

Abstract
Aim
Protein kinase (PK) A anchoring protein (AKAP) 12 is a scaffolding protein that anchors PKA to compartmentalize cyclic AMP signalling. This study assessed the consequences of the downregulation or deletion of AKAP12 on endothelial cell migration and angiogenesis.
Methods
The consequences of siRNA‐mediated downregulation AKAP12 were studied in primary cultures of human endothelial cells as well as in endothelial cells and retinas from wild‐type versus AKAP12−/− mice. Molecular interactions were investigated using a combination of immunoprecipitation and mass spectrometry.
Results
AKAP12 was expressed at low levels in confluent endothelial cells but its expression was increased in actively migrating cells, where it localized to lamellipodia. In the postnatal retina, AKAP12 was expressed by actively migrating tip cells at the angiogenic front, and its deletion resulted in defective extension of the vascular plexus. In migrating endothelial cells, AKAP12 was co‐localized with the PKA type II‐α regulatory subunit as well as multiple key regulators of actin dynamics and actin filament‐based movement; including components of the Arp2/3 complex and the vasodilator‐stimulated phosphoprotein (VASP). Fitting with the evidence of a physical VASP/AKAP12/PKA complex, it was possible to demonstrate that the VEGF‐stimulated and PKA‐dependent phosphorylation of VASP was dependent on AKAP12. Indeed, AKAP12 colocalized with phospho‐Ser157 VASP at the leading edge of migrating endothelial cells.
Conclusion
The results suggest that compartmentalized AKAP12/PKA signalling mediates VASP phosphorylation at the leading edge of migrating endothelial cells to translate angiogenic stimuli into altered actin dynamics and cell movement.
Keywords: angiogenesis, protein kinase A, protein kinase A anchoring protein 12, retina, vasodilator‐stimulated phosphoprotein
1. INTRODUCTION
Protein kinase (PK) A anchoring proteins (AKAPs) represent a family of approximately 50 scaffolding proteins that anchor PKA to define and compartmentalize cyclic AMP signalling [for reviews see1, 2, 3, 4, 5]. AKAP12 (also referred to as Gravin and Src‐suppressed C kinase substrate or SSeCKS) binds PKA type II regulatory subunits and was originally identified as an auto‐antigen in myasthenia gravis.6 The name, however, does not do full justice to the AKAP’s ability to bind additional signalling proteins and thus integrate multiple signalling networks, eg, G protein coupled receptor‐mediated signalling, PKC, PKD, phosphatases, phosphodiesterases or cyclins.1, 2, 3, 4, 5
The expression of AKAP12 in endothelial cells was initially overlooked as an early analysis of different tissues from baboons concluded that AKAP12 was lacking from all endothelial cells, with the exception of endothelium of the hepatic sinusoids.7 However, since AKAP12 RNA could be detected in cells treated with phorbol esters, it was proposed that endothelial levels of AKAP12 may be regulated through protein‐dependent mRNA catabolism.7 Cell activation certainly seems to be determinant for AKAP12 expression as it can be induced by lipopolysaccharide,8 and lysophosphatidylcholine.9 Moreover, it seems that AKAP12 is downregulated in contact inhibited cells, but upregulated under conditions that promote migration.10 More recently, AKAP12 has been implicated in the control of vascular integrity11, 12, 13 and endothelial cell function [for reviews see references14, 15, 16]. Indeed, AKAP12 induces blood retinal barrier formation by increasing angiopoietin‐1 and decreasing vascular endothelial growth factor (VEGF) levels in astrocytes.17, 18 In the NIH3T3 cell line, AKAP12 was found to be a critical regulator of genes linked to angiogenesis.19 At the molecular level, this effect was attributed to the downregulation of matrix metalloproteinase 920 and hypoxia‐inducible factor‐1α (HIF‐1α), by enhancing the interaction of HIF‐1α with von Hippel‐Lindau tumour suppressor protein and prolyl hydroxylase 2.17 Also, in cultured endothelial cells, exposure to hypoxia was reported to increase AKAP12 expression and the protein was linked to vascular stability and the inhibition rather than stimulation of angiogenesis.21 Such observations fit with reports that AKAP12 has been reported to act as a tumour suppressor that blocks the cell cycle and inhibits oncogenic proliferation, invasion, chemotaxis and neovascularization.22, 23 However, the situation is not at all clear as the expression of the protein is higher in cancer cells which would tend to indicate a positive role in proliferation.24 Although the role of AKAP12 in endothelial cells has been studied, experiments have concentrated on dermal endothelial cells,13 as well as immortalized cells21 and the role of AKAP12 on permeability—little has been done to assess the consequences of AKAP12 on angiogenesis. Therefore, this study focused on correlating the consequences of the downregulation or deletion of AKAP12 on angiogenesis in vitro and in vivo.
2. RESULTS
2.1. Effect of AKAP12 on endothelial cell migration and sprouting in vitro and in vivo
In confluent primary cultures of human endothelial cells, AKAP12 was localized to the cytoplasm as well as to distinct clusters at cell‐cell borders (Figure 1A). A comparable localization was detected in vivo in en face preparations of mouse aorta and was absent in samples from AKAP12−/− mice (Figure 1B). In sparsely populated and actively migrating endothelial cells, however, AKAP12 was detected in the cytoplasmic compartment and at the leading edge of lamellipodia (Figure 1C), where it colocalized with actin filaments. Moreover, AKAP12 was colocalized with the vasodilator‐stimulated phosphoprotein (VASP), which is an important regulator of actin dynamics, membrane protrusions and cell motility.25, 26
Figure 1.
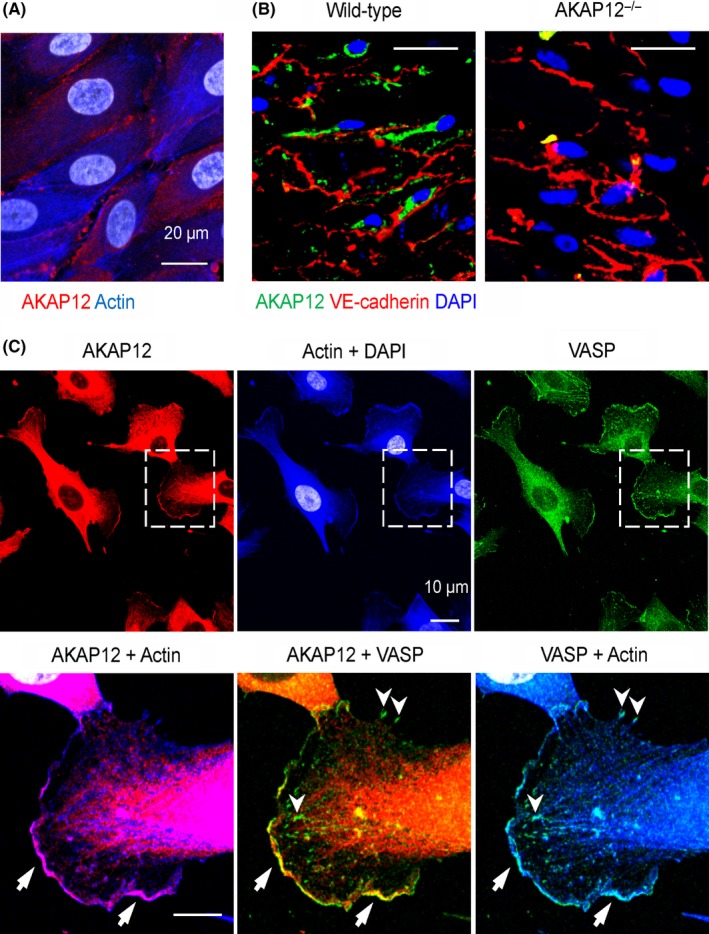
Localization of AKAP12 in human endothelial cells. (A) AKAP12 (red) and actin (blue) in confluent primary cultures of human umbilical vein endothelial cells; nuclei = grey, bar = 20 µm. (B) Localization of AKAP12 (green) and VE‐cadherin in en face preparations of wild‐type and AKAP12−/− mouse aortae. (C) AKAP12 (red) and VASP (green) in sparse/sub‐confluent primary cultures of human umbilical vein endothelial cells; actin = blue, nuclei = grey. Arrows and arrowheads indicate the leading edge of lamellipodia and focal adhesions respectively. Magnified areas are indicated by dashed boxes; Bars 20 µm, magnified views 10 µm. All images are representative of data obtained in 4‐5 independent cell batches or animals
To assess the importance of AKAP12 in angiogenesis, VEGF‐driven endothelial cell sprouting was studied in a modified spheroid assay. While VEGF elicited extensive sprouting in control endothelial cells, the small interfering RNA‐mediated downregulation of AKAP12 clearly attenuated the response (Figure 2A), even though the approach used only depleted ~50% of the endogenous AKAP12 protein (Figure 2B). As AKAP12−/− mice were available, endothelial cell sprouting was also assessed in isolated aortic rings. Endothelial cell sprouting under basal conditions was indistinguishable between rings from wild‐type and AKAP12−/− mice, however, VEGF‐induced sprouting was ablated in aortic rings from AKAP12−/− mice (Figure 2C and 2).
Figure 2.
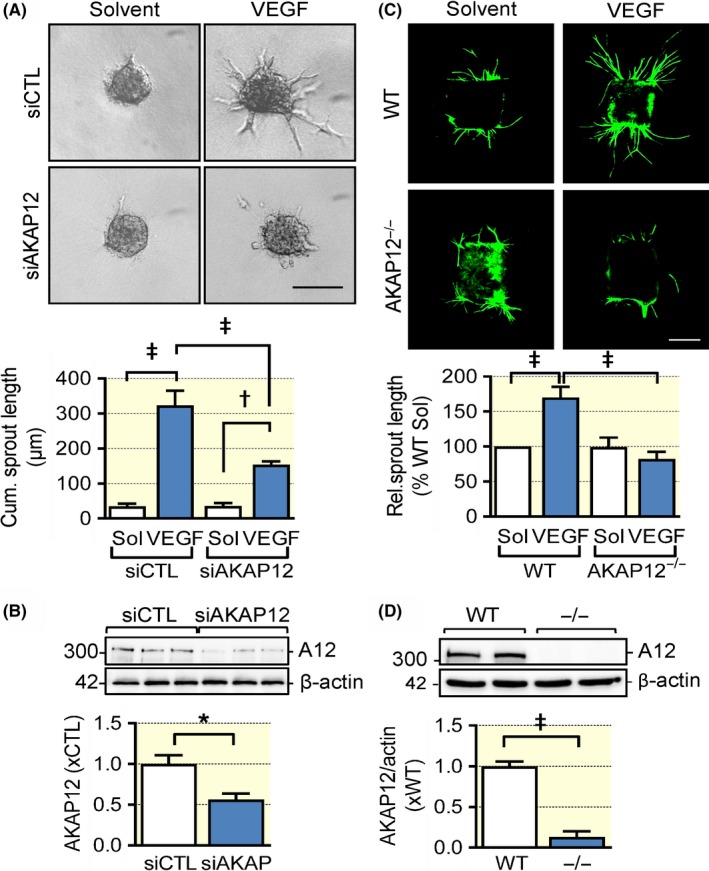
AKAP12 deletion impairs VEGF‐induced endothelial migration and sprouting in vitro. (A) Endothelial cell sprouting in a modified spheroid assay with control (CTL) or AKAP12 siRNA‐treated primary cultures of human endothelial cells. Experiments were performed in the absence and presence of VEGF (30 ng/mL); bar = 10 μm, n = 9 different cell preparations (two‐way ANOVA with Tukey's test). (B) siRNA‐mediated knockdown of AKAP12 (A12) in primary cultures of human endothelial cells (n = 6 different cell preparations, Students t‐test). (C) Aortic ring assay with rings from wild‐type (WT) or AKAP12−/− mice in the presence of solvent or VEGF (30 ng/mL). Seven days after embedding in collagen, endothelial cells were stained with CD31‐specific antibodies (green); bar, 500 μm, n = 5 different animals per genotype, 3 rings per animal and condition; (two‐way ANOVA with Tukey's test). (D) AKAP12 expression in wild‐type (WT) and AKAP12−/− (−/−) mice (n = 5 animals per group, Students t test). *P < 0.05, † P < 0.01, ‡ P < 0.001
To study AKAP12 in a more physiological context, retinal angiogenesis was monitored over the first postnatal week. When compared with retinas from wild‐type mice, AKAP12−/− retinas displayed a significantly delayed radial sprouting of the vascular plexus from the optic nerve to the periphery at postnatal days 2 (P2), 5 and 7 (Figure 3A). Endothelial cell proliferation at the vascular front was analysed by phospho‐histone 3 staining which revealed a significant reduction in the number of AKAP12−/− cells undergoing mitosis (Figure 3B). This fits well with the observation that on P5, AKAP12 expression was highest in endothelial cells at the leading edge of the angiogenic front (Figure 3C). Because of the lack of blood flow in the avascular area of the developing retina, the endothelial cells at the angiogenic front are subjected to hypoxia. Consistent with elevated AKAP12 levels at the vascular front, exposing subconfluent human endothelial cells to hypoxia increased AKAP12 expression in vitro (Figure S1). Interestingly, despite the decreased endothelial cell proliferation, the vascular network appeared denser at the leading front of AKAP12−/− retinas, indicating that the lack of AKAP12 attenuates endothelial cell migration. A more detailed analysis of the tip cells per visual field and the number of tip cell filopodia revealed that both were attenuated in retinas from AKAP12−/− mice compared to their wild‐type littermates (Figure 3D).
Figure 3.
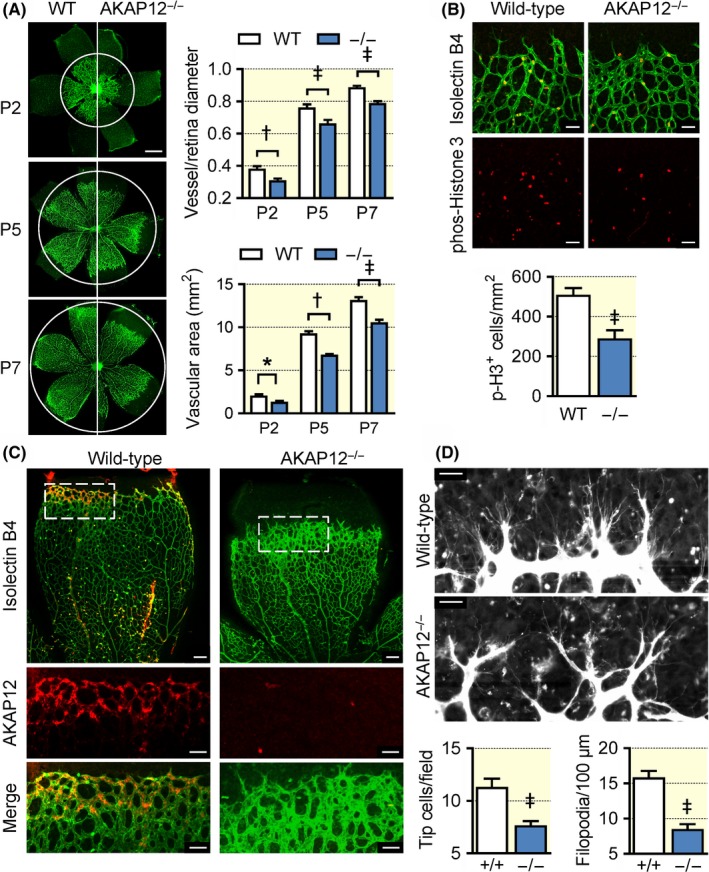
Consequences of AKAP12 deletion on retinal angiogenesis. (A) Isolectin B4 (green) staining of the vascular plexus from wild‐type (WT) and AKAP12−/− (−/−) littermates on postnatal days 2, 5 and 7. The white circles mark the limit of vascularization in WT retinas; n = 10‐12 animals per group (two‐way ANOVA with Tukey's test), bar = 500 µm. (B) Endothelial cell proliferation indicated using phospho‐histone 3 (red) at the angiogenic front of retinas from wild‐type and AKAP12−/− (−/−) mice on postnatal day 5, Isolectin B4 = green; n = 10 mice per group (Student's t test); bars = 50 µm. (C) Localization of AKAP12 (red) at the angiogenic from in retinas from wild‐type mice, ILB4 = green. Retinas from AKAP12−/− mice were included as a control. Similar images were obtained in an additional 5 mice per group; bars = 100 µm, bars in magnified views = 50 µm (D) Tip cell filopodia at the angiogenic front of retinas from wild‐type and AKAP12−/− (−/−) mice on postnatal day 5; bars = 20 µm, n = 4 animals per group (Student's t test). *P < 0.05, † P < 0.01, ‡ P < 0.001
2.2. Link between AKAP12 and key regulators of actin filament‐based movement
Tip cells are highly migratory, and the effects on retinal endothelial cells could be reproduced in murine pulmonary endothelial cells in vitro as the VEGF‐induced migration (in a scratch wound assay) that was detected in cells from wild‐type mice was largely absent in endothelial cells from AKAP12−/− mice (Figure 4A). Interestingly, endothelial cells from wild‐type and AKAP12−/− mice responded equally to FGF stimulation, indicating that specifically VEGF signalling was impaired in AKAP12‐deficient cells (Figure S2).
Figure 4.
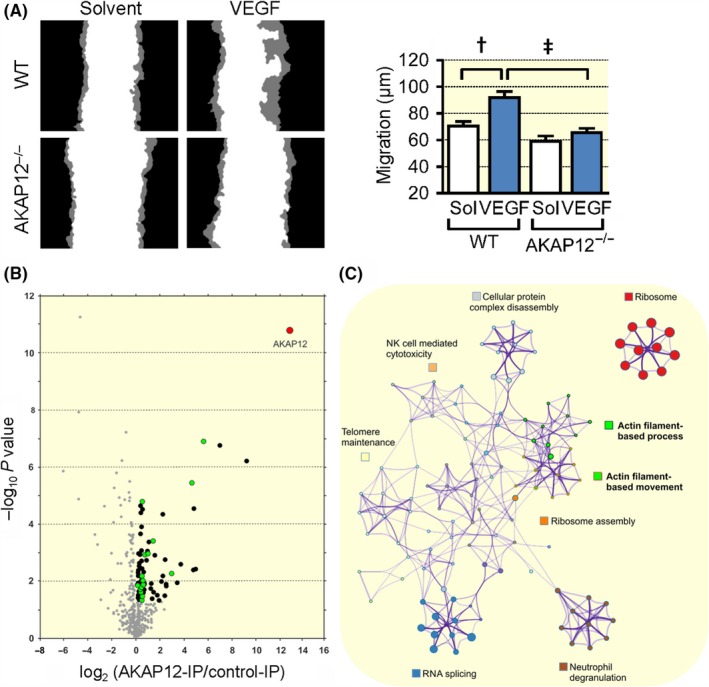
AKAP12 and endothelial cell migration. (A) Scratch wound assay with mouse lung endothelial cells from wild‐type (WT) or AKAP12−/− mice. Migration of cells at the wound edge (indicated in grey) in the presence or absence of VEGF (30 ng/mL) was followed by time‐lapse microscopy for up to 12 hours; n = 3 different cell preparations per genotype, 6 experiments per preparation; (two‐way ANOVA with Tukey's test) ***P < 0.001. (B) AKAP12 was immunoprecipitated from VEGF‐treated, migrating human endothelial cells and immunoprecipitates were subjected to mass spectrometry analysis. The volcano plot shows proteins significantly enriched in AKAP12‐IPs vs. control‐IPs (n = 6 independent cell batches; P < 0.05); proteins involved in actin‐based processes/movement are highlighted in green. (C) Metascape (http://metascape.org) protein‐protein interaction enrichment analysis of all proteins significantly enriched in AKAP12 immunoprecipitates. † P < 0.01, ‡ P < 0.001
Given that AKAP12 was apparently required to promote endothelial cell migration and angiogenesis in vitro and in vivo, mass spectrometry was used to detect proteins interacting with AKAP12 in migrating VEGF‐treated endothelial cells (Figure 4B, Table S1). Metascape (http://metascape.org) protein‐protein interaction enrichment analysis indicated an association of AKAP12 with several distinct protein complexes involved in RNA splicing, cellular protein complex disassembly, ribosome function and others (Figure 4C). The large numbers of ribosomal proteins associated with AKAP12 likely reflects the elevated AKAP12 expression in activated endothelial cells. More importantly, AKAP12 was found to associate with multiple key regulators of actin dynamics and actin filament‐based movement, including components of the Arp2/3 complex, caveolin‐1, coronin‐1C, tropomodulin‐3, tropomyosin α1/α3/α4, different myosin isoforms and the F‐actin capping protein subunit β. Indeed, it was possible to confirm the colocalization of AKAP12 with Arp3 at the leading edge of VEGF‐stimulated, migrating human endothelial cells (Figure 5A). Importantly, similar to the consequences of AKAP12 downregulation siRNAs directed against Arp3 also effectively attenuated VEGF induced endothelial cell sprouting in a modified spheroid assay (Figure 5B). Also, as our initial studies using VASP as a marker indicated that AKAP12 interacted with VASP, separate mass spectrometry experiments using VASP pull‐downs also indicated a direct interaction between the two proteins in human endothelial cells. Importantly, the PKA type II‐α regulatory subunit (PKA‐RII) was also identified in VASP pull‐downs, suggesting that AKAP12 forms complexes with VASP and PKA to translate kinase signalling pathways into actin cytoskeleton remodelling (Table S2, Figure S3). To link these studies with function, we first studied AKAP12 expression and its colocalization with VASP during the switch between static and migrating endothelial cells in a scratch wound assay. Consistent with our previous observations, AKAP12 expression remained consistently low in wound‐distant, non‐migrating endothelial cells. AKAP12 expression was, however, clearly elevated in migrating cells close to the wound where it colocalized with dynamic actin‐ and VASP‐enriched membrane protrusions but not stress fibres or the punctate staining characteristic of focal adhesions (Figure 5C). Moreover, similar to the impaired angiogenesis observed in AKAP12‐defieient endothelial cells, VEGF‐induced endothelial cell sprouting was attenuated in modified spheroids consisting of VASP‐deficient endothelial cells (Figure S4). VASP is reported to be required for the PKA‐mediated activation of Rac1, 27, 28, 29 however, no clear decrease in Rac activation was observed in subconfluent VEGF‐treated human endothelial cells following the downregulation of AKAP12 (Figure S5).
Figure 5.
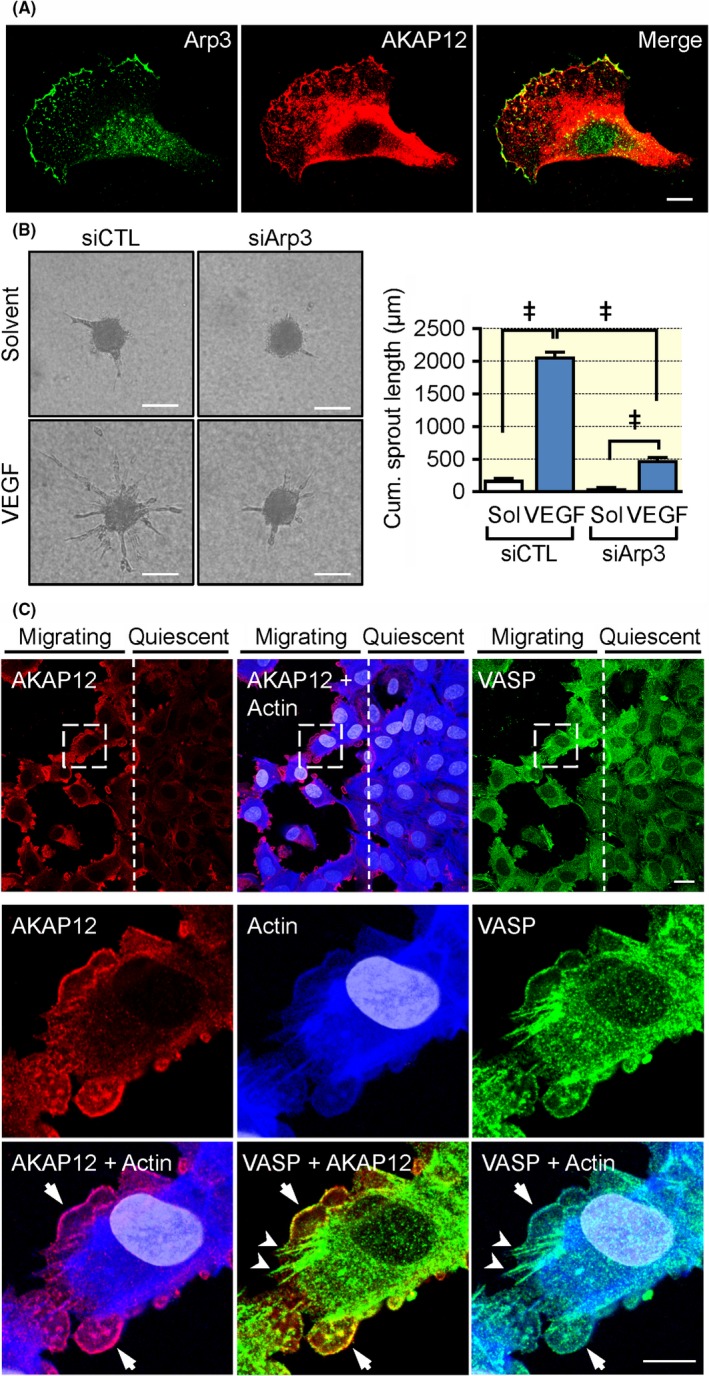
AKAP12 deletion impairs VEGF‐induced endothelial migration and sprouting in vitro. (A) Co‐localization of actin‐related protein 3 (Arp 3) and AKAP12 at the leading edge of lamellipodia in migrating human endothelial cells; bar = 10 µm. (B) Endothelial cell sprouting in a modified spheroid assay with control (CTL) or Arp3 siRNA treated primary cultures of human endothelial cells. Experiments were performed in the absence and presence of VEGF (30 ng/mL); bar = 50 μm, n = 4 different cell preparations (two‐way ANOVA with Tukey's test), ***P < 0.001. (C) A scratch wound together with VEGF (30 ng/mL) was applied to a confluent human endothelial cell monolayer to induce endothelial cell migration at the wound edge, whereas distant cells remained in a resting state. After 16 hours, cells were fixed and stained for AKAP12 (red), VASP (green) and actin (blue). Arrows and arrowheads in the magnified vies indicate co‐localization at the leading edge of lamellipodia and focal adhesions/stress fibres respectively. Magnified areas are indicated by dashed boxes. Bar = 20 μm, magnified views = 10 μm. Comparable results were obtained using three additional (biologically independent) cell batches. ‡ P < 0.001
2.3. Link between VEGF, AKAP12 and the PKA‐dependent phosphorylation of VASP
Our pull‐down experiments indicated VASP/AKAP12/PKA complex formation in endothelial cells. Given that AKAP12 is a PKA anchoring protein and VASP is a well‐established PKA target,26, 30 the next step was to address the link between VEGF‐stimulation, AKAP12 and VASP phosphorylation. In endothelial cells, VEGF stimulated the phosphorylation of VASP on Ser157, which induced a shift in apparent molecular weight from 46 to 50 kDa in SDS PAGE. Previous studies identified VASP Ser157 as the preferred PKA phosphorylation site26, 30 and consistent with this VEGF‐induced VASP phosphorylation on Ser157 was prevented by the kinase inhibitor H89 (Figure 6A and B), which inhibits PKA in addition to a number of other kinases.31 Also, fitting with a functional role of AKAP12 in VEGF‐induced VASP phosphorylation, AKAP12 co‐localized with phospho‐Ser157‐VASP at the leading edge of migrating endothelial cells (see arrows in Figure 6C). More importantly, VASP phosphorylation was largely prevented by the downregulation of AKAP12 (Figure 6D) as well as by blocking AKAP‐PKA complex formation (Figure 6E) using the cell‐penetrating peptide Ht‐31.32 The inactive control peptide Ht31P was without effect.
Figure 6.
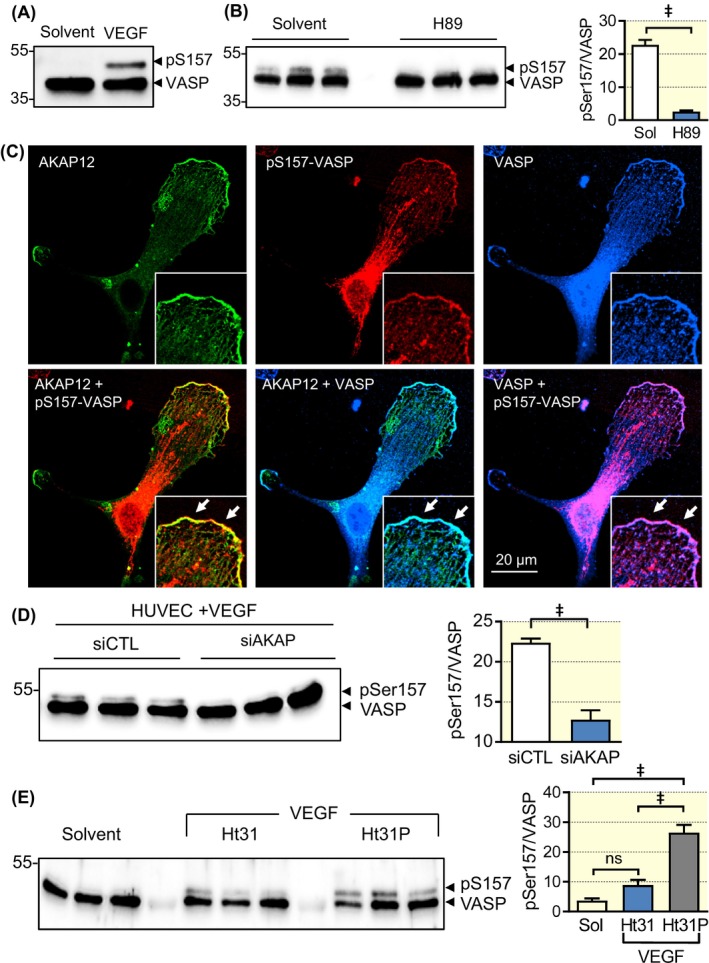
VEGF‐induced VASP phosphorylation is PKA and AKAP12 dependent. Human endothelial cells (~70% confluent) were starved overnight and then treated with either solvent (Sol) or VEGF (30 ng/mL) for 10 minutes in the absence and presence of the PKA inhibitor H89 (10 µM), Ht31 (10 µM), or control Ht31P (10 µM). In some experiments, cells were also treated with a control siRNA or siRNA directed against AKAP12. Lanes in the Western blots represent independent experiments (A) VEGF‐induced phosphorylation of VASP on Ser157. Please note that VASP Ser157‐phosphorylation induces a shift in the apparent molecular weight from 46 to 50 kDa. Comparable results were obtained in additional five independent experiments. (B) Effect of H89 on the VEGF‐induced phosphorylation of VASP on Ser157. Comparable results were obtained in additional four independent experiments. (C) Colocalization of AKAP12 (green), pSer157‐VASP (red) and VASP (blue) in a VEGF‐treated endothelial cell. Comparable results were obtained in additional two independent experiments. (D) Consequence of the siRNA‐mediated downregulation of AKAP12 on the phosphorylation of VASP on Ser157 in VEGF‐treated endothelial cells. n = 9 independent experiments (Student's t test). (E) Consequence of preventing PKA‐AKAP12 complex formation by the Ht31 inhibitory peptide (or control, Ht31P) on the phosphorylation of VASP on Ser157; n = 7 independent experiments (ANOVA and Tukey's multiple comparison). † P < 0.01, ‡ P < 0.001
3. DISCUSSION
The results of the current investigation indicate that AKAP12 is highly expressed in actively migrating, VEGF‐stimulated endothelial cells but is expressed at a much lower level in quiescent endothelial cells. Consistent with this, the downregulation of AKAP12 in vitro as well as its genetic deletion in vivo were associated with migration and proliferation defects as well as markedly attenuated angiogenesis. When expressed in subconfluent endothelial cells AKAP12 was localized to lamellipodia and associated with a number of proteins linked with the regulation of actin dynamics, lamellipodia function and thus cell migration, including the Arp2/3 complex and VASP. Moreover, the association of VASP with AKAP12 appeared to determine its PKA‐mediated phosphorylation downstream of VEGF stimulation.
AKAP12 was expressed at quite different levels in quiescent and actively migrating endothelial cells, a phenomenon more easily appreciated by looking at its expression in a scratch wound assays where the protein was clearly upregulated in the 2‐3 cells closest to the wound, while the cells further away were largely unaffected. This behaviour, which confirms an earlier observation,10 most likely accounts for early reports that AKAP12 was lacking from all endothelial cells.7 Whether AKAP12 is actively downregulated by the initiation of quiescence or upregulated by endothelial cell activation remains to be determined. However, the latter is the more likely given the fact that hypoxia, lipopolysaccharide,8 and lysophosphatidylcholine9 are able to induce AKAP12 expression. More recently the long noncoding RNA metastasis‐associated lung adenocarcinoma transcript 1 (MALAT1) was found to regulate the expression of miR‐145‐5p, which in turn targeted AKAP12.33 While these findings were made using prostate cancer cells, MALAT1,34, 35, 36 and miR‐14537, 38, 39 are both known to affect endothelial cell migration and angiogenesis, especially in cells exposed to hypoxia and in the tumour microenvironment.
In migrating cultured endothelial cells, AKAP12 was concentrated in lamellipodia but largely absent from actin stress fibres and focal adhesions. Consistent with its localization to lamellipodia, the downregulation of AKAP12 attenuated endothelial cell migration in the presence of VEGF, without affecting endothelial cell responses to bFGF. This was surprising since AKAP12 was itself proposed to regulate VEGF levels at least in prostate cancer cells.40 However, fitting with the consequences on migration, AKAP12 knockdown also abrogated VEGF‐induced endothelial cell sprouting in a modified spheroid assay in vitro as well as in an ex vivo aortic ring assay. These findings were also confirmed in the postnatal retina as the development of the vascular plexus was delayed in AKAP12−/− mice, a phenomenon that was also associated with decreased endothelial cell proliferation and a decrease in tip cell specification. Interestingly, the expression of AKAP12 in retinas from wild‐type mice was confined to the leading edge of the developing plexus where both hypoxia (which also increased AKAP12 expression in vitro) and the VEGF gradient are most pronounced. While our in vitro, ex vivo and in vivo observations were all consistent with a positive for of AKAP12 in the endothelial cell migration, and with effects observed in zebrafish,12 previous studies using cultured cells reached opposite conclusions. However, consistent with data from cancer cells where AKAP12 was reported to inhibit motility and cancer cell invasiveness,41 early reports of AKAP12 in endothelial cells linked it with the inhibition of angiogenesis via effects linked to HDAC7 42 and matrix metalloproteinase 9.20 Whether or not these contradictory effects can be explained by the overexpression models studied is unclear but the positive role of AKAP12 in migration reported in the present study was abolished both by its downregulation in vitro and genetic deletion in vivo, thus clearly linking AKAP12 with enhanced endothelial cell migration. This was reinforced by a study published during the preparation of this manuscript that clearly linked VASP with adhesion filopodia formation in osteosarcoma cells.43
Given that AKAP12 was apparently required to promote endothelial cell migration and angiogenesis in vitro and in vivo, we looked at its ability to interact with other proteins involved in regulating cell motility. The immunoprecipitation of AKAP12 from actively migrating cells revealed its interaction with multiple key regulators of actin dynamics and actin filament‐based movement, including components of the Arp2/3 complex. Certainly, AKAP12 co‐localized with Arp3 which nucleates the formation of actin filament arrays that are a hallmark of the lamellipodium at the leading edge of motile cells.44 Moreover, the downregulation of Arp3 had a similar inhibitory effect on endothelial cell sprouting in a modified spheroid assay to that observed following the downregulation of AKAP12. Since VASP is a PKA substrate and antagonist of actin filament capping45 that can enhance Arp2/3 complex‐based actin assembly and motility46, 47 and is involved in endothelial cell migration,48 we also assessed a link between VASP and AKAP12. The observation that VEGF‐induced VASP phosphorylation was largely prevented by the downregulation of AKAP12, as well as by blocking AKAP12‐PKA complex formation certainly tends to suggest that AKAP12 is required as a scaffold to permit the PKA‐dependent phosphorylation of VASP. This interaction would account for the comparable effects of VASP‐ and AKAP12‐downregulation on endothelial cell sprouting.
Because PKA‐mediated VASP phosphorylation is an crucial step for its translocation to the sites of high actin dynamics at the leading edge of endothelial cells,26 AKAP12‐based complex formation may be an important determinant to translate kinase signalling pathways into altered actin dynamics and angiogenic sprouting. There are of course additional molecules involved in the regulation of sprouting and migration, including Rac1, and the PKA/cAMP‐induced activation of Rac1 in endothelial cells was previously reported to require VASP,27, 28, 29 and be sensitive to the depletion of AKAP12 and AKAP220.49 However, we detected no clear decrease in VEGF‐induced Rac activation following the downregulation of AKAP12 in endothelial cells. One reason for this apparent inconsistency is that the experiments performed in the present study used exclusively subconfluent, actively migrating cells, while the cited studies reporting a clear link between PKA‐mediated Rac1 activation and either VASP or AKAP12 were performed using confluent cultures of human or immortalized murine endothelial cells, which would be expected to express little or no AKAP12. Also, Rac1 biology and activation are quite complex and while VASP was reported to be required for Rac1 activation by some authors,27, 28 others have reported that VASP can act as a Rac1 inhibitor.49 Although not studied here, it is interesting to speculate that the association of AKAP12 and VASP may regulate other cell functions. For example, both AKAP12 and VASP play a role in receptor internalization. AKAP12 is thought to be essential for the proper formation of signalling complexes with protein kinases/phosphatases, β‐arrestin and clathrin and thus for the agonist‐induced internalization and resensitization of G‐protein‐linked receptors.50 Ena/VASP proteins, on the contrary, interact with β2‐arrestin, and have been linked with the internalization of CCR251 and the epidermal growth factor receptor,52, 53 as well as the recycling of the transforming growth factor receptor.54 Therefore, it is tempting to speculate that the role of AKAP12 may not be limited to the integration of kinase signalling events, but AKAP12 may rather function as multi‐protein assembling machinery to coordinate actin dynamics as well as PKA‐dependent signalling at the angiogenic front.
4. MATERIALS AND METHODS
4.1. Animals
C57BL/6 mice (6‐8 weeks old) were purchased from Charles River (Sulzfeld, Germany). AKAP12‐deficient (AKAP12−/−) mice were kindly provided by Irwin Gelman (Buffalo, USA) and given their previously described reduced fertility,55 were bred as a heterozygous colony after backcrossing for 10 generations on to the C57BL/6 background. Mice were housed in conditions that conform to “European Convention for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes” (Council of Europe No 123, Strasbourg 1985). All experiments were approved by the governmental authorities (Regierungspräsidium Darmstadt F28/37). Littermates were used throughout this study. For the isolation of tumours and organs, mice were killed using 4% isoflurane in air and subsequent exsanguination.
4.2. Cell culture
Umbilical cords were obtained from a local hospital and umbilical vein endothelial cells were isolated and cultured as described,56 and used only up to passage 1. The use of human material in this study conforms to the principles outlined in the Declaration of Helsinki57 and the isolation of endothelial cells was approved in written form by the ethic committee of the Goethe University. Murine lung endothelial cells were isolated and cultured as described.58
4.3. Plasmids
Hexahistidine‐tagged human wild‐type VASP in pcDNA3 vector was previously described.26 The pCEP4‐Abi FL plasmid59 was a generous gift of Thomas Rudel (Berlin, Germany). pcDNA3‐Abl‐His6‐FLAG60 was a gift from Benjamin Turk (New Haven, CT) and subcloned into pcDNA3 with HindIII/NotI to remove the His6‐FLAG tag. The constitutively active GKP‐Abl mutant (“GKP” corresponds to the mutated amino acids, see below) was generated by site‐directed mutagenesis using the QuikChange Multi kit (Agilent Technologies, Waldbronn, Germany) to introduce the following activating mutations G2A, K70A, P242E and P249E.61 In addition to these mutations, kinase‐dead (KD)‐Abl included the K290M mutation.62 Primers for site‐directed mutagenesis (mutated DNA bases are highlighted in upper case letters):
| Primer | Sequence (5′‐3′) | |
|---|---|---|
| c_Abl_G2A | F | gctcggatccaccatggCgcagcagcctggaaaag |
| R | cttttccaggctgctgcGccatggtggatccgagc | |
| c‐Abl_K70A | F | gcagctcgatggaactccGCggaaaaccttcttgctgg |
| R | ccagcaagaaggttttccGCggagttccatcgagctgc | |
| c_Abl_PP | F | cagctcccaagcgcaacaagGAGactatctacggtgtgtccGAGaactacgacaagtgggaaatgg |
| R | ccatttcccacttgtcgtagttCTCggacacaccgtagatagtCTCcttgttgcgcttgggagctg | |
| c_ABL_K290M | F | gcctcactgtggccgtgATgaccttgaaggaggacac |
| R | gtgtcctccttcaaggtcATcacggccacagtgaggc |
4.4. Small interfering RNA (siRNA)
To silence AKAP12 expression, human endothelial cells were transiently transfected with siRNA directed against AKAP12 (5′ AGG‐UUA‐GUC‐ACG‐CCA‐AGA‐AdTdT 3′ and 5′ UUC‐UUG‐GCG‐UGA‐CUA‐ACC‐UdTdT 3′; Eurogentec, Cologne, Germany), or with control oligonucleotides (5′ UUC‐UCC‐GAA‐CGU‐GGC‐ACG‐A 3′ and 5′ UCG‐UGC‐CAC‐GUU‐CGG‐AGA‐AdTdT 3′; Eurogentec, Cologne, Germany). To silence ARP3 expression, cells were transfected with siRNA directed against ARP3 ((5′ UUG‐GUG‐AUG‐AAG‐CAA‐UAG‐AdTdT 3′ and 5′ UCU‐AUU‐GCU‐UCA‐UCA‐CCA‐AdTdT 3′). AKAP12, ARP3 and control transfections were performed using the Gene trans II kit (MoBiTec GmbH, Göttingen, Germany) according to the manufacturer's instructions. To silence VASP expression, Hs_VASP_4 and VASP_9 Flexi Tube siRNA were purchased from Qiagen (Hilden, Germany). VASP and control transfections were performed using Lipofectamine RNAiMAX (Thermo Fisher Scientific, Dreieich, Germany) according to the manufacturer's protocol.
4.5. Cell migration (scratch wound assay)
Endothelial cells were seeded on 96‐well plates (Ibidi, Martinsried, Germany) coated with fibronectin. Once confluent, a strip of cells was removed using a sterile 96‐well wound‐making device (Essen Bioscience, Ann Arbor, MI, USA) and migration followed for up to 24 hours by live cell imaging (IncuCyte). Percentage of wound closure was analysed using the IncuCyte 2010A software. Representative pictures in the results section show an overlay of the initial cell layer immediately after wounding (black area) and the endothelial cell layer after 24 hours (grey area) for each group.
4.6. Modified spheroid sprouting assay
Spheroids containing 500 cells were generated as described,63 in endothelial basal medium (EBM) supplemented with 2.5% FCS and either solvent or VEGF (30 ng/mL). After 24 hours in a collagen gel, angiogenesis was quantified by measuring the cumulative length of all the capillary like sprouts originating from an individual spheroid using a computer assisted microscope (Zeiss, Oberkochen, Germany) and ImageJ.64 At least five spheroids per experimental group and experiment were analysed.
4.7. Aortic ring assay
Aortae from wild‐type and AKAP12−/− mice were cleaned and dissected into 1 mm rings before embedding in a collagen gel (1.5 mg/mL collagen in Medium 199), seeded in a 48‐well plate and cultured for 7 days in EBM supplemented with 2.5% murine serum in the absence or presence of VEGF (10 ng/mL). After 7 days, the samples were fixed (4% formalin) and the outgrowth of capillaries was visualized using antibodies against CD31, as described.65 Sprout length was measured using Axiovision software (version 4.8, Zeiss).
4.8. Retina preparation and analysis
Retinas for whole‐mount immunohistochemistry were fixed in 4% PFA for 2 hours at room temperature, or overnight at 4 °C. After fixation, retinas were blocked and permeabilized in 1% BSA and 0.5% Triton X‐100 overnight at 4°C. Then retinas were washed three times in Pblec buffer (0.5% Triton X‐100, 1 mM CaCl2, 1 mM MgCl2, and 1 mM MnCl2 in PBS [pH 6.8]), and incubated overnight in Pblec containing Fitc‐labelled Isolectin B4 (1:100, Sigma). The following primary antibodies were diluted in 1% BSA and 0.5% Triton X‐100 and incubated overnight: sEH (1:250, provided by Michael Arand, Zurich, Switerland), GFAP (1:500; DAKO, Glostrup, Denmark or 1:1000; Chemicon, Temecula, CA, USA), NG‐2 (1:1000, Chemicon), aquaporin‐4 (1:500; Santa Cruz, Heidelberg, Germany), glutamine synthase (1:1000, Chemicon), vimentin (1:500; Nococastra, Berlin, Germany). For secondary detection, Alexa Fluor–coupled secondary antibodies (1:200) were used. Cell nuclei were visualized with DAPI (1:200; Molecular Probes). After antibody staining, retinas were post‐fixed in 4% PFA for 15 minutes before flat‐mounting in mounting medium (DAKO). Cryosections (10 µm) were cut after retinas were embedded within Tissue‐Tek OCT Compound (Sakura, Staufen, Germany). Samples were visualized with a confocal microscope (Leica SP8 confocal microscope and LASX software; Wetzlar, Germany or Carl Zeiss LSM‐780 and ZEN software) as described.66
4.9. Immunohistochemistry
Independent batches of endothelial cells were seeded on 1% bovine collagen coated 8‐well µ‐slides (Ibidi, Martinsried, Germany) at a density of 6 × 103/cm2 or 2.5 × 104/cm2 respectively. 24‐48 hours later, cells were briefly rinsed in PBS containing calcium and magnesium and then fixed for 10 minutes at room temperature (4% Histofix, Carl Roth, Karlsruhe, Germany). Following extensive washing with PBS, cells were blocked/permeabilized overnight at 4°C in 5% normal donkey serum with 0.5% Triton X‐100. After incubation with primary antibodies or fluorescent phalloidin in blocking solution overnight at 4°C, cells were washed with PBS, incubated with fluorescent secondary antibodies (Thermo Fischer Scientific) and mounted in PBS with 50% glycerol and 2.5% DABCO (Sigma‐Aldrich, Taufkirchen, Germany). Stained sections were investigated using a confocal laser scanning microscope (LSM 780, Zeiss) equipped with a 63‐fold oil immersion objective. Images were acquired and prepared for presentation using the ZEN software (Carl Zeiss, version 2.1, black).
For scratch wound assays, endothelial cells were grown to confluence in fibronectin‐coated 8‐well µ‐slides, starved overnight in MCDB 131 basal medium with 0.1% BSA before a scratch wound was applied with a yellow pipette tip. Detached cells were removed by replacing the medium supplemented with human VEGF (PeproTech, Hamburg, Germany) to a final concentration of 30 ng/mL. Cells were allowed to migrate for 13 hours before cells were fixed, stained and imaged as detailed above.
4.10. Antibodies and fluorescent probes
Anti‐AKAP12 (Abcam ab118365, goat; Abcam, ab49849, mouse; Abcam isogenic control antibody: ab91361, mouse), anti‐pSer157‐VASP (Nanotools, 5C6, mouse; Cell Signaling Technology, #3111, rabbit), anti‐Arp3 (Millipore 07‐272, rabbit), fluorescent phalloidin (Thermo Fischer Scientific, A22284). The monoclonal anti‐VASP was from Santa Cruz (SC46668) and the polyclonal anti‐VASP (M4) antibody used was generated as described.67
4.11. Immunoblotting
Cells were lysed in Triton X‐100 buffer and detergent‐soluble proteins were resuspended in SDS‐PAGE sample buffer. Proteins were separated by SDS‐PAGE and subjected to Western blotting and visualized by enhanced chemiluminescence using a commercially available kit (Amersham, Freiburg, Germany), as described 58. The antibodies directed against β‐actin were from Linaris (MAK6019, Dossenheim, Germany), and the VE‐cadherin antibody was from Santa Cruz (1:1000, sc‐9989).
4.12. AKAP12 immunoprecipitation
Six fibronectin‐coated 10 cm plates with confluent but not overgrown HUVEC cells from three different donors were split to twelve 12 cm dishes in normal growth medium. Thirteen hours after the split, HUVECs were stimulated with 30 ng/mL human VEGF for 1 hour. The medium was quickly aspirated, dishes were rapidly cooled down on ice‐water and lysed in 750 µL ice‐cold lysis buffer (50 mM Tris‐HCl pH 7.4, 100 mM NaCl, 1% Triton X‐100, 10 mM sodium pyrophosphate, 20 mM sodium fluoride, 2 mM activated sodium orthovanadate, 10 nM ocadaic acid and protease inhibitor mix) per plate. After scraping the cells, lysates were rotated for 10 minutes at 4°C and cleared by centrifugation for 10 minutes at 21 000 × g/4°C. Lysates of each donor were separated into equal halves and either 3 µg mouse anti‐AKAP12 (Abcam ab49849) or 3 µg istotype control antibody (Abcam ab91361) were added. After 2 hours of rotation in the cold‐room, lysates were spun down and transferred to new reaction tubes containing 20 µL pre‐equilibrated protein‐G sepharose (GE Healthcare, Freiburg, Germany). After two additional hours of incubation, the protein‐G resin with the immune complexes was washed three times with 800 µL lysis buffer each, using new reaction tubes after each washing step. The resin was resuspended in 1 mL 50 mM Tris‐HCl pH 7.4 with 100 mM NaCl without protease inhibitors, transferred to new reaction tubes, spun down and washed again in the same buffer. After completely removing the supernatant, the resin was snap‐frozen in liquid nitrogen and stored at ‐80°C until further processing for mass spectrometry. The whole procedure was repeated one week later with three additional, independent batches of endothelial cells, resulting in 6 AKAP12 and 6 control immunoprecipitations in total.
4.13. VASP pull‐downs
Two 10 cm dishes HEK cells each were triple‐transfected with His6‐VASP, Abi‐1 and GKP‐Abl or KD‐Abl using Lipofectamin 2000 according to the manufacturer's recommendations (ThermoFischer Scientific). Twenty‐three hours later, GKP cells were incubated with 20 µM DPH (Sigma‐Aldrich, Taufkirchen, Germany) and KD cells with 20 µM PP2 and 20 µM Glivec (Sigma‐Aldrich, Taufkirchen, Germany) for one hour. Cells were lysed in 1 mL lysis buffer each (40 mM HEPES‐NaOH pH 7.5, 500 mM NaCl, 1% Igepal‐CA630, 10 mM sodium pyrophosphate, 20 mM sodium fluoride, 10 nM ocadaic acid, and protease inhibitor mix) with (GKP) or without (KD) 2 mM‐activated sodium orthovanadate. After brief sonication, lysates were cleared by centrifugation and incubated with 20 µL anti‐6‐His Epitope Tag Affinity Matrix (#900501, Biolgegend, Fell, Germany) for 2.5 hours at 4°C. The matrix was washed 4 times with 600 µL of the corresponding lysis buffer, followed by 1 mL 5 mM HEPES pH 7.5. The purified VASP or pY‐VASP was eluted 3x with 100 µL 50 mM NaOH and immediately neutralized with 10 µL 1 M HEPES pH 6.8. Eluted proteins were pooled, purity and quantity were analysed by colloidal Coomassie staining (InstantBlue, Sigma‐Aldrich, Taufkirchen, Germany) and the tyrosine 16/39 phosphorylation status was confirmed by Western blotting with anti‐phosphotyrosine antibodies (4G10, Merck‐Millipore, Darmstadt, Germany) and mass spectrometry. Three microgram of tyrosine‐phosphorylated or non‐phosphorylated VASP were covalently linked to an affinity matrix (Affigel 10, Biorad, Munich, Germany) for 4 hours at 4°C and remaining binding sites were blocked with excessive amounts of ethanolamine. Five micrograms of purified bovine serum albumin coupled to Affigel 15 were used as control. For pull‐down experiments, two 10 cm dishes human endothelial cells per affinity matrix were lysed in 600 µL lysis buffer (1× TBS with 1% Triton X‐100, 10 mM sodium pyrophosphate, 20 mM sodium fluoride, 2 mM‐activated sodium orthovanadate, 10 nM ocadaic acid, and protease inhibitor mix). Lysates were cleared by centrifugation, incubated with the corresponding affinity matrix for 1.5 hours at 4°C and extensively washed in lysis buffer, using fresh reaction tubes after each washing step. After additional two rounds of washing with 5 mM HEPES‐NaOH pH 7.5, 100 mM NaCl and 0.2% Triton X‐100 (without protease inhibitors), pulled‐down proteins were eluted with 50 mM NaOH and immediately neutralized with 1 M HEPES‐NaOH pH 6.8, snap frozen in liquid nitrogen and stored at −80°C until mass spec analysis. The experiment was repeated with independent cell batches on a different day to yield four samples from VASP‐, pY‐VASP, or BSA‐affinity matrix, each.
4.14. Rac activity assay
Human endothelial cells were grown to a density of approximately 70% and transfected with a control siRNA or siRNA directed against AKAP12. After 24 hours, cells were detached from the culture dishes using accutase (Merck) and split in a ratio of 1:2 (to keep cells sub‐confluent) and after a further 24 hours were treated with VEGF (50 ng/mL, 30 minutes). Cells were washed with ice‐cold PBS and samples were further processed using the Rac1 Pull‐down Activation Assay Biochem Kit (Cytoskeleton, Denver, USA) according to the manufacturer’s protocol. 1200 µg cell lysate per sample was used to pull down active Rac1.
4.15. Proteomics
4.15.1. Sample preparation
Beads from AKAP12 pulldown were resuspended in 50 µL 6 M GdmCl, 10 mM TCEP, 50 mM Tris/HCl, pH 8.5 and incubated at 95°C for 5 min. Sample were diluted with 25 mM Tris/HCl, pH 8.5, 10% acetonitrile to obtain a final GdmCl concentration of 0.6 M. Thiols were alkylated with 10 mM chloroacetamid. Eluates from VASP pull down were diluted in 50 mM ammonium bicarbonate, reduced with 10 mM DTT and alkylated with 30 mM iodoacetamide. Proteins were digested with 1 µg trypsin (sequencing grade, Promega) overnight at 37°C under gentle agitation. Digestion was stopped by adding trifluoroacetic acid to a final concentration of 0.5%. Peptides were loaded on multi‐stop‐and‐go tip (StageTip) containing six C18 discs. Purification and elution of peptides was performed as described in.68 Peptides were eluted in wells of microtiter plates and peptides were dried and resolved in 1% acetonitrile, 0.1% formic acid.
4.15.2. Mass spectrometry
Liquid chromatography/mass spectrometry (LC/MS) was performed on a Thermo Scientific Q Exactive Plus equipped with an ultra‐high performance liquid chromatography unit (Thermo Scientific Dionex Ultimate 3000) and a Nanospray Flex Ion‐Source (Thermo Fisher Scientific). Peptides were loaded on a C18 reversed‐phase precolumn (Thermo Fisher Scientific) followed by separation on a with 2.4 µm Reprosil C18 resin (Dr. Maisch GmbH) in‐house packed picotip emitter tip (diameter 100 µm, 15 cm long from New Objectives) using an gradient from eluent A (4% acetonitrile, 0.1% formic acid) to 50% eluent B1 (99% acetonitrile, 0.1% formic acid) for 90 minutes (AKAP12 enrichment), or to 40% eluent B2 (80% acetonitrile, 0.1% formic acid) for 60 minutes followed by a second gradient to 80% B2 for 30 minutes (VASP pull down). MS data were recorded by data‐dependent acquisition Top10 method selecting the most abundant precursor ions in positive mode for HCD fragmentation. The Full MS scan range was 300 to 2000 m/z with resolution of 70 000, and an automatic gain control (AGC) value of 3 × 106 total ion counts with a maximal ion injection time of 160 ms. Only higher charged ions (2+) were selected for MS/MS scans with a resolution of 17 500, an isolation window of 2 m/z and an automatic gain control value set to 105 ions with a maximal ion injection time of 150 ms. Fullscan data were acquired in profile and fragments in centroid mode by Xcalibur software.
For data analysis MaxQuant (v1.6.0.1),69 Perseus 1.5.6.070 and Excel (Microsoft Office 2013) were used. N‐terminal acetylation (+42.01) and oxidation of methionine (+15.99) were selected as variable modifications and carbamidomethylation (+57.02) on cysteines as a fixed modification. The VASP pull‐down analysis included phosphorylation as modification and the option to match features between runs. The human reference proteome set (Uniprot, 2/2017, 71567 entries) was used to identify peptides and proteins with a false discovery rate (FDR) less than 1%. Minimal ratio count for label‐free quantification (LFQ) was 1. Reverse identifications and common contaminants were removed and the data‐set was reduced to proteins that were quantified in at least 4 of 6 samples (AKAP12 pulldown) or at least 3 of 4 sample (VASP pull down) in one experimental group. Missing LFQ values were replaced by random background values. Significant interacting proteins were determined by Students t test. Only VASP‐interacting proteins with a FDR < 0.05 (q value of permutation‐based false discovery rate) against BSA control were selected for the volcano plot.
4.16. Statistical analysis
Data are expressed as mean ± SEM. Statistical evaluation was performed using Student's t test for unpaired data, one‐way ANOVA followed by a Newman‐Keuls test, two‐way ANOVA with Tukey's test or ANOVA for repeated measures where appropriate. Values of P < 0.05 were considered statistically significant.
GOOD PUBLISHING PRACTICE
The authors confirm that the material submitted is conform with Good Publishing Practice in Physiology: Acta Physiol (Oxf). 2015;215(4):163‐4.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHORS' CONTRIBUTIONS
PMB, AEL and IF designed the research proposal and provided funding. PMB, YD, HS, AEL, JZ, IW, RP conducted the research. PMB, HS, IW analysed the data. PMB and IF wrote the paper. All authors read and approved final draft of the manuscript.
Supporting information
ACKNOWLEDGEMENTS
The authors are indebted to Isabel Winter, Mechtild Piepenbrock‐Gyamfi, Katharina Herbig and Jana Meisterknecht for expert technical assistance.
Benz PM, Ding Y, Stingl H, et al. AKAP12 deficiency impairs VEGF‐induced endothelial cell migration and sprouting. Acta Physiol. 2020;228:e13325 10.1111/apha.13325
Funding information
This work was supported by the Deutsche Forschungsgemeinschaft (SFB 834/1 A4 to A.E.L and I.F. and SFB 834/2 A8 to P.B., SFB 815/3 Z1 to I.W., and Exzellenzcluster 147 “Cardio‐Pulmonary Systems”).
See Editorial Commentary: Walker‐Gray, R., Klussmann, E. 2020. The role of AKAP12 in coordination of VEGF induced endothelial cell motility. Acta Physiol. 228, e13359.
REFERENCES
- 1. Han B, Poppinga WJ, Schmidt M. Scaffolding during the cell cycle by A‐kinase anchoring proteins. Pflugers Arch. 2015;467:2401‐2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Calejo AI, Tasken K. Targeting protein‐protein interactions in complexes organized by A kinase anchoring proteins. Front Pharmacol. 2015;6:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dema A, Perets E, Schulz MS, Deák VA, Klussmann E. Pharmacological targeting of AKAP‐directed compartmentalized cAMP signalling. Cell Signal. 2015;27:2474‐2487. [DOI] [PubMed] [Google Scholar]
- 4. Diviani D, Reggi E, Arambasic M, Caso S, Maric D. Emerging roles of A‐kinase anchoring proteins in cardiovascular pathophysiology. Biochim Biophys Acta. 2016;1863:1926‐1936. [DOI] [PubMed] [Google Scholar]
- 5. Torres‐Quesada O, Mayrhofer JE, Stefan E. The many faces of compartmentalized PKA signalosomes. Cell Sig. 2017;37:1‐11. [DOI] [PubMed] [Google Scholar]
- 6. Gordon T, Grove B, Loftus JC, et al. Molecular cloning and preliminary characterization of a novel cytoplasmic antigen recognized by myasthenia gravis sera. J Clin Invest. 1992;90:992‐999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grove BD, Bowditch R, Gordon T, Zoppo GD, Ginsberg MH. Restricted endothelial cell expression of gravin in vivo. Anat Rec. 1994;239:231‐242. [DOI] [PubMed] [Google Scholar]
- 8. Kitamura H, Okita K, Fujikura D, Mori K, Iwanaga T, Saito M. Induction of Src‐suppressed C kinase substrate (SSeCKS) in vascular endothelial cells by bacterial lipopolysaccharide. J Histochem Cytochem. 2002;50:245‐255. [DOI] [PubMed] [Google Scholar]
- 9. Sato N, Kokame K, Shimokado K, Kato H, Miyata T. Changes of gene expression by lysophosphatidylcholine in vascular endothelial cells: 12 up‐regulated distinct genes including 5 cell growth‐related, 3 thrombosis‐related, and 4 others. J Biochem. 1998;123:1119‐1126. [DOI] [PubMed] [Google Scholar]
- 10. Grove BD, Bruchey AK. Intracellular distribution of gravin, a PKA and PKC binding protein, in vascular endothelial cells. J Vasc Res. 2001;38:163‐175. [DOI] [PubMed] [Google Scholar]
- 11. Lee S‐W, Kim WJ, Choi YK, et al. SSeCKS regulates angiogenesis and tight junction formation in blood‐brain barrier. Nat Med. 2003;9:900‐906. [DOI] [PubMed] [Google Scholar]
- 12. Kwon H‐B, Choi YK, Lim J‐J, et al. AKAP12 regulates vascular integrity in zebrafish. Exp Mol Med. 2012;44:225‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Radeva MY, Kugelmann D, Spindler V, Waschke J. PKA compartmentalization via AKAP220 and AKAP12 contributes to endothelial barrier regulation. PLoS One. 2014;9:e106733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gelman IH. Emerging roles for SSeCKS/Gravin/AKAP12 in the control of cell proliferation, cancer malignancy, and barriergenesis. Genes Cancer. 2010;1:1147‐1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gelman IH. Suppression of tumor and metastasis progression through the scaffolding functions of SSeCKS/Gravin/AKAP12. Cancer Metastasis Rev. 2012;31:493‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee HS, Han J, Bai HJ, Kim KW. Brain angiogenesis in developmental and pathological processes: regulation, molecular and cellular communication at the neurovascular interface. FEBS J. 2009;276:4622‐4635. [DOI] [PubMed] [Google Scholar]
- 17. Choi YK, Kim JH, Kim WJ, et al. AKAP12 regulates human blood‐retinal barrier formation by downregulation of hypoxia‐inducible factor‐1α. J Neurosci. 2007;27:4472‐4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Choi YK, Kim KW. AKAP12 in astrocytes induces barrier functions in human endothelial cells through protein kinase Cζ. FEBS J. 2008;275:2338‐2353. [DOI] [PubMed] [Google Scholar]
- 19. Liu Y, Gao L, Gelman IH. SSeCKS/Gravin/AKAP12 attenuates expression of proliferative and angiogenic genes during suppression of v‐Src‐induced oncogenesis. BMC Cancer. 2006;6:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee SW, Jung KH, Jeong CH, et al. Inhibition of endothelial cell migration through the down‐regulation of MMP‐9 by A‐kinase anchoring protein 12. Mol Med Rep. 2011;4:145‐149. [DOI] [PubMed] [Google Scholar]
- 21. Weissmüller T, Glover LE, Fennimore B, et al. HIF‐dependent regulation of AKAP12 (gravin) in the control of human vascular endothelial function. FASEB J. 2014;28:256‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin X, Nelson PJ, Frankfort B, Tombler E, Johnson R, Gelman IH. Isolation and characterization of a novel mitogenic regulatory gene, 322, which is transcriptionally suppressed in cells transformed by src and ras. Mol Cell Biol. 1995;15:2754‐2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Suren D, Yildirim M, Alikanoglu AS, et al. Lack of relation of AKAP12 with p53 and Bcl‐2 in colorectal carcinoma. Asian Pac J Cancer Prev. 2014;15:3415‐3418. [DOI] [PubMed] [Google Scholar]
- 24. Yildirim M, Suren D, Yildiz M, et al. AKAP12/Gravin gene expression in colorectal cancer: clinical importance and review of the literature. J BUON. 2013;18:635‐640. [PubMed] [Google Scholar]
- 25. Bear JE, Gertler FB. Ena/VASP: towards resolving a pointed controversy at the barbed end. J Cell Sci. 2009;122:1947‐1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Benz PM, Blume C, Seifert S, et al. Differential VASP phosphorylation controls remodeling of the actin cytoskeleton. J Cell Sci. 2009;122:3954‐3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schlegel N, Burger S, Golenhofen N, Walter U, Drenckhahn D, Waschke J. The role of VASP in regulation of cAMP‐ and Rac 1‐mediated endothelial barrier stabilization. Am J Physiol Cell Physiol. 2008;294:C178‐C188. [DOI] [PubMed] [Google Scholar]
- 28. Schlegel N, Waschke J. VASP is involved in cAMP‐mediated Rac 1 activation in microvascular endothelial cells. Am J Physiol Cell Physiol. 2009;296:C453‐C462. [DOI] [PubMed] [Google Scholar]
- 29. Rath S, Liebl J, Fürst R, Vollmar AM, Zahler S. Regulation of endothelial signaling and migration by v‐ATPase. Angiogenesis. 2014;17:587‐601. [DOI] [PubMed] [Google Scholar]
- 30. Butt E, Abel K, Krieger M, et al. cAMP‐ and cGMP‐dependent protein kinase phosphorylation sites of the focal adhesion vasodilator‐stimulated phosphoprotein (VASP) in vitro and in intact human platelets. J Biol Chem. 1994;269:14509‐14517. [PubMed] [Google Scholar]
- 31. Lochner A, Moolman JA. The many faces of H89: a review. Cardiovasc Drug Rev. 2006;24:261‐274. [DOI] [PubMed] [Google Scholar]
- 32. Vijayaraghavan S, Goueli SA, Davey MP, Carr DW. Protein kinase A‐anchoring inhibitor peptides arrest mammalian sperm motility. J Biol Chem. 1997;272:4747‐4752. [DOI] [PubMed] [Google Scholar]
- 33. Xue D, Lu H, Xu H‐Y, Zhou C‐X, He X‐Z. Long noncoding RNA MALAT1 enhances the docetaxel resistance of prostate cancer cells via miR‐145‐5p‐mediated regulation of AKAP12. J Cell Mol Med. 2018;22:3223‐3237. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34. Michalik KM, You X, Manavski Y, et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res. 2014;114:1389‐1397. [DOI] [PubMed] [Google Scholar]
- 35. Huang J‐K, Ma L, Song W‐H, et al. LncRNA‐MALAT1 promotes angiogenesis of thyroid cancer by modulating tumor‐associated macrophage FGF2 protein secretion. J Cell Biochem. 2017;118:4821‐4830. [DOI] [PubMed] [Google Scholar]
- 36. Tee AE, Liu B, Song R, et al. The long noncoding RNA MALAT1 promotes tumor‐driven angiogenesis by up‐regulating pro‐angiogenic gene expression. Oncotarget. 2016;7:8663‐8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zou C, Xu Q, Mao F, et al. MiR‐145 inhibits tumor angiogenesis and growth by N‐RAS and VEGF. Cell Cycle. 2012;11:2137‐2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Climent M, Quintavalle M, Miragoli M, et al. TGFβ triggers miR‐143/145 transfer from smooth muscle cells to endothelial cells, thereby modulating vessel stabilization. Circ Res. 2015;116:1753‐1764. [DOI] [PubMed] [Google Scholar]
- 39. Thuringer D, Jego G, Berthenet K, Hammann A, Solary E, Garrido C. Gap junction‐mediated transfer of miR‐145‐5p from microvascular endothelial cells to colon cancer cells inhibits angiogenesis. Oncotarget. 2016;7:28160‐28168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Su B, Zheng Q, Vaughan MM, Bu Y, Gelman IH. SSeCKS metastasis‐suppressing activity in MatLyLu prostate cancer cells correlates with vascular endothelial growth factor inhibition. Cancer Res. 2006;66:5599‐5607. [DOI] [PubMed] [Google Scholar]
- 41. Su B, Bu Y, Engelberg D, Gelman IH. SSeCKS/Gravin/AKAP12 inhibits cancer cell invasiveness and chemotaxis by suppressing a PKC‐RAF/MEK/ERK pathway. J Biol Chem. 2009;285:4578‐4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Turtoi A, Mottet D, Matheus N, et al. The angiogenesis suppressor gene AKAP12 is under the epigenetic control of HDAC7 in endothelial cells. Angiogenesis. 2012;15:543‐554. [DOI] [PubMed] [Google Scholar]
- 43. Young LE, Latario CJ, Higgs HN. Roles for Ena/VASP proteins in FMNL3‐mediated filopodial assembly. J Cell Sci. 2018;131:jcs220814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Svitkina TM, Verkhovsky AB, McQuade KM, Borisy GG. Analysis of the actin‐myosin II system in fish epidermal keratocytes: mechanism of cell body translocation. J Cell Biol. 1997;139:397‐415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Ann Rev Cell Develop Biol. 2003;19:541‐564. [DOI] [PubMed] [Google Scholar]
- 46. Skoble J, Auerbuch V, Goley ED, et al. Pivotal role of VASP in Arp2/3 complex‐mediated actin nucleation, actin branch‐formation, and Listeria monocytogenes motility. J Cell Biol. 2001;155:89‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Havrylenko S, Noguera P, Abou‐Ghali M, et al. WAVE binds Ena/VASP for enhanced Arp2/3 complex‐based actin assembly. Mol Biol Cell. 2014;26:55‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Price CJ, Brindle N. Vasodilator‐stimulated phosphoprotein is involved in stress‐fiber and membrane ruffle formation in endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:2051‐2056. [DOI] [PubMed] [Google Scholar]
- 49. Jennissen K, Siegel F, Liebig‐Gonglach M, et al. A VASP‐Rac‐soluble guanylyl cyclase pathway controls cGMP production in adipocytes. Sci Signal. 2012;5:ra62. [DOI] [PubMed] [Google Scholar]
- 50. Lin F, Hy W, Malbon CC. Gravin‐mediated formation of signaling complexes in β2‐adrenergic receptor desensitization and resensitization. J Biol Chem. 2000;275:19025‐19034. [DOI] [PubMed] [Google Scholar]
- 51. Laban H, Weigert A, Zink J, et al. VASP regulates leukocyte infiltration, polarization, and vascular repair after ischemia. J Cell Biol. 2018;217:1503‐1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Philippar U, Roussos ET, Oser M, et al. A Mena invasion isoform potentiates EGF‐induced carcinoma cell invasion and metastasis. Dev Cell. 2008;15:813‐828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vehlow A, Soong D, Vizcay‐Barrena G, et al. Endophilin, Lamellipodin, and Mena cooperate to regulate F‐actin‐dependent EGF‐receptor endocytosis. EMBO J. 2013;32:2722‐2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tu K, Li J, Verma VK, et al. Vasodilator‐stimulated phosphoprotein promotes activation of hepatic stellate cells by regulating Rab11‐dependent plasma membrane targeting of transforming growth factor beta receptors. Hepatology. 2014;61:361‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Akakura S, Huang C, Nelson PJ, Foster B, Gelman IH. Loss of the SSeCKS/gravin/AKAP12 gene results in prostatic hyperplasia. Cancer Res. 2008;68:5096‐5103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Busse R, Lamontagne D. Endothelium‐derived bradykinin is responsible for the increase in calcium produced by angiotensin‐converting enzyme inhibitors in human endothelial cells. Naunyn Schmiedebergs Arch Pharmacol. 1991;344:126‐129. [DOI] [PubMed] [Google Scholar]
- 57. World Medical Association . Declaration of Helsinki. Recommendations guiding physicians in biomedical research involving human subjects. Cardiovasc Res. 1997;35:2‐3. [PubMed] [Google Scholar]
- 58. Fleming I, Fisslthaler B, Dixit M, Busse R. Role of PECAM‐1 in the shear‐stress‐induced activation of Akt and the endothelial nitric oxide synthase (eNOS) in endothelial cells. J Cell Sci. 2005;118:4103‐4111. [DOI] [PubMed] [Google Scholar]
- 59. Campa F, Machuy N, Klein A, Rudel T. A new interaction between Abi‐1 and betaPIX involved in PDGF‐activated actin cytoskeleton reorganisation. Cell Res. 2006;16:759‐770. [DOI] [PubMed] [Google Scholar]
- 60. Deng Y, Couch BA, Koleske AJ, Turk BE. A peptide photoaffinity probe specific for the active conformation of the Abl tyrosine kinase. ChemBioChem. 2012;13:2510‐2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hantschel O, Nagar B, Guettler S, et al. A myristoyl/phosphotyrosine switch regulates c‐Abl. Cell. 2003;112:845‐857. [DOI] [PubMed] [Google Scholar]
- 62. Balan V, Nangia‐Makker P, Jung YS, Wang YI, Raz A. Galectin‐3: a novel substrate for c‐Abl kinase. Biochim Biophys Acta. 2010;1803:1198‐1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Korff T, Dandekar G, Pfaff D, et al. Endothelial ephrinB2 is controlled by microenvironmental determinants and associates context‐dependently with CD31. Arterioscler Thromb Vasc Biol. 2006;26:468‐474. [DOI] [PubMed] [Google Scholar]
- 64. Rueden CT, Schindelin J, Hiner MC, et al. Image J2: ImageJ for the next generation of scientific image data. BMC Bioinf. 2017;18:529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zippel N, Ding Y, Fleming I. A modified aortic ring assay for the assessment of angiogenic potential in vitro. Methods Mol Biol. 2016;1430:205‐219. [DOI] [PubMed] [Google Scholar]
- 66. Hu J, Popp R, Frömel T, et al. Müller glia cells regulate Notch signaling and retinal angiogenesis via the generation of 19,20‐dihydroxydocosapentaenoic acid. J Exp Med. 2014;211:281‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Benz PM, Blume C, Moebius J, et al. Cytoskeleton assembly at endothelial cell‐cell contacts is regulated by αII‐spectrin‐VASP complexes. J Cell Biol. 2008;180:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Minimal, encapsulated proteomic‐sample processing applied to copy‐number estimation in eukaryotic cells. Nat Methods. 2014;11:319‐324. [DOI] [PubMed] [Google Scholar]
- 69. Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotech. 2008;26:1367‐1372. [DOI] [PubMed] [Google Scholar]
- 70. Tyanova S, Temu T, Sinitcyn P, et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Meth. 2016;13:731‐740. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials