

Abstract
[NiFe] hydrogenases are complex model enzymes for the reversible cleavage of dihydrogen (H2). However, structural determinants of efficient H2 binding to their [NiFe] active site are not properly understood. Here, we present crystallographic and vibrational‐spectroscopic insights into the unexplored structure of the H2‐binding [NiFe] intermediate. Using an F420‐reducing [NiFe]‐hydrogenase from Methanosarcina barkeri as a model enzyme, we show that the protein backbone provides a strained chelating scaffold that tunes the [NiFe] active site for efficient H2 binding and conversion. The protein matrix also directs H2 diffusion to the [NiFe] site via two gas channels and allows the distribution of electrons between functional protomers through a subunit‐bridging FeS cluster. Our findings emphasize the relevance of an atypical Ni coordination, thereby providing a blueprint for the design of bio‐inspired H2‐conversion catalysts.
Keywords: biocatalysis, crystal structure, hydrogen activation, [NiFe] hydrogenase, vibrational spectroscopy
Where does the H2 go? Using an F420‐reducing [NiFe]‐hydrogenase from Methanosarcina barkeri as a model enzyme, it is shown that the protein backbone provides a strained chelating scaffold that tunes the [NiFe] active site for efficient H2 binding and conversion. The protein matrix also directs H2 diffusion to the [NiFe] site via two gas channels and allows the distribution of electrons between functional protomers through a subunit‐bridging FeS cluster.

Introduction
Mapping out strategies for future energy storage and conversion represents one of the central challenges of the 21st century. Molecular hydrogen (H2) is an ideally clean fuel whose combustion releases large amounts of free energy but no greenhouse gases. To exploit it to its full potential, however, we require efficient and sustainable approaches for catalytic H2 cleavage and formation. [NiFe] hydrogenases are valuable model enzymes that catalyze H2 conversion at rates comparable to platinum electrodes by using a heterobimetallic active site containing cheap and earth‐abundant base metals only.1 Their rational utilization as biotechnological targets or blueprints for bio‐inspired catalysts, however, requires a thorough understanding of the structural and mechanistic determinants of their reactivity. Here, we employ a bidirectional F420‐reducing [NiFe] hydrogenase from the archaeon Methanosarcina barkeri MS (MbFRH) as a unique model system to yield structural and spectroscopic insights into a scarcely explored reaction intermediate that is the initial target for H2 binding to the active site of the enzyme. The structure of this central catalytic species is analyzed in detail to explore its relevance for the mechanism and performance of [NiFe] hydrogenases.
Results and Discussion
The crystal structure of MbFRH was refined using reflections up to d min=1.84 Å (Supporting Information, Table S1). Each asymmetric unit contains three subunits, FRH‐A, FRH‐B, and FRH‐G, and a total of four [4Fe4S] clusters as well as a flavin adenine dinucleotide (FAD) cofactor and the [NiFe] active site (detailed below). The FAD and the heterobimetallic [NiFe] center enable the redox conversion of the two substrates, coenzyme F420 and H2, respectively, while the [4Fe4S] clusters mediate intramolecular electron transfer (ET) between the two reaction sites (Figures 1 A and S1). These features are shared with the related [NiFe] hydrogenase from Methanothermobacter marburgensis (Mm), and both enzymes exhibit a dodecameric overall architecture in a spherical shape (Figure S1).2 Compared to the latter enzyme, however, MbFRH contains two additional cofactors that are both ET‐accessible and solvent‐exposed: a [2Fe2S] cluster bridging two FRH‐G subunits (Figures 1 A and S2) and a mononuclear Fe site in FRH‐A (Figures 1 A and S3).
Figure 1.
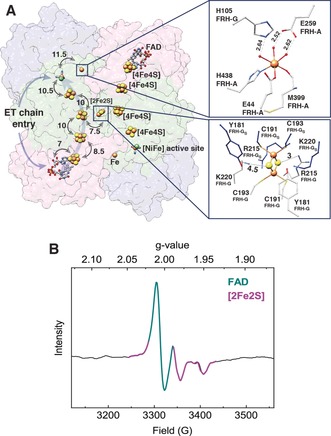
A) Reversible ET (indicated by arrows) between the FAD and the [NiFe] active site is enabled by a chain of [4Fe4S] clusters. The [NiFe] center could also exchange electrons with a mononuclear Fe site (upper inset), and the presence of a [2Fe2S] cluster (lower inset) allows electrons to commute between antiparallel ET chains of two heterotrimers. Distances given in Å; selected atoms and amino acids shown as spheres (Fe: orange, S: yellow, Ni: green) and sticks, respectively. B) EPR spectra of an MbFRH solution recorded at 80 K with 1 mW microwave power and 9.3 GHz microwave frequency. Signals from FAD and a [2Fe2S] cluster are highlighted in dark cyan and violet, respectively.
Based on element‐specific anomalous scattering, the single Fe ion has also been assigned in [NiFeSe] hydrogenases,3 while Ca2+ 4 or Mg2+ 5 ions are typically found at the same position in other [NiFe] hydrogenases (Table S2). The [2Fe2S] cluster likely contributes to the biological function of MbFRH by enabling the distribution of electrons among individual protomers of the dodecameric enzyme. In line with this proposal, solution‐phase electron paramagnetic resonance (EPR) experiments reveal signals from FAD, [2Fe2S] clusters (Figures 1 B and S4), and [4Fe4S] clusters (Figure S4), corroborating the extended electron‐transfer pathway of native MbFRH.
In the following, we will focus on the [NiFe] active site of MbFRH, which exhibits the consensus structural properties observed for other [NiFe] hydrogenases.6 Specifically, this heterobimetallic cofactor features two metal ions, Ni and Fe, that are coordinated by four strictly conserved cysteinate (Cys) residues and three Fe‐bound diatomic ligands. Based on infrared (IR) spectroscopic analyses, the latter constituents are generally assigned to one CO and two CN− ligands.7 Consistently, one CO and two CN stretching bands can be observed in IR spectra of MbFRH crystals, which confirms the presence of a standard set of inorganic ligands (Figure 2 A, black trace). Since these stretching vibrations are highly sensitive towards structural and electronic changes at the [NiFe] center,8 the observation of a single set of three IR absorption bands in the relevant spectral range also demonstrates that the active site resides in a homogenous redox‐structural state. In contrast to crystal structures of many other [NiFe] hydrogenases, no electron density can be detected in the third bridging position between the two metals for MbFRH (Figure 2 B).6a–6c This excludes the presence of oxygen‐containing ligands that would be indicative of inactive enzyme residing, for example, in the Niu‐A or Nir‐B states.6a, 9 Notably, a vacant third bridging site between the two metals has long been assumed to be a key feature of Nia‐S (also termed Ni‐SIa), the H2‐binding intermediate of [NiFe] hydrogenases.6a, 10 This assumption has been recently supported by spectroscopic analyses,11 but detailed structural insights into this central catalytic intermediate have been so far unavailable. In the following, we will shed light on these catalytic key aspects by using a joint approach of crystallographic analysis and vibrational spectroscopy.
Figure 2.
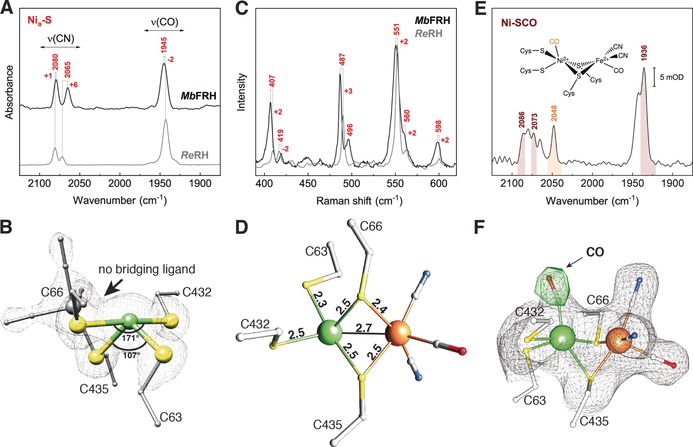
A) IR spectra of an MbFRH single crystal (at 80 K, black) and a protein solution of ReRH (10 °C, gray). Spectra were normalized with respect to the CO stretching‐band intensity. B) Crystal structure of the [NiFe] active site, exhibiting a distorted seesaw coordination geometry of the Ni(Cys)4 moiety and a vacant coordination site between the Ni and Fe ions. The 2 F obs−F calc electron density map after full refinement is shown as a gray mesh (1.8 σ). C) RR spectrum of an MbFRH single crystal (black) compared to that of a protein solution of ReRH.11b RR spectra were recorded at 80 K using 568‐nm laser excitation and normalized with respect to the most intense signal at 551/553 cm−1. D) [NiFe] active‐site crystal structure. Selected interatomic distances are given in Å. E) IR absorbance spectrum of an MbFRH single crystal, recorded at 80 K. Bands corresponding to the intrinsic, Fe‐bound diatomic ligands and the extrinsic, Ni‐bound CO of the Ni‐SCO redox‐structural state are highlighted in brown and orange, respectively. F) X‐ray structure of the CO‐inhibited [NiFe] active site. The 2 F obs−F calc electron density map after full refinement (1 σ) and the residual F obs−F calc map (5.5 σ) before CO‐modeling are shown as a gray mesh and green surface, respectively. Selected atoms and amino acid residues are shown as spheres (Ni in green, Fe in gray (B) and orange (D and F), S in yellow) and sticks, respectively.
While the presence or absence of bridging oxygen ligands can be clearly established from crystal‐structure analyses, H2‐derived (hydride) ligands are only observable in sub‐atomic‐resolution structures,12 and available crystal structures lacking detectable bridging ligands most likely represent hydride species.9e, 13 Since crystals of MbFRH have been prepared in a mildly reducing atmosphere containing up to 5 % H2, the derived structural data could reflect Nia‐S or a hydride species. Thus, before analyzing the active‐site structure of MbFRH in more detail, we studied the underlying crystals using different vibrational‐spectroscopic techniques to firmly identify the [NiFe] redox‐structural state.
IR spectra of MbFRH crystals reveal CO and CN stretching bands at 1945 and 2065/2080 cm−1, respectively, which resemble the IR fingerprint of the Nia‐S state of several [NiFe] hydrogenases (Figure 2 A).6a In particular, these vibrational frequencies are close to those observed for the as‐isolated regulatory hydrogenase from Ralstonia eutropha (ReRH), which is a spectroscopically valuable yet structurally unexplored reference system for the Nia‐S state.11 Based on this finding, contributions from the Nia‐C hydride intermediate and its photo‐inducible congener, Nia‐L, can be excluded since these catalytic species would give rise to higher and lower CO stretching frequencies, respectively.6a The fully reduced Nia‐SR state, however, cannot be excluded on the basis of IR‐spectroscopic data alone since its dominating sub‐species exhibits an IR fingerprint similar to that of Nia‐S in several [NiFe] hydrogenases.6a Thus, we next recorded resonance Raman (RR) spectra of MbFRH crystals to probe Fe−CO and Fe−CN metal–ligand vibrations as structural markers of the [NiFe] active site (Figure 2 C).11b, 14 In these measurements, Nia‐SR would be photo‐converted to Nia‐L, while Nia‐S would remain unaffected in terms of structural and electronic properties.11b, 14 Again, the obtained vibrational signature is very similar to that of the Nia‐S state as observed for ReRH and a membrane‐bound hydrogenase from the same organism (ReMBH).11b, 14b In particular, a structurally sensitive vibrational mode with considerable Fe−CO stretching character can be detected at 596–598 cm−1, which excludes contributions from Nia‐SR, since its photoproduct Nia‐L would give rise to an Fe−CO stretching frequency above 600 cm−1.11b, 14a Moreover, the intensity of the RR spectrum was found to increase with the excitation wavelength of the Raman probe laser, which is in line with previous observations for Nia‐S11b, 14 and contrary to expectations for reduced [NiFe] hydrogenases.11b
Finally, we also explored the effect of treating MbFRH crystals with CO, a typical inhibitor of [NiFe] hydrogenases.6a, 15 We performed these experiments to check for interaction of CO with the [NiFe] active site, which is expected for Nia‐S (yielding Ni‐SCO) but not for Nia‐C or Nia‐SR.15d Binding of extrinsic CO to the terminal vacant coordination site at the Ni ion of MbFRH is evident from the electron density at this position (Figures 2 F and S6) and a high‐frequency CO stretching band at 2048 cm−1 in the corresponding IR spectrum (Figure 2 E), as also observed for other [NiFe] hydrogenases in the Ni‐SCO state.15a, 15c, 15d Both structural and spectroscopic data show that this inhibited species accumulated to at least 50 % (Figure S6), while the remainder can be assigned to the Nia‐S parent state. In total, the above experiments show that the crystal structure of untreated MbFRH reflects a homogenous Nia‐S state, allowing a detailed analysis of this H2‐binding intermediate.
Structural and electronic properties of Nia‐S have been proposed to be essential for efficient H2 binding and hydride formation in [NiFe] hydrogenases.11b, 16 In the following, we will revisit these proposals to evaluate their validity based on the crystal structure. While there is wide agreement regarding the overall catalytic mechanism of [NiFe] hydrogenases, details about the central steps of H2 binding and activation are, so far, elusive. In particular, the site of initial H2 binding is not known, and either of the two metal ions may be involved in the formation of a (side‐on) H2 σ‐bond complex from Nia‐S. Experimental data on this first catalytic step are not yet available, but recent theoretical studies favor the Ni ion as the initial site of H2 binding.16, 17 According to these studies, the coordination geometry of this metal ion represents a key to the energetically favorable interaction of H2 with the [NiFe] active site. Specifically, a peculiar seesaw‐shaped geometry with trans S−Ni−S angles approaching 120° and 180° was postulated to be mandatory for thermodynamically favorable binding of H2 to Nia‐S.16 In line with this proposal, the [NiFe] active site of MbFRH exhibits trans S−Ni−S angles of 107° and 171°, thereby structurally confirming this unusual geometry of the Nia‐S state (Figure 2 B). This finding also agrees with previous RR studies on ReRH,11b highlighting the merit of combining crystallographic, spectroscopic, and theoretical methods.
Notably, four‐coordinate NiII sites, as found in Nia‐S, typically exhibit (distorted) square‐planar or tetrahedral coordination geometries. This indicates that the atypical seesaw geometry of this [NiFe] intermediate is dictated by the four‐cysteinate coordination pattern and the protein matrix of [NiFe] hydrogenases.16, 18 Remarkably, trans S−Ni−S angles in the Nia‐S state of MbFRH resemble those found in computationally optimized Nia‐S geometries of previous theoretical studies (124° and 151°), but comparison with the underlying crystal‐structure‐derived starting geometries (109° and 166°) yields an even better agreement.16 This indicates that the magnitude and relevance of the structural constraints imposed by the protein matrix is even more pronounced than previously anticipated. Further evidence for the relevance of structural constraints comes from comparing the experimental Ni−Fe distance of Nia‐S to values obtained in theoretical studies. Calculations on small‐ to medium‐size models typically report Ni−Fe distances of up to 3.3 Å,6a, 11b, 19 while models including larger parts of the protein matrix yield smaller values,19 close to those we observe in the crystal structure (2.7 Å; Figure 2 D). This indicates that the protein matrix compresses the Ni−Fe distance to values close to those observed for other catalytic intermediates, including hydride species, Nia‐C and Nia‐SR (2.6 Å),12 and presumed metal–H2 adducts (2.6–2.8 Å).16, 17b This effect likely minimizes structural reorganization during H2 turnover, thereby adding to the remarkable catalytic efficiency of [NiFe] hydrogenases.11b Additionally, a short Ni−Fe distance may also be relevant for metal–metal bond formation, as previously proposed for catalytic intermediates of [NiFe] hydrogenases.20 Remarkably, Ni−Fe distances obtained from large computational models only reproduce the experimental value observed for MbFRH if a low‐spin (S=0) configuration of the [NiFe] active site is assumed in the calculations,16, 19 supporting a singlet ground state of Nia‐S in MbFRH.
To further explore the initial interaction of MbFRH with H2, we also investigated possible intramolecular H2 transfer pathways. To this end, MbFRH crystals were derivatized with xenon to explore hydrophobic gas channels connecting the exterior of the enzyme with the [NiFe] active site (Figures 3 and S7 A). These experiments revealed a second H2‐transfer channel that has not been observed in similar studies on other [NiFe] hydrogenases.21 Surprisingly, the unrelated [NiFeSe] hydrogenase from Desulfovibrio vulgaris Hildenborough features a similar hydrophobic tunnel (Figure S7B), indicating analogous developments in certain hydrogenases that operate bidirectionally in vivo.22 While this narrow channel appears obstructed in crystal structures of other [NiFe] hydrogenases (Figure S7 B),21a, 21b dynamically enhanced H2 transfer via this route remains as a far‐reaching possibility and a target for future studies.
Figure 3.
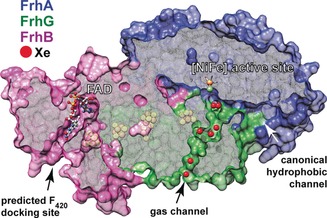
Seven Xe atoms (shown as red spheres) were detected within the noncanonical channel cutting through the FRH‐G subunit. FRH‐A, FRH‐B, and FRH‐G are represented by surfaces and colored navy blue, violet, and green, respectively.
Conclusion
In the current account we have experimentally explored the unique structural properties of the H2‐binding catalytic intermediate of [NiFe] hydrogenases. Besides revealing additional electron and H2 pathways, the spectroscopically validated crystal structure of a pure Nia‐S state unraveled two key determinants of efficient H2 cycling: a peculiar seesaw‐shaped coordination geometry of the Ni ion and a short Ni−Fe distance that is indicative of a low‐spin electronic ground state. Both structural observations contradict expectations for unconstrained low‐molecular‐weight transition‐metal compounds, thereby illustrating the central role of chelating ligand scaffolds and outer coordination layers in biocatalytic H2 cycling. These findings expand our understanding of [NiFe] hydrogenases and, thus, provide valuable guidance for the future design of bio‐inspired catalysts for H2‐based energy‐conversion approaches.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors would like to acknowledge Kathryn Perez (EMBL Heidelberg) for initial measurements as well as Klaus Fiebig (FU Berlin) and Oliver Lenz (TU Berlin) for helpful discussions. Funded by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008/1 – 390540038, (“Unifying Systems in Catalysis – UniSysCat”), the SPP 1927 (DO 785/7‐1, ZE 510/2‐1, SH 87/1‐1), the German excellence initiative (EXC 314 – “Unifying concepts in Catalysis – UniCat”). U. a. Gefördert durch die Deutsche Forschungsgemeinschaft (DFG) im Rahmen der Exzellenzstrategie des Bundes und der Länder – EXC 2008/1 – 390540038. Furthermore, it has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 810856. M.H. thanks the Leverhulme Trust (RPG‐2018‐188) for financial support.
Y. Ilina, C. Lorent, S. Katz, J.-H. Jeoung, S. Shima, M. Horch, I. Zebger, H. Dobbek, Angew. Chem. Int. Ed. 2019, 58, 18710.
Contributor Information
Dr. Marius Horch, Email: marius.horch@york.ac.uk.
Dr. Ingo Zebger, Email: ingo.zebger@tu-berlin.de.
Prof. Dr. Holger Dobbek, Email: holger.dobbek@biologie.hu-berlin.de.
References
- 1. Jones A. K., Sillery E., Albracht S. P., Armstrong F. A., Chem. Commun. 2002, 866–867. [DOI] [PubMed] [Google Scholar]
- 2. Vitt S., Ma K., Warkentin E., Moll J., Pierik A. J., Shima S., Ermler U., J. Mol. Biol. 2014, 426, 2813–2826. [DOI] [PubMed] [Google Scholar]
- 3. Garcin E., Vernede X., Hatchikian E. C., Volbeda A., Frey M., Fontecilla-Camps J. C., Structure 1999, 7, 557–566. [DOI] [PubMed] [Google Scholar]
- 4. Wagner T., Koch J., Ermler U., Shima S., Science 2017, 357, 699–703. [DOI] [PubMed] [Google Scholar]
- 5. Higuchi Y., Yagi T., Yasuoka N., Structure 1997, 5, 1671–1680. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Lubitz W., Ogata H., Rudiger O., Reijerse E., Chem. Rev. 2014, 114, 4081–4148; [DOI] [PubMed] [Google Scholar]
- 6b. Ogata H., Lubitz W., Higuchi Y., J. Biochem. 2016, 160, 251–258; [DOI] [PubMed] [Google Scholar]
- 6c. Shomura Y., Higuchi Y., Rev. Inorg. Chem. 2013, 33, 173–192; [Google Scholar]
- 6d. Fritsch J., Lenz O., Friedrich B., Nat. Rev. Microbiol. 2013, 11, 106–114; [DOI] [PubMed] [Google Scholar]
- 6e. Horch M., Lauterbach L., Lenz O., Hildebrandt P., Zebger I., FEBS Lett. 2012, 586, 545–556; [DOI] [PubMed] [Google Scholar]
- 6f. Shafaat H. S., Rudiger O., Ogata H., Lubitz W., Biochim. Biophys. Acta Bioenerg. 2013, 1827, 986–1002. [DOI] [PubMed] [Google Scholar]
- 7. Happe R. P., Roseboom W., Pierik A. J., Albracht S. P., Bagley K. A., Nature 1997, 385, 126. [DOI] [PubMed] [Google Scholar]
- 8. Darensbourg M. Y., Lyon E. J., Smee J. J., Coord. Chem. Rev. 2000, 206–207, 533–561. [Google Scholar]
- 9.
- 9a. Volbeda A., Martin L., Cavazza C., Matho M., Faber B. W., Roseboom W., Albracht S. P., Garcin E., Rousset M., Fontecilla-Camps J. C., J. Biol. Inorg. Chem. 2005, 10, 239–249; [DOI] [PubMed] [Google Scholar]
- 9b. Volbeda A., Martin L., Barbier E., Gutierrez-Sanz O., De Lacey A. L., Liebgott P. P., Dementin S., Rousset M., Fontecilla-Camps J. C., J. Biol. Inorg. Chem. 2015, 20, 11–22; [DOI] [PubMed] [Google Scholar]
- 9c. Frielingsdorf S., Fritsch J., Schmidt A., Hammer M., Lowenstein J., Siebert E., Pelmenschikov V., Jaenicke T., Kalms J., Rippers Y., Lendzian F., Zebger I., Teutloff C., Kaupp M., Bittl R., Hildebrandt P., Friedrich B., Lenz O., Scheerer P., Nat. Chem. Biol. 2014, 10, 378–385; [DOI] [PubMed] [Google Scholar]
- 9d. Ogata H., Kellers P., Lubitz W., J. Mol. Biol. 2010, 402, 428–444; [DOI] [PubMed] [Google Scholar]
- 9e. Fritsch J., Scheerer P., Frielingsdorf S., Kroschinsky S., Friedrich B., Lenz O., Spahn C. M., Nature 2011, 479, 249–252. [DOI] [PubMed] [Google Scholar]
- 10. Bleijlevens B., van Broekhuizen F. A., De Lacey A. L., Roseboom W., Fernandez V. M., Albracht S. P., J. Biol. Inorg. Chem. 2004, 9, 743–752. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Roncaroli F., Bill E., Friedrich B., Lenz O., Lubitz W., Pandelia M. E., Chem. Sci. 2015, 6, 4495–4507; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Horch M., Schoknecht J., Mroginski M. A., Lenz O., Hildebrandt P., Zebger I., J. Am. Chem. Soc. 2014, 136, 9870–9873. [DOI] [PubMed] [Google Scholar]
- 12. Ogata H., Nishikawa K., Lubitz W., Nature 2015, 520, 571–574. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Higuchi Y., Ogata H., Miki K., Yasuoka N., Yagi T., Structure 1999, 7, 549–556; [DOI] [PubMed] [Google Scholar]
- 13b. Abou-Hamdan A., Ceccaldi P., Lebrette H., Gutierrez-Sanz O., Richaud P., Cournac L., Guigliarelli B., De Lacey A. L., Leger C., Volbeda A., Burlat B., Dementin S., J. Biol. Chem. 2015, 290, 8550–8558; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Shomura Y., Yoon K. S., Nishihara H., Higuchi Y., Nature 2011, 479, 253–256; [DOI] [PubMed] [Google Scholar]
- 13d. Volbeda A., Amara P., Darnault C., Mouesca J. M., Parkin A., Roessler M. M., Armstrong F. A., Fontecilla-Camps J. C., Proc. Natl. Acad. Sci. USA 2012, 109, 5305–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Siebert E., Horch M., Rippers Y., Fritsch J., Frielingsdorf S., Lenz O., Velazquez Escobar F., Siebert F., Paasche L., Kuhlmann U., Lendzian F., Mroginski M. A., Zebger I., Hildebrandt P., Angew. Chem. Int. Ed. 2013, 52, 5162–5165; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5267–5270; [Google Scholar]
- 14b. Siebert E., Rippers Y., Frielingsdorf S., Fritsch J., Schmidt A., Kalms J., Katz S., Lenz O., Scheerer P., Paasche L., Pelmenschikov V., Kuhlmann U., Mroginski M. A., Zebger I., Hildebrandt P., J. Phys. Chem. B 2015, 119, 13785–13796. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Bagley K. A., Van Garderen C. J., Chen M., Duin E. C., Albracht S. P., Woodruff W. H., Biochemistry 1994, 33, 9229–9236; [DOI] [PubMed] [Google Scholar]
- 15b. Purec L., Krasna A. I., Rittenberg D., Biochemistry 1962, 1, 270–275; [DOI] [PubMed] [Google Scholar]
- 15c. Ogata H., Mizoguchi Y., Mizuno N., Miki K., Adachi S., Yasuoka N., Yagi T., Yamauchi O., Hirota S., Higuchi Y., J. Am. Chem. Soc. 2002, 124, 11628–11635; [DOI] [PubMed] [Google Scholar]
- 15d. Pandelia M. E., Ogata H., Currell L. J., Flores M., Lubitz W., Biochim. Biophys. Acta Bioenerg. 2010, 1797, 304–313. [DOI] [PubMed] [Google Scholar]
- 16. Bruschi M., Tiberti M., Guerra A., De Gioia L., J. Am. Chem. Soc. 2014, 136, 1803–1814. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Lill S. O., Siegbahn P. E., Biochemistry 2009, 48, 1056–1066; [DOI] [PubMed] [Google Scholar]
- 17b. Dong G., Phung Q. M., Hallaert S. D., Pierloot K., Ryde U., Phys. Chem. Chem. Phys. 2017, 19, 10590–10601; [DOI] [PubMed] [Google Scholar]
- 17c. Qiu S., Azofra L. M., MacFarlane D. R., Sun C., Phys. Chem. Chem. Phys. 2016, 18, 15369–15374. [DOI] [PubMed] [Google Scholar]
- 18. Horch M., Schoknecht J., Wrathall S. L. D., Greetham G. M., Lenz O., Hunt N. T., Chem. Sci. 2019, 10, 8981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jayapal P., Robinson D., Sundararajan M., Hillier I. H., McDouall J. J., Phys. Chem. Chem. Phys. 2008, 10, 1734–1738. [DOI] [PubMed] [Google Scholar]
- 20. Kampa M., Pandelia M. E., Lubitz W., van Gastel M., Neese F., J. Am. Chem. Soc. 2013, 135, 3915–3925. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Montet Y., Amara P., Volbeda A., Vernede X., Hatchikian E. C., Field M. J., Frey M., Fontecilla-Camps J. C., Nat. Struct. Biol. 1997, 4, 523–526; [DOI] [PubMed] [Google Scholar]
- 21b. Kalms J., Schmidt A., Frielingsdorf S., van der Linden P., von Stetten D., Lenz O., Carpentier P., Scheerer P., Angew. Chem. Int. Ed. 2016, 55, 5586–5590; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 5676–5680; [Google Scholar]
- 21c. Kalms J., Schmidt A., Frielingsdorf S., Utesch T., Gotthard G., von Stetten D., van der Linden P., Royant A., Mroginski M. A., Carpentier P., Lenz O., Scheerer P., Proc. Natl. Acad. Sci. USA 2018, 115, E2229–E2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Michel R., Massanz C., Kostka S., Richter M., Fiebig K., Eur. J. Biochem. 1995, 233, 727–735; [DOI] [PubMed] [Google Scholar]
- 22b. Valente F. M., Oliveira A. S., Gnadt N., Pacheco I., Coelho A. V., Xavier A. V., Teixeira M., Soares C. M., Pereira I. A., J. Biol. Inorg. Chem. 2005, 10, 667–682; [DOI] [PubMed] [Google Scholar]
- 22c. Marques M. C., Coelho R., Pereira I. A. C., Matias P. M., Int. J. Hydrogen Energy 2013, 38, 8664–8682. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary