination impairment, such as developmental coordination disorder (DCD)1, 2 and hypotonia of central nervous system origin,3, 4 is important for selecting the correct diagnostic algorithm, predicting familial recurrence risk, and treating the child. However, in young children with motor coordination difficulties, clinical recognition of mild EOA features may be challenging.
Ataxia is literally translated from the Greek as ‘without order’, denoting impaired coordinative neurological output, which is mostly associated with disturbed functioning of the corticobasal ganglia, cerebellar network, and/or gnostic sensory afferents.5 Ataxia can be phenotypically recognized by a lack of smoothly performed goal‐directed movements, resulting in dysdiadochokinesia, dysmetria, overshoot, impaired gait and posture, intention tremor, oculomotor dysfunction, and speech abnormalities. EOA refers to a group of rare heterogeneous disorders, mostly of recessively heritable origins, associated with the presentation of ataxic features before the age of 25 years.6 Individual patients with EOA may reveal heterogeneous ‘ataxic’ motor phenotypes,7 presenting ataxia as: (1) a solitary main symptom; (2) a main symptom in association with other comorbid movement disorder features; (3) a comorbid symptom, dominated by other prevailing movement disorder features, such as dystonia, chorea, or myoclonus; and (4) a stable and hardly discernible feature.
According to DSM‐5 criteria,8 DCD is a developmental motor disorder characterized by non‐progressive motor incoordination, which interferes with daily activities or academic achievement, that is not attributable to a neurological, intellectual, or visual condition.1 The descriptive diagnosis of the motor dysfunction is ‘motor dyspraxia’. When the latter is due to DCD, it can be associated with other disorders such as attention‐deficit/hyperactivity disorder,2 and also with mildly ataxic,9 dystonic, or choreatic10 features that are not excluded from the diagnosis.11 Physiologically delayed motor coordination in very young children is excluded from the DCD diagnosis, since it reflects typical brain development.12, 13 The underlying aetiology of DCD is still unclear, but dysfunctional activation within the cerebellar, basal ganglia, and corticospinal networks has been suggested.11, 14, 15
Central hypotonia is characterized by a subgroup of children with negative motor signs due to lack of muscle tone and/or joint hyperlaxity.3, 4 In general, lack of muscle tone can be associated with kinetic inaccuracy (sloppiness) and balance problems, which may mimic cerebellar dysfunction.16, 17
In the absence of criterion standards18 and in the context of ambiguous clinical descriptions, an insight into the reliability of phenotypic differentiation between EOA and other disorders with impaired coordination (DCD and central hypotonia) is important. In the present study, we aimed to: (1) investigate interobserver agreement on phenotypic assessment of EOA, DCD, and central hypotonia; (2) compare Scale for Assessment and Rating of Ataxia (SARA)19 and Paediatric Balance Scale (PBS)20 scores between phenotypic assignments; and (3) explore the effect of three scientifically validated, standardized EOA features on phenotypic consensus.
Method
The present study was observational in nature. The Medical Ethics Committee of the University Medical Center Groningen approved the study (approval no. METc 2015/01053). According to Dutch medical ethical law, all parents provided informed written consent; children older than 12 years provided informed written consent and children younger than 12 years provided informed assent.
Patients
Between 2013 and 2016, we enrolled 32 children clinically diagnosed with EOA (n=11; mean age=11y, range=6–17y), DCD (n=10; mean age=9y, range=6–13y), and central hypotonia (n=11; mean age=9y, range=5–14y); see Table 1 for a fuller breakdown of patient characteristics.
Table 1.
Patient characteristics
|
Total n=32 |
EOA n=11 |
DCD n=10 |
Central hypotonia n=11 |
p | |
|---|---|---|---|---|---|
| Age, y:moa | |||||
| Mean (SD) | 9:10 (3:3) | 11:4 (3:6) | 8:8 (2:4) | 9:4 (3:6) | 0.170 |
| Range | 4:0–17:0 | 6:0–17:0 | 6:0–13:0 | 4:0–14:0 | |
| Disease onset, y:mo | |||||
| Median (IQR) | 1:6 (0:6–3:9) | 2:0 (0:6–4:0) | 1:6 (0:6–3:3) | 1:6 (0:6–3:0) | 0.914 |
| Range | 0:0–8:0 | 0:0–8:0 | 0:0–5:0 | 0:0–5:0 | |
| Disease duration, y:moa | |||||
| Mean (SD) | 7:11 (3:1) | 8:11 (3:11) | 7:4 (2:5) | 7:7 (2:11) | 0.449 |
| Range | 2:6–15:0 | 3:0–15:0 | 4:0–11:0 | 2:6–12:6 | |
| Phenotypic severityb | |||||
| Median (IQR) | 1 (1–2) | 2 (1–2) | 1 (1–2) | 1 (0–1) | 0.001c |
| Range | 0–3 | 1–3 | 1–2 | 0–1 | |
| SARATOTAL | |||||
| Median (IQR) | 3.0 (0.0–8.0) | 9.5 (8.0–14.0) | 2.8 (0.4–3.6) | 0.0 (0.0–2.3)d | <0.001c |
| Range | 0.0–15.5d | 4.5–15.5 | 0.0–8.0d | 0.0–3.0d | |
| SARAGAIT | |||||
| Median (IQR) | 2.0 (0.0–3.9) | 5.5 (3.5–7) | 1.3 (0.0–2.5)d | 0.0 (0.0–0.5)d | <0.001c |
| Range | 0.0–8.0d | 2.0–8.0 | 0.0–2.5d | 0.0–2.0d | |
| SARAFINGER‐NOSE/FINGER‐CHASE | |||||
| Median (IQR) | 0.75 (0.0–2.0) | 2.0 (1.0–2.5) | 0.3 (0.0–1.0) | 0.0 (0.0–0.5) | <0.001c |
| Range | 0.0–3.5 | 0.5–3.5 | 0.0–2.0 | 0.0–1.25 | |
| SARAKINETIC | |||||
| Median (IQR) | 1.1 (0.0–3.4) | 4.5 (2.5–6.0) | 0.8 (0.0–2.1)d | 0.0 (0.0–1.0)d | <0.001c |
| Range | 0.0–6.5d | 0.5–6.5 | 0.0–4.5d | 0.0–2.0d | |
| SARASPEECH | |||||
| Median (IQR) | 0.0 (0.0–2.0) | 2.0 (2.0–3.0) | 0.0 (0.0–0.0) | 0.0 (0.0–0.0) | <0.001c |
| Range | 0.0–3.5 | 0.0–3.5 | 0.0–1.0 | 0.0–0.0 | |
| PBS | |||||
| Median (IQR) | 54.1 (50.5–55.9) | 48.9 (43.3–53.5) | 54.3 (53.0–55.4) | 56.0 (55.9–56.0)e | 0.001c |
| Range | 23.9–56.0e | 23.9–56.0e | 44.0–55.9 | 51.3–56.0e | |
Normal distribution.
Phenotypic severity of primary feature (0=normal, 1=mild, 2=moderate, 3=severe).
Significantly different. Although most p‐values reached the level of significance, early‐onset ataxia (EOA) and developmental coordination disorder revealed a quantitative and phenotypic overlap. From this perspective, one cannot clinically use absolute Scale for Assessment and Rating of Ataxia (SARA) scores for group assignment.
Negative age‐corrected SARA scores are expressed as 0 (i.e. optimal value).
Age‐corrected Paediatric Balance Scale (PBS) scores >56 are expressed as 56 (i.e. optimum values). IQR, interquartile range.
EOA
Before inclusion, all patients with EOA were clinically identified at the paediatric neurology outpatient clinic at the University Medical Center Groningen. All 11 patients fulfilled the ‘classical’ definition of EOA.6 In 9 out of 11 patients, EOA was genetically confirmed by the underlying diagnosis including: Friedreich’s ataxia (n=2); Poretti–Bolthauser syndrome (n=1); 2‐methyl‐3‐hydroxybutyryl‐CoA dehydrogenase deficiency (n=1); Niemann–Pick disease type C (n=1); Joubert syndrome‐23 (n=1); spinocerebellar ataxia type 5 (n=1); spinocerebellar ataxia type 13 (n=1); EBF3 gene mutation (hypotonia, ataxia, and delayed development syndrome [n=1]); and unknown (n=2). The ataxia phenotype of the two genetically undiagnosed children with EOA was confirmed by the multidisciplinary movement disorder team. Both children had cerebellar hypoplasia and/or atrophy on magnetic resonance imaging. In one patient, next‐generation sequencing revealed a de novo variant of unknown significance (PPP1R3F gene mutation); in the other patient, we did not record a potential genetic association. For further clinical information, see Appendix S1 (online supporting information).
DCD
All included DCD phenotypes fulfilled the official motor criteria for DCD.2, 8 Before inclusion, all patients with DCD had received an independent neurological examination at the outpatient clinic, including radiological, metabolic, and/or genetic workup. After exclusion of an underlying neurological cause, patients were referred to the rehabilitation centre. Independent rehabilitation specialists diagnosed the DCD motor criteria according to DSM‐5 criteria, including Movement Assessment Battery for Children scores under the 15th centile. DCD comorbidity concerned impaired concentration, attention‐deficit/hyperactivity disorder or mild learning problems (6 out of 10), social and emotional problems (5 out of 10), and pervasive developmental disorder not otherwise specified (1 out of 10). After the study was finalized, one patient with DCD was identified by next‐generation sequencing as having a KLF7 gene mutation. For further patient information, see Appendix S1.
Central hypotonia
All children from the central and/or mixed peripheral hypotonia3, 4 group were clinically described with isolated features of hypotonia and hyperlaxity (in the absence of comorbid movement disorder features).3, 4 Neurological disorders were excluded at the outpatient clinic, before enrolment in the study group. However, after the study was completed, one child supposedly diagnosed with ‘central’ hypotonia was eventually diagnosed with limb‐girdle muscular dystrophy type 2I. Post hoc analysis revealed that retrospective exclusion of this child from the central hypotonia study group would not have changed the study outcomes.
Exclusion criteria
The three study groups were clinically investigated for exclusion criteria concerning underlying acquired disorders, such as infection, trauma, tumour, intoxication, inflammation, ischaemia, haemorrhage, and/or para‐infectious causes. Magnetic resonance imaging scans were available for all 11 children with EOA, 8 out of 10 children with DCD, and 5 out of 11 children with central hypotonia. We also excluded children with cognitive and/or behavioural problems that could potentially interfere with the performance of the SARA and PBS motor tasks.
Assessments
For the procedures followed in the study, see Figure S1 (online supporting information).
Phenotypic assessment
Phenotypic assessments were performed according to previously published studies, using standardized videotaped SARA subscore tasks and guidelines.19 Three independent, not clinically informed, paediatric neurologists carried out the phenotypic assessments using standardized instructions and assessment forms (Fig. S2). In addition to the phenotype, assessors indicated the SARA motor domains (gait/posture, speech, kinetics) where they perceived the indicated phenotype, and the subjectively perceived severity of the coordination impairment (mild, moderate, or severe). Six of 32 children included in the study were patients of one of the assessors. In these children, we checked whether the initially indicated phenotype (as noted in the outpatient records) concurred with the presently indicated phenotype by the same neurologist; this appeared to be the case in all six.
We calculated the phenotypic interobserver agreement and stratified the outcomes for EOA, DCD, and central hypotonia. We characterized phenotypic assessment as ‘homogeneous’ when all three observers unanimously indicated the same phenotype as the clinical diagnosis.
Quantitative assessment
After a time interval of more than 6 weeks, the three assessors quantified the videotaped SARA performances according to the guidelines.19 Assessors were not allowed to review, compare, or discuss the preceding phenotypic assessments. In each patient, we determined the median score (obtained from the three assessors) for the SARATOTAL, SARAGAIT/POSTURE, SARAKINETIC, and SARAFINGER‐NOSE/FINGER‐CHASE (sub)scores and the relative SARA subscore percentage, that is, the contributions of SARAGAIT/POSTURE and SARAKINETIC to SARATOTAL (SARAGAIT/POSTURE/SARATOTAL×100% and SARAKINETIC/SARATOTAL×100% respectively). The PBS20 scores were provided by another independent investigator, blinded to the phenotypic assignments and SARA scores by the other assessors. In children, the reliability of this method is very high (intraclass correlation coefficient=0.997).20 We accounted for age‐related influences on the SARA and PBS scores21 by using age‐corrected SARA12 and PBS22 values. For rating scale information, an explanation of the age corrections, and age‐corrected SARA score values see Appendix S2 and Table S1 (online supporting information).
Phenotypic reassessment
After a time interval of 6 months, we redetermined the phenotypic homogeneity between incompletely differentiated patients with EOA and DCD. For this purpose, the same assessors were provided with three scientifically validated features for EOA recognition. These features were derived from previous phenotypic18 and quantitative ‘machine learning’ data using inertial measurement units.23, 24 The features used to identify EOA were: (1) marked irregularity of finger‐to‐nose movements, both between and within the kinetic trajectory of the finger‐to‐nose movement;24, 25, 26 (2) bow‐shaped trajectories in kinetic movement performances (in any spatial plane);24, 25, 26 and (3) ataxia features in more than one SARA domain, implicating more generalized cerebellar dysfunction.18 Before rescoring, assessors were not allowed to review, compare, or discuss previous assessments. Assessors did not receive clinical information and they were not informed about any interobserver agreement on their previous scores.
Statistical analysis
We determined the normality of age, disease onset, and disease duration, as well as severity of the primary phenotypic appearance, SARATOTAL, SARAGAIT/POSTURE, SARAKINETIC, SARAFINGER‐NOSE/FINGER‐CHASE, and PBS scores both graphically and using the Shapiro–Wilk test. In case of normality, the mean and standard deviation were reported for the entire group, as well as each of the separate groups. Otherwise, the median and interquartile ranges were reported. The range was reported as minimum and maximum values. We used Gwet’s agreement coefficient to determine interobserver agreement.27 The results from Gwett’s agreement coefficient were interpreted using the criteria set out by Landis and Koch (<0.20 slight; 0.21–0.40 fair; 0.41–0.60 moderate; 0.61–0.80 substantial; >0.81 almost perfect).28 We tested the effect of standardized EOA features on the homogeneity of incompletely separated phenotypes using McNemar’s test. A Kruskal–Wallis test (one‐way analysis of variance in the case of negative age‐corrected SARA scores) was used to test quantitative discernibility between the EOA, DCD, and central hypotonia groups. For the post hoc Mann–Whitney U test, see the ‘Results’ (post hoc analyses were Bonferroni‐corrected for multiple comparisons, resulting in a significance level of α≤0.002). All statistical tests were two‐sided. The significance level was set at α=0.05. Statistical analysis was performed using SPSS Statistics v22.0 (IBM Corp., Armonk, NY, USA).
Results
The three assessors phenotypically recognized the diagnostic groups as 90% (80–100%) for EOA, 70% (40–90%) for DCD, and 40% (10–50%) for central hypotonia. In 13 out of 32 children, at least one assessor had scored an alternative primary phenotype instead of the clinical phenotype EOA, DCD, or central hypotonia, including: normal and/or delayed (DCD, n=3; central hypotonia, n=8 by one to three assessors); upper motor neuron dysfunction/spasticity (central hypotonia, n=1 by one assessor); dystonia (DCD, n=1 by one assessor); and chorea (EOA, n=1 by one assessor) (Fig. 1; for the phenotypic assessment form see Fig. S2). Individual phenotypic assessments are shown in Table S2.
Figure 1.
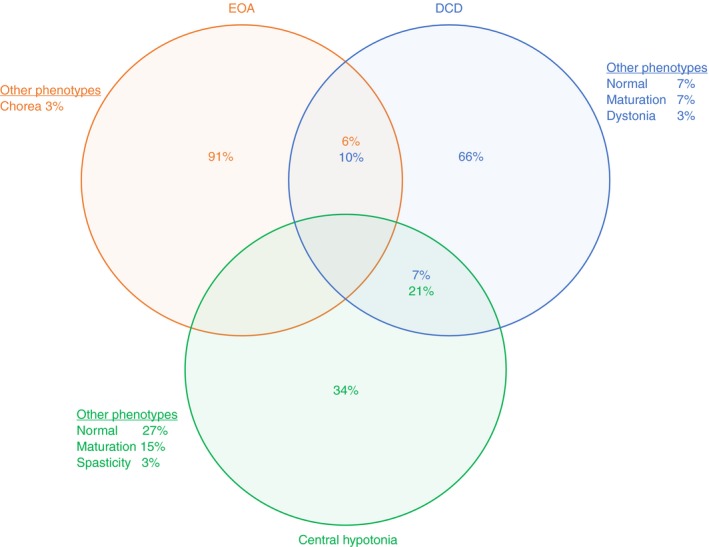
Phenotypic outcomes of children with early‐onset ataxia (EOA), developmental coordination disorder (DCD), and central hypotonia. The circles (EOA, orange; DCD, blue; central hypotonia, green) indicate the underlying diagnosis. The percentages indicate the perceived primary feature of the motor behaviour by the three observers.
Homogeneous phenotypic discrimination of EOA and interobserver agreement
Complete homogeneous phenotypic agreement (by all three assessors), in accordance with the clinical diagnosis, occurred in eight, two, and one patients in the clinical EOA, DCD, and central hypotonia phenotypes respectively (Table S3). The phenotypic interobserver agreements for the EOA, DCD, and central hypotonia (sub)groups (Gwet’s agreement coefficient) were: EOA=0.801 (p<0.001; substantial); DCD=0.327 (p=0.037; fair); central hypotonia=0.415 (p=0.005; moderate). Analysis of EOA and central hypotonia separation revealed complete phenotypic discrimination in all the patients. Analysis of EOA and DCD separation revealed complete phenotypic discrimination in 16 patients. In 5 out of 21 patients, one of the observers had assigned a patient to the other group (2 out of 11 patients with EOA as DCD and 3 out of 10 patients with DCD as EOA).
(Semi)quantitative subgroup analysis
Perceived severity of coordination problems
The assessors perceived more severe motor coordination impairment in the EOA and DCD phenotypes than in the central hypotonia phenotype (p=0.001 and p=0.009 respectively; Mann–Whitney U test). The indicated severity of coordination impairment did not statistically differ between patients with EOA and DCD (p=0.086; Mann–Whitney U test), although patients with EOA tended to reveal higher scores (Table 1).
SARA score comparison between EOA and DCD
For a comparison of SARA subscores between EOA, DCD, and central hypotonia, see Table 1. Comparing the age‐corrected SARATOTAL, SARAGAIT/POSTURE, SARAKINETIC, and SARAFINGER‐NOSE/FINGER‐CHASE scores between the EOA and DCD groups, revealed significantly higher outcomes in EOA (p<0.001, p<0.001, p=0.003, and p=0.002 respectively; Mann–Whitney U test) (Table S4). The five children with the EOA and DCD phenotypes assessed inhomogeneously consisted of two patients with EOA with SARATOTAL scores below the EOA group median and three patients with DCD with SARATOTAL scores at or above the DCD group median. These patients also revealed overlapping SARATOTAL scores (Fig. 2).
Figure 2.
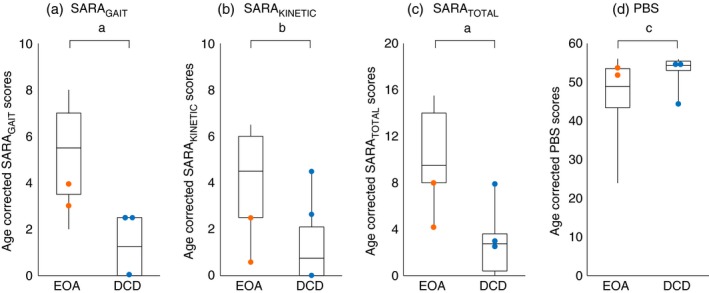
Motor profiles of age‐corrected Scale for Assessment and Rating of Ataxia (SARA) and Paediatric Balance Scale (PBS) scores for children with early‐onset ataxia (EOA) and developmental coordination disorder (DCD). The orange and blue dots indicate patients with an inhomogeneous phenotype (EOA, orange; DCD, blue). a–d: patients with an inhomogeneous phenotype tended to reveal overlapping SARAGAIT (a), SARAKINETIC (b), SARATOTAL (c), and PBS scores (d). Patients with EOA revealed the highest SARA (sub)scores. The box plots represent the median and first and third quartiles of the scores; the whiskers represent the minimum and maximum of the scores (negative age‐corrected SARA values are indicated as 0 and age‐corrected PBS scores >56 are represented as 56). a p<0.001, b p<0.01, c p<0.05.
SARA subscore comparison between patients with EOA and DCD
In all (11/11) children with EOA, both SARAGAIT/POSTURE and SARAKINETIC subscores contributed to the total SARA score. In contrast, in seven of 10 children with DCD either the SARAGAIT/POSTURE or SARAKINETIC subscores contributed to the total SARA score. Comparing the SARA subscore distribution over two versus one motor domain, revealed a significant difference between EOA and DCD groups (p=0.001; Mann–Whitney U test). This is illustrated in Figure 3.
Figure 3.
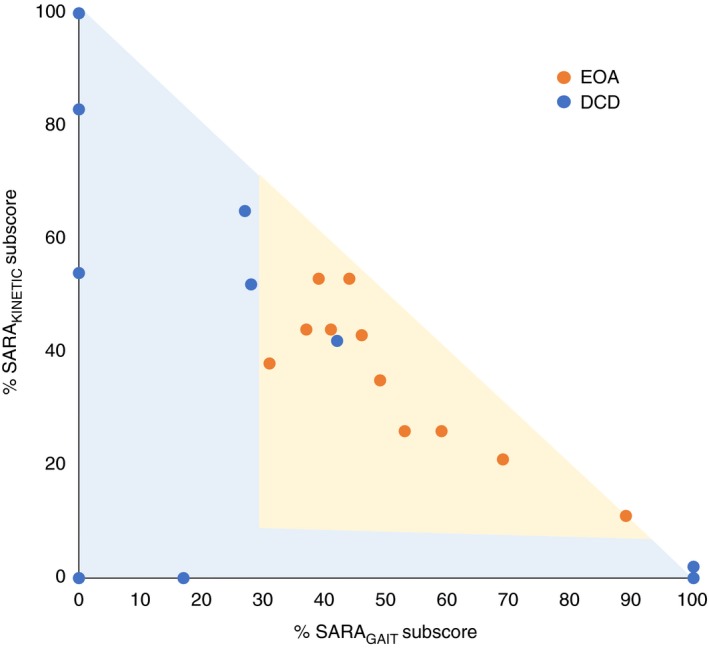
Age‐corrected median percentage of Scale for Assessment and Rating of Ataxia (SARA) subscores of SARA total scores in patients with early‐onset ataxia (EOA) and developmental coordination disorder (DCD). Patients with EOA revealed subscores in both gait and kinetic domains, whereas patients with DCD were mostly in one domain. The orange dots indicate patients with EOA and the blue dots indicate patients with DCD.
Effect of scientifically validated, standardized EOA features on rescored homogeneity
Reassessment with three scientifically validated EOA features seemingly increased the phenotypic homogeneity of the EOA and DCD groups (EOA: from eight to 10 patients; DCD: from two to seven patients). Completely homogeneous phenotypic discrimination between EOA and DCD phenotypes increased from 16 to 18 patients (first and second assessment respectively; p<0.02). In the majority (38/48) of reassessments, the assessors indicated that the phenotypic features supported their decision. The three features did not significantly differ regarding their estimated effect on phenotypic homogeneity.
Discussion
We investigated the diagnostic accuracy of phenotypic EOA recognition among DCD and central hypotonia phenotypes.
Phenotypic EOA recognition was reliable. EOA and central hypotonia phenotypes were homogeneously discerned, but phenotypic consensus on the separation between mildly affected EOA and severely affected DCD phenotypes was incomplete. In about a quarter of EOA and DCD phenotypes, one of three observers incorrectly assigned a patient to the opposite group. The inhomogeneously phenotyped children consisted of mildly affected EOA and severely affected DCD phenotypes with overlapping SARATOTAL scores. These findings suggest that signs and symptoms may overlap between the two phenotypes. For instance, patients with EOA who are diagnosed with spinocerebellar ataxia type 29 (ITPR1 gene mutation), may present with a broad clinical spectrum, including subtle phenotypes with mild, non‐progressive coordination impairment and cognitive disabilities.29 Furthermore, some patients with Joubert syndrome may present with mild developmental abnormalities, whereas others are severely ataxic.30 Conversely, features of ataxia are sometimes recognizable in patients with DCD. Furthermore, at the outpatient clinic, parents cannot always tell whether their child’s motor incoordination is progressive or not. From a conceptual point of view, DCD motor impairment affects daily tasks requiring sensorimotor integration, coordination, balance, motor learning, strategic planning, timing, sequencing, and visuospatial processing.11 Although often more severely affected, patients with EOA may face similar limitations during daily tasks. Previous investigations have attributed DCD motor impairment to dysfunctional signalling within the network connecting the cerebellum, basal ganglia, parietal lobe, dorsolateral prefrontal cortex, corpus callosum, and medial orbitofrontal cortex,14, 15, 31 which (at least partly) overlaps with that of EOA. Altogether, these findings suggest that one cannot always distinguish patients with EOA and DCD by the motor phenotype alone. In children with severely affected DCD motor phenotypes, these findings have consequences for diagnostic testing, since one may consider testing with an EOA algorithm (including magnetic resonance imaging and next‐generation sequencing with EOA, and perhaps even with dystonia or developmental disability gene panels). Since a DCD diagnosis depends on the exclusion of an underlying neurological disorder, further diagnostic testing could provide a different diagnosis in a proportion of DCD phenotypes.
When considering the incomplete phenotypic separation between EOA and DCD phenotypes, we explored the effect of previously proven,18, 23, 24 standardized phenotypic EOA features on phenotypic homogeneity. One validated standardized phenotypic EOA feature is that patients with EOA are likely to reveal assembled SARA motor (sub)scores from different SARA domains, reflecting global cerebellar involvement.18 In patients with EOA, this feature is understandable by the exclusion of acquired cerebellar lesions from the group. The remaining underlying genetic and metabolic disorders are likely to induce global cerebellar dysfunction. The other validated standardized phenotypic EOA features are derived from previous machine learning research.23, 24 To distinguish between EOA, DCD, and physiologically delayed motor behaviour, Martinez‐Manzanera et al. used automatic random‐forest classifiers on quantitative inertial measurement unit data.24 Results indicated that EOA kinetic trajectories are more irregular, both within and between intentional movement trajectories, such as the finger‐to‐nose test.24 The third validated standardized phenotypic EOA feature concerns the presence of a bow‐shaped intentional movement trajectory in any spatial plane.25, 26 Application of these three features resulted in a higher phenotypic consensus regarding the separation of EOA and DCD phenotypes. However, despite the modestly increased phenotypic homogeneity (from 16 to 18 out of 21 patients) full phenotypic consensus was not obtained. In addition to a real overlap between EOA and DCD phenotypes, this may also reflect the fact that the motor phenotype alone is insufficient to make a phenotypic decision since other diagnostic clues (such as family history, oculomotor findings, neurological examination findings, and data from radiological or laboratory investigations) are not taken into account.
The study has several limitations. First, it was observational in nature and the sample size was small. Second, videotaped scoring of the motor phenotype is an incomplete approach. On the other hand, a positive family history alone, or isolated cerebellar oculomotor features, cannot automatically be taken as pathognomonic for EOA either.32 Third, due to the study design, all patients had been clinically diagnosed before their inclusion in the study. Although the assessors were technically blinded to the clinical data, previous professional encounters at the outpatient clinic may have reduced the complexity of the phenotypic tasks. This suggests that the actual phenotypic consensus might be worse than already indicated. If so, the implications would remain the same and underline the need for: (1) supportive EOA features; and (2) an adequate diagnostic strategy for severe DCD phenotypes, especially when some ataxic features are present. Fourth, we reassessed the effect of EOA features on phenotypic homogeneity within the same assessor group. However, since: (1) the interval between the first and second phenotypic assessment was more than 6 months; (2) the assessors were not informed about the accuracy of their previous assessments; and (3) the assessors were allowed to select their phenotype among a large variety of possibilities (Fig. S2), we would not anticipate a large effect from this limitation. Finally, the reassessments with the EOA features were only performed in the inhomogeneously separated EOA and DCD phenotypes. However, since the EOA and central hypotonia phenotypes had been completely separated during the first assessment (in all patients by all three assessors), we did not anticipate a significant effect from reassessment. Future studies will hopefully make these points clearer.
In conclusion, the paediatric motor phenotype is not sufficient to distinguish between EOA and DCD. We hope that future standardized phenotypic EOA features, based on scientifically validated data, may help to improve the phenotypic consensus on EOA recognition.
Supporting information
Appendix S1: Clinical description of the patient groups and underlying diagnoses.
Appendix S2: Rating scale information.
Figure S1 : Flow diagram of followed procedure.
Figure S2 : Phenotypic assessment form.
Table S1 : Age‐related Scale for Assessment and Rating of Ataxia score values obtained in typically developing children
Table S2 : Individual phenotypic assessments
Table S3 : Percentage of homogeneously phenotyped children per clinical group
Table S4 : Differences in quantitative scores between early‐onset ataxia and developmental coordination disorder groups
Acknowledgements
We thank all the patients and their families for participating in the study. We thank all our colleagues from the Departments of Pediatrics, Neurology, Genetics, and Radiology of the University Medical Center Groningen for providing anonymous clinical data care to the study participants. The research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- 1. Harris SR, Mickelson ECR, Zwicker JG. Diagnosis and management of developmental coordination disorder. CMAJ 2015; 187: 659–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Blank R, Barnett AL, Cairney J, et al. International clinical practice recommendations on the definition, diagnosis, assessment, intervention, and psychosocial aspects of developmental coordination disorder. Dev Med Child Neurol 2019; 61: 242–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sanger TD, Chen D, Delgado MR, et al. Definition and classification of negative motor signs in childhood. Pediatrics 2006; 118: 2159–67. [DOI] [PubMed] [Google Scholar]
- 4. Ghibellini G, Brancati F, Castori M. Neurodevelopmental attributes of joint hypermobility syndrome/Ehlers–Danlos syndrome, hypermobility type: update and perspectives. Am J Med Genet C Semin Med Genet 2015; 169C: 107–16. [DOI] [PubMed] [Google Scholar]
- 5. Prudente CN, Hess EJ, Jinnah HA. Dystonia as a network disorder: what is the role of the cerebellum? Neuroscience 2014; 260: 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Harding AE. Clinical features and classification of inherited ataxias. Adv Neurol 1993; 61: 1–14. [PubMed] [Google Scholar]
- 7. Brandsma R, Lawerman TF, Kuiper MJ, Lunsing RJ, Burger H, Sival DA. Reliability and discriminant validity of ataxia rating scales in early onset ataxia. Dev Med Child Neurol 2017; 59: 427–32. [DOI] [PubMed] [Google Scholar]
- 8. American Psychiatry Association . Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Washington, DC: Author, 2013. [Google Scholar]
- 9. Mariën P, Wackenier P, De Surgeloose D, De Deyn PP, Verhoeven J. Developmental coordination disorder: disruption of the cerebello‐cerebral network evidenced by SPECT. Cerebellum 2010; 9: 405–10. [DOI] [PubMed] [Google Scholar]
- 10. Visser J. Developmental coordination disorder: a review of research on subtypes and comorbidities. Hum Mov Sci 2003; 22: 479–93. [DOI] [PubMed] [Google Scholar]
- 11. Vaivre‐Douret L, Lalanne C, Golse B. Developmental coordination disorder, an umbrella term for motor impairments in children: nature and co‐morbid disorders. Front Psychol 2016; 7: 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lawerman TF, Brandsma R, Burger H, Burgerhof JGM, Sival DA, the Childhood Ataxia and Cerebellar Group of the European Pediatric Neurology Society . Age‐related reference values for the pediatric Scale for Assessment and Rating of Ataxia: a multicentre study. Dev Med Child Neurol 2017; 59: 1077–82. [DOI] [PubMed] [Google Scholar]
- 13. Brandsma R, Spits AH, Kuiper MJ, et al. Ataxia rating scales are age‐dependent in healthy children. Dev Med Child Neurol 2014; 56: 556–63. [DOI] [PubMed] [Google Scholar]
- 14. Zwicker JG, Missiuna C, Harris SR, Boyd LA. Brain activation associated with motor skill practice in children with developmental coordination disorder: an fMRI study. Int J Dev Neurosci 2011; 29: 145–52. [DOI] [PubMed] [Google Scholar]
- 15. Zwicker JG, Missiuna C, Harris SR, Boyd LA. Developmental coordination disorder: a review and update. Eur J Paediatr Neurol 2012; 16: 573–81. [DOI] [PubMed] [Google Scholar]
- 16. Galli M, Cimolin V, Vismara L, et al. The effects of muscle hypotonia and weakness on balance: a study on Prader‐Willi and Ehlers‐Danlos syndrome patients. Res Dev Disabil 2011; 32: 1117–21. [DOI] [PubMed] [Google Scholar]
- 17. Horlings CG, Küng UM, van Engelen BG, et al. Balance control in patients with distal versus proximal muscle weakness. Neuroscience 2009; 164: 1876–86. [DOI] [PubMed] [Google Scholar]
- 18. Lawerman TF, Brandsma R, van Geffen JT, et al. Reliability of phenotypic early‐onset ataxia assessment: a pilot study. Dev Med Child Neurol 2016; 58: 70–6. [DOI] [PubMed] [Google Scholar]
- 19. Schmitz‐Hübsch T, du Montcel ST, Baliko L, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 2006; 66: 1717–20. [DOI] [PubMed] [Google Scholar]
- 20. Franjoine MR, Gunther JS, Taylor MJ. Pediatric balance scale: a modified version of the Berg Balance Scale for the school‐age child with mild to moderate motor impairment. Pediatr Phys Ther 2003; 15: 114–28. [DOI] [PubMed] [Google Scholar]
- 21. Kuiper MJ, Brandsma R, Vrijenhoek L, et al. Physiological movement disorder‐like features during typical motor development. Eur J Paediatr Neurol 2018; 22: 595–601. [DOI] [PubMed] [Google Scholar]
- 22. Franjoine MR, Darr N, Held SL, Kott K, Young BL. The performance of children developing typically on the Pediatric Balance Scale. Pediatr Phys Ther 2010; 22: 350–9. [DOI] [PubMed] [Google Scholar]
- 23. Mannini A, Martinez‐Manzanera O, Lawerman TF, et al. Automatic classification of gait in children with early‐onset ataxia or developmental coordination disorder and controls using inertial sensors. Gait Posture 2017; 52: 287–92. [DOI] [PubMed] [Google Scholar]
- 24. Martinez‐Manzanera O, Lawerman TF, Blok HJ, et al. Instrumented finger‐to‐nose test classification in children with ataxia or developmental coordination disorder and controls. Clin Biomech 2018; 60: 51–9. [DOI] [PubMed] [Google Scholar]
- 25. Bodranghien F, Bastian A, Casali C, et al. Consensus paper: revisiting the symptoms and signs of cerebellar syndrome. Cerebellum 2016; 15: 369–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Manto M, Bower JM, Conforto AB, et al. Consensus paper: roles of the cerebellum in motor control—the diversity of ideas on cerebellar involvement in movement. Cerebellum 2012; 11: 457–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gwet KL, editor. Handbook of Inter‐rater Reliability: The Definitive Guide to Measuring the Extent of Agreement Among Raters, 4th ed. Gaithersburg, MD: Advanced Analytics, LLC, 2014: 101–27. [Google Scholar]
- 28. Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics 1977; 33: 159–74. [PubMed] [Google Scholar]
- 29. Klar J, Ali Z, Farooq M, et al. A missense variant in ITPR1 provides evidence for autosomal recessive SCA29 with asymptomatic cerebellar hypoplasia in carriers. Eur J Hum Genet 2017; 25: 848–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vilboux T, Doherty DA, Glass IA, et al. Molecular genetic findings and clinical correlations in 100 patients with Joubert syndrome and related disorders prospectively evaluated at a single center. Genet Med 2017; 19: 875–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Biotteau M, Chaix Y, Blais M, Tallet J, Péran P, Albaret JM. Neural signature of DCD: a critical review of MRI neuroimaging studies. Front Neurol 2016; 7: 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gaymard B, Giannitelli M, Challes G, et al. Oculomotor impairments in developmental dyspraxia. Cerebellum 2017; 16: 411–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Clinical description of the patient groups and underlying diagnoses.
Appendix S2: Rating scale information.
Figure S1 : Flow diagram of followed procedure.
Figure S2 : Phenotypic assessment form.
Table S1 : Age‐related Scale for Assessment and Rating of Ataxia score values obtained in typically developing children
Table S2 : Individual phenotypic assessments
Table S3 : Percentage of homogeneously phenotyped children per clinical group
Table S4 : Differences in quantitative scores between early‐onset ataxia and developmental coordination disorder groups