

Abstract
SHIP2 (Src homology 2 domain‐containing inositol 5′‐phosphatase 2) belongs to the family of 5′‐phosphatases. It regulates the phosphoinositide 3‐kinase (PI3K)‐mediated insulin signalling cascade by dephosphorylating the 5′‐position of PtdIns(3,4,5)P3 to generate PtdIns(3,4)P2, suppressing the activity of the pathway. SHIP2 mouse models and genetic studies in human propose that increased expression or activity of SHIP2 contributes to the pathogenesis of the metabolic syndrome, hypertension and type 2 diabetes. This has raised great interest to identify SHIP2 inhibitors that could be used to design new treatments for metabolic diseases. This review summarizes the central mechanisms associated with the development of diabetic kidney disease, including the role of insulin resistance, and then moves on to describe the function of SHIP2 as a regulator of metabolism in mouse models. Finally, the identification of SHIP2 inhibitors and their effects on metabolic processes in vitro and in vivo are outlined. One of the newly identified SHIP2 inhibitors is metformin, the first‐line medication prescribed to patients with type 2 diabetes, further boosting the attraction of SHIP2 as a treatment target to ameliorate metabolic disorders.
Keywords: diabetes, diabetic kidney disease, insulin resistance, insulin signalling, lipid phosphatase, podocyte
1. DIABETES AND DIABETIC KIDNEY DISEASE
1.1. Diabetes and its complications
Diabetes is a heterogeneous group of diseases characterized by chronic hyperglycemia. It has classically been divided into type 1 (T1D) and type 2 diabetes mellitus (T2D) and a few rare forms of the disease. T1D is characterized by little or no production of insulin by the pancreatic beta‐cells, whereas T2D is characterized by both impaired insulin secretion and insulin resistance, leading to the inability of insulin‐sensitive muscle and adipose tissues to take up glucose. Chronic hyperglycemia leads to damage of blood vessels and consequently, diabetes is associated with both macrovascular and microvascular complications that are major causes for morbidity and mortality.1 The macrovascular complications include coronary artery disease, peripheral arterial disease and stroke, and the microvascular complications include diabetic kidney disease, neuropathy and retinopathy.1
1.2. Diabetic kidney disease
This review concentrates on diabetic kidney disease (DKD), the potentially life‐threatening complication of diabetes and the most common cause of chronic kidney disease in the developed world. As the incidence of diabetes is escalating, also the number of patients suffering from DKD is increasing.2 DKD is characterized by gradual increase in albumin excretion into urine, a decrease in glomerular filtration rate, elevated arterial blood pressure and development of glomerulosclerosis. It develops to 20%‐40% of patients with T1D and to even up to 50% of patients with T2D.3 The development of DKD is multifactorial involving hyperglycemia‐induced metabolic and hemodynamic processes that are further influenced by genetic and environmental factors. The non‐modifiable risk factors include genetics, sex, age at onset and duration of diabetes.3 Also a number of modifiable factors, including glycemic control, blood pressure, lipid abnormalities, smoking, chronic low‐grade inflammation, advanced glycation end products and lack of physical activity, affect the development of DKD.3
The pathophysiological changes in the kidney upon development of DKD involve both the tubular and glomerular compartments as well as the vasculature. An early feature of DKD is kidney hypertrophy, occurring in both glomerular and tubular compartments.4 The greatest hypertrophic change is observed in the proximal tubules. The growth of the tubules and enhanced Na+‐glucose cotransport lead to increased proximal tubule reabsorption and an increase in the glomerular filtration rate through tubuloglomerular feedback.4 Increased exposure to different diabetes‐associated metabolites may trigger various pathological pathways associated with tubulointerstitial fibrosis, inflammation, hypoxia and apoptosis that may contribute to the progression of DKD.4
1.3. Changes in the glomerular filtration barrier in DKD
Glomerulus is the central site of kidney injury in diabetes. Podocytes, the highly specialized epithelial cells, form together with the fenestrated endothelial cells and the glomerular basement membrane (GBM) the glomerular filtration barrier that prevents the passage of proteins of the size of albumin and larger from the capillary lumen to the urinary space while allowing water and small molecules to freely pass (Figure 1A). Podocytes wrap around the capillaries, with their large cell bodies extending to the urinary space, and their foot processes attaching to the underlying GBM. The neighbouring podocyte foot processes are interconnected by special cell adhesion structures called slit diaphragms (Figure 1B,C), composed of a set of transmembrane proteins and cytosolic adapter molecules linking the slit diaphragm to the underlying actin cytoskeleton and signalling networks.5 One of the central molecules of the slit diaphragm is the immunoglobulin superfamily member nephrin, shown to be essential for an intact glomerular filtration barrier as mutations in it lead to the congenital nephrotic syndrome of the Finnish type.6 The compartmentalization of the apical and basolateral domains of the podocytes by the slit diaphragm allows the expression of distinct sets of proteins in a polarized manner. As an example, podocalyxin is expressed in the apical domain of podocytes and with its negative charge, created by a glycocalyx, apparently maintains the glomerular filtration barrier open.7 More detailed molecular characterizations of podocytes and the slit diaphragm are provided in recent reviews.5, 8
Figure 1.
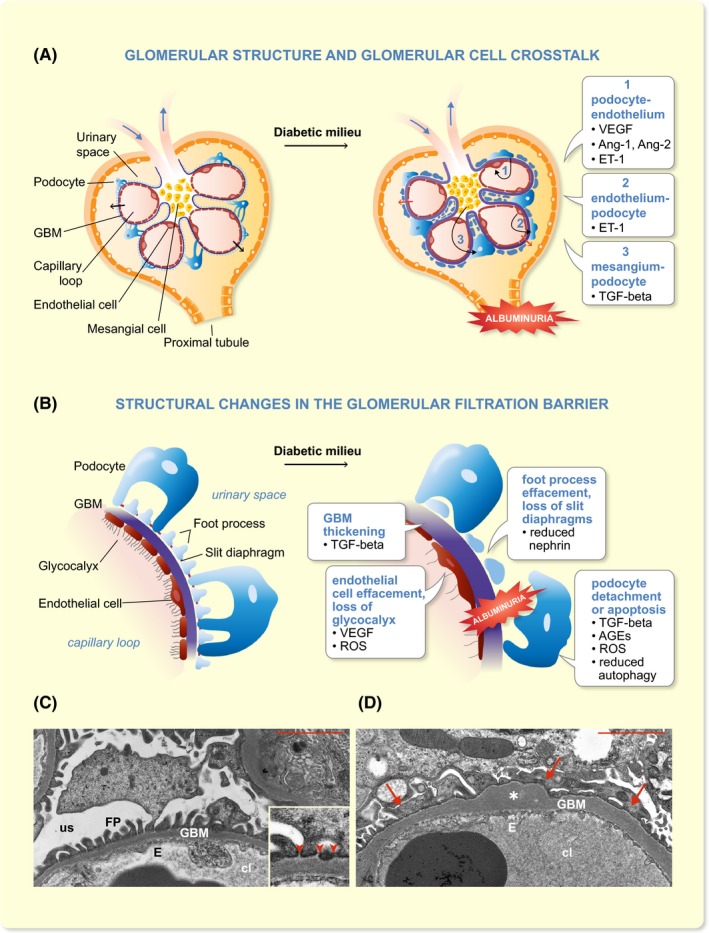
Glomerular filtration barrier and changes observed in diabetic milieu. A, A schematic cartoon of the glomerulus in health and in diabetic milieu. The cartoon also depicts the crosstalk (black, curved arrows in diabetic milieu) between podocytes and endothelium (1), endothelium and podocytes (2) and mesangial cells and podocytes (3). Ang‐1/2, angiopoietin‐1/2; ET‐1, endothelin‐1; GBM, glomerular basement membrane; TGF‐beta, transforming growth factor beta; VEGF, vascular endothelial growth factor. The black arrows indicate size‐ and charge‐selective filtration of plasma through the glomerular filtration barrier in health. Red arrows indicate leakage of albumin (albuminuria) through the damaged glomerular filtration barrier in diabetic milieu. B, A schematic cartoon of the glomerular filtration barrier and factors associated with the indicated changes in diabetic kidney disease. The glomerular filtration barrier consists of three layers, the endothelial cells with glycocalyx, the glomerular basement membrane (GBM) and the podocytes, with their foot processes interconnected with slit diaphragms. In diabetic milieu, the glomerular filtration barrier shows distinct changes associated with the development of albuminuria. These changes include glomerular basement membrane thickening, podocyte foot process and endothelial cell effacement, loss of slit diaphragms and endothelial glycocalyx, and detachment or apoptosis of podocytes. Examples of the factors involved in these changes are indicated. AGEs, advanced glycation end‐products; ROS, reactive oxygen species. C‐D, Electron microscopic images visualizing the glomerular filtration barrier in health and in diabetic kidney disease. C, An electron micrograph of the glomerular filtration barrier in a healthy mouse. The inset shows a higher magnification of the filtration barrier. Arrowheads indicate slit diaphragms. D, An electron micrograph of the glomerular filtration barrier in a mouse model of diabetes (E1‐DN mouse; the image is similar to fig. 6b,c in21), showing irregular thickening and bulging of the GBM and foot process effacement. cl, capillary loop; E, endothelial cell; FP, foot process; GBM, glomerular basement membrane; us; urinary space; asterisk, GBM thickening and bulging; arrow, podocyte foot process effacement; arrowhead, slit diaphragm. Scale bar in C and D, 2 µM
The diabetic milieu, exposing glomerular cells to high glucose, fatty acids, growth factors, cytokines and hormones, leads to glomerular hypertrophy and distinct changes in the structure and function of the glomerular filtration apparatus (Figure 1A,B). Histologically, DKD is characterized by mesangial matrix expansion and eventually glomerulosclerosis. At the electron microscopic level, the first sign of DKD is the thickening of the GBM. This is followed by effacement of the podocyte foot processes, loss of slit diaphragms and loss of podocytes by detachment or apoptosis, and progressive albuminuria (Figure 1B‐D).9 Podocytes, endothelial cells and mesangial cells are engaged in multidirectional crosstalk (illustrated in Figure 1A), and this has been shown to affect the development and progression of DKD, involving a number of pathways, as summarized below.
1.4. Podocyte‐mesangial cell axis in DKD
A central contributor to glomerular damage is hyperglycemia‐induced activation of the intrarenal renin‐angiotensin‐aldosterone system (RAAS) and production of excess angiotensin II.10 Angiotensin II can damage podocytes, endothelial cells and mesangial cells by activating pathways associated with extracellular matrix accumulation, fibrosis, inflammation and generation of reactive oxygen species (ROS).10 Increased generation of intracellular ROS leads to podocyte apoptosis.11 Podocyte loss is deleterious as podocytes are terminally differentiated cells and they lack the capacity to proliferate. The absolute number of podocytes can predict the progressive decline in renal function and proteinuria12; when podocyte loss exceeds 40%, sustained high‐grade proteinuria ensues and renal function declines.13
A central regulator of the synthesis of extracellular matrix proteins is transforming growth factor beta (TGF‐β), produced by mesangial cells.14 TGF‐β‐induced stimuli contribute to glomerular basement membrane thickening, mesangial matrix production and loss of podocytes.14 TGF‐β‐ induced podocyte loss may occur via apoptosis15 caused by activation of the mammalian target of rapamycin (mTOR) pathway, stimulation of mitochondrial oxidative phosphorylation and generation of ROS.16 TGF‐β may also induce detachment of viable podocytes via epithelium‐to‐mesenchyme transition (EMT),17 that is, by inducing downregulation of epithelial and upregulation of mesenchymal proteins, which may lead to podocyte detachment.17 Podocyte apoptosis may also be caused by hyperglycemia‐induced generation of advanced glycation end products (AGEs), which induce generation of intracellular ROS.18 Also reduction of autophagy, the lysosomal degradation pathway that maintains cellular homoeostasis, leads to podocyte apoptosis.19, 20 Of the slit diaphragm proteins, nephrin, for example, has been shown to be either downregulated21, 22 or mislocalized23, 24 in animal models of DKD, and the apical domain protein podocalyxin has been shown to be downregulated in the kidneys of diabetic rats.25 These changes contribute to podocyte foot process effacement and loss of slit diaphragms. As visualized by the above examples, a multitude of mechanisms contribute to podocyte injury and dysfunction (summarized in Figure 1B), as described in more detail in recent reviews.26, 27
1.5. Endothelial cell‐podocyte axis in DKD
Abnormal angiogenesis and endothelial cell damage in glomeruli are also central contributors to the development of DKD. New blood vessel formation has been observed in both T1D and T2D, and this contributes to glomerular hypertrophy in the early stages of diabetic kidney injury.28, 29 Morphological studies have revealed a reduction in glomerular capillary endothelial fenestrations and damage of the endothelial glycocalyx (Figure 1B).30, 31, 32 The proangiogenic factor vascular endothelial growth factor (VEGF) plays a key role in abnormal angiogenesis in the glomeruli in diabetes, and its expression correlates with new blood vessel formation in glomeruli.33 VEGF isoforms VEGFA and VEGFC are both produced by podocytes and their receptors are expressed in endothelial cells.34, 35 The expression of both VEGFA and its receptor, VEGF receptor 2, is increased in experimental diabetes.36 VEGFA is associated with glomerular endothelial cell damage and vascular dysfunction but interestingly, various VEGFA isoforms confer differing effects on the kidney.37 In line with this, a recent study revealed that upregulation of VEGFA165b isoform in mouse podocytes in vivo protects against diabetic kidney injury and albuminuria by phosphorylating VEGF receptor 2 in endothelial cells and restoring the endothelial glycocalyx damaged by diabetes.37 In experimental diabetes, VEGFC enhances the synthesis of endothelial glycocalyx and reduces albumin permeability in glomeruli, and thereby reduces the development of diabetic kidney injury.38 While animal experiments have suggested promise for antiangiogenic therapies, and especially for anti‐VEGF antibodies for treating DKD, findings in human, demonstrating development of renal injury and proteinuria as summarized in a recent review,39 have raised a concern about blocking VEGF signalling. Concomitantly, it appears that either too much or too little of VEGFA can lead to pathological changes in the kidney.39 Interestingly, inhibition of VEGFB signalling in experimental DKD models appears to protect against DKD—via non‐angiogenic mechanisms—by ameliorating renal lipotoxicity and thereby reducing pathological changes in the kidney, sensitizing podocytes to insulin and preventing renal dysfunction.40
The actions of VEGF are modulated by angiopoietin‐1, which is expressed in podocytes, while its receptor Tie2 is found in glomerular endothelium.41 In experimental diabetes, angiopoietin‐1 level is decreased concomitant to increased VEGFA and signs of diabetic kidney injury, including aberrant angiogenesis.42 Some of these changes, including proliferation of endothelial cells and albuminuria, could be reverted by targeted overexpression of angiopoietin‐1 in podocytes.42 Furthermore, the expression of angiopoietin‐2, a natural antagonist of angiopoietin‐1 signalling,43 was shown to be upregulated in podocytes and glomerular endothelial cells in an experimental model of diabetes concomitant with low expression of angiopoietin‐1.44 As podocyte‐specific overexpression of angiopoietin‐2 causes apoptosis of endothelial cells,45 the reduced angiopoietin‐1/angiopoietin‐2 ratio may be a contributing factor in the development of DKD.44 A recent study revealed increased mitochondrial dysfunction specifically in glomerular endothelial cells, but not in podocytes, in experimental streptozotocin‐induced diabetes in a mouse strain susceptible to diabetic kidney injury.46 The endothelial cells of these mice showed signs of injury and loss of fenestrae at an early stage of the disease. The mice also showed podocyte loss and albuminuria, and increased expression of endothelin‐1 receptor type A in the glomeruli, and increased circulating endothelin‐1.46 Notably, reducing mitochondrial stress pharmacologically or blocking endothelin‐1 receptor type A reduced injury of the endothelial cells and ameliorated podocyte loss and albuminuria.46
Crosstalk between the different cell types is also essential in the tubulointerstitial compartment and may play an important role in renal disease progression.47 More detailed discussion on the crosstalk between the different renal cell types is beyond the scope of this review, but the given examples underscore the complexity of the regulation of the glomerular filtration and emphasize the role of all glomerular cell types in maintaining the normal kidney function, as well as in contributing to the dysfunction of the filtration barrier in renal diseases, including DKD. The topic of glomerular cell crosstalk has been extensively covered by several recent reviews.47, 48, 49, 50
1.6. Mitochondrial dysfunction and hypoxia in DKD
The function of mitochondria and health of kidney are closely interlinked due to high energy and oxygen demands of the kidney, and thereby mitochondrial dysfunction is considered as a central contributor to the development and progression of DKD.51, 52, 53 Mitochondria regulate the levels of reactive oxygen species (ROS), cytosolic calcium and apoptosis and produce ATP for basic cellular functions as well as for cellular repair. An experimental model of diabetes in rat revealed decreased ATP content and fragmentation of mitochondria in proximal tubule cells prior to the onset of proteinuria, supporting a role for mitochondrial dysfunction as a driver in the development of DKD.54 While low levels of ROS, such as superoxide anions, are beneficial for cellular functions, excessive generation of ROS is a general sign of mitochondrial dysfunction in the kidney and toxic to the mitochondria and the cells.51, 52 Oxidative stress contributes to increased apoptosis and fibrosis, leading to declined renal function. A recent study revealed that inhibition of apoptosis signal‐regulating kinase 1 (ASK1), which induces apoptotic, fibrotic and inflammatory signalling in states of oxidative stress, reduces inflammation and fibrosis and prevents the reduction of the glomerular filtration rate in rodent models of DKD.55 This supports the hypothesis of beneficial effects of reducing oxidative stress in DKD. Another recent study searched for factors that protect from DKD and found that pyruvate kinase M2 (PKM2), a glycolytic enzyme, was elevated in patients with over 50 years duration of diabetes without signs of DKD.56 Activation of PKM2 in rodent models of diabetes normalized metabolic parameters and mitochondrial dysfunction and ameliorated kidney pathology, suggesting that increasing the glycolytic flux and mitochondrial biogenesis protects glomeruli from the toxicity caused by hyperglycemia.56 Mitochondrial function has also been linked with insulin signalling.57 The researchers showed that podocyte‐specific depletion of mitochondrial fusion‐associated protein prohibitin‐2 (PHB2) in mice results in proteinuria, renal failure and death of the animals.57 Strikingly, however, depletion of PHB2 in mice and cultured podocytes indicated that despite of the morphological changes of the mitochondria, the oxidative phosphorylation system was not impaired and also the oxygen consumption of podocytes lacking PHB2 remained normal. However, depletion of PHB2 resulted in insulin receptor‐mediated hyperactivity of mTOR signalling. In line with this, knocking out insulin receptor either alone or in combination with IGF‐1 receptor in mice depleted of PHB2 delayed the onset of renal failure without affecting the mitochondrial phenotype. The triple knockout animals remained proteinuric, though, indicating that an mTOR‐independent pathway regulates the proteinuria.57 Nevertheless, this study provides compelling evidence interconnecting mitochondrial dysfunction and insulin signalling, and emphasizes the importance of maintaining the activity of the insulin signalling cascade at an appropriate level.
Mitochondrial function and ROS production together with increased oxygen consumption are intimately linked to tissue hypoxia.53, 58 In diabetes, hyperglycemia increases the oxygen demand of the kidney and when not matched with increased oxygen delivery, intrarenal hypoxia emerges.59 Concomitantly, Franzén et al.60 measured intrarenal oxygen tension in diabetic mice, revealing that kidney hypoxia precedes albuminuria. This suggests that improving oxygen homoeostasis in the kidney could provide a way to treat DKD. Of note, however, specific targets may have highly cell‐specific consequences. Activating, for example, hypoxia inducible factor (HIF) in kidney tubules is beneficial for mitochondrial function and protects the kidney, whereas HIF activation in glomeruli leads to glomerulosclerosis and proteinuria.58 Altogether, the maintenance of mitochondrial homoeostasis, that is, mitochondrial biogenesis, fission and fusion as well as elimination of damaged, non‐functional mitochondria, is essential for maintaining normal kidney function.51 Studies have revealed that several antidiabetic drugs affect various mitochondrial functions independent of their hypoglycemic effects, including both protective and harmful effects, as highlighted by a recent review.61
1.7. Endoplasmic reticulum stress in DKD
Various diabetes‐associated factors, including hyperglycemia, proteinuria and increased levels of free fatty acids and advanced glycosylation end products, contribute to the development of endoplasmic reticulum (ER) stress that advances the progression of DKD.62, 63 ER stress markers have been shown to be upregulated in the kidneys of experimental animals as well as of patients with T2D.63 In DKD, protein misfolding and ER stress are evident in both podocytes and the tubular compartment, and can lead to apoptosis and extracellular matrix production.62 The 78‐kDa glucose‐regulated protein (GRP78) is a classical ER chaperone and a central regulator of protein folding processes, and, thus, it plays a key role in maintaining protein homoeostasis.64 A recent study revealed that high glucose treatment induces translocation of GRP78 to the plasma membrane in mesangial cells, where it binds to integrin β1, activates focal adhesion kinase and the downstream PI3K/AKT signalling.64 This enhances the synthesis of extracellular matrix proteins and glomerular fibrosis.64 Also other studies have linked insulin signalling to ER stress in diabetes. Madhusudhan et al.65 found that impaired insulin signalling together with hyperglycemia prevents the nuclear translocation of transcription factor spliced X‐box binding protein 1 (sXBP1), which resulted in maladaptive ER stress signalling in podocytes and worsened DKD. Concomitantly, the same team found that coagulation protease activated protein C (aPC) maintains ER homoeostasis via sXBP1 in podocytes.66 Specifically, they revealed that aPC can compensate for the absence of insulin receptor in podocytes in mice with experimental diabetes. This occurred by activation of the PI3K p85‐subunit‐dependent nuclear translocation of sXBP1, which prevented the maladaptive ER stress.66
1.8. Epigenetic changes in DKD
In addition to transcription factors and classical signalling cascades, genes associated with DKD are regulated by epigenetic mechanisms.67 Epigenetic changes do not alter the underlying DNA sequence but affect the accessibility of chromatin and expression of genes via DNA methylation, chromatin histone modifications and non‐coding RNAs. Environmental factors play a key role in the development of DKD, and the epigenetic mechanisms mediate the crosstalk between the genes and the environment. Epigenetic changes are also apparently involved in the so called metabolic memory, in which the gene expression pattern induced by high glucose persists even though the glycemic control has been achieved.67 The emerging developments in the epigenetic research in DKD may open new therapy options for DKD. A recent study revealed increased expression of DNA methyltransferase 1 in peripheral blood mononuclear cells in patients with DKD.68 This altered the methylation of the promoter regions of the regulators of the mTOR signalling pathway, leading to activation of the pathway and increased inflammation in the diabetic kidneys.68 Majumder et al.69 showed that modifying histone methylation in podocytes affects the course of glomerular diseases. They found that histone H3 lysine 27 trimethylation (H3K27me3) is diminished and the expression of demethylase UTX is increased in patients with DKD. This induces dedifferentiation of podocytes and accelerates the development of glomerular injury. Concomitantly, inhibiting the demethylase in diabetic db/db mice ameliorated kidney disease, further supporting the hypothesis that targeting the epigenetic events may be beneficial in treating DKD.69
1.9. Insulin signalling in DKD
Understanding the complexity of the molecular and cellular interaction networks and the exact molecular mechanisms involved in renal cell crosstalk will aid in designing new intervention strategies to slow down or stop the progression of renal diseases. At the same time, it has become apparent that the heterogeneous nature of diabetes makes treating it challenging, and the available treatment strategies often fail to stop the progression of the disease and its complications. Many of the studies above point out the potential of targeting a specific molecule or certain cellular modifications to prevent the development of DKD, yet we lack the personalized treatment options, called for due to the complex nature of the disease. A recent study describes a data‐driven cluster analysis of six variables in patients with newly characterized diabetes. The authors identified five clusters of patients with diabetes, each subgroup specified by distinct disease progression and risk of diabetic complications. Interestingly, one of the clusters, the patients most resistant to insulin, showed a high risk to develop DKD.70 In addition, previous studies have revealed that insulin resistance is associated with microalbuminuria in patients with either T1D71 or T2D72, 73 as well as in healthy people without diabetes,74 and that insulin resistance precedes and predicts the development of both microalbuminuria and nephropathy in T1D.75, 76 These data highlight the importance of insulin signalling in maintaining normal kidney function and insulin resistance as a central contributor to the development of kidney disease and albuminuria. It is also interesting to note that several of the mechanisms that participate in driving the development of DKD, as presented in the paragraphs above, converge with the insulin signalling pathway, further underlining the importance of this pathway as a central contributor to kidney health.
2. DEFECTS IN INSULIN SIGNALLING AND GLUCOSE TRANSPORTER TRAFFICKING LEAD TO THE DEVELOPMENT OF INSULIN RESISTANCE
Understanding the mechanisms leading to the development of insulin resistance is essential as insulin resistance is a key factor in the pathogenesis of diabetes and DKD, as described above.70, 77 Development of insulin resistance is caused by either reduced activity of the insulin signalling cascade or impaired trafficking of the insulin responsive glucose transporter 4 (GLUT4) to the plasma membrane.78 Insulin signalling is initiated by binding of insulin to insulin receptor leading to its activation, the consequent phosphorylation of a number of downstream substrates and activation of the phosphatidylinositol 3‐kinase (PI3K) pathway (Figure 2A). PI3K phosphorylates phosphatidylinositol 4,5‐bisphosphate (PtdIns(4,5)P2) to phosphatidylinositol 3,4,5‐trisphosphate (PtdIns(3,4,5)P3), which is a key lipid second messenger in various metabolic effects of insulin. To enhance glucose uptake, PtdIns(3,4,5)P3 transfers signals to downstream molecules including serine/threonine kinase Akt.78 In the basal state, GLUT4 resides mainly in the cytoplasm and only a small proportion is detected at the plasma membrane, but insulin‐induced activation of Akt induces rapid translocation of GLUT4‐containing vesicles to the cell surface.79, 80 The trafficking is controlled at various steps with distinct sets of proteins. Specifically, the GLUT4‐containing vesicles contain vesicle‐SNAREs (soluble N‐ethylmaleimide‐sensitive factor attachment protein receptors), which interact with target‐SNAREs on the plasma membrane. These together, further aided by sets of accessory proteins, facilitate the translocation, docking and fusion of the GLUT4‐containing vesicles with the plasma membrane leading to uptake of glucose into cells (Figure 2A).79, 80
Figure 2.
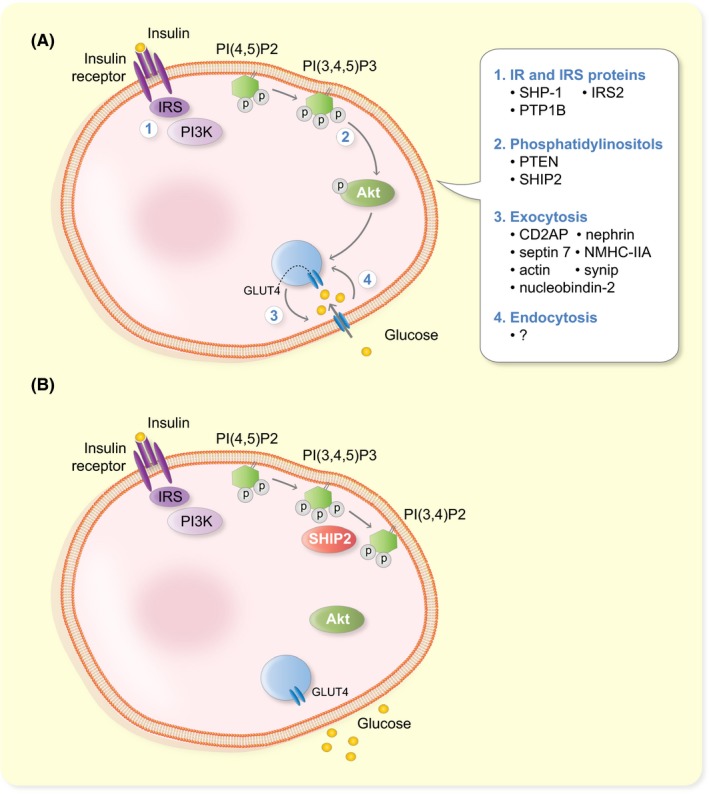
SHIP2 regulates the insulin signalling pathway. A, Simplified cartoon of the insulin signalling pathway indicating various points of regulation of its activity. Binding of insulin to its receptor leads to activation of phosphatidylinositol 3‐kinase (PI3K), phosphorylation of phosphatidylinositol 4,5‐bisphosphate (PI(4,5)P2) to phosphatidylinositol 3,4,5‐trisphosphate (PI(3,4,5)P3) and subsequent activation (phosphorylation, P) of Akt. This leads to translocation of insulin responsive glucose transporter 4 (GLUT4) to the plasma membrane and uptake of glucose into cells. The activity of the pathway can be modulated at the level of (1) the insulin receptor and insulin receptor substrate (IRS) proteins, (2) phosphorylation/dephosphorylation of phosphatidylinositols and (3) exocytosis or (4) endocytosis of GLUT4. Examples of proteins regulating the different steps in podocytes are indicated. B, SHIP2 suppresses the insulin signalling pathway by hydrolysing PI(3,4,5)P3 to PI(3,4)P2. This reduces glucose uptake
Defects either in the insulin receptor itself, for example, changes in its expression level or activity, in its downstream substrates or molecular regulators of the signalling cascade may lead to impaired activation of the insulin signalling pathway and insulin resistance (Figure 2A).78 However, the numbers of individuals with genetic defects in the insulin receptor are rare, proposing that peripheral insulin resistance is rather caused by defects downstream of the insulin receptor. It is plausible that the cause of insulin resistance is multifactorial, a combination of genetic defects and environmental factors, or that it is caused by simultaneous lower functional input of multiple components of the pathway, which in the end is insufficient to lead to full activation of the signalling cascade and uptake of glucose into cells.78 The complex regulation of the trafficking of GLUT4‐containing vesicles via various accessory molecules at different steps makes also the trafficking process a potential site for alterations, which may lead to impaired translocation of GLUT4 to the plasma membrane and consequent insulin resistance (Figure 2A).79, 80
3. INSULIN RESISTANCE AND DIABETIC KIDNEY DISEASE
3.1. Insulin resistance of podocytes and DKD
The seminal study by Ahlqvist et al.70 shows that peripheral insulin resistance plays a central role in the development of DKD, but it has remained open whether insulin resistance of podocytes could directly contribute to the disease progression. Studies in animal models of diabetes and DKD, supplemented by studies on cultured podocytes, have revealed that podocytes can develop insulin resistance. Podocytes express all the central components of the insulin signalling pathway as well as glucose transporters GLUT4 and GLUT1, both shown to be insulin responsive in podocytes, and can increase their glucose uptake upon insulin stimulation.81 Subsequent studies revealed that podocytes isolated from diabetic db/db mice show reduced level of Akt activation compared to their lean controls. Also podocytes isolated from the db/db mice, before the mice had developed albuminuria, were unable to phosphorylate Akt in response to insulin stimulation.82 The indication that insulin resistance of podocytes drives kidney dysfunction was provided by a study revealing that knocking out insulin receptor specifically in podocytes in mice leads to the development of DKD even under normoglycaemia.83 These mice showed typical structural features of DKD, including thickening of the GBM, podocyte foot process effacement, signs of podocyte loss by apoptosis and glomerulosclerosis and started developing albuminuria at 5 weeks of age.83 A recent study utilizing cultured podocytes revealed that high concentrations of insulin lead to downregulation of insulin receptor via proteasome‐dependent lysosomal degradation pathway, and this leads to insulin resistance of podocytes.84
3.2. Regulation of insulin signalling in podocytes
Several proteins that regulate the activity of the insulin signalling pathway and glucose uptake in podocytes have been identified, and examples of them, and the steps they functionally control, are presented in Figure 2A. At the level of the insulin receptor (Figure 2A, step 1), the protein tyrosine phosphatase Src homology‐2 domain containing phosphatase‐1 (SHP‐1), that is upregulated by high glucose, suppresses insulin signalling by dephosphorylating insulin receptor‐β.85 Another regulator of the insulin receptor is protein tyrosine‐phosphatase 1B (PTP1B). Inhibition of PTP1B was shown to have anti‐diabetic effects by improving insulin and leptin signalling in a model of diet‐induced obesity.86 Furthermore, knocking down PTP1B activates the PI3K pathway and protects podocytes from endoplasmic reticulum (ER) stress, which associates with podocyte apoptosis and proteinuria in DKD.87 Activated insulin receptor phosphorylates its downstream mediators insulin receptor substrates 1 and 2 (IRS1, IRS2). In podocytes, insulin activates IRS2 rather than IRS1.88 Irs2‐depleted podocytes show insulin resistance, which is caused by upregulation of a protein called phosphatase and tensin homolog deleted on chromosome 10 (PTEN). PTEN dephosphorylates the 3′‐position of PtdIns(3,4,5)P3 to generate PtdIns(4,5)P2, and consequently, upregulation of PTEN diminishes phosphorylation of Akt and reduces the activity of the insulin signalling pathway (Figure 2A, step 2). Knockdown of PTEN leads to phosphorylation of Akt and thus activation of the pathway. However, instead of protecting from ER stress, as in the case of PTP1B, this sensitizes podocytes to ER stress and apoptosis reflecting the complexity of the regulation of ER stress in podocytes.87 Another lipid phosphatase that regulates insulin signalling at the level of phosphoinositides (Figure 2A, step 2) is Src homology 2 domain‐containing inositol 5′‐phosphatase 2 (SHIP2). It acts, similarly as PTEN, on PtdIns(3,4,5)P3, but hydolyses its 5′‐position generating PtdIns(3,4)P2 and thereby activates the insulin signalling pathway.89 The role of SHIP2 as a suppressor of insulin signalling is described in more detail in paragraph 4.
3.3. Regulation of glucose transporter trafficking in podocytes
Insulin‐stimulated glucose uptake can be regulated either during the exocytosis of the GLUT4 containing vesicles (GCVs) from the cytoplasmic pool to the plasma membrane (Figure 2A, step 3) or during their endocytosis (Figure 2A, step 4) to the endosomal compartment for recycling. For exocytosis, GLUT4 needs to be sorted first into GCVs, and this is controlled by adapter protein CD2‐associated protein (CD2AP) in podocytes.90 When CD2AP is missing, GLUT4 remains as perinuclear clusters, and insulin‐stimulated glucose uptake is reduced.90 Translocation of GCVs to the plasma membrane requires intact actin cytoskeleton, and in podocytes, insulin stimulates reorganization of the cortical actin cytoskeleton in an insulin receptor‐dependent manner.83 The docking and fusion of GCVs with the plasma membrane is facilitated by nephrin, which associates with the GCV‐protein vesicle‐associated membrane protein 2 (VAMP‐2).91 Nephrin also associates with the small cytoskeletal GTPase septin 7, and the protein complex containing septin 7 plays a central role in controlling the vesicle docking and fusion.92, 93, 94 It has been proposed that septin 7 forms a filamentous barrier to hinder GCV translocation to the plasma membrane thereby reducing glucose uptake.93 Depletion of septin 7 enhances the interaction between nephrin and VAMP2 to facilitate the docking and fusion of GCVs with the plasma membrane, leading to increased glucose uptake.93 In addition to nephrin, also non‐muscle myosin heavy chain IIA (NMHC‐IIA) and the membrane protein SNAP23 are found in the septin 7 complex and, interestingly, septin 7 regulates the activity of non‐muscle myosin IIA in this complex.92 Accordingly, depletion of septin 7 leads to enhanced activity of non‐muscle myosin IIA in the complex, and this increases GCV docking and fusion and uptake of glucose into cells.92 Knockdown of NMHC‐IIA or nucleobindin‐2, yet another interaction partner of septin 7, both reduce glucose uptake into podocytes.92, 94 Also phosphorylation of synip on serine 99 is necessary for GCV translocation and glucose uptake in podocytes.95 Thus far, no regulators of GCV endocytosis in podocytes have been identified (Figure 2A, step 4).
3.4. Targeting insulin resistance—a way to personalized medication in DKD?
Defining whether podocytes in patients with DKD are insulin resistant is challenging, and the rarity of normoglycaemic patients who show insulin resistance and develop DKD suggest that development of DKD involves a combination of multiple factors in addition to insulin resistance.96 Recent reviews summarize in more detail the molecular mechanisms involved in the development of the kidney complication in diabetes, including the role of insulin resistance in the context of the insulin signalling and glucose transporters.80, 97, 98 The significance of insulin signalling in maintaining the kidney function is supported by intervention studies in both patients with diabetes99, 100, 101 and experimental animal models of diabetes102, 103 showing that insulin sensitizers are renoprotective. These data together support the potential of targeting the components and regulators of the insulin signalling pathway as a strategy for developing new therapeutic interventions to preserve the kidney function in diabetes. Furthermore, the new subclassification of diabetes, identifying insulin resistance as a central risk factor for the development of DKD, allows to better predict the disease progression and outcome, and to design personalized treatments for patients in each subclass, including the severely insulin resistant ones at high risk for DKD.104
4. SHIP2 SUPPRESSES THE INSULIN SIGNALLING PATHWAY
As described above, one of the potential therapeutic target molecules that regulates the insulin signalling pathway is the lipid phosphatase SHIP2 (Figure 2B). SHIP2 is widely expressed in different tissues, with high mRNA expression in human skeletal muscle, heart and placenta.89 In mouse, SHIP2 mRNA was detected in all tissues analysed,105 and a LacZ reporter gene analysis (LacZ reporter inserted into the Inppl1 locus) in the Inppl1−/− (SHIP2‐depleted) mice revealed high expression of the LacZ reporter in brain, skeletal muscle and heart and lower in liver, lung and kidney.106 SHIP1, the close homologue of SHIP2 that shares approx. 38% sequence identity with SHIP2, is expressed mainly in cells of hematopoietic origin and during spermatogenesis.107 SHIP2 has several conserved domains, including the NH3‐terminal Src homology 2 (SH2) domain, the central 5′‐phosphatase domain and the COOH‐terminal proline‐rich and sterile ⍺ motif (SAM) domains (Figure 3). The SH2 domain is missing from the short isoform of SHIP2, generated by alternative splicing. In addition, SHIP2 harbours several tyrosine, serine and threonine phosphorylation sites (reviewed in 108). As SHIP2 suppresses the insulin signalling pathway by dephosphorylating PtdIns(3,4,5)P3 to PtdIns(3,4)P2,89 leads elevated expression level or increased activity of SHIP2 to reduced activation of Akt and reduced uptake of glucose into cells (Figure 2B), as shown in various cell culture models.109, 110, 111 It has been proposed that the catalytic activity of SHIP2 is controlled by its tyrosine phosphorylation,112, 113 but this has remained controversial and, in addition, it has been proposed that serine and threonine phosphorylation could play a role in regulating SHIP2 activity.114 The ability of SHIP2 to negatively regulate Akt activation necessitates the translocation of SHIP2 to the plasma membrane, where its central substrate, PtdIns(3,4,5)P3, resides.115 In addition to its catalytic phosphatase activity, SHIP2 has other independent functions mediated via its numerous interaction partners. Thereby SHIP2 regulates various cellular processes, including PI3K‐mediated insulin signalling, FGF‐ and EGF‐mediated signalling, cell spreading, migration and adhesion, actin cytoskeleton dynamics, endocytosis and apoptosis. Mutations in INPPL1 (gene encoding SHIP2) have been revealed in opsismodysplasia, a disease of bone maturation,116, 117 and via its various activities, SHIP2 associates with many other diseases, including diabetes, cancer, atherosclerosis and neurodegenerative diseases (for reviews, see, for example 108, 118, 119). This review concentrates on SHIP2 in diabetes.
Figure 3.
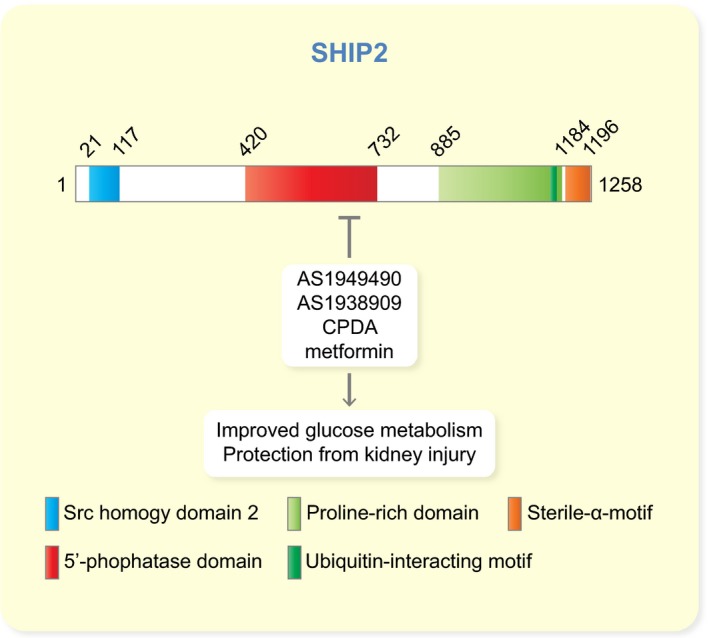
Schematic cartoon of the domain structure of SHIP2 and SHIP2 inhibitors that have been shown to have metabolic effects. The numbers indicate the start and end amino acids of the different domains and motifs
5. GENETIC STUDIES OF SHIP2 IN DIABETES AND DIABETIC KIDNEY DISEASE
SHIP2 is encoded by the INPPL1 gene located on human chromosome 11.89 Several genetic studies link SHIP2 to metabolic disorders, showing that polymorphisms in INPPL1 may contribute to the pathogenesis of the metabolic syndrome, hypertension and T2D (Table 1). The first piece of evidence was provided by finding a mutation (R1142C) in the proline‐rich region of the protein in the Goto‐Kakizaki rats, a model for T2D, and in spontaneously hypertensive rats, apparently affecting the binding of SH3‐domain containing proteins to this region.120 Overexpression of the mutated form of SHIP2 in CHO cells overexpressing the insulin receptor (CHO‐IR) resulted in decreased Akt activation, slightly impairing insulin signalling.120 Further work on patients with T2D identified a patient with a 16‐base pair deletion in the 3‐prime untranslated region of INPPL1, potentially affecting protein synthesis. Indeed, reporter gene studies with the mutated region indicated that the deletion results in increased mRNA and protein expression, and could thus contribute to increased SHIP2 expression and insulin resistance.120 Further analysis of patients with T2D of Caucasian origin in the UK and Belgium revealed a significant association of the 16‐base pair deletion and T2D, proposing that INPPL1 is a candidate gene contributing to the genetic susceptibility of T2D.120 A study in Japanese patients with T2D revealed a number of single nucleotide polymorphisms (SNPs), several of which were located in the 5′‐phosphatase catalytic region of SHIP2. However, many of the SNPs were detected more frequently in the controls than in patients with T2D.121 In vitro overexpression studies in CHO‐IR cells revealed that overexpression of a construct including a particular SNP located in the 5′‐phosphatase region inhibited insulin‐induced phosphorylation of Akt2 less potently than overexpression of wild‐type SHIP2, supporting a protective role for this SNP against insulin resistance in this cohort of patients with T2D.121 Another study in a Japanese cohort identified SNPs in the promoter and 5‐prime untranslated region of INPPL1 and investigated their association with impaired fasting glycaemia.122 Overexpression of one of the identified SNPs in cultured cells caused an increase in the promoter activity, suggesting that SNPs in this region could at least partly account for impaired fasting glycaemia at least in the patient cohort in question.122 SNPs in INPPL1 have also been found to be associated with the metabolic syndrome. A study with Finnish patients with T1D identified an association of two SNPs in INPPL1 with the metabolic syndrome in men,123 and another study on British patients with T2D found an association of SNPs and haplotypes of INPPL1 with hypertension and other components of the metabolic syndrome.124 The metabolic effects of the mutations and single nucleotide polymorphisms in INPPL1 are summarized in Table 1.
Table 1.
Mutations and single nucleotide polymorphisms (SNPs) in the SHIP2 gene INPPL1 and their metabolic consequences
| SNP/mutation | Effect | Species | Reference |
|---|---|---|---|
|
R1142C |
|
Rat | Marion et al.120 |
| 16‐bp deletion, 3′‐UTR |
|
Human | Marion et al.120 |
| L632I |
|
Human | Kagawa et al.121 |
| +334 C/T |
|
Human | Ishida et al.122 |
|
rs2276048 rs2276047 |
|
Human | Hyvönen et al.123 |
|
snp8 rs2276047 rs9886 |
|
Human | Kaisaki et al.124 |
In the above studies, control subjects showed in INPPL1 SNPs that are protective against T2D. Patients with T2D, on the other hand, had in INPPL1 SNPs that are associated with and could predispose to the metabolic syndrome, hypertension and T2D. In studies with mice, SHIP2 protein in diabetic db/db mice has been shown to be upregulated in insulin‐sensitive tissues, including skeletal muscle, adipose tissue125 and podocytes,126 apparently contributing to insulin resistance. Interestingly, SHIP2 was upregulated in podocytes prior to the development of albuminuria, and this observation was confirmed in insulin‐resistant, obese Zucker fatty rats.126 These data accord with the genetic studies suggesting a role for SHIP2 in the development of insulin resistance and DKD.
6. SHIP2 MOUSE MODELS SUPPORT A ROLE FOR SHIP2 IN METABOLIC DISORDERS
6.1. SHIP2 knockout mouse models
Both long‐ and short‐term mouse models have been utilized to analyse the role of SHIP2 as a regulator of metabolism, including both knockout and overexpression strategies (Table 2). Two different whole‐body knockout mouse models for SHIP2 have been generated. The first model targeted exons 19‐29 of Inppl1 (mouse SHIP2 gene).127 The mice showed increased sensitivity to insulin and reduced expression of hepatic gluconeogenic enzymes, leading to neonatal hypoglycemia and death within 3 days after birth.127 Compared to the wild type mice, the adult heterozygous mice showed greater insulin sensitivity, higher expression of GLUT4 at the plasma membrane, and responded to insulin stimulation with higher glycogen synthesis rate in the skeletal muscle. However, the genomic fraction deleted in this model included not only exons 19‐29 of Inppl1, but also the third exon of another gene, Phox2a.127 It therefore remains uncertain whether the phenotype of the mice is caused by absence of Inppl1, Phox2a or both, or by expression of a truncated form of SHIP2.
Table 2.
SHIP2 (encoded by Inppl1) mouse models and their phenotypes. The mouse model generated by Clément et al.127 is not included as it remains uncertain whether deletion of Phox2a affects the phenotype
| SHIP2 knockout mouse models | |||
|---|---|---|---|
| Model | Generation strategy | Phenotype | Reference |
| Inppl1 knockout | Deletion of first 18 exons of Inppl1 |
|
Sleeman et al.106 |
| Catalytic inactivation of SHIP2 | Deletion of exons 18 and 19 of Inppl1 |
|
Dubois et al.128 |
| Catalytic inactivation of SHIP2 in proximal tubules | Deletion of exons 18 and 19 of Inppl1 in proximal tubules |
|
Sayyed et al.129 |
| catalytic inactivation of one SHIP2 allele in endothelium | deletion of exons 18 and 19 of Inppl1 in endothelium |
|
Watt et al.130 |
| SHIP2 overexpression mouse models | |||
|---|---|---|---|
| Model | Generation strategy | Phenotype | Reference |
| Overexpression of wild‐type SHIP2 in liver in db/+ mice | Adenovirus‐mediated gene transfer in the liver, 4 d |
|
Fukui et al.131 |
| Overexpression of 5′‐phosphatase defective SHIP2 in liver in db/db mice | Adenovirus‐mediated gene transfer in the liver, 4 d |
|
Fukui et al.131 |
| Overexpression of 5′‐phosphatase defective SHIP2 in liver in KKAy mice | Adenovirus‐mediated gene transfer in the liver, 5 d |
|
Grempler et al.132 |
| SHIP2 transgenic mice | Overexpression of SHIP2 using modified β‐actin promoter |
|
Kagawa et al.133 |
The second Inppl1 knockout strategy deleted the first 18 exons of Inppl1 including the translation‐initiating ATG, leading to total lack of Inppl1 mRNA and SHIP2 protein.106 These mice lived till adulthood, but showed truncated facial features due to abnormal skeletal growth and reduced body length and weight, despite a tendency of increased food intake. The Inppl1−/− mice showed normal glucose and insulin levels as well as normal glucose and insulin tolerance, but decreased serum triglycerides, non‐esterified free fatty acids, cholesterol and leptin levels at 12 weeks of age compared to wild‐type littermates. In the basal state, Inppl1−/− mice showed no difference in the activation of Akt compared to wild‐type mice, but in response to insulin, the null mice showed increased activation of Akt in liver and muscle. Interestingly, the null mice gained less weight when placed on high‐fat diet for 6 weeks, and did not show an increase in serum lipids, insulin or glucose. The Inppl1 null mice on high‐fat diet also showed a greater basal metabolic rate and increased energy expenditure.106 Thus, the lack of Inppl1 protects the mice against diet‐induced obesity.
A more recent study describes generation of a mouse model with catalytically inactive SHIP2, deleting exons 18 and 19.128 These mice had reduced body length and weight, defects in the development of the skull and the female genital tract, and reduced skeletal muscle weight and adiposity. Especially, the males were hyperphagic, their serum triglycerides and cholesterol levels as well as insulin secretion from the mutant islets were lower. No difference was observed in glucose tolerance and insulin sensitivity or signalling,128 similar to the Inppl1 knockout described above.106 Further studies on this model revealed increased microvilli formation in the proximal tubules of the kidney due to activation of the ezrin/radixin/moesin family of proteins, leading to increased reabsorption by the proximal tubules.129 Specifically, reabsorption of low‐molecular‐weight proteins, glucose and phosphate was increased by the proximal tubule cells. Essentially similar kidney phenotype was observed when Inppl1 was catalytically inactivated specifically in proximal tubules only.129 Interestingly, there was a trend of increased uptake of intravenously injected, fluorescently labelled albumin by proximal tubules after catalytic inactivation of Inppl1 in proximal tubules.129 Watt et al. 130 generated mice with catalytic inactivation of one Inppl1 allele in endothelium‐specific manner to define the role of SHIP2 in vascular function. They found that the mice developed increased vascular oxidative stress and systemic insulin resistance because of impaired glucose uptake in adipose tissue and skeletal muscle. Thus, the study proposes that normal vascular SHIP2 is necessary for maintaining systemic glucose homoeostasis and preventing endothelial dysfunction induced by oxidative stress.130
6.2. SHIP2 overexpression mouse models
Several studies have addressed the role of SHIP2 in the regulation of metabolism via overexpression strategies, inspired by the finding that SHIP2 is upregulated in peripheral insulin sensitive tissues in insulin resistant, diabetic db/db mice.125, 126 Transient, adenovirus‐mediated overexpression of wild‐type SHIP2 in non‐diabetic db/+ mice led to overexpression of SHIP2 in the liver but not in other tissues.131 This decreased insulin‐induced activation of Akt in the liver and increased the expression of gluconeogenic genes, leading to hyperinsulinemia and potentiated the increase in blood glucose after oral glucose intake. This indicates that overexpression of SHIP2 in liver leads to systemic insulin resistance.131 Adenovirus‐mediated overexpression of 5′‐phosphatase‐defective SHIP2 (dominant‐negative SHIP2) in diabetic db/db mice improved the ability of insulin to induce Akt activation in the liver, and partly reduced the enhanced expression of gluconeogenic genes.131 This lowered fasting blood glucose and reduced elevated glucose and insulin levels after oral glucose intake, indicating that inhibition of SHIP2 in liver is effective in ameliorating hyperglycemia. Insulin signalling in skeletal muscle or adipose tissue was not affected in either model.131 In another study, mutated form of SHIP2 lacking the 5′‐phosphatase activity was introduced via adenoviral infection into KKAy mice, a model of hyperglycemia and hyperinsulinemia.132 This resulted in increased basal and insulin‐stimulated Akt activation in liver, decreased glucose production after puryvate challenge, and increased hepatic glycogen content. Hepatic SHIP2 inhibition also increased glycolysis and serum triglycerides, improved glucose tolerance and reduced prandial blood glucose levels after feeding ad libitum, supporting the idea that SHIP2 inhibition improves glucose metabolism.132
In addition to the above two short‐term models, Kagawa et al.133 generated transgenic mice overexpressing SHIP2 using a modified β‐actin promoter, leading to SHIP2 overexpression in liver, skeletal muscle, white adipose tissue, pancreas, brown adipose tissue and brain. The transgenic mice gained more body weight and showed increased fasting plasma insulin, whereas fasting glucose, leptin, adiponectin, total cholesterol, triglycerides and non‐esterified fatty acids did not differ from the wild‐type littermates under normal diet. However, the transgenic mice showed impaired glucose tolerance and insulin sensitivity. This could be caused by reduced activity of the PI3K pathway, as insulin‐stimulated activation of Akt was reduced in adipose tissue, skeletal muscle and liver in the SHIP2 overexpressing mice. The expression of gluconeogenic genes in the liver was increased and glycogen content decreased. These data indicate that increased expression of SHIP2 impairs glucose metabolism and insulin sensitivity.133
7. SHIP2 INHIBITORS AMELIORATE METABOLIC DISORDERS
The in vitro and in vivo studies described above, revealing that SHIP2 overexpression suppresses insulin signalling and debilitates glucose metabolism, have increased the interest to develop SHIP2 inhibitors as a potential therapeutic approach to treat obesity‐induced insulin resistance and T2D. A number of inhibitors have been identified, but for most of them, no data on metabolic effects are available and some inhibit also SHIP1 (see discussion on the beneficial metabolic effects of SHIP1 inhibition in paragraph 10).134, 135, 136, 137, 138 A few studies describe the effects of SHIP2 inhibition on metabolic parameters either in cells in vitro or in vivo.139, 140, 141, 142 Interestingly, we recently identified metformin, the first‐line medication used to treat T2D, as a novel SHIP2 inhibitor.140 The following section focuses on the SHIP2 inhibitors whose roles in metabolic disorders have been defined (Figure 3). The metabolically less well‐characterized SHIP2 or SHIP1/SHIP2 inhibitors are summarized in recent reviews.108, 119
7.1. Small molecule SHIP2 inhibitors
Using malachite green phosphate assay for measuring SHIP2 activity, Suwa et al.142 screened in a high‐throughput approach a chemical library for inhibitors of the phosphatase domain of SHIP2. This led to the identification of AS1949490, which inhibited human SHIP2 with an IC50 value of 0.62 µmol/L and a Ki value of 0.44 µmol/L, but did not inhibit other phosphatases, such as SHIP1, PTEN, synaptojanin or myotubularin. In L6 myotubes, AS1949490 increased insulin‐induced activation of Akt in a dose‐dependent manner, and increased both glucose consumption and glucose uptake. In FAO hepatoma cells, AS1949490 reduced glucose production, and acute administration of the inhibitor to normal mice reduced the expression of gluconeogenic genes in the liver. Furthermore, administration of AS1949490 to db/db mice for 7‐10 days reduced both fasting and non‐fasting blood glucose and reduced the area under the curve in oral glucose tolerance test. This was accompanied by increased phosphorylation of GSK3β in the liver, an indication of the activation of insulin signalling.142 These findings supported the idea that inhibition of SHIP2 has therapeutic potential as a treatment for T2D.
The same group further identified another novel SHIP2 inhibitor, AS1938909, with an IC50 value of 0.57 µmol/L and a Ki value of 0.44 µmol/L for human SHIP2.141 This inhibitor was also highly selective for SHIP2, and increased Akt activation, glucose consumption and glucose uptake in L6 myotubes. Both AS1938909 and the previously identified AS1949490 increased the expression level of GLUT1 mRNA in L6 myotubes, which could contribute to the increased glucose uptake.141 The effects of AS1938909 in vivo were not defined.
Ichihara et al.142 utilized in silico ligand‐based drug design combining the structures of AS1949490 and NGD‐61338, another novel SHIP2 inhibitor with an IC50 value of 1.1 µmol/L,134 to design new SHIP2 inhibitor candidates.139 The designed compounds and their derivatives were tested for their ability to activate Akt as an indirect measure of SHIP2 inhibition. Compound N‐[4‐(4‐chlorobenzyloxy)pyridine‐2‐yl]‐2‐(2,6‐difluorophenyl)‐acetamide (CPDA), the most potent of these compounds, was further tested in db/db mice. Treatment of the mice with CPDA for 10‐13 days decreased the fasting blood glucose, reduced hepatic expression of gluconeogenic genes and ameliorated glucose intolerance, and the effects were comparable to those of AS1949490.139
7.2. Antidiabetic drug metformin inhibits SHIP2
We utilized in silico structure‐based virtual screening of small molecule chemical libraries to identify SHIP2 inhibitors, and one of the identified molecules was metformin.140 This supports the proposition that metformin not only acts via reducing gluconeogenesis in the liver, but also enhances insulin sensitivity and glucose uptake in peripheral tissues. The early studies on the mechanisms of action of metformin revealed that mitochondria are central for the effects of metformin in liver.143, 144 To produce ATP, mitochondria carry out oxidation of NADH, produced by glycolysis or β‐oxidation of fatty acids, generating NAD+ and electrons. The electrons are transferred through the respiratory chain complexes I, II, III and IV to O2, eventually generating H2O. The protons generated by oxidation of NADH are pumped to the intermitochondrial membrane through respiratory complexes I, III and IV, and the proton gradient drives ATP synthase to generate ATP from ADP. Metformin has been shown to inhibit the complex I (NADH:ubiquinone oxidoreductase) leading to reduced cellular respiration.143, 144 The reduction in cellular ATP concentration and increase in ADP/ATP and AMP/ATP ratios activates the energy sensor AMP‐activated protein kinase (AMPK). Indeed, Zhou et al.145 revealed that metformin activates AMPK and thereby suppresses hepatic glucose production and expression of lipogenic enzymes, induces fatty acid oxidation, and also enhances glucose transport to muscle, leading to reduced plasma glucose and triglycerides. Metformin also upregulates PPARγ coactivator 1⍺ (PGC‐1⍺) via AMPK, and selectively downregulates transcription factors that facilitate PGC‐1⍺‐mediated gluconeogenesis, thus suppressing gluconeogenesis.146 Metformin can affect hepatic metabolism also via AMPK‐independent pathways, as metformin reduced gluconeogenesis in mice deficient of AMPK, leading to the proposition that metformin inhibits hepatic gluconeogenesis via decreased energy state.147 Notably, the effects of metformin on AMPK activity are apparently concentration dependent, as low concentrations of metformin, as detected in the portal vein, suppressed gluconeogenesis via activation of AMPK, without increasing the cellular AMP/ATP ratio.148
Metformin has also been shown to induce the expression of fibroblast growth factor 21 (FGF21), an endocrine hormone with anti‐obesity and anti‐diabetes properties, in the liver in AMPK‐independent manner by inhibiting mitochondrial complex I.149 Also, metformin‐induced accumulation of AMP abrogates the glucagon‐induced activation of adenylate cyclase.150 This leads to reduced level of cyclic AMP (cAMP) and reduced activation of protein kinase A (PKA) and its targets, and, thus, inhibition of gluconeogenesis.150 In addition to affecting mitochondrial complex I, metformin has been shown to directly bind to the redox shuttle enzyme mitochondrial glycerophosphate dehydrogenase.151 Inhibition of this enzyme by metformin leads to altered hepatic redox state, reduction of the conversion of lactate and glycerol to glucose, and reduced hepatic gluconeogenesis.151 However, the finding that metformin causes an acute inhibition of glycerophosphate dehydrogenase has recently been challenged.152 In addition, metformin exerts some of its beneficial metabolic effects via altering the gut microbiota by affecting genes encoding metalloproteins or metal transporters,153 another facet of the multiple modes of action of the drug.
As introduced above, in the liver the only direct target of metformin is glycerophosphate dehydrogenase,151 although this has been challenged.152 SHIP2 represents the second direct molecular target identified for metformin.140 In in vitro experiments with purified recombinant proteins, metformin was found to directly bind to and inhibit the catalytic activity of the recombinant SHIP2 phosphatase domain with an IC50 value of 6.0 µmol/L, but it did not inhibit SHIP1 or PTEN confirming the specificity of metformin for SHIP2.140 Metformin also inhibited the activity of SHIP2 in cultured L6 myotubes and podocytes and increased glucose uptake in both cell types. In L6 myotubes, metformin increased the amount of GLUT4 at the plasma membrane by inhibiting endocytosis of GLUT4 and thereby enhanced glucose uptake, without affecting Akt activity. In vivo, metformin inhibited SHIP2 activity in muscle and kidney tissue of db/db mice after 12 days of treatment. This short‐term metformin treatment enhanced insulin sensitivity of the db/db mice, visualized by reduced glucose area under the curve in insulin tolerance test, but hyperglycemia was not reduced. This could be due to the compensatory gluconeogenesis in the kidney.140 Even though the major site of metformin action has previously shown to be inhibition of gluconeogenesis in the liver,154 and the 12‐day metformin treatment reduced the expression of gluconeogenesis genes in the liver, metformin did not inhibit the activity of SHIP2 in cultured hepatoma cells or in the liver of db/db mice.140 It thus appears that metformin has tissue‐specific direct targets, mitochondrial glycerophosphate dehydrogenase in the liver151 (see, however152) and SHIP2 in muscle and kidney tissue.140 In addition to direct binding to SHIP2, various other indirectly activated pathways have been shown to be involved in metformin‐induced glucose uptake in muscle tissue and as in liver, they occur via both AMPK‐dependent and ‐independent mechanisms. For example, metformin has been shown to activate atypical protein kinase C (aPKC) via AMPK‐, ERK‐ and PDK1‐dependent pathway to enhance glucose uptake in muscle.155 A later study revealed that metformin increases glucose uptake independent of AMPK via novel and conventional PKC isoforms.156 Metformin has also been shown to increase the abundance of GLUT1 at the plasma membrane157 and to decrease GLUT4 endocytosis in muscle cells.140, 158
One of the challenges in metformin research has been the great variability in experimental concentrations and treatment times, making comparisons between studies difficult. Of note, cells in culture tend to lose the receptors via which metformin enters the cells.159 Therefore, the concentrations of metformin required in the in vitro experiments may greatly vary depending on the cell type, whether the line is a primary cell line or an immortalized cell line maintained in culture for a long period of time, and on the exposure time to metformin.
Collectively, several SHIP2 inhibitors have been identified but only four of them have been analysed in vitro or in vivo and shown to improve metabolic parameters (Figure 3). Identification of metformin as a SHIP2 inhibitor and the findings described above, indicating that SHIP2 inhibition improves glucose metabolism, indicate that SHIP2 is an excellent target to design new treatments to ameliorate insulin resistance and T2D.
8. DOES SHIP2 CONVEY THE RENOPROTECTIVE EFFECTS OF METFORMIN?
In addition to its major action in reducing hyperglycaemia, metformin has been shown to have renoprotective effects: It reduces albuminuria in patients with T2D160, 161 and in a rat model of T2D,162 and also increases the estimated glomerular filtration rate in patients with T1D.163 Various mechanisms have been proposed. In rats with T2D, metformin increases the expression of nephrin164 and podocalyxin,165 both known to be downregulated or mislocalized in DKD.21, 22, 23, 24, 164, 165 Treatment of primary rat podocytes with metformin activates protein deacetylase sirtuin 1 (SIRT1) and AMPK, and prevents high glucose‐induced downregulation of SIRT1 expression.166 Metformin also reduces high glucose‐induced apoptosis of podocytes in vitro.167 Interestingly, podocyte loss was found to be more pronounced in patients with T2D receiving non‐metformin medication as compared to patients receiving metformin or people without diabetes, supporting a renoprotective role for metformin.140 This, coupled to the finding that SHIP2 activity is elevated in the kidneys of patients with T2D receiving other than metformin medication proposes that metformin could protect podocytes from injury via inhibiting SHIP2 activity.140 This proposition was supported by in vitro studies on immortalized human podocytes, in which SHIP2 overexpression has been shown to enhance apoptosis coupled with reduced Akt activation.126 Metformin restored SHIP2 overexpression‐induced reduction in Akt activation and normalized the level of apoptosis.140 An earlier study revealed that upregulation of SHIP2 in podocytes in diabetic animal models occurred already before the reported age of onset of proteinuria, and before any histological changes in the glomeruli were observed.126 This indicates that upregulation of SHIP2 is not a secondary effect of podocyte damage but rather contributes to it, and raises an intriguing question whether SHIP2 inhibition at an early stage of diabetes could prevent podocyte injury and development of DKD. Answering this question calls for carrying out a randomized controlled trial with metformin.
The above examples and the previous paragraph illustrate the great complexity of the mechanisms of action of metformin. Clearly, more work is necessary to understand the exact molecular mechanisms via which metformin acts in different tissues, and specifically, to define the central pathways via which metformin acts in patients with T2D in long‐term treatments. A great amount of work needs to be carried out with the new SHIP2 inhibitors to define their exact mechanisms of action in different tissues, and to identify the potential SHIP2‐independent mechanisms of action. This will aid in the drug development process when designing derivatives of the best candidate molecules to further improve their characteristics. The mechanistic understanding of how the inhibitor works will also help in identifying the potential adverse effects.
9. BENEFITS AND CAVEATS OF INHIBITING SHIP2
As described in the above paragraph, the effects of SHIP2 inhibition on metabolic parameters in vitro and in vivo point out a clear beneficial role for SHIP2 inhibition in reducing insulin resistance and hyperglycemia and improving glucose metabolism.139, 140, 141, 142 In addition, SHIP2 inhibition by metformin protects podocytes from injury at a stage when SHIP2 expression or activity is elevated, such as in DKD,140 proposing that SHIP2 inhibition could be a potent approach to prevent DKD. This is a fascinating aspect as the subgroup of T2D‐patients with severe insulin resistance is more susceptible to develop DKD,70 raising a question whether intensive treatment of these patients with metformin, or in future, with more potent SHIP2 inhibitors, could provide an effective means to prevent the development and progression of this fierce complication. Further studies are also needed to define whether the renoprotective effects of SHIP2 inhibition are due to reduction of general peripheral insulin resistance or direct protective effects of SHIP2 inhibition on podocytes.
The genetic mouse models to a large part support a protective role for reduced expression or lowered catalytic activity of SHIP2 against metabolic disorders, with the exception of catalytic inactivation of one Inppl1 allele in vasculature.130 This led to increased oxidative stress, endothelial dysfunction and systemic insulin resistance, and the authors raised a concern for SHIP2 inhibition in endothelium.130 Previously, no such effects were described in the whole body Inppl1 knockout,106 and Watt et al.168 do not speculate what could be the reason for such a difference in the metabolic outcome in these two models. The studies with SHIP2 inhibitors, described above, do not raise concerns on systemic insulin resistance but rather report contrary findings. Furthermore, expression of catalytically inactive SHIP2 in hepatic HepG2 cells has been shown to reduce ROS generation. Also, inhibition of SHIP2 with AS1949490 in CD2AP‐deficient podocytes, showing increased oxidative stress, reduced ROS generation.169 Despite this, increased apoptosis, caused by lack of CD2AP, was not reduced but aggravated by SHIP2 inhibition, indicating that when CD2AP is missing, SHIP2 inhibition may have unexpected consequences.169 Uncovering the mechanisms involved calls for further studies using in vivo models. The studies in cultured cells or short‐term administration of SHIP2 inhibitors in vivo may not reflect the outcome of long‐term inhibitor administration, and there may also be cell‐type specific differences in the effects of SHIP2 inhibition. Also, the uptake of the various inhibitors to different cell types very likely varies, which may result in distinct outcomes compared to genetic inactivation of SHIP2. SHIP2 has also functions that are independent of its catalytic activity, relying on its protein‐protein interactions.118 It may thus be that in certain cases the genetic knockout models produce a phenotype that does not directly reflect the outcome of reducing only the catalytic activity of SHIP2 with inhibitors, leaving the protein interaction domains still functional.
An interesting beneficial aspect of the catalytic inactivation of SHIP2 in mice, either generally or specifically in kidney proximal tubules, is the increased reabsorption ability of the proximal tubules.129 The study showed both enhanced reabsorption of low‐molecular‐weight proteins as well as a trend of increased uptake of injected albumin.129 The authors proposed that SHIP2 could represent a new therapeutic target for renal Fanconi syndrome, characterized by urinary loss of low molecular weight proteins and metabolites due to either acquired or inherited dysfunction of proximal tubules.129 The authors were able to mimic some of the aspects of the genetic model with a SHIP2 inhibitor in cultured cells in vitro,129 but the effects of SHIP2 inhibition with small molecule inhibitors in vivo on proximal tubule structure and functional properties await further studies. It will also be interesting to define whether SHIP2 inhibition in glomerular diseases presenting with increased leakage of albumin, such as DKD, show increased tubular uptake of albumin achieved by SHIP2 inhibition.
SHIP2 inhibition has also been addressed in the field of cancer research as a potential treatment for specific types of cancer. This is a reflection of a paradigm shift in the cancer field as it comes to phosphatases, including SHIP2.135 Phosphatases were earlier considered rather as tumour suppressors, but the finding that they can activate signalling pathways proposes that they could actually be potential oncogenes and thereby targets for cancer treatment. In the case of SHIP2, its hydrolysis product, PtdIns(3,4)P2, binds to Akt more potently than PtdIns(3,4,5)P3.170 Furthermore, both PtdIns(3,4)P2 and PtdIns(3,4,5)P3 are required for the high‐level activation of Akt.171 It has been proposed that both PtdIns(3,4)P2 and PtdIns(3,4,5)P3 are necessary for cancer cell survival and malignancy, and both their absolute amount and ratio control cell death.172 In line with this “two PIP hypothesis”, studies revealing SHIP2 as a target to treat cancer and showing beneficial effects of SHIP2 inhibitors are emerging. In clinical specimens of breast,173 colorectal,136, 174 non‐small cell lung175 and hepatocellular176 cancer, the expression level of SHIP2 has been shown to be elevated and to correlate with decreased patient survival. Beneficial effects of SHIP2 inhibition have been proposed in breast135, 177 and colorectal cancer.136 In gastric cancer specimens, however, SHIP2 expression is frequently downregulated, and this enhances the malignant behaviour of gastric cancer cells.178 Accordingly, it appears that the effects of SHIP2 in different cells are context dependent. It is important to note, that mice that express catalytically inactive SHIP2 survive longer than 18 months and show no signs of tumour development,128 and neither do mice depleted of SHIP2.106 These data increase the prospects of SHIP2 as a potential cancer treatment target in specific types of cancer in which SHIP2 is overexpressed or overactivated. The benefits and caveats of SHIP2 inhibition are summarized in Table 3.
Table 3.
Benefits and caveats of inhibiting the catalytic activity of SHIP2. See the text for details and references
| Benefits | Caveats |
|---|---|
| General metabolism | |
|
|
| Kidney | |
|
|
| Cancer | |
|
|
10. FUTURE PROSPECTS
The SHIP2 inhibition studies carried out thus far with AS1949490 and CPDA in vivo in mice have covered relatively short‐term treatments, remaining shorter than 2 weeks.139, 142 The long‐term use of SHIP2 inhibitor metformin is safe, except that metformin is contraindicated when kidney function is declining. Long‐term treatments of diabetic rodent models with AS1949490, CPDA and other new SHIP2 inhibitors described above are warranted to confirm that these inhibitors per se do not cause harmful side effects. New SHIP2 inhibitors with better drug‐like characteristics (for example, solubility and bioavailability) than those of the previously characterized SHIP2 inhibitors would be welcome as leads for drug development. The recent classification of patients with diabetes to five distinct groups with specific profiles of disease progression and risks for complications70 offers, for the first time, possibilities for precision medicine in diabetes. In terms of the kidney complication, the patients who are severely insulin resistant are at high risk to develop DKD.70 The potential of SHIP2 inhibitors to act as insulin sensitizers highlights their potential to provide new leads for personalized medications for this subgroup of patients.
Some of the developed inhibitors are panSHIP inhibitors that have been tested in cancer cells,135 but their effects on metabolic disorders are thus far uncharacterized. At least inhibition of SHIP1 with a SHIP1‐specific inhibitor K118 can be highly beneficial, as K118 improves the metabolic parameters of diet‐induced obese mice by attenuating inflammation in the visceral adipose tissue.179 The K118 treatment was relatively short, 4 weeks; a longer treatment would be needed to confirm that no side effects occur upon time. This, and treatment of diabetic rodent models with either panSHIP inhibitors or with a combination of SHIP1 and SHIP2 inhibitors will be needed to define the possible superiority of the combined inhibition over inhibition of each protein alone.
The finding that patients with T2D receiving metformin exhibit less podocyte loss than patients with other medication140 raises several fundamental research lines. To show that metformin acts renoprotectively in patients with T2D, a randomized controlled trial is needed. The other research line to pursue is the identification of SHIP2 inhibitors more potent than metformin, especially in terms of better inhibition of SHIP2 in podocytes, followed by defining whether these molecules provide better renoprotection than metformin in diabetic animal models with kidney disease. One could envisage that highly insulin resistant patients at high risk to develop DKD70 would greatly benefit from treatments targeting SHIP2 with an inhibitor enhancing insulin sensitivity as well as protecting kidneys from injury. Furthermore, knowing that SHIP2 is upregulated in several types of cancer and that this correlates with decreased patient survival, raises an intriguing question whether SHIP2 inhibitors could provide potential means also for new personalized treatment options for cancer patients.
CONFLICTS OF INTEREST
The author declares no conflicts of interest.
ACKNOWLEDGEMENTS
Business Finland, the Jane and Aatos Erkko Foundation and the Jusélius Foundation are acknowledged for financial support. Eero Lehtonen (University of Helsinki, Finland) is thanked for the electron microscopy images and Vincent Dumont, Laura Hautala, Eero Lehtonen and Zydrune Polianskyte‐Prause (University of Helsinki, Finland) for critical comments on the text and figures.
Lehtonen S. SHIPping out diabetes—Metformin, an old friend among new SHIP2 inhibitors. Acta Physiol. 2020;228:e13349 10.1111/apha.13349
REFERENCES
- 1. Fowler MJ. Microvascular and macrovascular complications of diabetes. Clin Diab. 2008;26:77‐82. [Google Scholar]
- 2. de Boer IH, Rue TC, Hall YN, Heagerty PJ, Weiss NS, Himmerfarb J. Temporal trends in the prevalence of diabetic kidney disease in the United States. J Am Med Assoc. 2011;305:2532‐2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harjutsalo V, Groop P‐H. Epidemiology and risk factors for diabetic kidney disease. Adv Chronic Kidney Dis. 2014;21:260‐266. [DOI] [PubMed] [Google Scholar]
- 4. Vallon V, Thomson SC. Renal function in diabetic disease models: the tubular system in the pathophysiology of the diabetic kidney. Annu Rev Physiol. 2012;74:351‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grahammer F, Schell C, Huber TB. The podocyte slit diaphragm ‐ from a thin grey line to a complex signalling hub. Nat Rev Nephrol. 2013;9:587‐598. [DOI] [PubMed] [Google Scholar]
- 6. Kestilä M, Lenkkeri U, Männikkö M, et al. Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575‐582. [DOI] [PubMed] [Google Scholar]
- 7. Takeda T, Go WY, Orlando RA, Farquhar MG. Expression of podocalyxin inhibits cell‐cell adhesion and modifies junctional properties in Madin‐Darby canine kidney cells. Mol Biol Cell. 2000;11:3219‐3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Perico L, Conti S, Benigni A, Remuzzi G. Podocyte‐actin dynamics in health and disease. Nat Rev Nephrol. 2016;12:692‐710. [DOI] [PubMed] [Google Scholar]
- 9. Wolf G, Chen S, Ziyadeh FN. From the periphery of the glomerular capillary wall toward the center of disease, Podocyte injury comes of age in diabetic nephropathy. Diabetes. 2005;54:1626‐1634. [DOI] [PubMed] [Google Scholar]
- 10. Siragy HM, Carey RM. Role of the intrarenal renin‐angiotensin‐aldosterone system in chronic kidney disease. Am J Nephrol. 2010;31:541‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Susztak K, Raff AC, Schiffer M, Böttinger EP. Glucose‐induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. [PubMed] [Google Scholar]
- 12. Meyer TW, Bennett PH, Nelson RG. Podocyte number predicts long‐term urinary albumin excretion in Pima indians with type II diabetes and microalbuminuria. Diabetologia. 1999;42:1341‐1344. [DOI] [PubMed] [Google Scholar]
- 13. Wharram BL, Goyal M, Wiggins JE, et al. Podocyte depletion causes glomerulosclerosis: diphteria toxin‐induced podocyte depletion in rats expressing human diphteria toxin receptor transgene. J Am Soc Nephrol. 2005;16:2941‐2952. [DOI] [PubMed] [Google Scholar]
- 14. Lee HS. Pathogenic role of TGF‐b in diabetic nephropathy. J Diabetes Metab. 2013;S9:008. [Google Scholar]
- 15. Schiffer M, Bitzer M, Roberts ISD, et al. Apoptosis in podocytes induced by TGF‐β and Smad7. J Clin Invest. 2001;108:807‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Abe Y, Sakairi T, Beeson C, Kopp JB. TGF‐β1 stimulates mitochondrial oxidative phosphorylation and generation of reactive oxygen species in cultured mouse podocytes, mediated in part by the mTOR pathway. Am J Physiol Renal Physiol. 2013;305:F1477‐F1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Y, Kang YS, Dai C, Kiss LP, Wen X, Liu Y. Epithelial‐to‐mesenchymal transition is a potential pathway leading to podocyte dysfunction and proteinuria. Am J Pathol. 2008;172:299‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chuang PY, Yu Q, Fang W, Uribarri J, He JC. Advanced glycation endproducts induce podocyte apoptosis by activation of the FOXO4 transcription factor. Kidney Int. 2007;72:965‐976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tagawa A, Yasuda M, Kume S, et al. Impaired podocyte autophagy exacerbates proteinuria in diabetic nephropathy. Diabetes. 2016;65:755‐767. [DOI] [PubMed] [Google Scholar]
- 20. Lenoir O, Jasiek M, Hénique C, et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes‐induced glomerulosclerosis. Autophagy. 2015;11:1130‐1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hyvönen ME, Dumont V, Tienari J, et al. Early‐onset diabetic E1‐DN mice develop albuminuria and glomerular injury typical of diabetic nephropathy. BioMed Res Int. 2015;2015:102969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bonnet F, Cooper ME, Kawachi H, Allen TJ, Boner G, Cao Z. Irbesartan normalizes the deficiency in glomerular nephrin expression in a model of diabetes and hypertension. Diabetologia. 2001;44:874‐877. [DOI] [PubMed] [Google Scholar]
- 23. Dumont V, Tolvanen TA, Kuusela S, et al. PACSIN2 accelerates nephrin trafficking and is upregulated in diabetic kidney disease. FASEB J. 2017;31:3978‐3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Inoki K, Mori H, Wang J, et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest. 2011;121:2181‐2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wasik AA, Koskelainen S, Hyvönen ME, et al. Ezrin is down‐regiulated in diabetic kidney glomeruli and regulates actin organization and glucose uptake via GLUT1 in cultured podocytes. Am J Pathol. 2014;184:1727‐1739. [DOI] [PubMed] [Google Scholar]
- 26. Lin JS, Susztak K. Podocytes: the weakest link in diabetic kidney disease? Curr Diab Rep. 2016;16:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kumar PA, Welsh GI, Saleem MA, Menon RK. Molecular and cellular events mediating glomerular podocyte dysfunction and depletion in diabetes mellitus. Front Endocrinol. 2014;5:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Osterby R, Nyberg G. New vessel formation in the renal corpuscles in advanced diabetic glomerulopathy. J Diab Compl. 1987;1:122‐127. [DOI] [PubMed] [Google Scholar]
- 29. Østerby R, Tapia J, Nyberg G, et al. Renal structures in type 2 diabetic patients with elevated albumin excretion rate. APMIS. 2001;109:751‐761. [DOI] [PubMed] [Google Scholar]
- 30. Nieuwdorp M, Mooij HL, Kroon J, et al. Endothelial glycocalyx damage coincides with microalbuminuria in type 1 diabetes. Diabetes. 2006;55:1127‐1132. [DOI] [PubMed] [Google Scholar]
- 31. Toyoda M, Najafian B, Kim Y, Caramouri ML, Mauer M. Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Diabetes. 2007;56:2155‐2160. [DOI] [PubMed] [Google Scholar]
- 32. Weil EJ, Lemley KV, Mason CC, et al. Podocyte detachment and reduced glomerular capillary endothelial fenestrations promote kidney disease in type 2 diabetic nephropathy. Kidney Int. 2012;82:1010‐1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kanesaki Y, Suzuki D, Uehara G, et al. Vascular endothelial growth factor gene expression is correlated with glomerular neovascularization in human diabetic nephropathy. Am J Kidney Dis. 2005;45:288‐294. [DOI] [PubMed] [Google Scholar]
- 34. Foster RR, Hole R, Anderson K, et al. Functional evidence that vascular endothelial growth factor may act as an autocrine factor on human podocytes. Am J Physiol Renal Physiol. 2003;284:F1263‐F1273. [DOI] [PubMed] [Google Scholar]
- 35. Foster RR, Satchell SC, Seckley J, et al. VEGF‐C promotes survival in podocytes. Am J Physiol Renal Physiol. 2006;291:F196‐F207. [DOI] [PubMed] [Google Scholar]
- 36. Cooper ME, Vranes D, Youssef S, et al. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR‐2 in experimental diabetes. Diabetes. 1999;48:2229‐2239. [DOI] [PubMed] [Google Scholar]
- 37. Oltean S, Qiu Y, Ferguson JK, et al. Vascular endothelial growth factor‐A165b is protective and restores endothelial glycocalyx in diabetic nephropathy. J Am Soc Nephrol. 2015;26:1889‐1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Onions KL, Gamez M, Buckner NR, et al. VEGFC reduces glomerular albumin permeability and protects against alterations in VEGF receptor expression in diabetic nephropathy. Diabetes. 2019;68:172‐187. [DOI] [PubMed] [Google Scholar]
- 39. Tanabe K, Maeshima Y, Wada J. Antiangiogenic therapy for dibetic nephropathy. BioMed Res Int. 2017;2017:5724069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Falkevall A, Mehlem A, Palombo I, et al. Reducing VEGF‐B signaling ameliorates ranal lipotoxicity and protects against diabetic kidney disease. Cell Metab. 2017;25:713‐726. [DOI] [PubMed] [Google Scholar]
- 41. Satchell SC, Harper SJ, Tooke JE, Kerjaschki D, Saleem MA, Mathieson PW. Human podocytes express angiopoietin 1, a potential regulator of glomerular vascular endothelial growth factor. J Am Soc Nephrol. 2002;13:544‐550. [DOI] [PubMed] [Google Scholar]
- 42. Dessapt‐Baradez C, Woolf AS, White KE, et al. Targeted glomerular angiopoietin‐1 therapy for early diabetic kidney disease. J Am Soc Nephrol. 2014;25:33‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Maisonpierre PC, Suri C, Jones PF, et al. Angiopoietin‐2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55‐60. [DOI] [PubMed] [Google Scholar]
- 44. Rizkalla B, Forber JM, Cao Z, Boner G, Cooper ME. Temporal renal expression of angiogenic growth factors and their receptors in experimental diabetes: role of the renin‐angiotensin system. J Hypertension. 2005;23:153‐164. [DOI] [PubMed] [Google Scholar]
- 45. Davis B, Dei Cas A, Long DA, et al. Podocyte‐specific expression of angiopoietin‐2 causes proteinuria and apoptosis of glomerular endothelia. J Am Soc Nephrol. 2007;18:2320‐2329. [DOI] [PubMed] [Google Scholar]
- 46. Qi H, Casalena G, Shi S, et al. Glomerular endothelial mitochondrial dysfunction is essential and characteristic of diabetic kidney disease susceptibility. Diabetes. 2017;66:763‐778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ebefors K, Nyström J. New insights into crosstalk in the kidney. Curr Opin Nephrol Hypertens. 2017;26:143‐147. [DOI] [PubMed] [Google Scholar]
- 48. Daehn IS. Glomerular endothelial cell stress and cross‐talk with podocytes in early diabetic kidney disease. Frontiers Med. 2018;6:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maestroni S, Zerbini G. Glomerular endothelial cells versus podocytes as the cellular target in diabetic nephropathy. Acta Diab. 2018;55:1105‐1111. [DOI] [PubMed] [Google Scholar]
- 50. Fu J, Lee K, Chuang PY, Liu Z, He JC. Glomerular endothelial cell injury and cross talk in diabetic kidney injury. Am J Physiol Renal Physiol. 2015;308:F287‐F297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nature Rev Nephrol. 2017;13:629‐646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Forbes JM, Thorburn DR. Mitochondrial dysfunction in diabetic kidney disease. Nature Reviews Nephrol. 2018;14:291‐312. [DOI] [PubMed] [Google Scholar]
- 53. Schiffer TA, Friederich‐Persson M. Mitochondrial reactive oxygen species and kidney hypoxia in the development of diabetic nephropathy. Front Physiol. 2017;8:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Coughlan MT, Nguyen T‐V, Penfold SA, et al. Mapping time‐course mitochondrial adaptations in the kidney in experimental diabetes. Clin Sci. 2016;130:711‐720. [DOI] [PubMed] [Google Scholar]
- 55. Liles JT, Corkey BK, Notte GT, et al. ASK1 contributes to fibrosis and dysfunction in models of kidney disease. J Clin Invest. 2018;128:4485‐4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Qi W, Keenan HA, Li Q, et al. Pyruvate kinase M2 activation may protect against the progresison of diabetic glomerular pathology and mitochondrial dysfunction. Nature Med. 2017;23:753‐762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ising C, Koehler S, Brahler S, et al. Inhibition of insulin/IGF‐1 receptor signaling protects from mitochondria‐mediated kidney failure. EMBO Mol Med. 2015;7:275‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Persson P, Palm F. Hypoxia‐inducible factor activation in diabetic kidney disease. Curr Opin Nephrol Hypertens. 2017;26:345‐350. [DOI] [PubMed] [Google Scholar]
- 59. Hansell P, Welch WJ, Blantz RC, Palm F. Determinants of kidney oxygen consumption and their relationship to tissue exygen tension in diabetes and hypertension. Clin Exp Pharmacol Physiol. 2013;40:123‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Franzén L, Pihl L, Khan N, Gustafsson H, Palm F. Pronounced kidney hypoxia precedes albuminuria in type 1 diabetic mice. Am J Physiol renal Physiol. 2016;310:F807‐F809. [DOI] [PubMed] [Google Scholar]
- 61. Yaribeygi H, Atkin SL, Sahebgar A. Mitochondrial dysfunction in diabetes and the regulatory roles of antidiabetic agents on the mitochondrial function. J Cell Physiol. 2019;234:8402‐8410. [DOI] [PubMed] [Google Scholar]
- 62. Fan Y, Lee K, Wang N, He JC. The role of endoplasmic reticulum stress in diabetic nephropathy. Curr Diab Rep. 2017;17:17. [DOI] [PubMed] [Google Scholar]
- 63. Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nature Rev Nephrol. 2017;13:681‐696. [DOI] [PubMed] [Google Scholar]
- 64. Van Krieken R, Mehta N, Wang T, et al. Cell surface expression of 78‐kDa glucose‐regulates protein (GRP78) mediates diabetic nephropathy. J Biol Chem. 2019;294:7755‐7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Madhusudhan T, Wang H, Dong W, et al. Defective podocyte insulin signalling through p85‐XBP1 promotes ATF6‐dependent maladaptive ER‐stress response in diabetic nephropathy. Nature Comm. 2015;6:6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Madhusudhan T, Wang H, Ghosh S, et al. Signal integration at the PI3K‐p85‐XBP1 hub endows coagulation protease activated protein C with insulin function. Blood. 2017;130:1445‐1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kato M, Natarajan R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nature Rev Nephrol. 2019;15:327‐345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen G, Chen H, Ren S, et al. Aberrant DNA methylation of mTOR pathway genes promotes inflammatory activation of immune cells in diabetic kidney disease. Kidney Int. 2019;96:409‐420. Epub ahead of print:pii: S0085‐2538(0019)30273‐X. [DOI] [PubMed] [Google Scholar]
- 69. Majumder S, Thieme K, Batchu SN, et al. Shifts in podocyte histone H3K27me3 regulate mouse and human glomerular disease. J Clin Invest. 2018;128:483‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ahlqvist E, Storm P, Käräjämäki A, et al. Novel subgroups of adult‐onset diabetes and their association with outcomes: a data‐driven cluster analysis of six variables. Lancet Diab Endocrinol. 2018;6:361‐369. [DOI] [PubMed] [Google Scholar]
- 71. Yip J, Mattock MB, Morocutti A, Sethi M, Trevisan R, Viberti G. Insulin resistance in insulin‐dependent diabetic patients with microalbuminuria. Lancet. 1993;342:883‐887. [DOI] [PubMed] [Google Scholar]
- 72. Parvanova AI, Trevisan R, Iliev IP, et al. Insulin resistance and microalbuminuria: a cross‐sectional, case‐control study of 158 patients with type 2 diabetes and different degrees of urinary albumin excretion. Diabetes. 2006;55:1456‐1462. [DOI] [PubMed] [Google Scholar]
- 73. Groop L, Ekstrand A, Forsblom C, et al. Insulin resistance, hypertension and microalbuminuria in patients with type 2 (non‐insulin‐dependent) diabetes mellitus. Diabetologia. 1993;36:642‐647. [DOI] [PubMed] [Google Scholar]
- 74. Pilz S, Rutters F, Nijpels G, et al. Insulin sensitivity and albuminuria: the RISC study. Diab Care. 2014;37:1597‐1603. [DOI] [PubMed] [Google Scholar]
- 75. Orchard TJ, Chang Y‐F, Ferrell RE, Petro N, Ellis DE. Nephropathy in type 1 diabetes: a manifestation of insulin resistance and multiple genetic susceptibilities? Kidney Int. 2002;62:963‐970. [DOI] [PubMed] [Google Scholar]
- 76. Ekstrand A, Groop P‐H, Grönhagen‐Riska C. Insulin resistance precedes microalbuminuria in patients with insulin‐dependent diabetes mellitus. Nephrol Dial Transplant. 1998;13:3079‐3083. [DOI] [PubMed] [Google Scholar]
- 77. Lay AC, Coward R. The evolving importance of insulin signaling in podocyte health and disease. Frontiers Endocrinol. 2018;9:693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pessin JE, Saltiel AR. Signaling pathways in insulin action: molecular targets of insulin resistance. J Clin Invest. 2000;106:165‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jaldin‐Fincati JR, Pavarotti M, Frendo‐Cumbo S, Bilan PJ, Klip A. Update on GLUT4 vesicle traffick: a cornerstone of insulin action. Trend Endocrinol Metab. 2017;28:597‐611. [DOI] [PubMed] [Google Scholar]
- 80. Wasik AA, Lehtonen S. Glucose transporters in diabetic kidney disease‐ friends or foes? Frontiers Endocrinol. 2018;9:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Coward RJ, Welsh GI, Yang J, et al. The human glomerular podocyte is a novel target for insulin action. Diabetes. 2005;54:3095‐3102. [DOI] [PubMed] [Google Scholar]
- 82. Tejada T, Catanuto P, Ijaz A, et al. Failure to phosphorylate AKT in podocytes from mice with early diabetic nephropathy promotes cell death. Kidney Int. 2008;73:1385‐1393. [DOI] [PubMed] [Google Scholar]
- 83. Welsh GI, Hale LJ, Eremina V, et al. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 2010;12:329‐340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lay AC, Hurcombe JA, Betin VMS, et al. Prolonged exposure of mouse and human podocytes to insulin induces insulin resistance through lysosomal and proteasomal degradation of the insulin receptor. Diabetologia. 2017;60:2299‐2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Drapeau N, Lizotte F, Denhez B, Guay A, Kennedy CR, Geraldes P. Expression of SHP‐1 induced by hyperglycemia prevents insulin actions in podocytes. Am J Physiol Endocrinol Metab. 2013;304:E1188‐E1198. [DOI] [PubMed] [Google Scholar]
- 86. Krishnan N, Konidaris KF, Gasser G, Tonks NK. A potent, selective, and orally bioavailable inhibitor of the protein‐tyrosine phosphatase PTP1B improves insulin and leptin signaling in animal models. J Biol Chem. 2018;293:1517‐1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Garner KL, Betin VMS, Pinto V, et al. Enhanced insulin receptor, but not PI3K, signalling protects podocytes from ER stress. Sci Rep. 2018;8:3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Santamaria B, Marquez E, Lay A, et al. IRS2 and PTEN are key molecules controlling insulin sensitivity in podocytes. Biochim Biophys Acta. 2015;1853:3224‐3234. [DOI] [PubMed] [Google Scholar]
- 89. Pesesse X, Deleu S, De Smedt F, Drayer L, Erneux C. Identification of a second SH2‐domain‐containing protein closely related to the phosphatidylinositol polyphosphate 5‐phosphatase SHIP. Biochem Biophys Res Commun. 1997;239:697‐700. [DOI] [PubMed] [Google Scholar]
- 90. Tolvanen TA, Dash SN, Polianskyte‐Prause Z, Dumont V, Lehtonen S. Lack of CD2AP disrupts Glut4 trafficking and attenuates glucose uptake in podocytes. J Cell Sci. 2015;128:4588‐4600. [DOI] [PubMed] [Google Scholar]
- 91. Coward RJ, Welsh GI, Koziell A, et al. Nephrin is critical for the action of insulin on human glomerular podocytes. Diabetes. 2007;56:1127‐1135. [DOI] [PubMed] [Google Scholar]
- 92. Wasik AA, Dumont V, Tienari J, et al. Septin 7 reduces nonmuscle myosin IIA activity in the SNAP23 complex and hinders GLUT4 storage vesicle docking and fusion. Exp Cell Res. 2017;350:336‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wasik AA, Polianskyte‐Prause Z, Dong M‐Q, et al. Septin 7 forms a complex with CD2AP and nephrin and regulates glucose transporter trafficking. Mol Biol Cell. 2012;23:3370‐3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Saito T, Yamada E, Okada S, et al. Nucleobindin‐2 is a positive regulator for insulin‐stimulated glucose transporter 4 translocation in fenofibrate treated E11 podocytes. Endocr J. 2014;61:933939. [DOI] [PubMed] [Google Scholar]
- 95. Yamada E, Saito T, Okada S, et al. Synip phosphorylation is required for insulin‐stimulated Glut4 translocation and glucose uptake in podocytes. Endocrine J. 2014;61:523‐527. [DOI] [PubMed] [Google Scholar]
- 96. Filippone EJ, Gupta A, Farber JL. Normoglycemic diabetic nephropathy: the role of insulin resistance. Case Rep Nephrol Urol. 2014;4:137‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gnudi L, Coward R, Long DA. Diabetic nephropathy: perspective on novel molecular mechanisms. Trens Endocrinol Metab. 2016;27:820‐830. [DOI] [PubMed] [Google Scholar]
- 98. Lay AC, Coward R. The evolving importance of insulin signaling in podocyte health and disease. Front Endocrinol. 2018;9:693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Miyazaki Y, Cersosimo E, Triplitt C, DeFronzo RA. Rosiglitazone decreases albuminuria in type 2 diabetic patients. Kidney Int. 2007;72:1367‐1373. [DOI] [PubMed] [Google Scholar]
- 100. Schernthaner G, Matthews DR, Charbonnel B, Hanefeld M, Brunetti P. Efficacy and safety of pioglitazone versus metformin in patients with type 2 diabetes mellitus: a double‐blind, randomized trial. J Clin Endocrinol Metab. 2004;89:6068‐6076. [DOI] [PubMed] [Google Scholar]
- 101. Bakris G, Viberti G, Weston WM, Heise M, Porter LE, Freed MI. Rosiglitazone reduces urinary albumin excretion in type II diabetes. J Hum Hypert. 2003;17:7‐12. [DOI] [PubMed] [Google Scholar]
- 102. Ohtomo S, Izuhara Y, Takizawa S, et al. Thiazolidinediones provide better renoprotection than insulin in an obese, hypertensive type II diabetic rat model. Kidney Int. 2007;72:1512‐1519. [DOI] [PubMed] [Google Scholar]
- 103. Zhang H, Saha J, Byun J, et al. Rosiglitazone reduces renal and plasma markers of oxidative injury and reverses urinary metabolite abnormalities in the amelioration of diabetic nephropathy. Am J Physiol Renal Physiol. 2008;295:F1071‐1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Prasad RB, Groop L. Precision medicine in type 2 diabetes. J Intern Med. 2018;285:40‐48. [DOI] [PubMed] [Google Scholar]
- 105. Schurmans S, Carrio R, Behrends J, Pouillon V, Merino J, Clément S. The mouse SHIP2 (Inppl1) gene: Complementary DNA genomic structure, promoter analysis, and gene expression in the embryo and adult mouse. Genomics. 1999;62:260‐271. [DOI] [PubMed] [Google Scholar]
- 106. Sleeman MW, Wortley KE, Lai K‐M, et al. Absence of the lipid phosphatase SHIP2 confers resistance to dietary obesity. Nature Med. 2005;11:199‐205. [DOI] [PubMed] [Google Scholar]
- 107. Liu Q, Shalaby F, Jones J, Bouchard D, Dumont DJ. The SH2‐containing inositol polyphosphate 5‐phosphatase, Ship, is expressed during hematopoiesis and spermatogenesis. Blood. 1998;91:2753‐2759. [PubMed] [Google Scholar]
- 108. Thomas MP, Erneux C, Potter B. SHIP2: Structure, function and inhibition. ChemBioChem. 2017;18:233‐247. [DOI] [PubMed] [Google Scholar]
- 109. Blero D, De Smedt F, Pesesse X, et al. The SH2 domain containing inositol 5‐phosphatase SHIP2 controls phosphatidylinositol 3,4,5‐trisphosphate levels in CHO‐IR cells stimulated by insulin. Biochem Biophys Res Commun. 2001;282:839‐843. [DOI] [PubMed] [Google Scholar]
- 110. Wada T, Sasaoka T, Funaki M, et al. Overexpression of SH2‐containing inositol phosphatase 2 results in negative regulation of insulin‐induced metabolic actions in 3T3‐L1 adipocytes via its 5'‐phosphatase catalytic activity. Mol Cell Biol. 2001;21:1633‐1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Sasaoka T, Hori H, Wada T, et al. SH2‐containing inositol phosphatase 2 negatively regulates insulin‐induced glycogen synthesis in L6 myotubes. Diabetologia. 2001;44:1258‐1267. [DOI] [PubMed] [Google Scholar]
- 112. Batty I, van der Kaay J, Gray A, Telfer J, Dixon M, Downes C. Downes CP. The control of phosphatidylinositol 3,4‐bisphosphate concentrations by activation of the Src homology 2 domain containing inositol polyphosphate 5‐phosphatase 2, SHIP2. Biochem J. 2007;407:255‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Pesesse X, Dewaste V, De Smedt F, et al. The Src homology 2 domain containing inositol 5‐phosphatase SHIP2 is recruited to the epidermal growth factor (EGF) receptor and dephosphorylates phospatidylinositol 3,4,5‐trisphosphate in EGF‐stimulated COS‐7 cells. J Biol Chem. 2001;276:28348‐28355. [DOI] [PubMed] [Google Scholar]
- 114. Edimo WE, Janssens V, Waelkens E, Erneux C. Reversible Ser/Thr SHIP phosphorylation: a new paradigm in phosphoinositide signalling? BioEssays. 2012;34:634‐642. [DOI] [PubMed] [Google Scholar]
- 115. Ishihara H, Sasaoka T, Ishiki M, et al. Membrane localization of Src homology 2‐containing inositol 5'‐phosphatase 2 via Shc association is required for the negative regulation of insulin signaling in rat1 fibroblasts overexpressing insulin receptors. Mol Endocrinol. 2002;16:2371‐2381. [DOI] [PubMed] [Google Scholar]
- 116. Huber C, Faqeih E, Bartholdi D, et al. Exome sequencing identifies INPPL1 mutations as a cause of opsismodysplasia. Am J Hum Genet. 2013;92:144‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Below J, Earl D, Shively K, et al. Whole‐genome analysis reveals that mutations in inositol polyphosphate phosphatase‐like 1 cause opsismodysplasia. Am J Hum Genet. 2013;92:137‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Erneux C, Edimo WE, Deneubourg L, Pirson I. SHIP2 multiple functions: a balance between a negative control of PtdIns(3,4,5)P3 level, a positive control of PtdIns(3,4)P2 production, and intrinsic docking properties. J Cell Chem. 2011;112:2203‐2209. [DOI] [PubMed] [Google Scholar]
- 119. Viernes DR, Choi LB, Kerr WK, Chrisholm JD. Discovery and development of small molecule SHIP phosphatase modulators. Med Res Rev. 2013;34:795‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Marion E, Kaisaki PJ, Pouillon V, et al. The gene INPPL1, encoding the lipid phosphatase SHIP2, is a candidate for type 2 diabetes in rat and man. Diabetes. 2002;51:2012‐2017. [DOI] [PubMed] [Google Scholar]
- 121. Kagawa S, Sasaoka T, Yaguchi S, et al. Impact of src homology 2‐containing inositol 5′‐phosphatase 2 gene polymorphisms detected in a japanese population on insulin signaling. J Clin Endocrinol Metab. 2005;90:2911‐2919. [DOI] [PubMed] [Google Scholar]
- 122. Ishida S, Funakoshi A, Miyasaka K, Shimokata H, Ando F, Takiguchi S. Association of SH‐2 containing inositol 5′‐phosphatase 2 gene polymorphisms and hyperglycemia. Pancreas. 2006;33:63‐67. [DOI] [PubMed] [Google Scholar]
- 123. Hyvönen ME, Ihalmo P, Forsblom C, et al. INPPL1 is associated with the metabolic syndrome in men with type 1 diabetes, but not with diabetic nephropathy. Diabet Med. 2012;12:1589‐1595. [DOI] [PubMed] [Google Scholar]
- 124. Kaisaki PJ, Delepine M, Woon PY, et al. Polymorphisms in type II SH2 domain‐containing inositol 5‐phosphatase (INPPL1, SHIP2) are associated with physiological abnormalities of the metabolic syndrome. Diabetes. 2004;53:1900‐1904. [DOI] [PubMed] [Google Scholar]
- 125. Hori H, Sasaoka T, Ishihara H, et al. Association of SH2‐containing inositol phosphatase 2 with the insulin resistance of diabetic db/db mice. Diabetes. 2002;51:2387‐2394. [DOI] [PubMed] [Google Scholar]
- 126. Hyvönen ME, Saurus P, Wasik A, et al. Lipid phosphatase SHIP2 downregulates insulin signalling in podocytes. Mol Cell Endocrinol. 2010;328:70‐79. [DOI] [PubMed] [Google Scholar]
- 127. Clément S, Krause U, Desmedt F, et al. The lipid phosphatase SHIP2 controls insulin sensitivity. Nature. 2001;409:92‐97. [DOI] [PubMed] [Google Scholar]
- 128. Dubois E, Jacoby M, Blockmans M, et al. Developmental defects and rescue from glucose intolerance of a catalytically‐inactive novel SHIP2 mutant mouse. Cell Signal. 2012;24:1971‐1980. [DOI] [PubMed] [Google Scholar]
- 129. Sayyed SG, Jouret F, Vermeersch M, Pérez‐Morga D, Schurmans S. The lipid 5‐phosphatase SHIP2 controls renal brush border ultrastructure and function by regulating the activation of ERM proteins. Kidney Int. 2017;92:125‐139. [DOI] [PubMed] [Google Scholar]
- 130. Watt NT, Gage MC, Patel PA, et al. Endothelial SHIP2 suppresses Nox2 NADPH oxidase‐dependent vascular oxidative stress, endothelial dysfunction, and systemic insulin resistance. Diabetes. 2017;66:2808‐2821. [DOI] [PubMed] [Google Scholar]
- 131. Fukui K, Wada T, Kagawa S, et al. Impact of the liver‐specific expression of SHIP2 (SH2‐containing inositol 5′‐phosphatase 2) on insulin signaling and glucose metabolism in mice. Diabetes. 2005;54:1958‐1967. [DOI] [PubMed] [Google Scholar]
- 132. Grempler R, Zibranova D, Schoelch C, van Marle A, Rippmann JF, Redemann N. Normalization of prandial blood glucose and improvement of glucose tolerance by liver‐specific inhibition of SH2 domain‐containing inositol phosphatase 2 (SHIP2) in diabetic KKAy mice. Diabetes. 2007;56:2235‐2241. [DOI] [PubMed] [Google Scholar]
- 133. Kagawa S, Soeda Y, Ishihara H, et al. Impact of transgenic overexpression of SH2‐containing inositol 5′‐phosphatase 2 on glucose metabolism and insulin signaling in mice. Endocrin. 2008;149:642‐650. [DOI] [PubMed] [Google Scholar]
- 134. Annis DA, Cheng CC, Chuang C‐C, et al. Inhibitors of the lipid phosphatase SHIP2 discovered by high throughput affinity selection‐mass spectrometry screening of combinatorial libraries. Comb Chem & High Throughput. Screen. 2009;12:760‐771. [DOI] [PubMed] [Google Scholar]
- 135. Fuhler GM, Brooks R, Toms B, et al. Therapeutic potential of SH2 domain‐containing inositol‐5'‐phosphatase 1 (SHIP1) and SHIP2 inhibition in cancer. Mol Med. 2012;18:65‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Hoekstra E, Das AM, Willemsen M, et al. Lipid phosphatase SHIP2 functions as oncogene in colorectal cancer by regulating PKB activation. Oncotarget. 2016;7:73525‐73540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Vandeput F, Combettes L, Mills SJ, et al. Biphenyl 2,3’,4,5’,6‐pentakisphosphate, a novel inositol polyphosphate surrogate, modulates Ca2+ responses in rat hepatocytes. FASEB J. 2007;21:1481‐1491. [DOI] [PubMed] [Google Scholar]
- 138. Rowe T, Hale C, Zhou A, et al. A high‐throughput microfluidic assay for SH2 domain‐containing inositol 5‐phosphatase 2. ASSAY Drug Dev Technologies. 2006;4:175‐183. [DOI] [PubMed] [Google Scholar]
- 139. Ichihara Y, Fujimura R, Tsuneki H, et al. Rational design and synthesis of 4‐substituted 2‐pyrin‐2‐ylamides with inhibitory effects on SH2 domain‐containing inositol 5'‐phosphatase 2 (SHIP2). Eur J Med Chem. 2013;62:649‐660. [DOI] [PubMed] [Google Scholar]
- 140. Polianskyte‐Prause Z, Tolvanen TA, Lindfors S, et al. Metformin increases glucose uptake and acts renoprotectively by reducing SHIP2 activity. FASEB J. 2019;33:2858‐2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Suwa A, Kurama T, Yamamoto T, Sawada A, Shimokawa T, Aramori I. Glucose metabolism activation by SHIP2 inhibitors via up‐regulation of GLUT1 gene in L6 myotubes. Eur J Pharmacol. 2010;642:177‐182. [DOI] [PubMed] [Google Scholar]
- 142. Suwa A, Yamamoto T, Sawada A, et al. Discovery and functional characterization of a novel small molecule inhibitor of the intracellular phosphatase, SHIP2. British J Pharmacol. 2009;158:879‐887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. El‐Mir M‐Y, Nogueira V, Fontaine E, Avéret N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex 1. J Biol Chem. 2000;275:223‐228. [DOI] [PubMed] [Google Scholar]
- 144. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti‐diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochemical J. 2000;348:607‐614. [PMC free article] [PubMed] [Google Scholar]
- 145. Zhou G, Myers R, Li Y, et al. Role of AMP‐activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167‐1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Aatsinki S‐M, Buler M, Salomäki H, Koulu M, Pavek P, Hakkola J. Metformin induces PGC‐1a expression and selectively affects hepatic PGC‐1a functions. British J Pharmacol. 2014;171:2351‐2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Foretz M, Hébrard S, Leclerc J, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355‐2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Cao J, Meng S, Chang E, et al. Low concentrations of metformin suppress glucose production in hepatocytes through AMP‐activated protein kinae (AMPK). J Biol Chem. 2014;289:20435‐20446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Kim KH, Jeong YT, Kim SH, et al. Metformin‐induced inhibition of the mitochondrial respiratory chain increases FGF21 expression via ATF4 activation. Biochem Biophys Res Commun. 2013;440:76‐81. [DOI] [PubMed] [Google Scholar]
- 150. Miller RA, Chu Q, Xie J, Fotetz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494:256‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Madiraju AK, Erion DM, Rahimi Y, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510:542‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Calza G, Nyberg E, Mäkinen M, et al. Lactate-induced glucose output is unchanged by metformin at a therapeutic concentration - A mass spectrometry imaging study of the perfused rat liver. Front Pharmacol. 2018;9:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Wu H, Esteve E, Tremaroli V, et al. Metformin alters the gut microbiome of individuals with treatment‐naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nature Med. 2017;23:850‐858. [DOI] [PubMed] [Google Scholar]
- 154. Hundal RS, Krssak M, Dufour S, et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes. 2000;49:2063‐2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Sajan MP, Bandyopadhyay G, Miura A, et al. AICAR and metformin, but not exercise, increase muscle glucose transport through AMPK‐, ERK‐, and PDK1‐dependent activation of atypical PKC. Am J Physiol Endocrinol Metab. 2010;298:E179‐E192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Turban S, Stretton C, Drouin O, et al. Defining the controbution of AMP‐activated protein kinase (AMPK) and protein kinase C (PKC) in regulation of glucose uptake by metformin in skeletal muscle cells. J Biol Chem. 2012;287:20088‐20099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Hundal HS, Ramlal T, Reyes R, Leiter LA, Klip A. Cellular mechanism of metformin action involves glucose transporter translocation from an intracellular pool to the plasma membrane in L6 muscle cells. Endocrinol. 1992;131:1165‐1173. [DOI] [PubMed] [Google Scholar]
- 158. Yang J, Holman GD. Long‐term metformin treatment stimulates cardiomyocyte glucose transport through an AMP‐activated protein kinase‐dependent reduction in GLUT4 endocytosis. Endocrin. 2006;147:2728‐2736. [DOI] [PubMed] [Google Scholar]
- 159. Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci. 2012;122:253‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Amador‐Licona N, Guízar‐Mendoza J‐M, Vargas E, Sánchez‐Camargo G, Zamora‐Mata L. The short‐term effect of a switch from glibenclamide to metformin on blood pressure and microalbuminuria in patients with type 2 diabetes mellitus. Arch Med Res. 2000;31:571‐575. [DOI] [PubMed] [Google Scholar]
- 161. Pan Q, Xu Y, Yang N, et al. Comparison of acarbose and metformin on albumin excretion in patients with newly diagnosed type 2 diabetes. Medicine. 2016;95:e3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Kim J, Shon E, Kim C‐S, Kim JS. Renal podocyte injury in a rat model of type 2 diabetes is prevented by metformin. Exp Diab Res. 2012;2012: Article ID 210821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Petrie JR, Chaturvedi N, Ford I, et al. Cardiovascular and metabolic effects of metformin in patients with type 1 diabetes (REMOVAL): a double‐blind, randomized, placebo‐controlled trial. Lancet Diabetes Endocrinol. 2017;5:597‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Zhai L, Gu J, Yang D, Hu W, Wang W, Ye S. Metformin ameliorates podocyte damage by restoring renal tissue nephrin expression in type 2 diabetic rats. J Diab. 2017;9:510‐517. [DOI] [PubMed] [Google Scholar]
- 165. Zhai L, Gu J, Yang D, Wang W, Ye S. Metformin ameliorates podocyte damage by restoring renal tissue podocalyxin expression in type 2 diabetic rats. J Diab Res. 2015;2015:231825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Rogacka D, Audzeyenka I, Rychłowski M, et al. Metformin overcomes high glucose‐induced insulin resistance of podocytes by pleiotropic effects on SIRT1 and AMPK. BBA Mol Basis Dis. 2018;1864:115‐125. [DOI] [PubMed] [Google Scholar]
- 167. Langer S, Kreutz R, Eisenreich A. Metformin modulates apoptosis and cell signaling of human podocytes under high glucose conditions. J Nephrol. 2016;29:765‐773. [DOI] [PubMed] [Google Scholar]
- 168. Gorgani‐Firuzjaee S, Meshkani R. SH2 domain‐containing inositol 5‐phosphatase (SHIP2) inhibition ameliorates high glucose‐induced de‐novo lipogenesis and VLDL production through regulating AMPK/mTOR/SREBP1 pathway and ROS production in HepG2 cells. Free Radic Biol Med. 2015;89:679‐689. [DOI] [PubMed] [Google Scholar]
- 169. Saurus P, Tolvanen TA, Lindfors S, et al. Inhibition of SHIP2 in CD2AP‐deficient podocytes ameliorates reactive oxygen species generation but aggravates apoptosis. Sci Rep. 2017;7:10731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto‐oncogene product by phosphatidylinostitol‐3,4‐bisphosphate. Science. 1997;275:665‐668. [DOI] [PubMed] [Google Scholar]
- 171. Alessi DR, Andelkovic M, Caudwell B, et al. Mechanism of activation of protein kinase B by insuin and IGF‐1. EMBO J. 1996;15:6541‐6551. [PMC free article] [PubMed] [Google Scholar]
- 172. Kerr WK. Inhibitor and activator: dual functions for SHIP in immunity and cancer. Ann NY Acad Sci. 2011;1217:1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Prasad NK, Tandon M, Handa A, et al. High expression of obesity‐linked phosphatase SHIP2 in invasive breast cancer correlates with reduced disease‐free survival. Tumour Biol. 2008;29:330‐341. [DOI] [PubMed] [Google Scholar]
- 174. Yang JU, Fu M, Ding Y, et al. High SHIP2 expression indicates poor survival in colorectal cancer. Dis Markers. 2014;218968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Fu M, Fan W, Pu X, et al. Elevated expression of SHIP2 correlates with poor prognosis in non‐small cell lung cancer. Int J Clin Exp Pathol. 2013;6:2185‐2191. [PMC free article] [PubMed] [Google Scholar]
- 176. Fu M, Gu X, Ni H, et al. High expression of inositol polyphosphate phosphatase like‐1 associates with unfavorable survival in hepatocellular carcinoma Int . J Clin Exp Pathol. 2013;6:2515‐2522. [PMC free article] [PubMed] [Google Scholar]
- 177. Ghosh S, Scozzaro S, Ramos AR, et al. Inhibition of SHIP2 activity inhibits cell migration and could prevent metastasis in breast cancer cells. J Cell Sci. 2018;131:jcs216408. [DOI] [PubMed] [Google Scholar]
- 178. Ye Y, Ge YM, Xiao MM, et al. Suppression of SHIP2 contributes to tumorigenesis and proliferation of gastric cancer cells via activation of Akt. J Gastroenterol. 2016;51:230‐240. [DOI] [PubMed] [Google Scholar]
- 179. Srivastava N, Iyer S, Sudan R, et al. A small‐molecule inhibitor of SHIP1 reverses age‐ and diet‐associated obesity and metabolic syndrome. JCI Insight. 2016;1:e88544. [DOI] [PMC free article] [PubMed] [Google Scholar]