

Abstract
Interactions on the molecular level control structure as well as function. Especially interfaces between innocent alkyl groups are hardly studied although they are of great importance in larger systems. Herein, London dispersion in conjunction with solvent interactions between linear alkyl chains was examined with an azobenzene‐based experimental setup. Alkyl chains in all meta positions of the azobenzene core were systematically elongated, and the change in rate for the thermally induced Z→E isomerization in n‐decane was determined. The stability of the Z‐isomer increased with longer chains and reached a maximum for n‐butyl groups. Further elongation led to faster isomerization. The origin of the intramolecular interactions was elaborated by various techniques, including 1H NOESY NMR spectroscopy. The results indicate that there are additional long‐range interactions between n‐alkyl chains with the opposite phenyl core in the Z‐state. These interactions are most likely dominated by attractive London dispersion. This work provides rare insight into the stabilizing contributions of highly flexible groups in an intra‐ as well as an intermolecular setting.
Keywords: azobenzenes, London dispersion, molecular probes, solvent effects, spectroscopy
An azobenzene setup was used to examine London dispersion in conjunction with solvent interactions between linear alkyl chains. 1H NOESY NMR analysis indicated additional long‐range interactions with the opposite phenyl core in the Z‐isomer. This work provides rare insight into the stabilizing contributions of highly flexible groups in an intra‐ as well as an intermolecular setting.
The interaction of chemical entities on the molecular level is crucial for their structure and, therefore, for their properties as well as function. While stronger interactions, such as hydrogen bonds or Lewis pairs, are well‐established, weak ones such as van der Waals interactions still have not been studied in depth and are usually neglected in more complex systems. London dispersion represents the attractive part of the van der Waals potential and was first introduced by Fritz London in 1930.1 While London dispersion plays a dominant role in the interaction of hydrocarbons, these non‐covalent interactions are omnipresent and not limited to a certain type of functional group.2 In synthesis, large and polarizable moieties—in most cases rigid alkyl groups—can be utilized as dispersion energy donors if placed at the correct distance.3 They allow the stabilization of extreme bonding situations,4 therefore overcompensating repulsive forces to generate, for example, the longest carbon–carbon bond in alkanes reported to date.5 Biological systems6 are also stabilized by the significant contributions of dispersion forces. In addition, they dominate the interaction energy in frustrated Lewis pairs (FLPs)7 or in the aggregation of aromatic species.8 Other studies have demonstrated their substantial influence on the stability of organometallic complexes9 and in catalysis.10 In recent years, numerous computational methods were developed that give access to a comprehensive toolbox for efficiently evaluating the dispersion interactions in molecular systems with high accuracy.11
Nevertheless, London dispersion is often regarded as negligible in solution. However, studies have been conducted towards addressing this statement and investigating the influence of the solvent on conformer or dimer stability.12, 13 In these studies, it was tried to dissect the different contributions to the total energy of the molecular system. The authors observed a very large attenuation of dispersion forces due to competitive dispersion interactions with the solvent, which were not completely cancelled out.
In contrast to rigid alkyl dispersion donors, such as adamantyl or tert‐butyl moieties, highly flexible n‐alkyl chains bear more complex challenges for estimating their dispersion donor abilities. Therefore, London dispersion interactions between linear alkyl chains have still only marginally been explored.14, 15 At elevated temperatures, a large number of different conformers have to be considered for these alkanes for a conclusive evaluation of the experimental findings. For n‐pentane, the gauche conformer is already the most abundant one at room temperature.16 Furthermore, the potential formation of hairpin structures for longer alkyl chains complicates meaningful interpretation.17 Computational analysis of the large number of possible conformers often turns out to be very challenging with standard resources. Therefore, experimental data is essential to understand these fundamental interactions. The azobenzene switch has been established as a powerful tool to investigate London dispersion forces and was chosen to address these open questions (Scheme 1).13, 18, 19
Scheme 1.
