

Abstract
Aggregation of amyloid‐β (Aβ) that leads to the formation of plaques in Alzheimer's disease (AD) occurs through the stepwise formation of oligomers and fibrils. An earlier onset of aggregation is obtained upon intracerebral injection of Aβ‐containing brain homogenate into human APP transgenic mice that follows a prion‐like seeding mechanism. Immunoprecipitation of these brain extracts with anti‐Aβ oligomer antibodies or passive immunization of the recipient animals abrogated the observed seeding activity, although induced Aβ deposition was still evident. Here, we establish that, together with Aβ monomers, Aβ oligomers trigger the initial phase of Aβ seeding and that the depletion of oligomeric Aβ delays the aggregation process, leading to a transient reduction of seed‐induced Aβ deposits. This work extends the current knowledge about the role of Aβ oligomers beyond its cytotoxic nature by pointing to a role in the initiation of Aβ aggregation in vivo. We conclude that Aβ oligomers are important for the early initiation phase of the seeding process.
Keywords: Alzheimer's disease, Aβ seeding, Aβ oligomers, amyloid‐β plaques
Abbreviations
- Aβ
amyloid‐β
- AD
Alzheimer's disease
- APP
amyloid precursor protein
- CSF
cerebrospinal fluid
- DCX
doublecortin
Introduction
Misfolding and aggregation of certain proteins are fundamental features of neurodegenerative diseases. In AD, genetic evidence from mutations in the amyloid precursor protein (APP) and Presenilins strongly implicate Aβ in the pathogenesis of familial AD 4, 10. Fibrillogenesis from Aβ monomers to oligomers, protofibrils and finally fibrils occurs via a nucleation dependent polymerization process. This cascade of Aβ aggregation can also be induced in vivo by a single inoculation of Aβ‐rich brain extract in mouse models of cerebral amyloidosis that follows a prion‐like seeding mechanism 12, 20. The spreading of Aβ pathology to other brain regions originates within the limbic connectome 35 and is thought to involve the transport of preformed Aβ aggregates that nucleate the formation of new amyloid deposits 11, suggesting an involvement of Aβ in the seeding process. There is a growing evidence that aggregated Aβ in the brain homogenate is essential for the amyloid‐inducing activity. Both, aggregate depletion of these brain extracts as well as immunizing the recipient‐seeded mice with an anti‐Aβ antibody prevented seeding 6, 20, 21, while synthetic Aβ preparations accelerated Aβ pathology but seemed to be less potent compared to complete brain homogenate 29. The amyloid‐inducing agent was shown to range in size from small, soluble to proteinase K‐resistant assemblies 15. Extended sonification of the brain extract increased the seeding capacity 15, whereas treatment with formaldehyde or boiling reduced the seeding potential 8, 20.
Although many features of the seed‐inducing factor have been uncovered in the past, the role of Aβ intermediates, in particular of oligomers, during seeding remains elusive. There is mounting evidence that oligomers are not only the most neurotoxic form of Aβ 14, 31, 34 but also an important intermediate form that might itself play a role in the aggregation process. Injection of Aβ oligomers into the lateral ventricle of nonhuman primates induced AD‐like pathology such as tau hyperphosphorylation or synaptic loss and accumulated in brain regions associated with memory and cognitive functions 7. Since passive immunization with antibodies against Aβ oligomers resulted in a reduction of plaque burden and an improvement of cognitive functions in APP transgenic mice 16, 27, 28, we further investigated the ability of oligomeric Aβ to influence the seeding process.
Material and Methods
Mice
We used heterozygous 5×FAD transgenic mice coexpressing human APPK670N/M671L (Sw)+I716V (Fl)+V717I(Lo) and PS1M146L+L286V under the control of the neuron‐specific Thy‐1 promoter 23 and heterozygous APP23 transgenic mice 30 expressing human APPK670N/M671L. We backcrossed heterozygous 5×FAD and APP23 mice to C57BL/6N mice to generate heterozygous 5×FAD or APP23 mice and non‐transgenic littermates. Animals were group‐housed under specific pathogen‐free conditions. Mice were kept under a 12‐h light, 12‐h dark cycle with food and water ad libitum. All animal experiments were carried out in accordance with the policies of the state of Baden‐Württemberg under license numbers G13‐093 and G16‐60.
Preparation of brain extract for intracerebral injections
Brains from aged (5×FAD 51‐week‐old/APP23 84‐week old) amyloid‐depositing transgenic mice or young 5×FAD mice (14‐week old) were used. After removal of the cerebellum, the brain samples were immediately fresh frozen on dry ice and stored at −80°C until use. Samples were homogenized at 10% (w/v) in sterile phosphate‐buffered saline (PBS, Lonza), sonicated 3 × 5 s (70% amplitude) and centrifuged at 3000 g for 5 minutes. The supernatant was stored until use at −80°C.
Immunoprecipitation of brain extract
For immunoprecipitation (IP), 400 µg of total protein from brain extract (10% w/v) was diluted in a total volume of 1000 µL. G sepharose beads (GE Healthcare) were washed 3× with PBS and 50 µL per 500‐µL diluted brain homogenate were incubated with 1 µL of Aβ antibody 3552 (kindly provided by E. Kremmer, Ludwig Maximilians University, Munich, Germany) 33 or amyloid oligomeric‐specific antibody A11 (A11, Millipore) overnight by 4°C shaking head over heel. The supernatant was reused. This experiment was repeated until the remaining oligomers resulted in greatly reduced band intensities as detected by western blot probed with Aβ antibody 6E10 (Covance). The cleared supernatant was used for the experiments.
CSF collection
CSF samples were taken as described previously, with slight modification 1, 5. CSF samples were taken from the cisterna magna of anesthetized mice using glass micropipettes (Stoelting). Samples were checked for blood contamination by visual examination, and those with blood contamination were excluded from the analysis. The samples were frozen immediately at −80°C.
Stereotaxic surgery
Mice were anesthetized via intraperitoneal injection of a mixture of ketamine (100 mg/kg body weight) and xylazine (10 mg/kg body weight) in saline. For bilateral stereotactic injections of brain homogenates, a Hamilton syringe needle was placed into the hippocampus (AP‐2.3 mm; lateral ± 2.0 mm; DV 2.0 mm) of 6‐week‐old male 5×FAD mice or 24‐week‐old male APP23 mice. A volume of 2.0 µL (or as indicated) was injected at an injection speed of 1.25 µL/min and the needle was kept in place for additional 2 minutes before withdrawal. The surgical site was cleaned with sterile saline and the incision sutured. Mice were monitored until recovery from anesthesia.
Passive immunization
For passive immunization 6‐week‐old male 5×FAD mice were stereotactically injected with 10% w/v aged brain homogenate. The weekly antibody treatment with either 100 µL of amyloid oligomeric antibody A11 (Millipore) or with 100‐µL rabbit IgG as control, started 2 weeks after stereotactic injections of brain homogenates. Mice were sacrificed 1 week after the last treatment.
Histology
Mice were deeply anesthetized with a mixture of ketamine (300 mg per kg) and xylazine (20 mg per kg) and transcardially perfused with ice‐cold PBS and 4% paraformaldehyde. Brains were removed and postfixed for 24 h in 4% paraformaldehyde (Roti®‐Histofix, Roth), followed by 48 h in 30% sucrose (in PBS). Frozen brains were cut into 25‐µm‐thick coronal serial sections on a sliding microtome (SM2000R, Leica Biosystems, Wetzlar, Germany) and collected in 5% Gycerol (in PBS). Immunofluorescence staining was performed using the following antibodies: rabbit polyclonal antibody 3552 specific for Aβ 1‐40 (kindly provided by E. Kremmer, Ludwig Maximilians University, Munich, Germany; diluted 1:3000), rabbit anti‐doublecortin (DCX; 1:5000; abcam), mouse monoclonal anti‐NeuN (1:200; Millipore) and DAPI (Roche) was used as a counterstain. Dense‐core plaques were stained with Thiazine Red (Sigma Aldrich; 2 µM solution in PBS for 5 minutes at RT followed by 3 × 5 minutes washes). Appropriate secondary antibodies conjugated to Alexa 488 or 555 (1:1500) were used.
Assessment of Aβ plaque load and cell analysis
Fluorescent images of brain slices were taken using a Zeiss Axio Imager M2M microscope with a CCD camera. Every 10th brain section was immunostained and Aβ load was determined as a fraction of the hippocampus immunoreactive for Aβ using the imaging software ImageJ (National Institutes of Health freeware). Cell number was quantified by counting the number of immunoreactive cells for DCX in the dentate gyrus in a 1 in 10 series of sections throughout the whole hippocampus; three to five animals per group and six sections per animal were analyzed. The hippocampal area was defined based on the mouse brain atlas 26. All analyses were conducted in a blinded manner. Confocal images of NeuN‐positive cells in the dentate gyrus were taken with an Olympus confocal microscope (Fluoview FV 1000).
Quantification of Aβ by immunoassay
Human Aβ was measured by a sandwich electrochemiluminescence (ECL)‐linked immunoassay using the Meso Scale Discovery Sector Imager 2400, as described previously 24. For more sensitive detection of Aβ species, the MSD Triplex sandwich immunoassay was used 25. All samples were measured in duplicates.
Immunoblot analysis of injected brain homogenates and CSF
CSF and Brain homogenates used for injection and immunoprecipitation were subjected to SDS‐PAGE using 10%–20% Tris‐tricine gels (Invitrogen). Proteins were transferred onto a nitrocellulose membrane (0.1‐µm pore size; Protran; Whatman) and probed with antibodies specific to human Aβ (6E10, Covance, 1:2000 dilution), and (3552; 1:2000 dilution) or with an antibody specific to oligomers (A11, Millipore, 1:1000 dilution) and visualized using Amersham ECL plus (GE Healthcare).
Dot blot assay
Two microliters of each sample were transferred onto a nitrocellulose membrane (0.1‐μm pore size; Whatman). Membranes were allowed to air dry and subsequently immunoblotted using antibodies 6E10 and A11 and corresponding HRP‐conjugated secondary antibodies. Antibody reactivity was visualized using the ECL reagent. Bioluminescence was assessed in a Chemidoc MP imaging system (Bio‐Rad, Munich, Germany).
Statistical analysis
Data sets were tested for normality with the Kolmogorov–Smirnov test and the appropriate parametric or nonparametric statistical comparison test was carried out using GraphPad Prism software, version 6.2. Depending on the outcome, the test used was either parametric or non‐parametric, i.e. one‐way ANOVA or the Kruskal–Wallis test, the t test or Mann–Whitney test. Significance level α was set at 0.05. Reported values are means ± S.E.M.
Results
Depletion of Aβ oligomers delays the initiation of exogenously induced amyloidosis
We used an indirect approach and injected 6‐week‐old predepositing 5×FAD mice 23 with Aβ‐containing brain homogenate (10% w/v) followed by weekly treatment with either the oligomer‐specific antibody A11 13 or a polyclonal IgG serum as control to determine the role of Aβ oligomers in the seed‐induced Aβ plaque development (Figure 1A). This passive immunization treatment regimen significantly reduced the seed‐induced plaque load, although amyloid induction was still observed and not completely abolished (Figure 1B). In an alternative approach, we injected 5×FAD mice either with Aβ‐containing brain homogenate from aged 5×FAD mice, with brain homogenate immunodepleted with A11 for Aβ oligomers or with brain homogenate immunodepleted for Aβ. Immunoblotting and dot blot assays confirmed the presence of total protein and monomeric Aβ in both inoculates but significantly less oligomeric Aβ in the A11 immunodepleted extract and almost no oligomeric and monomeric Aβ in the Aβ‐immunodepleted extract (Figure 1C). Again, quantification of Aβ immunoreactivity revealed that the A11‐immunodepleted extract yielded a significantly less induced Aβ deposition than the complete brain homogenate, whereas the uninjected or wt injected 5×FAD controls at the same age were free of Aβ deposits (Figure 1D). Similar findings were obtained when we injected APP23 mice 30 with aged APP23 transgenic brain extract or its immunodepleted versions (Figure 1E,F), suggesting that the almost complete absence of oligomeric Aβ intermediates limits Aβ seeding.
Figure 1.
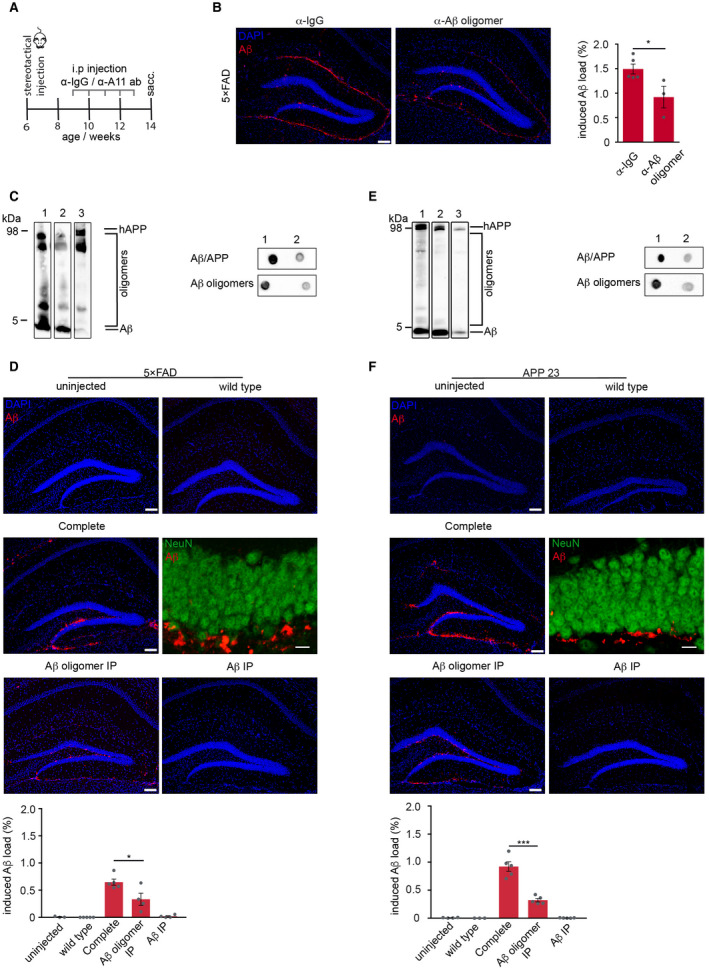
Reduction of Aβ oligomers diminishes exogenously induced Aβ deposition. A. Schematic representation of the passive immunization paradigm. 6‐week‐old 5×FAD transgenic mice were intracerebrally injected with Aβ‐containing brain homogenate (10% w/v) from aged mice, followed by weekly intraperitoneal administration of either A11 antibody or IgG for 5 weeks. B. Immunofluorescence staining against Aβ with antibody 3552 (red) and quantification of hippocampal‐induced Aβ load revealed significantly reduced Aβ load in mice passively immunized with A11 antibody compared to control immunized mice with IgG (n = 3–5 mice per group) Mann–Whitney test: *P = 0.036. C. Immunoblot analysis with Aβ‐specific antibody 6E10 of brain homogenate from 5×FAD that was used for injections. D. Reduction of Aβ oligomers in the injected brain homogenate significantly reduced seed‐induced Aβ deposition in 5×FAD tg mice. E. Immunoblot analysis with Aβ‐specific antibody 6E10 of brain homogenate from APP23 transgenic mice that was used for injections. Note that Aβ oligomers were significantly reduced in the aged brain homogenate after several rounds of immunoprecipitation with A11 antibody (lane 2) and total Aβ after immunoprecipitation with antibody 3552 (lane 3). Representative dot blots confirmed the low abundance of total Aβ/APP and Aβ oligomers in the A11‐depleted brain homogenate. F. Reduction of Aβ oligomers in the injected brain homogenate significantly reduced seed‐induced Aβ deposition in APP23 mice. Injection of wt brain homogenate or Aβ‐depleted brain homogenate failed to induce seeding in 5×FAD and APP23 transgenic mice. Confocal images of NeuN‐positive neurons (green) and Aβ (red) confirm that the seed‐induced Aβ deposits are located extracellularly. Unpaired t‐test for 5×FAD mice (n = 4–5 mice per group): *P = 0.03 and for APP23: ***P = 0.0002. Scale bar in (B, D and F) indicates 100 µm in the overview and 10 µm in the higher magnification images in (D and F).
Time course of Aβ seeding
Since seeding was shown to be a time‐dependent process 20, we next performed a time course analysis. The first seeded Aβ plaques were observed 6 weeks post‐injection in 80% of 5×FAD mice that had been injected with complete brain extract but were mostly absent in the ones injected with immunodepleted brain extract (Figure 2A, C and E) and completely absent in uninjected 5×FAD mice of the same age (Supplementary Figure 1A,B), indicating that the lack of Aβ oligomers in the inoculate delays the onset of seeding. Interestingly, at longer incubation times, the formerly significant difference in the amount of seeding was diminished and disappeared over time (Figure 2A,C). The same phenomenon was observed in APP23 transgenic mice (Figure 2B,C). To account for the differences in the amount of total Aβ in the brain extract measured by ELISA (5×FAD: ratio complete/Aβ oligomer IP: 4.5; APP23: ratio complete/Aβ oligomer IP: 3), we adjusted the volumes of the A11‐immunodepleted brain extract accordingly. However, this adjustment neither changed the amount of induced Aβ deposits (Figure 2D) nor did it reach the same percentage of visible seeding pattern (complete brain homogenate: 80% vs. increased volume of Aβ oligomer IP: 40%) at 6 weeks post‐injection (Figure 2E). Thus, larger volumes of A11‐immunodepleted brain homogenate failed to accelerate Aβ seeding.
Figure 2.
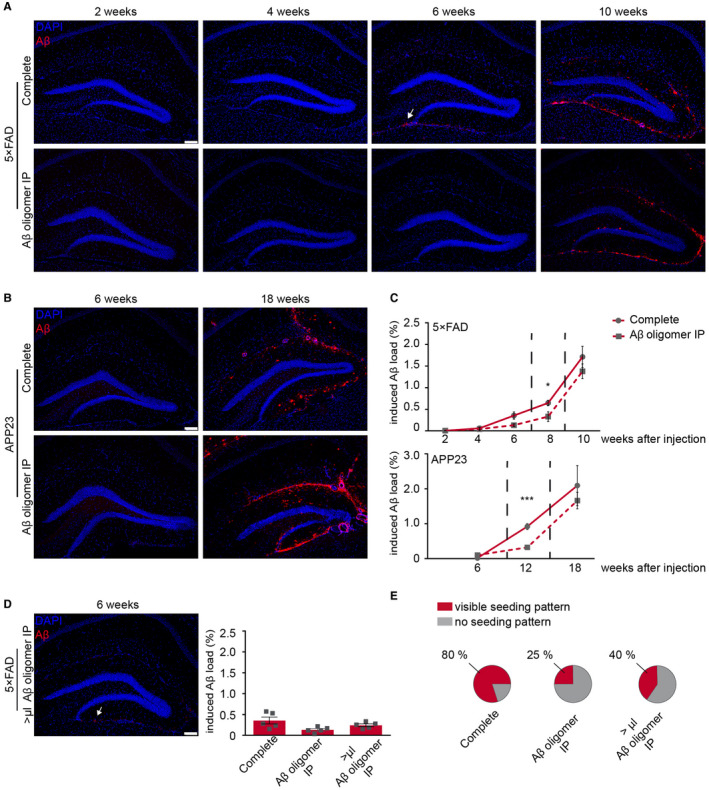
Seed‐induced Aβ load aligns with longer incubation time. A. 6‐week‐old 5×FAD transgenic mice were injected with complete brain homogenate or Aβ oligomer depleted brain homogenate and incubated for 2, 4, 6 or 10 weeks. First seed‐induced Aβ deposits appeared at 6 weeks post‐injection in mice injected with complete brain homogenate (white arrow). B. 24‐week‐old APP23 transgenic mice were injected with complete brain homogenate or Aβ oligomer‐depleted brain homogenate and incubated for 6 or 18 weeks. C. The induced Aβ load was nearly similar 10 weeks after injection in 5×FAD mice (upper graph) and was not significantly different 18 weeks after injection in APP23 mice (lower graph). The cohort of mice for the 8‐week time point is reused from Figure 1D,F as indicated by the dashed lines. D. The injection of a higher volume (>μL) of A11‐depleted brain homogenate shows an earlier onset of induced Aβ deposition (white arrow) and a slight but not significant increase at 6 weeks post‐injection (n = 5 mice per group; One‐way ANOVA: F(2,12) = 2.495 P = 0.1241; n.s.; Tukey's multiple comparisons test n.s.). E. In 80% of 5×FAD injected with complete brain homogenate a typical seeding pattern was visible compared to only 25% when A11‐depleted homogenate was injected and 40% when a higher volume (>μL) of the latter brain homogenate was intracerebrally injected. Scale bar in (A, B andD) represents 100 μm. Indicated is the mean ± S.E.M.
Seeding capacity of Aβ oligomers in CSF and young brain homogenate
Previous studies have proposed Aβ peptides, in particular soluble forms of Aβ as seeding factor 6, 15, 20, 29, but the exact Aβ seeding species still remains elusive. In order to circumvent the use of artificial synthetic assemblies, we focused on the cerebrospinal fluid (CSF) and brain homogenate of young APP transgenic mice 19 as natural sources for Aβ oligomers and assessed their potencies to seed Aβ plaques. We found excessive amounts of Aβ oligomers but only negligible monomeric Aβ in both preparations and especially in CSF (Figures 3A,C and 4A). However, despite the high abundance of Aβ oligomers in both inoculates, no seeded plaques were observed even when the injection volumes were doubled (Figures 3B,D and 4B), indicating that oligomeric Aβ alone fails to induce seeding, thus implying the importance of concomitant monomeric Aβ bioavailability.
Figure 3.
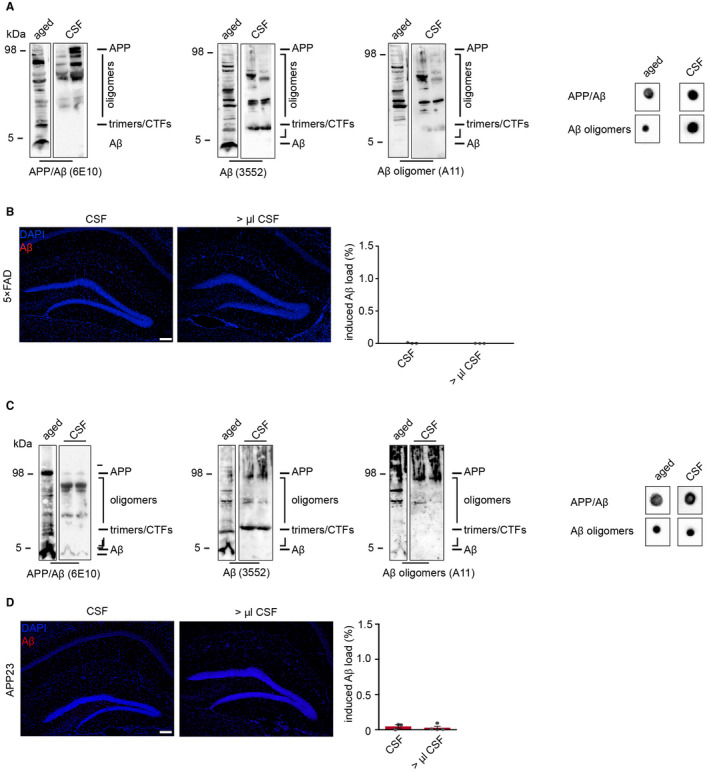
Seeding capacity of Aβ oligomers from murine CSF. A. CSF from young, 9‐week‐old 5×FAD mice revealed the presence of Aβ oligomers but no monomeric Aβ that was confirmed by dot blot analysis. B. Despite these high levels of oligomeric Aβ in the CSF of young mice, this preparation failed to induce Aβ deposition. C. Immunoblot and dot blot analyses confirmed the abundance of oligomeric Aβ in the CSF of 21‐week‐old APP23 transgenic mice as well (each lane represents a different CSF probe) when compared to aged brain homogenate. D. Undiluted murine CSF from APP23 mice was injected into young, predepositing APP23 transgenic mice and analyzed after a 3‐month incubation period. Neither CSF from APP23 mice nor higher volumes of the CSF revealed any seed‐induced Aβ depositions. Scale bar in (B, D) indicates 100 µm. (B) n(2.5 µL CSF) = 3; n(5 µL CSF) = 3. (D) n(2.5 µL CSF) = 3; n(5 µL CSF) = 4. Indicated is the mean ± S.E.M.
Figure 4.
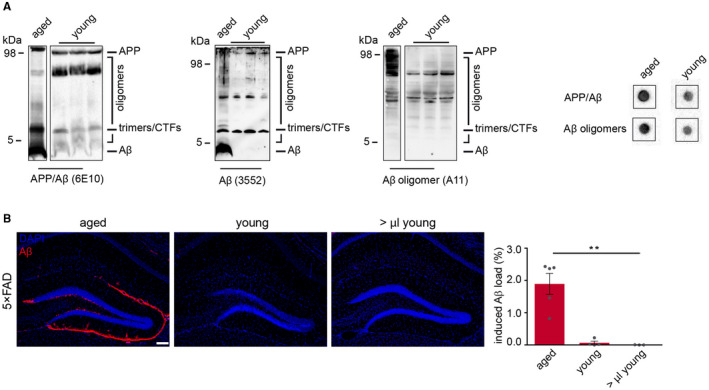
Seeding capacity of Aβ oligomers from young 5×FAD mice. A. Immunoblot analysis using SDS‐PAGE and dot blot assay (right panel) revealed the presence of Aβ oligomers but no monomeric Aβ in brain homogenates of young, 16‐week‐old 5×FAD transgenic mice. Membranes were probed with hAβ/APP‐specific antibody 6E10, 3552 or Aβ oligomer‐specific antibody A11. The presence of Aβ oligomers in young brain homogenate was also confirmed by dot blot analysis. B. Immunofluorescence staining against Aβ with antibody 3552 (red) showed robust induced Aβ deposition when 10% w/v of aged brain homogenate was injected, whereas no amyloid deposition was observed with young brain homogenate even with higher volumes. Scale bar in (B) represents 100 μm. One‐way ANOVA F(2,9) = 21.09; P = 0.0004; Bonferroni's multiple comparisons test **P< 0.0021). n(aged brain homogenate) = 5; n(2.5 μL young brain homogenate) = 4; n(5.0 μL young brain homogenate) = 3. Indicated is the mean ± S.E.M.
Spreading of induced Aβ deposits is independent of Aβ oligomers and has no effect on newborn neurons
Since exogenously induced cerebral β‐amyloidosis in APP transgenic mice was shown to spread within the limbic connectome and use neuronal pathways 35, we next compared the efficiency of complete vs. A11‐immunodepleted brain homogenate to induce Aβ spreading and the ability to induce dense‐core plaques. To address this question, we first analyzed the emergence of Aβ deposits in the dorsal subiculum and entorhinal cortex of 5×FAD mice that had been inoculated with Aβ seeds into the hippocampal formation. After 10 weeks of incubation, the amount of amyloid induction was remarkably similar and was not significantly different between both injection groups in the subiculum as well as in the entorhinal cortex (Supplementary Figure 2A). This was also true for the number of seeded Thiazine Red‐positive dense‐core plaques in the hippocampus (Supplementary Figure 2B), suggesting that the formation of compact plaques was independent of the presence of A11‐reactive oligomers in the inoculant.
Due to the cytotoxicity of Aβ oligomers 32, we finally assessed the impact of A11‐reactive oligomers in the inoculate on newborn neurons and examined the number of DCX‐positive cells at 2, 8 and 10 weeks post‐injection. As expected, we found a reduction of DCX‐positive cells over time 36, but there was no significant difference between the two brain homogenate preparations (Supplementary Figure 3), which is consistent with a recent publication describing little or no cytotoxic activity of high molecular weight oligomers 34.
Discussion
In summary, we here report the importance of Aβ oligomers for the early and rapid seed‐induced aggregation process as they represent the first nucleation step in the absence of Aβ fibrils. As soon as fibrils are formed in the diseased brain, further additional aggregation and the growth of Aβ deposits follows a pathway independent of A11‐reactive oligomers. The formation of these oligomers could therefore be determined as the time‐limiting step. In the present study, a significant reduction of these A11 oligomers in the brain homogenate and the immunization with A11 antibody‐limited Aβ seeding and was shown to influence the initial step of the Aβ polymerization process by delaying the development of seed‐induced Aβ plaques. However, A11‐depleted brain homogenate retained its seeding capacity, although to a lesser extent, suggesting that the remaining monomers are sufficient to induce Aβ deposition. However, due to our immunoprecipitation procedure, we can't rule out completely that Aβ fibrils might play a role as well. Interestingly, with longer incubation times, this difference in the amount of seeding disappeared over time indicating that once aggregation starts, Aβ oligomers might only play a minor role during the later aggregation phase. These results are in agreement with studies demonstrating that polymerization of Aβ aggregates can occur through a secondary pathway that overtakes primary nucleation as the relevant source of new oligomers 2, 18, supporting the notion that A11‐reactive oligomers seem to be important in the early initial phase of the seeding process. However, despite the high abundance of Aβ oligomers in the CSF and brain homogenate of young APP transgenic mice, those preparations failed to induce Aβ seeding. Thus, monomeric species of Aβ might be sufficient for the initiation of Aβ plaque formation which is in line with a previous finding by another group showing formaldehyde fixed brain homogenate that contains predominantly monomeric Aβ displayed plenty of seeding capacity 8. Nevertheless, we can't exclude the possibility that there is an as yet “unknown” factor and/or Aβ fibrils in the brain homogenate that is missing in the brain extract of young mice as well as in the CSF. Another study with similar results attributed the lack of N‐terminally truncated Aβ species and smaller Aβ particles in the CSF to its failure to induce Aβ seeding 9. Future studies should include a detailed biochemical investigation of both extracts in comparison to aged brain homogenate. To finally address the question if monomers alone are sufficient to induce Aβ seeding, it will be important to study brain homogenate with significantly reduced monomer content.
In vitro experiments showed that oligomer growth follows a defined mechanism that is distinct from fibril growth and independent of the monomer concentration by adding only one monomer at a time 3, 22. Thus, the missing part of preformed oligomers will most likely be replenished by newly generated oligomers, a process that is known to be less dependent on monomer concentration but on time, finally extending the lag phase and shifting the aggregation curve to the right. The fibrils formed afterward by nucleated conformational conversion of A11‐reactive oligomers might grow rapidly due to the increased access of Aβ monomers 17. The monomers, as the fundamental building blocks for all subsequent intermediate forms of Aβ peptides in the aggregation process on the way to insoluble mature fibrils and finally to amyloid plaques, should hence be able to form the initial nucleus that is needed for seed‐induced Aβ deposition.
Ethics approval
All animal studies were reviewed, approved and carried out in accordance with the policies of the state of Baden‐Württemberg under license numbers G13‐093 and G16‐60.
Authors contributions
N.K. and M.M.‐L. conceived and designed the study. N.K. contributed to all aspects of the experiments and data analysis. N.S., S.Z.‐W., C.W. and C.A.‐A. assisted with the experimental work. B.N. carried out the ELISA experiments. N.K., M.P., C.H. and M.M.‐L. discussed the results. M.M.‐L. wrote the manuscript with help from N.K. and further input from all co‐authors. The APP23 mice were a kind gift of Novartis, Basel, Switzerland. All authors read and approved the final manuscript.
Competing Financial Interests
C.H. is an advisor of F. Hoffmann‐La Roche and has a collaboration agreement with DENALI. All other authors declare that they have no conflict of interest.
Supporting information
Figure S1. Induced Aβ deposition. (A) Fluorescence microscopy of Aβ (red) and DAPI (blue) in 5×FAD mice either 6 weeks or (B) 10 weeks post‐injection with transgenic brain homogenate, or in uninjected control 5×FAD animals at the same age. (C) Fluorescence microscopy of Aβ (red) and DAPI (blue) in APP23 mice 18 weeks post‐injection with transgenic brain homogenate or in uninjected control APP23 mice at the same age. Scale bars represent 100 μm.
Figure S2. Formation of dense core plaques after injection of oligomer‐depleted brain homogenate. (A) Thiazine Red staining to detect dense core plaques revealed similar numbers of plaques in the hippocampus (a; insert i and iii) and entorhinal cortex (a; insert ii and iv) of 5×FAD mice 10 weeks after injection. Mann‐Whitney test: n.s. Scale bar in (A) represents 500 μm and in the inserts 100 μm. Indicated is the mean ± S.E.M per group. (B) The number of dense core plaques within the seeding pattern of the dentate gyrus was not significantly different between both injection groups (complete versus oligomer‐depleted brain homogenate; n = 4). Inserts in the middle panel confirmed the existence of dense‐core plaques within the seeding area by co‐labeling of Aβ plaques (with antibody 3552 in green) and thiazinred. Mann‐Whitney test: n.s. Scale bar in (B) represents 100 μm. Indicated is the mean ± S.E.M per group.
Figure S3. Impact of Aβ oligomers on neuronal precursor cells. Fluorescence microscopy of DCX (red) and DAPI (blue) at 2, 8 and 10 weeks post‐injection. Quantification of DCX‐positive cells in the dentate gyrus of 5×FAD transgenic mice either injected with complete homogenate or oligomer‐depleted brain homogenate revealed a decrease in DCX positive cells over time, but no significant difference between both injection groups was observed (complete brain homogenate n = 5 for all time points; Aβ oligomer IP: n(2, 10 weeks) = 4; n(8 weeks) = 3; Kruskal‐Wallis test ***0,0004; Dunn's multiple comparisons test). Scale bar represents 100 μm and in the inserts 20 μm. Indicated is the mean ± S.E.M per group.
Acknowledgements
We are particularly grateful to J. Göldner, D. Bleckmann and T. Bachhuber for technical assistance and G. Fritz for advice and discussions.
This work was supported by the Fill in the Gap fellowship (Medical Faculty Freiburg) to N.K., the Emmy Noether Program of the Deutsche Forschungsgemeinschaft (Grant number: ME 3542/1‐1 to M.M.L.) and a grant of the Deutsche Forschungsgemeinschaft (Grant number: ME 3542/2‐1 to M.M.L.).
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
- 1. Bachhuber T, Katzmarski N, McCarter JF, Loreth D, Tahirovic S, Kamp F et al (2015) Inhibition of amyloid‐beta plaque formation by alpha‐synuclein. Nat Med 21:802–807. [DOI] [PubMed] [Google Scholar]
- 2. Cohen SI, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L et al (2013) Proliferation of amyloid‐beta42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci U S A 110:9758–9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Collins SR, Douglass A, Vale RD, Weissman JS (2004) Mechanism of prion propagation: amyloid growth occurs by monomer addition. PLoS Biol 2:e321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. DeMattos RB, Bales KR, Parsadanian M, O'Dell MA, Foss EM, Paul SM, Holtzman DM (2002) Plaque‐associated disruption of CSF and plasma amyloid‐beta (Abeta) equilibrium in a mouse model of Alzheimer's disease. J Neurochem 81:229–236. [DOI] [PubMed] [Google Scholar]
- 5. Duran‐Aniotz C, Morales R, Moreno‐Gonzalez I, Hu PP, Fedynyshyn J, Soto C (2014) Aggregate‐depleted brain fails to induce Abeta deposition in a mouse model of Alzheimer's disease. PLoS One 9:e89014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Forny‐Germano L, e Silva NM, Batista AF, Brito‐Moreira J, Gralle M, Boehnke SE et al (2014) Alzheimer's disease‐like pathology induced by amyloid‐beta oligomers in nonhuman primates. J Neurosci 34:13629–13643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fritschi SK, Cintron A, Ye L, Mahler J, Buhler A, Baumann F et al (2014) Abeta seeds resist inactivation by formaldehyde. Acta Neuropathol 128:477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fritschi SK, Langer F, Kaeser SA, Maia LF, Portelius E, Pinotsi D et al (2014) Highly potent soluble amyloid‐beta seeds in human Alzheimer brain but not cerebrospinal fluid. Brain 137(Pt 11):2909–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353–356. [DOI] [PubMed] [Google Scholar]
- 10. Jucker M, Walker LC (2013) Self‐propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kane MD, Lipinski WJ, Callahan MJ, Bian F, Durham RA, Schwarz RD et al (2000) Evidence for seeding of beta ‐amyloid by intracerebral infusion of Alzheimer brain extracts in beta ‐amyloid precursor protein‐transgenic mice. J Neurosci 20:3606–3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300:486–489. [DOI] [PubMed] [Google Scholar]
- 13. Koffie RM, Meyer‐Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia‐Alloza M et al (2009) Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A 106:4012–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Langer F, Eisele YS, Fritschi SK, Staufenbiel M, Walker LC, Jucker M (2011) Soluble Abeta seeds are potent inducers of cerebral beta‐amyloid deposition. J Neurosci 31:14488–14495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee J, Culyba EK, Powers ET, Kelly JW (2011) Amyloid‐beta forms fibrils by nucleated conformational conversion of oligomers. Nat Chem Biol 7:602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee EB, Leng LZ, Zhang B, Kwong L, Trojanowski JQ, Abel T, Lee VM (2006) Targeting amyloid‐beta peptide (Abeta) oligomers by passive immunization with a conformation‐selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J Biol Chem 281:4292–4299. [DOI] [PubMed] [Google Scholar]
- 17. Liu P, Reed MN, Kotilinek LA, Grant MK, Forster CL, Qiang W et al (2015) Quaternary structure defines a large class of amyloid‐beta oligomers neutralized by sequestration. Cell Rep 11:1760–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maia LF, Kaeser SA, Reichwald J, Hruscha M, Martus P, Staufenbiel M, Jucker M (2013) Changes in amyloid‐beta and Tau in the cerebrospinal fluid of transgenic mice overexpressing amyloid precursor protein. Sci Transl Med 5:194re2. [DOI] [PubMed] [Google Scholar]
- 19. Meyer‐Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E et al (2006) Exogenous induction of cerebral beta‐amyloidogenesis is governed by agent and host. Science 313:1781–1784. [DOI] [PubMed] [Google Scholar]
- 20. Morales R, Bravo‐Alegria J, Duran‐Aniotz C, Soto C (2015) Titration of biologically active amyloid‐beta seeds in a transgenic mouse model of Alzheimer's disease. Sci Rep 5:9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nguyen PH, Li MS, Stock G, Straub JE, Thirumalai D (2007) Monomer adds to preformed structured oligomers of Abeta‐peptides by a two‐stage dock‐lock mechanism. Proc Natl Acad Sci U S A 104:111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J et al (2006) Intraneuronal beta‐amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci 26:10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Page RM, Baumann K, Tomioka M, Perez‐Revuelta BI, Fukumori A, Jacobsen H et al (2008) Generation of Abeta38 and Abeta42 is independently and differentially affected by familial Alzheimer disease‐associated presenilin mutations and gamma‐secretase modulation. J Biol Chem 283:677–683. [DOI] [PubMed] [Google Scholar]
- 24. Page RM, Gutsmiedl A, Fukumori A, Winkler E, Haass C, Steiner H (2010) Beta‐amyloid precursor protein mutants respond to gamma‐secretase modulators. J Biol Chem 285:17798–17810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paxinos GF, KBJ (2001) The Mouse Brain in Stereotaxic Coordinates, 2nd edn. Academic Press: San Diego, CA. [Google Scholar]
- 26. Rasool S, Albay R 3rd, Martinez‐Coria H, Breydo L, Wu J, Milton S et al (2012) Vaccination with a non‐human random sequence amyloid oligomer mimic results in improved cognitive function and reduced plaque deposition and micro hemorrhage in Tg2576 mice. Mol Neurodegener 7:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rasool S, Martinez‐Coria H, Wu JW, LaFerla F, Glabe CG (2013) Systemic vaccination with anti‐oligomeric monoclonal antibodies improves cognitive function by reducing Abeta deposition and tau pathology in 3xTg‐AD mice. J Neurochem 126:473–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stohr J, Watts JC, Mensinger ZL, Oehler A, Grillo SK, DeArmond SJ et al (2012) Purified and synthetic Alzheimer's amyloid beta (Abeta) prions. Proc Natl Acad Sci U S A 109:11025–11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. De Strooper B, Karran E (2016) The cellular phase of Alzheimer's disease. Cell 164:603–615. [DOI] [PubMed] [Google Scholar]
- 30. Sturchler‐Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S et al (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease‐like pathology. Proc Natl Acad Sci U S A 94:13287–13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS et al (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long‐term potentiation in vivo . Nature 416:535–539. [DOI] [PubMed] [Google Scholar]
- 32. Walsh DM, Selkoe DJ (2007) A beta oligomers—a decade of discovery. J Neurochem 101:1172–1184. [DOI] [PubMed] [Google Scholar]
- 33. Yamasaki A, Eimer S, Okochi M, Smialowska A, Kaether C, Baumeister R et al (2006) The GxGD motif of presenilin contributes to catalytic function and substrate identification of gamma‐secretase. J Neurosci 26:3821–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang T, Li S, Xu H, Walsh DM, Selkoe DJ (2017) Large soluble oligomers of amyloid beta‐protein from Alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate. J Neurosci 37:152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ye L, Hamaguchi T, Fritschi SK, Eisele YS, Obermuller U, Jucker M, Walker LC (2015) Progression of seed‐induced Abeta deposition within the limbic connectome. Brain Pathol 25:743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ziegler‐Waldkirch S, Sauer JF, Erny D, Savanthrapadian S, Loreth D, Katzmarski N et al (2018) Seed‐induced Abeta deposition is modulated by microglia under environmental enrichment in a mouse model of Alzheimer's disease. EMBO J 37:167–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Induced Aβ deposition. (A) Fluorescence microscopy of Aβ (red) and DAPI (blue) in 5×FAD mice either 6 weeks or (B) 10 weeks post‐injection with transgenic brain homogenate, or in uninjected control 5×FAD animals at the same age. (C) Fluorescence microscopy of Aβ (red) and DAPI (blue) in APP23 mice 18 weeks post‐injection with transgenic brain homogenate or in uninjected control APP23 mice at the same age. Scale bars represent 100 μm.
Figure S2. Formation of dense core plaques after injection of oligomer‐depleted brain homogenate. (A) Thiazine Red staining to detect dense core plaques revealed similar numbers of plaques in the hippocampus (a; insert i and iii) and entorhinal cortex (a; insert ii and iv) of 5×FAD mice 10 weeks after injection. Mann‐Whitney test: n.s. Scale bar in (A) represents 500 μm and in the inserts 100 μm. Indicated is the mean ± S.E.M per group. (B) The number of dense core plaques within the seeding pattern of the dentate gyrus was not significantly different between both injection groups (complete versus oligomer‐depleted brain homogenate; n = 4). Inserts in the middle panel confirmed the existence of dense‐core plaques within the seeding area by co‐labeling of Aβ plaques (with antibody 3552 in green) and thiazinred. Mann‐Whitney test: n.s. Scale bar in (B) represents 100 μm. Indicated is the mean ± S.E.M per group.
Figure S3. Impact of Aβ oligomers on neuronal precursor cells. Fluorescence microscopy of DCX (red) and DAPI (blue) at 2, 8 and 10 weeks post‐injection. Quantification of DCX‐positive cells in the dentate gyrus of 5×FAD transgenic mice either injected with complete homogenate or oligomer‐depleted brain homogenate revealed a decrease in DCX positive cells over time, but no significant difference between both injection groups was observed (complete brain homogenate n = 5 for all time points; Aβ oligomer IP: n(2, 10 weeks) = 4; n(8 weeks) = 3; Kruskal‐Wallis test ***0,0004; Dunn's multiple comparisons test). Scale bar represents 100 μm and in the inserts 20 μm. Indicated is the mean ± S.E.M per group.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.