

Abstract
Background
Clinical exome sequencing (CES) is rapidly becoming the diagnostic test of choice in patients with suspected Mendelian diseases especially those that are heterogeneous in etiology and clinical presentation. Reporting large CES series can inform guidelines on best practices for test utilization, and improves accuracy of variant interpretation through clinically‐oriented data sharing.
Methods
This is a retrospective series of 509 probands from Qatar who underwent singleton or trio CES either as a reflex or naïve (first‐tier) test from April 2014 to December 2016 for various clinical indications.
Results
The CES diagnostic yield for the overall cohort was 48.3% (n = 246). Dual molecular diagnoses were observed in 2.1% of cases; nearly all of whom (91%) were consanguineous. We report compelling variants in 11 genes with no established Mendelian phenotypes. Unlike reflex‐WES, naïve WES was associated with a significantly shorter diagnostic time (3 months vs. 18 months, p < 0.0001).
Conclusion
Middle Eastern patients tend to have a higher yield from CES than outbred populations, which has important implications in test choice especially early in the diagnostic process. The relatively high diagnostic rate is likely related to the predominance of recessive diagnoses (60%) since consanguinity and positive family history were strong predictors of a positive CES.
Keywords: Arab, clinical exome sequencing, consanguinity, Mendelian diseases, Middle East, Qatar
1. INTRODUCTION
Genetic heterogeneity, phenotypic variability, and disease rarity (with consequent lack of familiarity) are all factors that make the traditional diagnostic approach to genetic disorders, whereby a specific gene is selected for sequencing based on the clinical phenotype, very challenging. Whole exome sequencing, with its ability to interrogate many genes in both hypothesis‐driven and hypothesis‐free approaches, has revolutionized the diagnostic process, and clinical exome sequencing (CES) is now a widely adopted diagnostic test. For the purposes of manuscript terminology, CES can be performed as a first‐tier test without performing any prior diagnostic workup, which is commonly, referred to as a “naïve” CES. It can also be performed as a “reflex” CES following prior inconclusive diagnostic workup consisting of imaging studies and/or laboratory studies and/or other genetic tests (whole genome array CGH, single gene testing or NGS multigene panels).
The diagnostic yield of CES, commonly estimated at ~25%, is high compared to other diagnostic tools, including molecular karyotyping, which is officially endorsed by professional societies as a first‐tier test in patients with developmental delay and those with congenital anomalies. Although CES has not yet been endorsed by professional societies as a first‐tier test for patients with suspected Mendelian disorders, recent studies support such an indication for CES and suggest that earlier CES is associated with more favorable cost/benefit ratio (Stark et al., 2018).
In 2015, we reported a high diagnostic yield of CES based on our local experience with the first 149 patients undergoing this testing (Yavarna et al., 2015). The relatively high diagnostic yield appeared to correlate with the frequency of homozygous recessive etiologies, which accounted for the majority of cases. This is not surprising since the Middle Eastern population is characterized by a high level of consanguinity, which was found to strongly predict a positive CES result in that study. Since that study, several others have confirmed a relatively high molecular diagnostic yield of CES in Middle‐Eastern families (Alfares et al., 2017; Al‐Shamsi, Hertecant, Souid, & Al‐Jasmi, 2016; Anazi et al., 2017; Charng et al., 2016; Fattahi et al., 2016; Monies et al., 2017). In this follow‐up study, we expand the reporting of CES from more than 500 previously unpublished cases. In addition to confirming the trends observed in the smaller cohort, the large size of this cohort allowed us to report substantially more novel variants, including those in genes that we propose as novel candidate genes. Our expanded cohort also allowed us to observe the time‐saving advantage of “naïve” versus “reflex” CES.
2. PATIENTS AND METHODS
2.1. Patients
This study includes patients referred to the Clinical and Metabolic Genetics, Pediatrics Department at Hamad Medical Corporation (HMC) from April 2014 to December 2016. Patients were referred for a variety of reasons, including neurocognitive/neurodevelopmental or neuromuscular disorders, multiple congenital anomalies or disorders of other organ systems, such as immunologic, endocrine, or cardiac disorders (described below and in Table 2). Among the 509 patients, 359 underwent CES as a first‐tier test (naïve CES), while 150 patients had a prior diagnostic workup with imaging and/or laboratory studies, including other genetic tests such as whole genome array CGH, single gene testing or NGS multigene panels, which did not establish a diagnosis (reflex CES).
Table 2.
Distribution of diagnostic cases according to clinical presentations
| Categories | Total number (%) | Total number of positive cases (%) | Singleton CES | Trio and Quad CES | |
|---|---|---|---|---|---|
| Neurocognitive (NC) disorders | 229 | 45.0% | 106 (46.0%) | 63 | 43 |
| Neuromuscular (NM) disorders | 45 | 9.0% | 29 (64.4%) | 15 | 14 |
| Multiple congenital anomalies (MCAs) | 116 | 23.0% | 45 (38.8%) | 32 | 13 |
| Other system manifestations (OSMs)a | 119 | 23.0% | 66a (55.5%) | 44 | 22 |
| Total | 509 | 246 (48.3%) | 154 | 92 | |
Ten cases of each Endocrine and GI, and four cases of each nephrology, ophthalmology, Immunology, metabolic, pulmonology, rheumatology, dermatology, neurology.
Patients received pre‐ and post‐test counseling about the scope of CES and its potential to reveal information unrelated to their original disease; informed consent was obtained from all patients or guardians by local clinicians. Peripheral blood samples from patients and their parents were obtained for CES as available. Detailed clinical data including medical history, family pedigree, thorough physical and dysmorphology examination, and any clinically indicated tests, such as MRI or genetic/metabolic were collected. The CES was performed as a part of the diagnostic work up and standard of care. In the case of an inconclusive CES, reanalysis of CES was done at least 1 year after the date of completion of the original CES. This study was approved by the local IRB (Study protocol no: MRC‐01‐18‐273). The “solved” cases in our study were determined to be diagnostic based on molecular results of CES and reanalysis of CES and clinical correlation by a group of expert clinical geneticists, genetic counselors and clinical laboratory scientists.
2.2. CES and variant calling
DNA extracted from peripheral blood samples was sent to a molecular diagnostic laboratory and CES, bioinformatics analysis and variant confirmation and interpretation were performed as reported earlier; when ordered based on clinical indication, mitochondrial testing was performed via a separate mitochondrial genome sequencing and deletion testing assay, with final CES and mitochondrial results returned as a single, combined report (Yavarna et al., 2015). Briefly, all variants were classified according to the ACMG guidelines (Richards et al., 2015). To score variants for the allele frequency criteria in the guidelines, gnomAD and the GME databases were used. A proprietary method for copy number variant (CNV) analysis was used (Retterer et al., 2015). All reported CNVs, as well as other clinically reported variants, were orthogonally confirmed using an appropriate secondary method, such as exon‐level array CGH microarray.
2.3. Statistical analysis
The significance of the diagnostic rate of trio‐CES versus singleton‐CES, p values was calculated by 1‐tailed Fisher exact test. A p value of 0.05 was used as a significance threshold. Odds ratio and 95% confidence interval (CI) for the significance of difference in diagnostic rate were also calculated using SPSS.
3. RESULTS
3.1. Patient demographics
A total of 509 patients who presented with rare and diverse genetic disorders underwent CES. The male to female ratio was 1:1 (Female: N = 248, 49%; Male: N = 261, 51%). Patient ages ranged from prenatal to 53 years. Out of the 509 patients, 467 patients (92%) were children (18 years or younger); 265 (52%) were children younger than 5 years at the time of testing.
Trio (child‐parents) or quad (child, sibling, parents) analysis was used for CES in 34% of cases (159/467) in the child‐proband group, more than in the adult group (8/42 patients; 19%) (Odds ratio: 2.2 [95% CI, 0.99–4.8], p < 0.03), reflecting limited parental availability for adult patients. Males were slightly overrepresented in the childhood group (male: 252 of 467 patients, 54%; female: 215 of 467 patients, 46%; Odds ratio: 1.37 [95% CI, 1.0–1.7], p < 0.01) while the opposite was observed in the adult group (men: 9 of 42 patients, 21%; women: 33 of 42 patients, 79%; Odds ratio: 3.86 [95% CI, 1.887–7.901, p < 0.01]). Consanguinity or a positive family history was observed or reported in 65.0% and 32.0% of cases, respectively. The backgrounds of the probands included nationals from Qatar (58.0%), other Arab countries (27.0%) and the Indian subcontinent (15.0%; Table 1).
Table 1.
Patient demographics for 509 cases
| Group | Sub‐group | Number (%) |
|---|---|---|
| Gender | Male | 261 (51%) |
| Female | 248 (49%) | |
| Age | 0 ≤ 5 years | 265 (52%) |
| 5 ≤ 18 years | 202 (40%) | |
| >18 years | 42 (8%) | |
| Nationality | Qatari | 294 (58%) |
| Other Arab countries | 139 (27%) | |
| Indian subcontinent | 76 (15%) | |
| Parental consanguinity | Yes | 332 (65%) |
| No | 177 (35%) | |
| Family history | Positive | 162 (32%) |
| Negative | 347 (68%) |
3.2. Patients and their clinical presentations
Patients presented with diverse clinical phenotypes: 229 (45.0%) had neurocognitive (NC), 45 (9.0%) neuromuscular (NM) disorders, 116 (23.0%) multiple congenital anomalies (MCAs), and 119 (23.0%) had other system manifestations (OSMs), such as endocrine, GI, or immunologic features (Table 2). The clinical presentation in several patients showed atypical phenotype meaning that the clinical presentation deviates from the established gene‐linked phenotype as summarized in (Supporting Information Table S6).
3.3. Diagnostic yield of CES
The overall diagnostic yield by the CES in this study was 48.3% (n = 246), (naive 48.0%, n = 173/359 vs. reflex CES 48.6%, n = 73/150). The molecular diagnostic rate for each of the phenotypic groups described above is shown in Table 2.
The solved cases (diagnostic yield) were stratified according to the major clinical phenotypes, NC: 106 (46.0%); NM: 29 (64.4%); MCAs: 45 (38.8%); OSMs: 66 (55.5%; Table 2).
The overall diagnostic yield for children younger than 5 years was 132/265 (49.0%). Of the children younger than 5 years at the time of testing (n = 265), trio‐CES was performed for 32% (n = 87) of cases, and singleton‐CES for 68% (n = 178) cases, respectively. The diagnostic yield of trio‐CES was 53% (46/87), which was higher than for singleton‐CES (85/177; 48%). The parental consanguinity in this group was 96/132 cases (72%).
The overall diagnostic yield for children aged 5–18 years was 47% (96/202). Out of the total number children aged 5–18 years (n = 202), trio‐CES and singleton‐CES were performed for 72 (35.0%) and 130 (65%) of cases, respectively. The diagnostic rate for trio‐CES (43/72 cases; 59.0%) was significantly higher compared to singleton‐CES (53/130 cases; 40.0%) (Odds ratio: 2.15 [95% CI, 1.2–3.9], p < 0.01) (Table 3). The parental consanguinity in this group was 62/97 cases (65%). The presence of consanguinity and positive family history predicted a higher clinical sensitivity (56.3%) as compared with those who lacked both (37.0%; p = 0.02). (Table 4).
Table 3.
Molecular diagnosis rate of phenotypic subgroups by age group
| Categories | Age groups | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ≤5 years | 5 ≤ 18 years | >18 years | |||||||
| Total no | Singleton | Trio | Total no | Singleton | Trio and | Total no | Singleton | Trio and | |
| Neurocognitive (NC) only | 59 | 37 | 22 | 45 | 24 | 21 | 2 | 2 | 0 |
| Neuromuscular (NM) only | 12 | 6 | 6 | 16 | 9 | 7 | 2 | 1 | 1 |
| Multiple congenital anomalies (MCA) | 29 | 22 | 7 | 12 | 6 | 6 | 4 | 4 | 0 |
| Other system manifestations | 32 | 20 | 12 | 23 | 14 | 9 | 10 | 9 | 1 |
| Total | 132 | 85 | 47 | 96 | 53 | 43 | 18 | 16 | 2 |
Table 4.
Distribution of cases for parental consanguinity and family history
| Group | Parental consanguinity (PC) | Family history (FH) | Parental consanguinity and family history | |||
|---|---|---|---|---|---|---|
| Classification | Yes | No | Yes | No | Yes/positive | No/negative |
| Total no | 332 | 177 | 162 | 347 | 119 | 135 |
| Solved | 174 (52.4%) | 70 (39.5%) | 88 (54.3%) | 156 (44.9%) | 67 (56.3%) | 50 (37.0%) |
| Odds ratio: 1.68 (95% CI, 1.2–2.4) p = 0.003 | Odds ratio: 1.45 (95% CI, 1.0–2.1) p = 0.03 | Odds ratio: 2.19 (95% CI, 1.3–3.6) p = 0.01 | ||||
3.4. Characterization of CES molecular findings
Using CES, we identified 176 novel and 62 previously published pathogenic or likely pathogenic single nucleotide variants in known genes linked to the clinical phenotypes (Table 5, Supporting Information Tables S1–S3). More than half of the reported PV were homozygous variants in AR genes (60.0%) consistent with the high rate of consanguinity in the study population. Nevertheless, a considerable number of patients had PVs in AD (33%) or X‐linked genes (5%), or de novo CNVs (2%) (Figure 1).
Table 5.
Mutation analysis in diagnostic cases
| Group | Gene | ||||
|---|---|---|---|---|---|
| Novel, likely pathogenic variants in known disease genes | Autosomal recessive (AR) | Autosomal dominant (AD) | Semi dominant | X‐linked | |
|
PIEZO1x3 LRP5 PTH GJC2 ELOVL4 EDNRB COL7A1 CHRNAx2 ANK3 CUL7 AIPL1 ALDH7A1 DAG1 ACY1 CNTNAP1 B3GALNT2 x2 OCLN CENPF x2 LONP1 VPS13B LRBA GFPT1 GLE1 CWF19L1x3 XYLT1 ECEL1 ATP6V0A4 SLC6A3x2 DYM EIF2B2 SLC2A2 FKTN FBP1 GCLC PHKB NBEAL2 RAB27A SLC46A1 HPS1x2 HPS5 SLC25A15 IGHMBP2 ITPA CC2D2Ax2 KCTD7 LIPH MED17 MICU1x7 ATPAF2 RRM2B GNPTG MYBPC3 KLHL41 NEBx3 NHEJ1 NPC1 PDE6C PEX2 PGAP3 PLEC POMT1 LAMA1 SLC10A2x2 DNAH5 DNAH11 ASPM RYR1 LIFR TCTN3 TMPRSS4x2 CYP2R1 CYP2R1 EIF2AK3 WWOX POLH TBCD PEX13 RPIA x2 FANCA FBXL4 SPTA1 |
ACTA1 SLC4A1 PLA2G7 KIF1A EVC DES COL6A1 ABCB4 BSCL2 ANKRD11 ASXL3 BRAF CAMTA1 CHD2 ARID1Bx2 COL11A1 TBX18 RAD21 DSG2 DYNC1H1x2 COL5A2 COL3A1 SLC1A3 LDLR FGFR2 FOXP2 GATAD2B GRIN2Ax2 HNRNPU KMT2D KAT6Bx2 KCNT1x2 LDB3 EFTUD2 MED13Lx2 MEF2Cx2 AHDC1 NF1 NOTCH1 COL1A1 PIEZO1 PIK3CD PIK3R1 POGZ PURA RUNX2 SALL4 SCN2A SETD2 SHANK3 SLC9A9 ATL1 SPECC1L DNMT3A TP63 TCOF1 |
OPA1 |
COL4A5 ATRX DDX3X USP35 HUWE1x2 KDM5C KIAA2022 PDHA1x2 TAF1 |
||
| Variants in novel candidate genes |
HSPB11 CDK9 x2 KIAA0195 UNC13A TRAF3IP2 LAMTOR4 SCHIP1 and IQCJ‐SCHIP1 MAP4K4 |
TUBA3E CLCN3 GRM5 XPOT GALNT11 |
|||
| Previously reported P/LP variants in known disease genes |
SIX6 MYL2 COL7A1 CC2D2Ax2 TMIE VPS13B SRD5A3 MCEE LAMA2 x2 PCNT DDCx2 PLA2G6 ISCA2 UQCRQ C12orf57 RSPH9 SNX14 SEPN1 CLCN1 MKKS ABCB4 SEPSECS XYLT1 SGCA OTOF RNASEH2C BCS1L SLC24A5 TMPRSS3 PEX1 PEX5 GALC TRAPPC9 |
TCOF1 SMARCB1 SYNGAP1 MBD5 MYH3 PTPN11x3 TP53 RYR1 LDB3 TPM2 MAP2K1 CSNK2A1 FLG SATB2 TRPV4 NSD1 FGFR2 TNNI2 MYL2 TBCD PPP2R5C CDK13 |
LDLR |
FOXP3 MECP2 MECP2 MECP2 CLCN4 |
|
| Novel copy number variants |
|
||||
| Mitochondrial pathogenic findings |
|
||||
Figure 1.
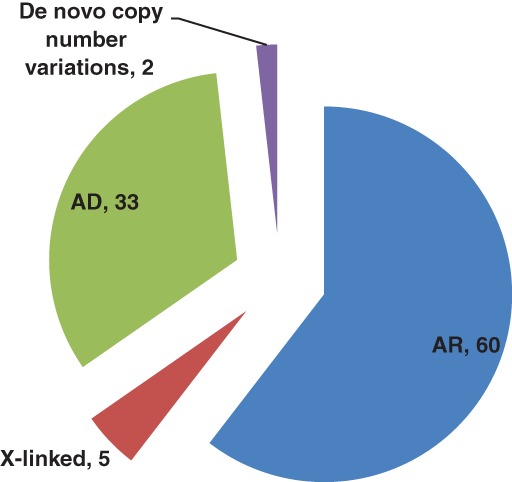
Distribution of cases based on mode of inheritance [Color figure can be viewed at http://wileyonlinelibrary.com]
Out of AR cases, (60%) had homozygous variants for AR traits, 12 (2.3%) patients were compound heterozygous (Table 6) and three (0.5%) had two compound homozygous variants (a complex allele) in the same gene, which was consistent with their original clinical presentations (Table 7).
Table 6.
Patients with compound heterozygous P/LP variants
| Patient | Clinical indication | Gene | Disease | Variant | cDNA |
|---|---|---|---|---|---|
| 1 | Intrahepatic cholestasis, elevated liver enzymes, vitamin D deficiency, and short stature | CYP2R1 | Vitamin D25‐hydroxylasedeficiency |
p.L257SfsX6 p.L89R |
c.266T>G |
| 3 | Seizures, hypomyelination, lactic acidosis, cryptorchidism, and a history of intrauterine growth retardation and premature birth | ALDH7A1 | Epilepsy, pyridoxine‐dependent |
p.V481E p.C154R |
c.1442T>A c.460T>C |
| 4 | Possible Joubert syndrome | CC2D2A | Joubert syndrome |
p.L457X p.E998V |
c.1370T>A c.2993 A>T |
| 5 | Cerebellar ataxia, cerebellar atrophy, and intellectual disability. The family history is significant for two siblings with similar features. | CWF19L1 | Spinocerebellar ataxia 17 |
IVS8‐2A>G p.V229F |
|
| 6 | Arthrogryposis, cerebral palsy, and developmental delay | DAG1 | Alpha‐dystroglycanopathy |
p.T531N p.A98V |
c.1592 C>A c.293 C>T |
| 7 | Elevated liver enzymes, elevated creatine kinase, and motor delay | MICU1 | MICU1‐related muscular dystrophy |
p.Q185X Partial gene duplication |
c.553 C>T NA |
| 8 | Congenital hydrocephalus and developmental delay | B3GALNT2 | Muscular dystrophy‐dystroglycanopathy (congenital with brain and eye anomalies, Type A, 11) |
IVS2‐1G>A p.M75I |
c.261‐1G>A c.225G>A |
| 9 | Slow progressive congenital myopathy | PLEC | Muscular dystrophy, limb‐girdle, Type 2Q |
p.A2110V p.R307C |
c.6329 C>T c.919 C>T |
| 10 | Cerebellar ataxia, cerebellar atrophy, and intellectual disability | CWF19L1 | Spinocerebellar ataxia 17 |
IVS8‐2A>G p.V229F |
c.850‐2A>G c.685G>T |
| 11 | Allergic colitis | SLC10A2 | Primary bile acid |
p.C106X p.P251L |
c.318 C>A c.752 C>T |
| 12 | Autism spectrum disorder | ANK3 | Autism spectrum disorder |
p.P1489S p.S2366P |
c.4465 C>T c.7096T>C |
Table 7.
Patients with two homozygous variants in the same gene
| SN | Clinical indication | Gene | Disease | Variant | cDNA |
|---|---|---|---|---|---|
| 1 | Skeletal dysplasia with spine and femur with Swedish key/monkey wrench appearance. Parental consanguinity | XYLT1 | Desbuquois dysplasia Type 2 |
p.A21GfsX173 IVS7‐3C>T |
c.62delC c.1588‐3C>T |
| 2 | Nonimmune hydrops | PIEZO1 | Lymphatic dysplasia with nonimmune hydrops fetalis |
p.E679X p.A1496V |
c.2035 G>T c.4487 C>T |
| 3 | Pericardial effusion, cardiomegaly, ascites, hepatomegaly, short limbs, hydrops, and echogenic bowel and kidney. Parental consanguinity | SPTA1 | Hereditary spherocytosis type 3 |
p.W279X p.N1934S |
c.836 G>A c.5801 A>G |
Two patients were homozygous for PV in genes currently only known to be associated with autosomal dominant disorders (Table 8).
Table 8.
Patients with homozygous variants in genes typically associated with AD traits
| Patient | Clinical indication | Gene | Disease | Variant | cDNA |
|---|---|---|---|---|---|
| 1 | Epilepsy, hemiparesis, and migrational anomalies | SLC1A3 | Episodic ataxia type 6 | p.K60Q | c.178 A>C |
| 2 | Failure to thrive, lung fibrosis, vasculitis, rash, recurrent angiodema, alopecia, and intermittent limb edema | TMEM173 | STING‐associated vasculopathy with onset in infancy | p.R281W | c.841 C>T |
Eleven (2.1%) patients had two molecularly‐identified genetic disorders consistent with their original clinical presentation (Table 9). In addition, 210 variants of uncertain clinical significance (VUS) were reported. These variants were not included in the diagnostic yield calculations.
Table 9.
Patients with dual molecular diagnoses
| SN | Inheritance | Gene | Disease |
|---|---|---|---|
| 1 |
AR AR |
NBEAL2 DNAH5 |
Gray platelet syndrome Primary Ciliary dyskinesia |
| 2 |
AD AD |
EFTUD2 NOTCH1 |
Mandibulofacial dysostosis Adams–Oliver syndrome 5 |
| 3 |
AD AR |
RYR1 PIEZO1 |
RYR1 related disorder (neuromuscular) Lymphatic dysplasia with nonimmune hydrops fetalis |
| 4 |
AR AD |
LRP5 PLA2G7 |
Van Buchem disease Type 2 Platelet‐activating factor acetylhydrolase deficiency |
| 5 |
AD Mitochondrial |
TPM2 MT‐CYB |
Arthrogryposis Leber hereditary optic neuropathy |
| 6 |
AD AR |
SLC1A3 MCEE |
Episodic ataxia Type 6 Methylmalonic aciduria |
| 7 |
AR AR |
GLE1 ATM |
Lethal congenital contracture Syndrome‐1 Ataxia telangiectasia |
| 8 |
AR AR |
LAMA2 TMPRSS4 |
Muscular dystrophy, congenital merosin‐deficient Cerebral atrophy |
| 9 |
AR AR |
NEB GALC |
Nemaline myopathy Krabbe disease |
| 10 |
AD AD (candidate) |
EVC TUBA3E |
Ellis–van Creveld syndrome Global developmental delay, primary microcephaly, lissencephaly, epilepsy (candidate gene) |
| 11 |
AR AR (candidate) |
RPIA LAMTOR4 |
Ribose‐5‐phosphate isomerase deficiency Candidate gene |
3.5. Novel disease gene discovery
Eleven novel candidate genes were reported as potential contributors to the clinical presentation in the respective patients. These were considered as part of the overall diagnostic yield, and results were managed and returned clinically, with appropriate counseling.
CLCN3 (p.G327A heterozygous, de novo): Two‐year‐old female who presented with optic atrophy and overweight status.
HSPB11 (p.L62AfsX14 homozygous, biparental): Two‐year‐old female with lung hypoplasia, polycystic kidneys, and cardiac hypertrophy.
MACF1 (p.A3721T homozygous, biparental): Deceased female infant with multiple brain abnormalities (severe hydrocephalus and cerebral atrophy).
XPOT (p.C877X heterozygous, inheritance unknown): Forty‐nine‐year‐old male with schizophrenia and a family history of dilated cardiomyopathy, psychiatric illness, and early Alzheimer disease. The reported variant was not detected in mother nor in brother with cardiomyopathy.
GRM5 (AD) (p.S266T heterozygous, de novo): Eleven‐year‐old female with complex vocal and motor tics, learning disability, and obsessive–compulsive disorder.
SCHIP1 (p.R452* homozygous): 4‐year‐old male of consanguineous parents with a history of hypotonia, macrocephaly, developmental delay, and abnormal MRI findings. This homozygous nonsense variant segregated in the proband's two similarly affected sisters.
TUBA3E (p.A150V and p.R215C compound heterozygous): Male with bilateral limb reduction, horseshoe kidney, undescended testes, and neuronal migration defect on brain MRI
UNC13A (p.V1619G homozygous, biparental): Male child with epileptic encephalopathy with intractable seizures and global developmental delay. The family history is significant for parental consanguinity.
LAMTOR4 (p.Q90GfsX42 homozygous, biparental): Female child with Microcephaly, developmental delay, hypertonia, cortical hypomyelination, abnormal white matter, and dysmorphic features.
MAP4K4 (p.S604T homozygous, biparental): Male child with microcephaly, speech delay, facial dysmorphism, developmental delay, and aggressive behavior. Family history is significant for a similarly affected sister who is also homozygous for this variant. The proband's two unaffected brothers were heterozygous for this variant.
GALNT11 (p.R384X Homozygous biparental) Male child withdisproportionate limb shortening, facial dysmorphism, short stature, failure to thrive
3.6. ACMG secondary findings
Fifteen percent of patients/families opted to receive secondary findings. Secondary findings were reported in three probands (3.9% of those who elected to receive these findings) in our cohort, and was performed per to the American College of Medical Genetics and Genomics (ACMG) recommendations (Green et al., 2013). One patient had an autosomal dominant heterozygous pathogenic variant (p.E22K) in the MYL2 gene associated with hypertrophic cardiomyopathy requiring medical action; one patient had an autosomal dominant heterozygous pathogenic variant (p.R248Q) in the TP53 gene for Li–Fraumeni syndrome; and one female patient had an autosomal dominant heterozygous pathogenic variant (c.4136_4137delCT) in the BRCA1 gene. The latter two patients were referred to a cancer center for further management.
3.7. CES reanalysis
CES was negative (nondiagnostic) for 27% of patients (136/509), with no reportable variants and/or where the reported variant did not explain the proband's phenotype. Nevertheless, reanalysis by CES revealed diagnostic results in 34 patients, corresponding to 25% of reanalysis cases. Supporting Information Table S4.
3.8. CES shortens time to diagnosis
The average time to a definitive diagnosis from the patient's first visit to a genetic clinic was significantly shorter (p ≤ 0.0001) for patients who had CES as a first‐tier test (3 months; 173/246 positive cases) compared to those who had other diagnostic workups first, and who then later reflexed to CES (18 months; 73/246 positive cases). An early diagnosis can inform medical management and potentially improve patient quality of life. Supporting Information Table S5 summarizes treatable diagnoses identified by CES. It is also important for defining patient prognoses for families and may enable a patient to participate in clinical trials earlier. Finally, earlier diagnoses can also lead to earlier “cascade” or familial testing. Supporting Information Table S5.
4. DISCUSSION
We previously reported that the diagnostic yield of CES was 60% in 149 patients from Qatar. In this larger study, as we continually enrolled a new cohort of 509 CES with rare and diverse disorders, the overall diagnostic yield of CES was 48.3% (n = 246). The lower diagnostic rate in this current study may be related to the strict use of ACMG guidelines for variant classification, which was not used in the first cohort, but may also involve other factors. Within our cohort, the highest diagnostic rate (64%) was observed for patients with neuromuscular disorders.
As expected, autosomal recessive variants accounted for the majority of identified causative variants. Consistent with our prior published experience, we note that this pattern extends to genes that had only been linked to human diseases in the autosomal dominant mode of inheritance (Monies, Abouelhoda, et al., 2017). In addition, enhanced autozygosity facilitated the co‐inheritance of more than one homozygous disease‐causing variant with resulting dual molecular diagnoses in several cases. The apparently lower incidence of these “multilocus” phenotypes compared to a previous estimate is likely related to our strict use of ACMG guidelines to call pathogenic variants. Indeed, our estimate is nearly identical to another study involving a comparable Middle Easter population that applied to the same criteria (Monies et al., 2017).
Eleven novel candidate genes were reported in this cohort of patients. These genes had not been reported in associated with human disease or the published data to support human disease association may not yet be definitive. While supporting data for the candidacy of these genes (e.g., model organism data, intolerance of the gene to sequence variation, tissue or developmental timing of expression, or knowledge of the gene function and pathway analysis) are suggestive, we emphasize that these remain candidates pending future confirmation through the reporting of similarly affected patients.
Others have noted the importance of reanalyzing CES to improve the diagnostic rate (Ewans et al., 2018; Shamseldin et al., 2017; Wright et al., 2018). However, it is likely that CES will never reach 100% diagnostic rate even in patients with a clearly genetic etiology because of technical limitations. Despite the encouraging early results from WGS in these “negative” cases, the full potential of WGS in patients who could not be diagnosed by CES remains to be seen (Alfares et al., 2018). Testing other tissue sources as well as additional modalities at the RNA, epigenetic, and multilocus levels will likely be necessary to resolve an even higher proportion of cases.
The value of an early diagnosis for most patients is to improve management and quality of life. It is also of critical importance to the patient's family as defining a diagnosis allows specific prognostic predictions, connecting to other families and patient support groups. Although there are limited treatments available for many patients, an earlier diagnosis may allow these patients to participate in available clinical trials.
A limitation of interpretation of variants found by CES in ME patients has been the lack of reliable control cohort data, such as estimating the minor allele frequencies of variants in healthy individuals from the Middle East. However, this obstacle can be overcome by establishing databases of normal variants and disease‐causing variants in Arab population. Examples of current efforts in Qatar and the Arabian gulf region include establishing the Qatar Genome Program (QGP) and the Saudi Human Genome Program (SHGP).
5. CONCLUSIONS
CES is a powerful tool for ending the diagnostic odyssey in cases with an unsolved/undiagnosed genetic disorder after traditional molecular diagnostic approaches have been exhausted, and may even be better deployed as a first step approach. CES has the potential to identify potential novel candidate genes and variants in multiple genes (dual diagnoses) resulting in a more complex disease phenotype. Our results support the adoption of CES as routine clinical diagnostic services locally in Qatar and perhaps to other populations with similar characteristics.
CONFLICT OF INTEREST
The authors declare that there are no conflict of interest.
Supporting information
Supplementary Table 1 Novel LP variants in diagnostic cases (VUS in novel candidate genes are in bold)
Supplementary Table 2: Previously reported P/LP variants in diagnostic cases
Supplementary Table 3: Detailed clinical features in diagnostic cases
Supplementary Table 4: Detailed CES reanalysis data
Supplementary Table 5: Directly treatable disorders identified by CES
Supplementary Table 6: List of cases with atypical presentation, thus broadening the phenotypic spectrum of the respective disorders
ACKNOWLEDGMENTS
The authors would like to thank patients and their families described in this article, the healthcare providers who were involved in their patient care and extend our thanks to Mr. Patricio Santos Ayroso and Ms. Magi Martin for their administrative support. The authors also acknowledge the work of the clinical exome team at GeneDx.
Al‐Dewik N, Mohd H, Al‐Mureikhi M, et al. Clinical exome sequencing in 509 Middle Eastern families with suspected Mendelian diseases: The Qatari experience. Am J Med Genet Part A. 2019;179A:927–935. 10.1002/ajmg.a.61126
REFERENCES
- Alfares, A. , Alfadhel, M. , Wani, T. , Alsahli, S. , Alluhaydan, I. , Al Mutairi, F. , … Zada, A. A. P. (2017). A multicenter clinical exome study in unselected cohorts from a consanguineous population of Saudi Arabia demonstrated a high diagnostic yield. Molecular Genetics and Metabolism, 121(2), 91–95. 10.1016/j.ymgme.2017.04.002 [DOI] [PubMed] [Google Scholar]
- Alfares, A. , Aloraini, T. , Al Subaie, L. , Alissa, A. , Al Qudsi, A. , Alahmad, A. , … Alfadhel, M. (2018). Whole‐genome sequencing offers additional but limited clinical utility compared with reanalysis of whole‐exome sequencing. Genetics in Medicine, 20, 1328–1333. 10.1038/gim.2018.41 [DOI] [PubMed] [Google Scholar]
- Al‐Shamsi, A. , Hertecant, J. L. , Souid, A. K. , & Al‐Jasmi, F. A. (2016). Whole exome sequencing diagnosis of inborn errors of metabolism and other disorders in United Arab Emirates. Orphanet Journal of Rare Diseases, 11(1), 94 10.1186/s13023-016-0474-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anazi, S. , Maddirevula, S. , Faqeih, E. , Alsedairy, H. , Alzahrani, F. , Shamseldin, H. E. , … Alkuraya, F. S. (2017). Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Molecular Psychiatry, 22(4), 615–624. 10.1038/mp.2016.113 [DOI] [PubMed] [Google Scholar]
- Charng, W.‐L. , Karaca, E. , Akdemir, C. , Zeynep, G. , Tomasz, A. , Mehmed, M. , … Lupski, J. R. (2016). Exome sequencing in mostly consanguineous Arab families with neurologic disease provides a high potential molecular diagnosis rate. BMC Medical Genomics, 9, 42 10.1186/s12920-016-0208-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewans, L. J. , Schofield, D. , Shrestha, R. , Zhu, Y. , Gayevskiy, V. , Ying, K. , … Roscioli, T. (2018). Whole‐exome sequencing reanalysis at 12 months boosts diagnosis and is cost‐effective when applied early in Mendelian disorders. Genetics in Medicine, 20, 1564–1574. 10.1038/gim.2018.39 [DOI] [PubMed] [Google Scholar]
- Fattahi, Z. , Kalhor, Z. , Fadaee, M. , Vazehan, R. , Parsimehr, E. , Abolhassani, A. , … Najmabadi, H. (2016). Improved diagnostic yield of neuromuscular disorders applying clinical exome sequencing in patients arising from a consanguineous population. Clinical Genetics, 91, 386–402. 10.1111/cge.12810 [DOI] [PubMed] [Google Scholar]
- Green, R. C. , Berg, J. S. , Grody, W. W. , Kalia, S. S. , Korf, B. R. , Martin, C. L. , … Ormond, K. E. (2013). ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genetics in Medicine, 15(7), 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monies, D. , Abouelhoda, M. , AlSayed, M. , Alhassnan, Z. , Alotaibi, M. , Kayyali, H. , … Alkuraya, F. S. (2017). The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Human Genetics, 136(8), 921–939. 10.1007/s00439-017-1821-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monies, D. , Maddirevula, S. , Kurdi, W. , Alanazy, M. H. , Alkhalidi, H. , Al‐Owain, M. , … Alkuraya, F. S. (2017). Autozygosity reveals recessive mutations and novel mechanisms in dominant genes: Implications in variant interpretation. Genetics in Medicine, 19(10), 1144–1150. 10.1038/gim.2017.22 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. , ACMG Laboratory Quality Assurance Committee (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retterer, K. , Scuffins, J. , Schmidt, D. , Lewis, R. , Pineda‐Alvarez, D. , Stafford, A. , … Haverfield, E. (2015). Assessing copy number from exome sequencing and exome array CGH based on CNV spectrum in a large clinical cohort. Genetics in Medicine, 17(8), 623–629. 10.1038/gim.2014.160 [DOI] [PubMed] [Google Scholar]
- Shamseldin, H. E. , Maddirevula, S. , Faqeih, E. , Ibrahim, N. , Hashem, M. , Shaheen, R. , & Alkuraya, F. S. (2017). Increasing the sensitivity of clinical exome sequencing through improved filtration strategy. Genetics in Medicine, 19(5), 593–598. 10.1038/gim.2016.155 [DOI] [PubMed] [Google Scholar]
- Stark, Z. , Schofield, D. , Martyn, M. , Rynehart, L. , Shrestha, R. , Alam, K. , … White, S. M. (2018). Does genomic sequencing early in the diagnostic trajectory make a difference? A follow‐up study of clinical outcomes and cost‐effectiveness. Genetics in Medicine, 21, 173–180. 10.1038/s41436-018-0006-8 [DOI] [PubMed] [Google Scholar]
- Wright, C. F. , McRae, J. F. , Clayton, S. , Gallone, G. , Aitken, S. , FitzGerald, T. W. , … Firth, H. V. (2018). Making new genetic diagnoses with old data: Iterative reanalysis and reporting from genome‐wide data in 1,133 families with developmental disorders. Genetics in Medicine, 20, 1216–1223. 10.1038/gim.2017.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavarna, T. , Al‐Dewik, N. , Al‐Mureikhi, M. , Ali, R. , Al‐Mesaifri, F. , Mahmoud, L. , … Ben‐Omran, T. (2015). High diagnostic yield of clinical exome sequencing in middle eastern patients with Mendelian disorders. Human Genetics, 134(9), 967–980. 10.1007/s00439-015-1575-0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 Novel LP variants in diagnostic cases (VUS in novel candidate genes are in bold)
Supplementary Table 2: Previously reported P/LP variants in diagnostic cases
Supplementary Table 3: Detailed clinical features in diagnostic cases
Supplementary Table 4: Detailed CES reanalysis data
Supplementary Table 5: Directly treatable disorders identified by CES
Supplementary Table 6: List of cases with atypical presentation, thus broadening the phenotypic spectrum of the respective disorders