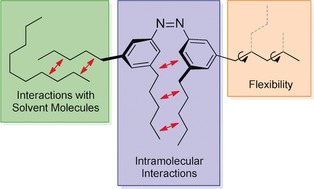
Intra‐ and intermolecular interactions, as well as flexibility in all‐meta n‐alkylated azobenzenes.
Azobenzenes are photoswitchable molecules, which can isomerize from the thermodynamically most stable E‐state to the metastable Z‐state, which is about 11.7 kcal mol−1 higher in energy (Scheme 2).20 An important change upon isomerization is the reduction of the distance between the carbon atoms in the para position of the rings from 9.1 Å21 to about 6.2 Å,22 bringing substituents on the azobenzene rings in close proximity. In a previous study, we had demonstrated that for azobenzene derivatives substituted in the meta positions, the substituents come into a distance where London dispersion donors stabilize the Z‐isomer of the azobenzene.18
Scheme 2.
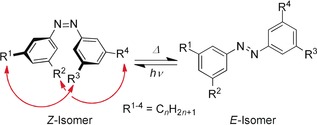
Isomerization of all‐meta n‐alkylated azobenzene systems.
For the current study, n‐alkyl substituents were systematically attached in the meta positions of the azobenzene scaffold with increasing lengths, ranging from methyl up to n‐octyl chains. The system was deliberately designed such that each of the two alkyl chains on a phenyl ring faces another one on the opposite ring in the Z‐isomer. Therefore, the overall interaction contacts are increased, leading to high sensitivity towards small changes in the system.
For the synthesis of the azobenzene probes, a highly flexible strategy was designed. The side chains of choice were introduced by a Wittig reaction using 5‐nitroisophthalaldehyde. Subsequent hydrogenation and oxidative azo coupling yielded the symmetric all‐meta n‐alkylated azobenzenes 1–8 (Scheme 3). The modular synthesis of this azobenzene probe can be conveniently altered to modify the substituents as desired.
Scheme 3.
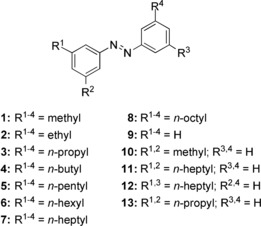
Overview of azobenzene derivatives investigated in this study.
To study the alkyl–alkyl interactions, all azobenzenes were switched from the E‐ to the Z‐state by irradiation at 302 nm. The thermally induced back‐isomerization at a given temperature was then followed by UV/Vis spectroscopy to directly correlate the chain length of the alkyl substituents with the stability of the Z‐isomer of the azobenzene.
The measurements were conducted in n‐decane to minimize solvophobic contributions to the thermally induced Z→E isomerization barrier. As can be seen in Figure 1, the half‐lives of the Z‐isomers of the all‐meta‐substituted azobenzenes at 40 °C increased dramatically at the beginning upon elongation of the alkyl substituents. This observation is in accordance with the expectation of increasing attractive London dispersion interactions with increasing chain length. For the n‐butyl chains, a maximum is reached with a half‐life that is about five times higher than that for the tetramethyl‐substituted derivative 1. Further elongation leads to a decrease in the half‐lives down to approximately 23 h. This decrease might be caused by competitive alkyl–alkyl interactions between alkyl chains on the same phenyl ring, which would reduce the overall interaction strength with alkyl groups on the opposite phenyl ring.
Figure 1.
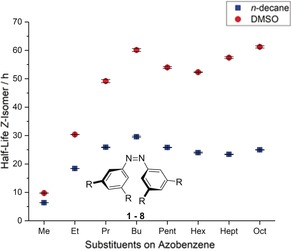
Half‐lives of all‐meta‐substituted azobenzenes 1–8 with different chain lengths at 40 °C in n‐decane and DMSO.
In an analogous study, van Craen and co‐workers had investigated the effect of n‐alkyl‐substituted ligands on the stabilization of dimers of titanium(IV) helicate complexes. They had also observed a higher degree of dimerization with increased chain length up to n‐heptyl.14 However, for solubility reasons, the dimerization of the titanium complexes had to be studied in THF, which should add solvophobic contributions to the attractive interactions in favor of the dimer formation.
It therefore seemed quite likely that in solvents such as THF or dimethyl sulfoxide (DMSO), our system would also show an increased half‐life for the Z‐isomer for the elongated chains, and this was indeed observed. When the measurements were conducted in DMSO, an overall increase in the Z‐isomer half‐lives was observed (Figure 1). This can be rationalized by a thermodynamic stabilization of the Z‐isomer in a more polar solvent. This was also demonstrated by Haberfield and co‐workers, who calorimetrically examined the enthalpies for the Z→E isomerization of Z‐azobenzene in cyclohexane versus cyclohexanone.23 In addition, they found a slight energetic increase in the transition state due to less favored solute–solvent interactions. This observation further helps explaining the changed course of the half‐lives for the azobenzenes 6–8 with n‐hexyl or longer chains. Here, strong solvophobic interactions contribute extensively to a higher‐energy transition state with increasing chain length and therefore a longer half‐life for these compounds. The preference for solvent–solvent over solute–solvent interactions corresponds also to the observed, drastically decreased solubility of these azobenzenes in DMSO.
At different temperatures, the overall trend of the isomerization rates stays essentially the same (Figure 2). The lowest isomerization rate at each temperature, and therefore the most stable Z‐isomer, was observed for azobenzene 4 with the n‐butyl substituents. Furthermore, the isomerization rate decreased again from 7 to 8. Although this did not seem to be the case at 25 °C, a higher error for this measurement might influence the result here. Overall, a threefold increase in the isomerization rates was observed upon increasing the temperature by 10 °C. This acceleration is independent of the chain length of the alkyl substituents and was observed for each azobenzene investigated.
Figure 2.
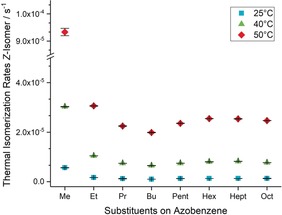
Thermal isomerization rates of all‐meta‐substituted azobenzenes 1–8 with different chain lengths in n‐decane at three different temperatures.
In total, isomerization rates were measured at five different temperatures. An Exner plot was constructed to exclude a change in the isomerization mechanism (see Figure S2 in the Supporting Information). The plot shows a high linear correlation indicating a constant mechanism for the investigated temperature range. The collected kinetic data allowed us to further evaluate the enthalpies and entropies of activation for the Z→E isomerization for all compounds (Table 1).
Table 1.
Experimental kinetic data of the thermal Z→E isomerization of 1–8 in n‐decane as the solvent. Energies are given in kcal mol−1, entropies in cal K−1 mol−1. Errors were calculated from the highest slope and y‐intercept error out of three Eyring–Polanyi fits.
|
Compound |
ΔH ≠ Z‐E |
ΔS ≠ Z‐E |
ΔG ≠ Z‐E [a] |
|---|---|---|---|
|
1 |
20.1±0.3 |
−12.5±1.0 |
24.6±0.6 |
|
2 |
21.5±0.2 |
−12.9±0.5 |
25.3±0.3 |
|
3 |
21.6±0.5 |
−13.3±1.7 |
25.5±1.1 |
|
4 |
21.9±0.3 |
−12.4±0.9 |
25.6±0.6 |
|
5 |
21.9±0.5 |
−12.0±1.6 |
25.5±1.0 |
|
6 |
21.9±0.2 |
−12.1±0.6 |
25.5±0.4 |
|
7 |
22.0±0.1 |
−11.8±0.4 |
25.5±0.2 |
|
8 |
21.7±0.4 |
−12.8±1.3 |
25.5±0.8 |
[a] At 25 °C, 1 atm.
The enthalpy of activation shows a large increase with longer chains from 1 to 3. For compounds 4–7, the values stay constant with a small decrease for 8. This is not surprising as elongation of the alkyl substituents increases the possible number of attractive contacts between the chains. The contributions to the increasing enthalpy of activation are dominated by London dispersion interactions. However, insertion of additional methylene units also enhances the degrees of rotational and vibrational freedom, leading to a compensation of the attraction for longer alkyl substituents. This also explains why for these linear alkyl chains, a plateau is reached. Interpretation of the experimentally determined entropic contributions to the isomerization barrier is complex. Extending the chains should increase the conformational freedom, and hence increase the destabilizing entropic contributions. However, it can be expected that London dispersion interactions will restrict the flexibility of the chains, counteracting the effect of larger substituents to the entropic contributions to a certain degree. Additionally, interactions with the solvent have to be considered. Solvophobic effects based on cohesive properties of the solvent should be less important for our experiments in n‐decane, but have to be taken into account for solvents with higher polarity such as DMSO. Although a qualitative estimation is reasonable, the calculated error of the entropies is too large to quantify and dissect individual contributions.
It is expected that the strength of London dispersion interactions should correlate with the number as well as the position of the substituents. Therefore, different substitution patterns were examined. By exchanging two n‐heptyl substituents in 7 for hydrogen atoms, either azobenzene 12 with alkyl chains on opposite phenyl rings or compound 11 with both substituents on the same phenyl ring were obtained. Additionally, the dimethyl analogue 10 was prepared. Hydrogen substituents are not isoelectronic to alkyl substituents. Hence, the overall electron distribution in the phenyl rings is different, which also influences the isomerization rates. Therefore, unsubstituted azobenzene (9) was also investigated for comparison. In Figure 3, the half‐lives of the Z‐isomers of these compounds are depicted.
Figure 3.
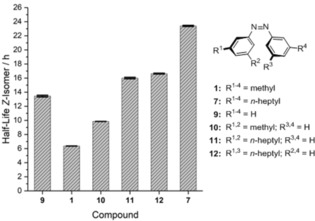
Half‐lives of azobenzenes with different substitution patterns and/or different chain lengths at 40 °C in n‐decane.
As expected, the substitution of two Me groups with H atoms in compound 10 led to an increase in the half‐life by approximately 3 h, illustrating the electronic difference of H versus alkyl substitution. When all alkyl substituents are replaced by hydrogen atoms, the half‐life further increases. This observation correlates well with the change in electron density. An increase in the electron density leads to faster isomerization, and therefore, to a shorter half‐life. The diheptyl derivatives 11 and 12 show substantially longer half‐lives than the Me derivatives 1 and 10 because of the increased attractive London dispersion interactions resulting from alkyl–alkyl as well as alkyl–aryl interactions. The similar half‐lives of azobenzenes 11 and 12 result from the same number of alkyl–aryl contacts, indicating that alkyl–alkyl contacts are of minor importance here. However, compound 11, with both heptyl chains on the same phenyl ring, only shows a marginally shorter half‐life, which can be rationalized by considering the altered electronic structure compared to compound 12.
To further elucidate the large difference between 10 and 11, 3,5‐di‐n‐propyl‐substituted azobenzene 13 was synthesized, and switched to the photostationary state under continuous irradiation at 302 nm. Afterwards, a 1H NOESY NMR spectrum was recorded (Figure 4). This technique can be used to identify hydrogen atoms that are up to 5 Å apart from each other. Between 3 and 5 Å, London dispersion interactions can contribute significantly to the stability of molecular entities. The marked cross‐peaks show that the methyl groups of the n‐propyl substituents are close enough to interact with the opposite phenyl ring. This long‐range interaction of the alkyl chains with the Ph moiety is attractive in character and significantly contributes to the longer half‐life of 11 compared to 10. This finding further indicates that attractive alkyl–aryl interactions have a considerable impact on the stability of the Z‐isomer.
Figure 4.
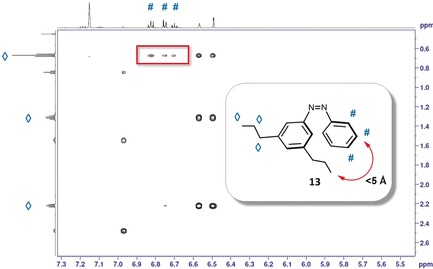
1H NOESY NMR spectrum of the Z‐isomer of compound 13 in benzene‐d 6 in the photostationary state. The marked cross‐peaks indicate the close proximity of the methyl groups with the adjacent phenyl ring, leading to attractive London dispersion.
An additional 1H NOESY NMR experiment with compound 11 also revealed the close proximity of the alkyl substituents and the opposite phenyl ring. Interestingly, here the interaction with the fourth methylene unit counted from the phenyl ring was clearly observed, which is in a similar distance range as for 13. The small deviation can be rationalized by a different conformation for 11.
In conclusion, we have observed subtle changes in London dispersion interactions by measuring the isomerization rates of n‐alkyl‐substituted azobenzenes. The presented experimental setup allowed us to determine the changes occurring upon stepwise elongation of the n‐alkyl chains with high precision and reproducibility. The Z→E isomerization decelerated from methyl to n‐butyl substituents, maximizing London dispersion from alkyl–alkyl and alkyl–aryl interactions. With longer alkyl substituents, the isomerization is again accelerated up to a certain threshold because of the higher entropic contributions that overcompensate the stabilization of the Z‐state by London dispersion. Furthermore, solvophobic contributions dominate in solvents with higher polarity such as DMSO for alkyl chains longer than n‐hexyl.
This work provides further evidence that London dispersion interactions in solution are still contributing in a relevant way even to chemical systems with a high degree of flexibility. These results provide deeper insight into how the subtle balance between steric repulsion, attractive London dispersion, and entropy can be adjusted with flexible dispersion energy donors. The excellent solubility of these azobenzenes in a broad range of solvents as well as their modular synthesis provides the possibility to extend the investigation also to other types of interactions, such as fluoroalkyl–fluoroalkyl or fluoroalkyl–alkyl interactions, as well as other solvent systems in the future.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support was provided by the Justus Liebig University Giessen and by the Deutsche Forschungsgemeinschaft (SPP1807). We thank Dr. Heike Hausmann and Anja Platt for NMR support and Jan M. Schümann for fruitful discussions.
M. A. Strauss, H. A. Wegner, Angew. Chem. Int. Ed. 2019, 58, 18552.
References
- 1. London F., Z. Phys. 1930, 63, 245–279. [Google Scholar]
- 2. London F., Trans. Faraday Soc. 1937, 33, 8b. [Google Scholar]
- 3.
- 3a. Wagner J. P., Schreiner P. R., Angew. Chem. Int. Ed. 2015, 54, 12274–12296; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12446–12471; [Google Scholar]
- 3b. Hwang J., Li P., Smith M. D., Shimizu K. D., Angew. Chem. Int. Ed. 2016, 55, 8086–8089; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8218–8221. [Google Scholar]
- 4.
- 4a. Rösel S., Balestrieri C., Schreiner P. R., Chem. Sci. 2017, 8, 405–410; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Rösel S., Quanz H., Logemann C., Becker J., Mossou E., Cañadillas-Delgado L., Caldeweyher E., Grimme S., Schreiner P. R., J. Am. Chem. Soc. 2017, 139, 7428–7431; [DOI] [PubMed] [Google Scholar]
- 4c. Rösel S., Becker J., Allen W. D., Schreiner P. R., J. Am. Chem. Soc. 2018, 140, 14421–14432; [DOI] [PubMed] [Google Scholar]
- 4d. Fokin A. A., Zhuk T. S., Blomeyer S., Pérez C., Chernish L. V., Pashenko A. E., Antony J., Vishnevskiy Y. V., Berger R. J. F., Grimme S., et al., J. Am. Chem. Soc. 2017, 139, 16696–16707. [DOI] [PubMed] [Google Scholar]
- 5. Schreiner P. R., Chernish L. V., Gunchenko P. A., Tikhonchuk E. Y., Hausmann H., Serafin M., Schlecht S., Dahl J. E. P., Carlson R. M. K., Fokin A. A., Nature 2011, 477, 308–311. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Wagner J. P., Schreiner P. R., J. Chem. Theory Comput. 2014, 10, 1353–1358; [DOI] [PubMed] [Google Scholar]
- 6b. Vondrásek J., Kubar T., Jenney F. E., Adams M. W. W., Kozísek M., Cerný J., Sklenár V., Hobza P., Chem. Eur. J. 2007, 13, 9022–9027; [DOI] [PubMed] [Google Scholar]
- 6c. Hanlon S., Biochem. Biophys. Res. Commun. 1966, 23, 861–867; [DOI] [PubMed] [Google Scholar]
- 6d. Kolář M., Kubař T., Hobza P., J. Phys. Chem. B 2011, 115, 8038–8046; [DOI] [PubMed] [Google Scholar]
- 6e. Nick Pace C., Scholtz J. M., Grimsley G. R., FEBS Lett. 2014, 588, 2177–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bistoni G., Auer A. A., Neese F., Chem. Eur. J. 2017, 23, 865–873. [DOI] [PubMed] [Google Scholar]
- 8. Fatima M., Steber A. L., Poblotzki A., Pérez C., Zinn S., Schnell M., Angew. Chem. Int. Ed. 2019, 58, 3108–3113; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3140–3145. [Google Scholar]
- 9.
- 9a. Löffler S., Lübben J., Wuttke A., Mata R. A., John M., Dittrich B., Clever G. H., Chem. Sci. 2016, 7, 4676–4684; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Liptrot D. J., Guo J.-D., Nagase S., Power P. P., Angew. Chem. Int. Ed. 2016, 55, 14766–14769; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14986–14989; [Google Scholar]
- 9c. Ahlquist M. S. G., Norrby P.-O., Angew. Chem. Int. Ed. 2011, 50, 11794–11797; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 11998–12001; [Google Scholar]
- 9d. Lin C.-Y., Guo J.-D., Fettinger J. C., Nagase S., Grandjean F., Long G. J., Chilton N. F., Power P. P., Inorg. Chem. 2013, 52, 13584–13593; [DOI] [PubMed] [Google Scholar]
- 9e. Song L., Schoening J., Wölper C., Schulz S., Schreiner P. R., Organometallics 2019, 38, 1640–1647. [Google Scholar]
- 10.
- 10a. Meyer T. H., Liu W., Feldt M., Wuttke A., Mata R. A., Ackermann L., Chem. Eur. J. 2017, 23, 5443–5447; [DOI] [PubMed] [Google Scholar]
- 10b. Procházková E., Kolmer A., Ilgen J., Schwab M., Kaltschnee L., Fredersdorf M., Schmidts V., Wende R. C., Schreiner P. R., Thiele C. M., Angew. Chem. Int. Ed. 2016, 55, 15754–15759; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15986–15991; [Google Scholar]
- 10c. Yatham V. R., Harnying W., Kootz D., Neudörfl J.-M., Schlörer N. E., Berkessel A., J. Am. Chem. Soc. 2016, 138, 2670–2677; [DOI] [PubMed] [Google Scholar]
- 10d. Lyngvi E., Sanhueza I. A., Schoenebeck F., Organometallics 2015, 34, 805–812; [Google Scholar]
- 10e. Detmar E., Müller V., Zell D., Ackermann L., Breugst M., Beilstein J. Org. Chem. 2018, 14, 1537–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Riplinger C., Pinski P., Becker U., Valeev E. F., Neese F., J. Chem. Phys. 2016, 144, 024109; [DOI] [PubMed] [Google Scholar]
- 11b. Grimme S., Hansen A., Brandenburg J. G., Bannwarth C., Chem. Rev. 2016, 116, 5105–5154; [DOI] [PubMed] [Google Scholar]
- 11c. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104; [DOI] [PubMed] [Google Scholar]
- 11d. Grimme S., Ehrlich S., Goerigk L., J. Comput. Chem. 2011, 32, 1456–1465; [DOI] [PubMed] [Google Scholar]
- 11e. Caldeweyher E., Bannwarth C., Grimme S., J. Chem. Phys. 2017, 147, 034112; [DOI] [PubMed] [Google Scholar]
- 11f. Grimme S., Bannwarth C., Caldeweyher E., Pisarek J., Hansen A., J. Chem. Phys. 2017, 147, 161708. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Pollice R., Bot M., Kobylianskii I. J., Shenderovich I., Chen P., J. Am. Chem. Soc. 2017, 139, 13126–13140; [DOI] [PubMed] [Google Scholar]
- 12b. Yang L., Adam C., Nichol G. S., Cockroft S. L., Nat. Chem. 2013, 5, 1006–1010; [DOI] [PubMed] [Google Scholar]
- 12c. Adam C., Yang L., Cockroft S. L., Angew. Chem. Int. Ed. 2015, 54, 1164–1167; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1180–1183; [Google Scholar]
- 12d. Hwang J., Dial B. E., Li P., Kozik M. E., Smith M. D., Shimizu K. D., Chem. Sci. 2015, 6, 4358–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Strauss M. A., Wegner H. A., Eur. J. Org. Chem. 2019, 295–302. [Google Scholar]
- 14. van Craen D., Rath W. H., Huth M., Kemp L., Räuber C., Wollschläger J. M., Schalley C. A., Valkonen A., Rissanen K., Albrecht M., J. Am. Chem. Soc. 2017, 139, 16959–16966. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Ray C., Brown J. R., Kirkpatrick A., Akhremitchev B. B., J. Am. Chem. Soc. 2008, 130, 10008–10018; [DOI] [PubMed] [Google Scholar]
- 15b. Snyder R. G., Strauss H. L., Elliger C. A., J. Phys. Chem. 1982, 86, 5145–5150; [Google Scholar]
- 15c. Tsuzuki S., Honda K., Uchimaru T., Mikami M., J. Phys. Chem. A 2004, 108, 10311–10316. [Google Scholar]
- 16. Salam A., Deleuze M. S., J. Chem. Phys. 2002, 116, 1296–1302. [Google Scholar]
- 17.
- 17a. Lüttschwager N. O. B., Wassermann T. N., Mata R. A., Suhm M. A., Angew. Chem. Int. Ed. 2013, 52, 463–466; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 482–485; [Google Scholar]
- 17b. Liakos D. G., Neese F., J. Chem. Theory Comput. 2015, 11, 2137–2143. [DOI] [PubMed] [Google Scholar]
- 18. Schweighauser L., Strauss M. A., Bellotto S., Wegner H. A., Angew. Chem. Int. Ed. 2015, 54, 13436–13439; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13636–13639. [Google Scholar]
- 19. Heindl A. H., Wende R. C., Wegner H. A., Beilstein J. Org. Chem. 2018, 14, 1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Dias A. R., Minas Da Piedade M. E., Martinho Simões J. A., Simoni J. A., Teixeira C., Diogo H. P., Meng-Yan Y., Pilcher G., J. Chem. Thermodyn. 1992, 24, 439–447; [Google Scholar]
- 20b. Adamson A. W., Vogler A., Kunkely H., Wachter R., J. Am. Chem. Soc. 1978, 100, 1298–1300. [Google Scholar]
- 21. Harada J., Ogawa K., Tomoda S., Acta Crystallogr. Sect. B 1997, 53, 662–672. [Google Scholar]
- 22. Mostad A., Rømming C., Acta Chem. Scand. 1971, 25, 3561–3568. [DOI] [PubMed] [Google Scholar]
- 23. Haberfield P., Block P. M., Lux M. S., J. Am. Chem. Soc. 1975, 97, 5804–5806. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary