

Abstract
Designing engineering materials with high stiffness and high toughness is challenging as stiff materials tend to be brittle. Many biological materials realize this objective through multiscale (i.e., atomic‐ to macroscale) mechanisms that are extremely difficult to replicate in synthetic materials. Inspired from the architecture of such biological structures, we here present flow‐assisted organization and assembly of renewable native cellulose nanofibrils (CNFs), which yields highly anisotropic biofibers characterized by a unique combination of high strength (1010 MPa), high toughness (62 MJ m−3) and high stiffness (57 GPa). We observed that properties of the fibers are primarily governed by specific ion characteristics such as hydration enthalpy and polarizability. A fundamental facet of this study is thus to elucidate the role of specific anion binding following the Hofmeister series on the mechanical properties of wet fibrillar networks, and link this to the differences in properties of dry nanostructured fibers. This knowledge is useful for rational design of nanomaterials and is critical for validation of specific ion effect theories. The bioinspired assembly demonstrated here is relevant example for designing high‐performance materials with absolute structural control.
Keywords: anions, Hofmeister series, mechanical properties, nanomaterials, self-assembly
A hydrodynamics‐based approach for the organization and assembly of fibrillar building blocks into hierarchical macrostructures with multiscale control over the nanostructure leads to exceptional mechanical properties. For the first time, structure–property relationships are understood depending on specific ion effects for wet and dry nanofibrillar systems.
Introduction
Nanoscale building blocks of biological origin exhibit outstanding mechanical properties due to strong interactions and directional assembly of the polymeric constituents.1 Natural provenance and low density destined them to be the best material choice for designing future lightweight renewable materials for a range of advanced applications. Controlled arrangement of these building blocks into hierarchical materials with an attractive combination of stiffness, strength and toughness is of tremendous interest for researchers in materials science and engineering.2 Fibers, in particular are desirable for a wide range of applications, such as lightweight construction in aerospace and automobiles, fuel cell electrodes and many others.3 Additionally, fibers have high economic importance as the production of synthetic fiber accounts for more than 20 % of overall plastic production which is valued at around $55 trillion in 2017.4
Multiple techniques such as wet or dry spinning have been developed to manufacture fibers from nanoparticles, however most of them just do not provide absolute control over the final nano‐ to macro structure.1b, 5 Bottom‐up assembly using microfluidics is a promising strategy for designing structures with defined morphology.1b, 6 Self‐assembly of colloids and nano‐colloids is typically induced by screening or neutralizing the surface charges through the introduction of electrolytes or acids, where the destabilization of colloidal solutions is achieved from a long‐known method of salting out or charge neutralization explained by the classical DLVO (Derjaguin–Landau–Vervey–Overbeek) theory.7
In the 1888, Franz Hofmeister described the capacity of different ionic species referred as kosmotropes or chaotropes to “create” or “break” the ordered structure of water based on the order of the anion and cation series, the so‐called “Hofmeister series”, following their ability to salt out proteins.8 Since then, Hofmeister effects have shown general implications on phenomenon associated with bulk solutions and at interfaces, which is important in the self‐assembly of proteins, polymers, and inorganic or organic nanoparticles.9 The large diversity of different ions provide numerous opportunities to rationally design engineering materials, such as regulating the gelation process of supramolecular hydrogels.
Herein, we apply the accumulated knowledge of Hofmeister effects, today known as specific ion effects, to the state‐of‐the‐art microfluidics‐based assembly process, where CNF dispersions are subjected to flow fields to orient the fibrils along the flow direction, followed by gelation to fabricate nanostructured materials with superior mechanical properties. This nanostructure is inspired by the aligned fibrillar structure in the S2 layer of wood fiber walls.
Experimentally, we show that the ion‐induced assembly of cellulose nanomaterials can moderately reproduce the trends in the mechanical properties as indicated for different ions in the Hofmeister series. First, our analysis of tensile properties shows that choice of ions results in variations in the strength, toughness and stiffness of dry nanostructured CNF biofibers. Furthermore, the dynamic structural features of hydrogels tested by rheology indicate variations in the storage modulus that could be linked to the making and breaking of the nanostructure under shear forces due to the effects of the specific ions. The properties of the hydrogel influence the nanostructure development during wet to dry transition, and these observations clarify the variations in properties of dry CNF fibers. In accordance with recent work from our group that studied the assembly of CNFs based on diffusion of protons,6b, 10 we here suggest that anions such as Cl−, Br−, ClO4 −, HSO4 −, H2PO4 − are included in or expelled from the supramolecular hydrogels to minimize the perturbation of water. Thus, these anions participate in the self‐assembly through direct or indirect influence on CNF interactions. Variations in the mechanical properties are related to the structural discrepancies induced by different ions, based on the most reliable explanations for the Hofmeister series and specific ion effects.
Results and Discussion
Macroscale bio‐based fibers (biofibers) are prepared from flow‐induced alignment of nanofibrils followed by the assembly through surface‐charge controlled sol to gel transition.6b In this hydrodynamics‐based approach, dispersed nanofibrils (Supporting Information, Figures S1 and S2) are aligned in the flow direction with the help of flow fields before inducing a self‐assembly to lock the nanostructure into a metastable colloidal glass.6b, 11 The goal is accomplished by using an elongational flow fields‐based double flow‐focusing geometry (Figures 1 a and S3) where the channel geometry consists of a core flow fluid and two sheath flow fluids6b, 12 In the core flow, a dispersion of surface charged CNFs is injected where the nanofibrils are free to translate due to electrostatic repulsions and Brownian rotary diffusion.13 The first sheath flow consists of deionized (DI) water which aligns the fibrils in the flow direction6b and also protects fibrils from shear close to the channel walls. The alignment of fibrils is quickly diminished due to the Brownian motion; hence it is important to lock the nanofibrils in the aligned state before the system approaches isotropy. The second sheath flow thus consists of a gel initiator that reduces the double layer repulsion between the nanofibrils mainly through a protonation of the carboxyl groups but also through an increased ionic strength. This induces the self‐assembly of the fibrils into a well‐packed colloidal glass with maximized CNF‐CNF contacts (Figure 1 b). Van der Waals forces and arrested translational and rotational movement are the presumed mechanism behind the gel formation. The obtained continuous gel threads are subsequently picked up from the DI water bath, held at their ends and air‐dried (Figure 1 c). Characterization of the CNFs fiber surface with scanning electron microscopy (SEM) (Figures 1 d and e) sampled in the longitudinal direction showed highly anisotropic and densely packed organization of fibrils.
Figure 1.
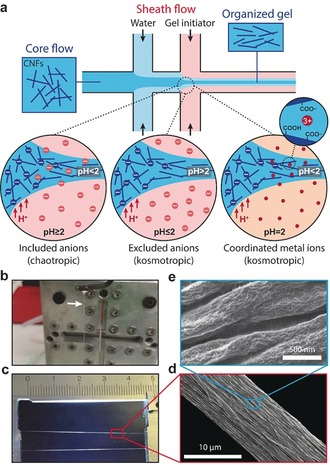
Biofiber assembly. a) Schematic of the channel setup used to achieve flow‐induced alignment and illustration of three alternative locking mechanism. b) Picture of the gel thread after the assembly. c) Picture of the dried CNF biofiber. d) and e) Electron micrographs of the fibers.
Using this assembly technique, we explore three alternative gel initiation mechanisms (Figure 1 a), including acids that are either chaotropic or kosmotropic, or a Lewis acid in the form of FeCl3 with the highest propensity to coordinate ligands on the surface of the CNFs to form complexes which crosslink fibrils in strong hydrogel networks.9d, 14 Chaotropic ions disrupt the order of water, have a low hydration enthalpy, are relatively larger, and generally more polarizable, whereas kosmotropic ions increase the order of water, have a high hydration enthalpy, are relatively small, and generally less polarizable. In the specific ion effect theories, the hydration enthalpy and the polarizability of ions have been suggested to explain the Hofmeister series by direct interactions with water and dispersed colloids.15
To understand how CNF hydrogels are affected by the gel initiation mechanisms, we must elucidate the properties of CNF surfaces. Cellulose should be considered chaotropic since kosmotropic polymers are typically soluble in water. Furthermore, most carbohydrate polymers have a positive chi‐parameter which means that their interaction with water is typically not stronger than interactions between water molecules.16 However, the added carboxyl groups on the surface of CNFs are strongly hydrated, but at a charge density of 0.6 mmol g−1, the coverage is moderate and at low pH, most of the carboxyl groups are protonated and the chaotropic properties dominate.17
Chaotropic ions accumulate near chaotropic surfaces, such as CNFs, in order to minimize the perturbation of water (Figure 2 a) or are adsorbed via ion‐surface dispersion interactions (Figure 2 b), whereas kosmotropic ions accumulate in the bulk water solution (Figure 2 a) or at kosmotropic surfaces for the same reason.9d
Figure 2.
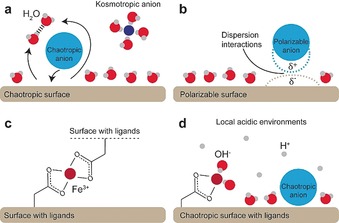
Specific ion effect schematics. a) Chaotropic or kosmotropic ions are accumulated at or excluded from surfaces. b) Polarizable ions adsorb to surfaces via dispersion interactions. c) Coordination complex formation. d) Local acidic environments induced by coordinated or adsorbed ions via balancing of ionic species similar to the Donnan equilibrium.
Fe3+ ions are highly hydrated and coordinate water, but the entropy of the system can be increased by exchanging the tightly bound water with something which has fewer degrees of freedom, such as carboxyl ligands on the CNF surface. Metal ions therefore accumulate inside the hydrogels by a combination of electrostatic attraction and dative covalent coordination bonds regardless of their kosmotropic nature.9d, 14b For this to conform to the specific ion effect theories which attempt to explain the Hofmeister series by the perturbation of water, we can no longer use average properties of the CNFs but must realize that specific groups, for example, carboxyl ligands, enable specific ion binding.
Furthermore, chaotropic and kosmotropic anions (Figure 2 d), or coordinated Fe3+ (Figure 2 c) induce a local environment (Figure 2 d).18 The cause of the local environment is similar to the Donnan equilibrium for fibers in pulp, with the fiber surface or the hydrogel surface acting as a semipermeable membrane through which ions migrate depending on their specific properties (Figure S4).18 Accumulation of chaotropic ions close to chaotropic CNF surfaces inside the hydrogel thus increase the local concentration of acid, whereas excluded kosmotropic ions result in a lower concentration of acid inside the hydrogel than in the bulk solution (Figure 1 a). An inevitable consequence of this Hofmeister effect is that the pH measurement using glass electrodes is ion‐specific since chaotropic acids accumulate at the surface of glass electrodes and induce a local pH.19 There are therefore slight uncertainties in pH measurements, which are indeed difficult to avoid and moreover diminish small specific ion effect variations since the pH is not exactly what we measure.
The above information allows us to predict how different gel initiators affect the properties of the hydrogels (illustrated in Figure 1 a). The biofiber properties can subsequently be related to the properties of the hydrogel in order to rationalize ion‐specific assemblies of CNFs using flow‐induced alignment.
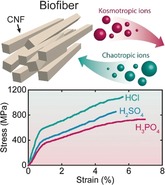
For a comprehensive understanding of how specific ions affect the assembly process and hence the mechanical properties of the fibers, we first prepared fibers using three different acids: HCl, H2SO4, and H3PO4 at pH 2 and a CNF concentration of 0.3 wt % (Figure 3 a). A linear elastic region followed by a plastic region is observed in all stress‐strain curves (Figures 3 a and b). The yield point in the stress‐strain curves highlights the transition from elastic to plastic deformation and is most probably associated with the relative sliding of nanofibrils.6b, 20 Some fibers have a secondary yield point at high strain which has been hypothesized to be governed by cooperative entanglements and release of nanofibrils from such structures.21
Figure 3.
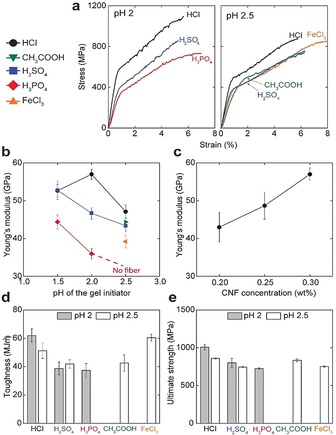
Tensile mechanical properties of CNF biofibers. a) Representative stress‐strain curves of fibers prepared at pH 2 and 2.5 of the gel initiator and 0.3 wt % CNF concentration. Young's modulus as a function of b) pH of the gel initiator and, c) CNF concentration for HCl. d) Toughness and e) ultimate strength of fibers prepared with different gel initiators at pH 2 and 2.5. The different acids are referred to by the color and shape of the data points. Error bars are 90 % confidence intervals based on at least 10 different measurements for each type of sample. All the measurements are done at 50 % RH.
CNF fibers prepared using HCl results in stiffer fibers compared to those prepared using H2SO4 and H3PO4, with a modulus and ultimate strength of ≈57 GPa and ≈1010 MPa, respectively (Figure 3 a). The obtained modulus and ultimate strength with H3PO4 is ≈38 GPa and ≈740 MPa (Figures 3 b and e). Surprisingly, despite significant variations in the elastic modulus of the fibers, the orientation of nanofibrils remain almost the same as determined by wide‐angle X‐ray scattering (WAXS) (Figure S5). The orientation index22 measured for HCl, H2SO4 and H3PO4 is 0.850±0.003, 0.867±0.007 and 0.847±0.012, respectively. The large variation in terms of mechanical properties highlights that the process of flow‐induced assembly results in a certain level of orientation of the nanofibrils,6b whereas the properties of the fibers are governed largely by the specific properties of the anions of acids that are used as gel initiators.
Infrared spectroscopy shows that a large portion of the carboxyl groups of CNFs are still in a dissociated state (1600 cm−1) in fibers prepared at pH 2 (Figure S6 a), which shows the importance of considering local environments.
For a thorough understanding on the ion‐specific properties of our materials, we proceeded to prepare fibers with four different gel initiators at pH 2.5 and a CNF concentration of 0.3 wt % (Figure 3 a). Fibers prepared using HCl resulted in the stiffest fibers similar to the case at pH 2. Among the acids HCl, H2SO4, and CH3COOH, the lowest stiffness was obtained with CH3COOH. Surprisingly, we did not manage to obtain fibers with H3PO4 at pH 2.5 since the gel threads were too weak to sustain air–water interfacial tension while lifted out of the water bath. The compiled data in Figure 3 b shows that a lower pH lead to stiffer and stronger fibers, with the exception of HCl at pH 1.5
Apart from anionic acids, fibers were also prepared using FeCl3 of pH 2.5 and an inferior modulus (39±2) but enhanced ultimate strength (834±21) and strain‐to‐failure (≈8 %) was obtained (Figure 3 a). This is related to the dative covalent coordination complexes which affect the relaxation and consolidation of the aligned CNF structure and hence the morphology of the fiber upon drying.23 Interestingly, unlike Fe3+ treated isotropic films, the carbonyl vibration of Fe3+ treated oriented fibers show complete dissociation of the carboxyl groups (Figure S6 b).9d Fibrils in parallel configuration favors complexation which pushes the equilibrium towards dissociation regardless of the local acidic environment.
To extend the property range of these fibers, we tuned the concentration of CNFs in the dispersions and observed that the stiffness increased proportionally to the solids content of the hydrogel, from a stiffness of ≈57 GPa and a strength of 1010 MPa at 0.3 wt % to a stiffness of 43 GPa and a strength of 770 at 0.2 wt % (Figure 3 c). The decrease in stiffness and ultimate strength is related to the decreased solids content of the gel thread that may lead to heterogeneously packed networks of the nanofibrils in the dried fiber. However, it was experimentally challenging to obtain reliable statistics on the density measurements of our materials. Nevertheless, these results show that the stiffest materials also tend to be the strongest by ensuring adequate load transfer and cohesion between the nanoscale constituents.1a
Apart from the strength and stiffness, we also evaluated the work of fracture (toughness) defined as the area under stress‐strain curves (Figure 3 d). The CNF fibers prepared with HCl at pH 2 has an exceptionally high toughness of 62±6 MJ m−3. Such high toughness has not yet been reported for any cellulosic material prepared with other known fabrication techniques (Table 1). In a wider perspective, the mechanical properties of our fibers are superior to all commercially available cellulose fibers (Viscose, Cordenka, Lyocell, and Ioncell‐F fibers) and all other nanocellulose fibers prepared with dry or wet spinning techniques (Table S1).6b, 24 Notably, our CNF fibers also outperform the mechanical performance of dragline spider silk, which is often consider as a gold standard for biopolymeric materials, which has a similar strength and toughness but up to 10 times lower stiffness.25
Table 1.
Comparison of the toughness of our fibers with the cellulose fibers prepared with other techniques.
|
Cellulose Fiber/Material Type |
Preparation Method |
Toughness [MJ m−3] |
Reference |
|---|---|---|---|
|
Viscose |
Dissolution and regeneration |
33 |
Adusumali, et. al24c |
|
Modal |
Dissolution and regeneration |
37 |
Adusumali, et. al24c |
|
Lyocell |
Dissolution and regeneration |
35 |
Adusumali, et. al24c |
|
Rayon |
Dissolution and regeneration |
41 |
Adusumali, et. al24c |
|
Flax |
Liberation from the stem |
16.7 |
Adusumali, et. al24c |
|
Bacterial Cellulose |
Wet twisting |
45 |
Gao, et. al26 |
|
CNF |
Wet extrusion (Syringe) |
31 |
Mohammadi, et. al27 |
|
Bacterial Cellulose Sheets |
Surface selective dissolution |
13 |
Soykeabkaew, et. al28 |
|
CNF Ribbon |
Hydrogel stretching |
19 |
Tang, et. al22 |
|
TEMPO‐oxidized CNF |
Wet extrusion (Syringe) |
8.7 |
Walther, et. al29 |
|
Carboxymethylated CNF |
Microfluidics |
62 |
Present Work |
We proceeded to study the rheological properties of hydrogels formed by the different gel initiators (procedure depicted in Figure 4 a) in order to relate the stiffness and strength of the CNF fibers to the ion‐specific properties of the hydrogels. The hydrogels have the typical oscillatory frequency response of self‐supporting hydrogels (Figure 4 b) and showed a strain stiffening behavior (Figure 4 c). A lower pH of the gel initiation solution results in a greater storage modulus of the hydrogels (Figure 4 b) and stronger fibers (Figure 3 b), which in turn suggests that there is a strong relationship between the properties of the hydrogel and the dry fiber, even though the rheological investigations are made for an unoriented CNF gels and the fibers are indeed oriented. There might be several explanations to this correlation which merits further investigations.
Figure 4.
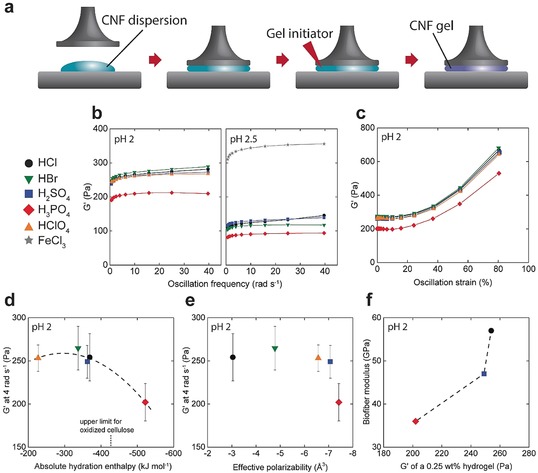
Rheological properties of CNF hydrogels. a) The sample preparation and instrumental setup. b) Frequency sweeps at pH 2 and 2.5. c) Amplitude sweep at pH 2. d) Relationship between storage modulus and absolute hydration enthalpy of the anion. e) Relationship between storage modulus and effective polarizability of the anion. f) Relationship between the biofiber modulus and the storage modulus of the hydrogel. The different acids are referred to by the color and shape of the data points. Error bars are 90 % confidence intervals based on at least 4 different measurements for each type of sample.
H3PO4‐initiated gels showed the lowest storage modulus which is related to an inferior stiffness of the dried fibers (Figure 2 b). Thus, at high pH, we were unable to form hydrogels strong enough to be lifted from the water bath when using H3PO4. FeCl3, on the other hand, greatly increases the stiffness of the hydrogel due to the coordination with carboxyl groups and other detailed molecular interactions described elsewhere,9d, 30 but interestingly this elevated storage modulus of FeCl3 initiated gels was not translated into fiber stiffness. It appears that excessive strength of the gel networks is not beneficial in the wet to dry transition, and can for example prevent further alignment of fibrils or create a more porous structure of the dry fibers. A further example of this discrepancy is the reduced stiffness of the fibers prepared by HCl at pH 1.5 compared to those prepared at pH 2 (Figure 3 b), although pH 1.5 gave stronger hydrogels. The fibers formed by FeCl3 show the highest toughness (Figure 3 d) and it can be suggested that this is due to a more porous network with hidden length scales, the hydration of Fe3+ (Figure S6 b), or directly related the sacrificial energy of breaking coordination complexes since this toughness was not achieved with HCl at pH 1.5.
In order to characterize the specific ion effects, we have related the properties of the hydrogels to the inherent properties of the different ions. Starting with ion hydration as a measure of water perturbation, we use acids with a broad range of hydration enthalpies (Figure 4 d).31 H2PO4 −, the most hydrated and hence most kosmotropic anion, is according to the theory excluded from the hydrogel and the following charge balance (opposite to Figure S1) leads to a higher pH inside the gel which is associated with less stiff hydrogels (Figure 4 a) and fibers (Figure 3 b). This is supported by elemental analysis which shows that the portions of Cl or S that migrates into fibers are similar, whereas a smaller portion of the available P enters the fibers (Table S2).
For the other ions, no significant relationship between storage modulus and hydration enthalpy is observed, and this can be related to the state of hydration of the CNFs. Acetate or formate, as models for carboxyl groups on the CNFs, have hydration enthalpies on the order of 420–430 kJ mol−1, which is hence the upper limit for the hydration of the CNFs. Ions with hydration enthalpy below this thus accumulate inside the hydrogels and reduce the local pH which translates into stronger hydrogels. The same argument can be adapted based on the hydration entropy of the anions (Figure S7), which follow a similar trend as the hydration enthalpy. The hydration enthalpy and the pK a of the acids are strongly related (Figure S8), with H3PO4 having the highest pK a of 2 and HClO4 the lowest of close to 10. The balance between associated and dissociated acids is also important in terms of water perturbation and ion migration, and in this respect H3PO4 solutions at pH 2 have the greatest portion of associated acids (Table S2).
Moisture sorption into dried hydrogel samples, which reduces the stiffness of CNF materials,24b also follows the hydration of the different ions (Figure S9 and Table S3). However, the subtle differences in water sorption cannot alone explain the differences in mechanical properties of the biofibers.
We continued to study the polarizability of the anion as a measure of ion‐surface dispersion interactions. Although the introduced anions have variations in effective polarizability,15c, 32 we found no relationship with regards to the storage modulus of the hydrogels (Figure 4 e). For example, H2PO4 − and HSO4 − have close to the same polarizability, but H2PO4 − initiate significantly softer hydrogels which implies that the dispersion is not decisive in these systems. Ion‐surface dispersion interactions are mediated by the difference in dielectric response of the surface and the medium.15c Surfaces with low permittivity, such as oil (2) or cellulose (3–7), in relation to water (80) result in strong adsorption of polarizable ions, and we therefore assume that most of these anions to certain degree adsorbs to CNFs. Instead the hydration‐controlled accumulation and exclusion of ions dictates if adsorption is possible.
The general trend is that a stiffer hydrogel result in a stiffer fiber (Figure 4 f). However, H2SO4 produces weaker fibers than expected based on gel stiffness, which indicates that there are other mechanisms involved. A property that differentiate H2SO4 from the other acids is the secondary pK a just above 2, and hence a significant amount of SO4 2− exists in the gel initiation solution (Figure S8). SO4 2− is significantly more hydrated than HSO4 − and the intricate balance between monovalent and divalent anions, such as their accumulation or exclusion, or the lower salt concentration should be considered as a cause of the inferior stiffness of these fibers.
To gain further insight of how accumulation of different ions in between CNFs affect the fibril‐fibril contacts, we prepared isotropic hydrogel networks using vacuum filtration.33 We added the ions to these networks in two ways: soaking dried network in the gel initiation solution (Figure 5 a) or soaking the filter cake before drying (Figure 5 b). These isotropic CNF networks were only marginally affected by the specific ions (Table S4). This shows that the structure in the oriented fibers is more sensitive to specific ions, which is reasonable since the fiber properties are determined by contact between fibrils surfaces, whereas random CNF gels are primarily determined by the properties of the network.30 Thus accumulated or adsorbed ions which disrupt fibril contacts is detrimental for oriented fibers (Figure 5 c) while random CNF networks retain a fixed interlocking regardless of gel initiator (Figure 5 d). Consequently, the sizes, hydration and other properties of adsorbed or accumulated ions are important since, for instance, larger ions are reasonably more disruptive (Figure 5 c) which has been shown for bulky counter‐ions.34 This can potentially also explain why H2SO4 lead to less stiff fibers compared to HCl despite the observation that these hydrogels have more or less the same storage modulus (Figure 4 f and Figure 3 b). The same reasoning is valid in the case of H3PO4 and FeCl3 where considerable amounts of ions block fibril contacts (Table S2).
Figure 5.
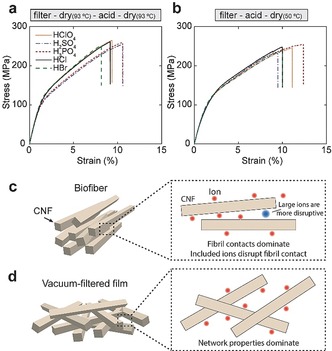
Mechanical properties of isotropic CNF films. a) and b) Representative stress‐strain curves for films with different treatments referred to by color and structure of the line. c) and d) Illustrations of the organization in a biofiber (c) and a vacuum filtered film (d) and how specific ions presumably affect these materials. Data is averaged for at least 4 samples of each type. All the measurements are performed at 50 % RH.
To evaluate the extractability of the ions as a measure how tightly they are bound inside the fibril network, we used ion chromatography. In this experiment, ions are extracted with water from the dry films in Figure 5 b, and Table 2 shows that the extractability in moles per weight of Br−, Cl−, and H2PO4 − follow the Hofmeister series and the thermodynamics of hydration, that is, Cl− is easier to extract than Br−. The amount of HSO4 − is lower than the other due to the divalent nature of sulfuric acid (Table S2).
Table 2.
Extractability of ions measured by ion chromatography.
|
Ion type |
% wt/wt |
% mol/wt |
|---|---|---|
|
Bromide |
0.29 |
0.0036 |
|
Chloride |
0.16 |
0.0045 |
|
Sulfates |
0.15 |
0.0015 |
|
Phosphates |
0.58 |
0.0060 |
Conclusion
In conclusion, our CNF biofibers stand out from other known biofibers based on their superior mechanical performance. Our results show that the process of flow‐induced alignment and assembly lead to a certain level of orientation of the nanofibrils and that the divergence in mechanical properties of the biofibers is governed largely by a combination of pH of the gel initiator, CNF concentration, and specific ion effects. The direct influence of different ions is evident from WAXS results, which shows no difference in the orientation of crystal planes of biofibers prepared using different gel initiators. It is thus important to consider the accumulation of ions in proximity to the nanofibrils and the resulting local pH as well as disrupted fibril contact. The common description of how the presence of anions or cations perturb water molecules and how this dictate the accumulation of ions in the wet nanoparticle networks, invoke some notion of nanostructure making or breaking,35 although a thorough understanding of this remains elusive. Here, we have tried to provide an understanding of these ideas by describing the variations in the mechanical properties of both hydrogels and CNF fibers with the choice of ions that potentially is coupled to the differences in the nanoscale networks. Though with the limitations of the experimental tools, it is challenging to determine the exact state of ions inside the network structure. Another picture that emerges from the present results is that ions potentially do not disrupt isotropic networks with larger pore sizes, rather the effect is more observable in oriented structures where the cohesion is mainly governed by fibril contacts. Nevertheless, our results show that a Hofmeister series to certain degree, explain wet and dry fibrillar material systems following the direct interactions of ions with the surface of CNFs. Although more experimental efforts are needed to parameterize better the use specific ion effects in the rational assembly of high‐performance nanostructures.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This study is funded by Knut and Alice Wallenberg Foundation through Wallenberg Wood Science Center. The authors are grateful to Prof. William D. Nix for fruitful discussions. N.M. acknowledge the kind support of Dr. Irene Linares Arregui from Department of Solid Mechanics at KTH with tensile test experiments.
N. Mittal, T. Benselfelt, F. Ansari, K. Gordeyeva, S. V. Roth, L. Wågberg, L. D. Söderberg, Angew. Chem. Int. Ed. 2019, 58, 18562.
Contributor Information
Dr. Nitesh Mittal, Email: niteshm@mit.edu.
Dr. Tobias Benselfelt, Email: bense@kth.se.
Prof. L. Daniel Söderberg, Email: dansod@kth.se.
References
- 1.
- 1a. Wegst U. G. K., Bai H., Saiz E., Tomsia A. P., Ritchie R. O., Nat. Mater. 2015, 14, 23; [DOI] [PubMed] [Google Scholar]
- 1b. Ling S., Kaplan D. L., Buehler M. J., Nat. Rev. Mater. 2018, 3, 18016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Meyers M. A., Chen P.-Y., Lin A. Y. M., Seki Y., Prog. Mater. Sci. 2008, 53, 1–206. [Google Scholar]
- 2. Huang W., Restrepo D., Jung J.-Y., Su F. Y., Liu Z., Ritchie R. O., McKittrick J., Zavattieri P., Kisailus D., Adv. Mater. 2019, 1901561. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Gui Z., Zhu H., Gillette E., Han X., Rubloff G. W., Hu L., Lee S. B., ACS Nano 2013, 7, 6037–6046; [DOI] [PubMed] [Google Scholar]
- 3b. Li Y., Guo F., Hao Y., Gupta S. K., Hu J., Wang Y., Wang N., Zhao Y., Guo M., Proc. Natl. Acad. Sci. USA 2019, 116, 9245–9250; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Ma H., Burger C., Hsiao B. S., Chu B., J. Mater. Chem. 2011, 21, 7507–7510. [Google Scholar]
- 4. Lebreton L. C. M., van der Zwet J., Damsteeg J. W., Slat B., Andrady A., Reisser J., Nat. Commun. 2017, 8, 15611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Behabtu N., Young C. C., Tsentalovich D. E., Kleinerman O., Wang X., Ma A. W. K., Bengio E. A., ter Waarbeek R. F., de Jong J. J., Hoogerwerf R. E., Fairchild S. B., Ferguson J. B., Maruyama B., Kono J., Talmon Y., Cohen Y., Otto M. J., Pasquali M., Science 2013, 339, 182–186. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Wang C. W., Sinton D., Moffitt M. G., J. Am. Chem. Soc. 2011, 133, 18853–18864; [DOI] [PubMed] [Google Scholar]
- 6b. Mittal N., Ansari F., K. Gowda V , Brouzet C., Chen P., Larsson P. T., Roth S. V., Lundell F., Wågberg L., Kotov N. A., Söderberg L. D., ACS Nano 2018, 12, 6378–6388. [DOI] [PubMed] [Google Scholar]
- 7. Ninham B. W., Adv. Colloid Interface Sci. 1999, 83, 1–17. [Google Scholar]
- 8.
- 8a. Zhang Y., Furyk S., Bergbreiter D. E., Cremer P. S., J. Am. Chem. Soc. 2005, 127, 14505–14510; [DOI] [PubMed] [Google Scholar]
- 8b. Jungwirth P., Cremer P. S., Nat. Chem. 2014, 6, 261; [DOI] [PubMed] [Google Scholar]
- 8c. Hofmeister F., Arch. Exp. Pathol. Pharmakol. 1888, 24, 247–260. [Google Scholar]
- 9.
- 9a. Nebot V. J., Ojeda-Flores J. J., Smets J., Fernández-Prieto S., Escuder B., Miravet J. F., Chem. Eur. J. 2014, 20, 14465–14472; [DOI] [PubMed] [Google Scholar]
- 9b. Carnegie R. S., Gibb C. L. D., Gibb B. C., Angew. Chem. Int. Ed. 2014, 53, 11498–11500; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11682–11684; [Google Scholar]
- 9c. Du R., Hu Y., Hübner R., Joswig J. O., Fan X., Schneider K., Eychmüller A., Sci. Adv. 2019, 5, eaaw4590; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9d. Benselfelt T., Nordenström M., Hamedi M. M., Wågberg L., Nanoscale 2019, 11, 3514–3520; [DOI] [PubMed] [Google Scholar]
- 9e. Benítez A. J., Walther A., Biomacromolecules 2017, 18, 1642–1653; [DOI] [PubMed] [Google Scholar]
- 9f. Shimizu M., Saito T., Isogai A., J. Membr. Sci. 2016, 500, 1–7. [Google Scholar]
- 10. Mittal N., Jansson R., Widhe M., Benselfelt T., Håkansson K. M. O., Lundell F., Hedhammar M., Söderberg L. D., ACS Nano 2017, 11, 5148–5159. [DOI] [PubMed] [Google Scholar]
- 11. Nordenström M., Fall A., Nyström G., Wågberg L., Langmuir 2017, 33, 9772–9780. [DOI] [PubMed] [Google Scholar]
- 12. Kamada A., Mittal N., Söderberg L. D., Ingverud T., Ohm W., Roth S. V., Lundell F., Lendel C., Proc. Natl. Acad. Sci. USA 2017, 114, 1232–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Brouzet C., Mittal N., Lundell F., Söderberg L. D., Macromolecules 2019, 52, 2286–2295; [Google Scholar]
- 13b. Brouzet C., Mittal N., Söderberg L. D., Lundell F., ACS Macro Lett. 2018, 7, 1022–1027. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Dong H., Snyder J. F., Williams K. S., Andzelm J. W., Biomacromolecules 2013, 14, 3338–3345; [DOI] [PubMed] [Google Scholar]
- 14b. Williams K. S., Andzelm J. W., Dong H., Snyder J. F., Cellulose 2014, 21, 1091–1101. [Google Scholar]
- 15.
- 15a. Sivan U., Curr. Opin. Colloid Interface Sci. 2016, 22, 1–7; [Google Scholar]
- 15b. Parsons D. F., Bostrom M., Nostro P. L., Ninham B. W., Phys. Chem. Chem. Phys. 2011, 13, 12352–12367; [DOI] [PubMed] [Google Scholar]
- 15c. Ninham B. W., Nostro P. L., Molecular Forces and Self Assembly: In Colloid, Nano Sciences and Biology, Cambridge University Press, Cambridge, 2010. [Google Scholar]
- 16. Kocherbitov V., Carbohydr. Polym. 2016, 150, 353–358. [DOI] [PubMed] [Google Scholar]
- 17. Sthoer A., Hladílková J., Lund M., Tyrode E., Phys. Chem. Chem. Phys. 2019, 21, 11329–11344. [DOI] [PubMed] [Google Scholar]
- 18. Grignon J., Scallan A. M., J. Appl. Polym. Sci. 1980, 25, 2829–2843. [Google Scholar]
- 19. Boström M., Craig V. S. J., Albion R., Williams D. R. M., Ninham B. W., J. Phys. Chem. B 2003, 107, 2875–2878. [Google Scholar]
- 20. Henriksson M., Berglund L. A., Isaksson P., Lindström T., Nishino T., Biomacromolecules 2008, 9, 1579–1585. [DOI] [PubMed] [Google Scholar]
- 21. Benítez A. J., Torres-Rendon J., Poutanen M., Walther A., Biomacromolecules 2013, 14, 4497–4506. [DOI] [PubMed] [Google Scholar]
- 22. Tang H., Butchosa N., Zhou Q., Adv. Mater. 2015, 27, 2070–2076. [DOI] [PubMed] [Google Scholar]
- 23. Benselfelt T., Engström J., Wågberg L., Green Chem. 2018, 20, 2558–2570. [Google Scholar]
- 24.
- 24a. Lundahl M. J., Klar V., Wang L., Ago M., Rojas O. J., Ind. Eng. Chem. Res. 2017, 56, 8–19; [Google Scholar]
- 24b. Benítez A. J., Walther A., J. Mater. Chem. A 2017, 5, 16003–16024; [Google Scholar]
- 24c. Adusumali R.-B., Reifferscheid M., Weber H., Roeder T., Sixta H., Gindl W., Macromol. Symp. 2006, 244, 119–125; [Google Scholar]
- 24d. Harish S., Michael D. P., Bensely A., Lal D. M., Rajadurai A., Mater. Charact. 2009, 60, 44–49; [Google Scholar]
- 24e. Ashby M. F., Gibson L. J., Wegst U., Olive R., Proc. R. Soc. London Ser. A 1995, 450, 123–140. [Google Scholar]
- 25.
- 25a. Liu Y., Sponner A., Porter D., Vollrath F., Biomacromolecules 2008, 9, 116–121; [DOI] [PubMed] [Google Scholar]
- 25b. Vehoff T., Glišović A., Schollmeyer H., Zippelius A., Salditt T., Biophys. J. 2007, 93, 4425–4432; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25c. Liu Y., Shao Z., Vollrath F., Nat. Mater. 2005, 4, 901–905; [DOI] [PubMed] [Google Scholar]
- 25d. Bowen C. H., Dai B., Sargent C. J., Bai W., Ladiwala P., Feng H., Huang W., Kaplan D. L., Galazka J. M., Zhang F., Biomacromolecules 2018, 19, 3853–3860. [DOI] [PubMed] [Google Scholar]
- 26. Gao H. L., Zhao R., Cui C., Zhu Y. B., Chen S. M., Pan Z., Meng Y. F., Wen S.-M., Liu C., Wu H. A., Yu S. H., Natl. Sci. Rev. 2019, 10.1093/nsr/nwz077. [DOI] [Google Scholar]
- 27. Mohammadi P., Toivonen M. S., Ikkala O., Wagermaier W., Linder M. B., Sci. Rep. 2017, 7, 11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Soykeabkaew N., Sian C., Gea S., Nishino T., Peijs T., Cellulose 2009, 16, 435–444. [Google Scholar]
- 29. Walther A., Timonen J. V. I., Díez I., Laukkanen A., Ikkala O., Adv. Mater. 2011, 23, 2924–2928. [DOI] [PubMed] [Google Scholar]
- 30. Benselfelt T., Nordenström M., Lindström S. B., Wågberg L., Adv. Mater. Interfaces 2019, 6, 1900333. [Google Scholar]
- 31.
- 31a. Barrett J., Inorganic chemistry in aqueous solution, Vol. 21, Royal Society of Chemistry, London, 2003; [Google Scholar]
- 31b. Marcus Y., J. Chem. Soc. Faraday Trans. 1 1987, 83, 339–349; [Google Scholar]
- 31c. Smith D. W., J. Chem. Educ. 1977, 54, 540. [Google Scholar]
- 32. Parsons D. F., Ninham B. W., J. Phys. Chem. A 2009, 113, 1141–1150. [DOI] [PubMed] [Google Scholar]
- 33. Zhao M., Ansari F., Takeuchi M., Shimizu M., Saito T., Berglund L. A., Isogai A., Nanoscale Horiz. 2018, 3, 28–34. [DOI] [PubMed] [Google Scholar]
- 34. Guerrero-García G. I., González-Mozuelos P., Olvera de la Cruz M., ACS Nano 2013, 7, 9714–9723. [DOI] [PubMed] [Google Scholar]
- 35. Ding Y., Hassanali A. A., Parrinello M., Proc. Natl. Acad. Sci. USA 2014, 111, 3310–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary