

Abstract
Mutations in the centrosomal protein 290 (CEP290) gene cause various ciliopathies involving retinal degeneration. CEP290 proteins localize to the ciliary transition zone and are thought to act as a gatekeeper that controls ciliary protein trafficking. However, precise roles of CEP290 in photoreceptors and pathomechanisms of retinal degeneration in CEP290-associated ciliopathies are not sufficiently understood. Using conditional Cep290 mutant mice, in which the C-terminal myosin-tail homology domain of CEP290 is disrupted after the connecting cilium is assembled, we show that this domain is essential for protein confinement between the inner and the outer segments. Upon disruption of the myosin-tail homology domain, inner segment plasma membrane proteins, including syntaxin 3 (STX3), synaptosome-associated protein 25 (SNAP25), and interphotoreceptor matrix proteoglycan 2 (IMPG2), rapidly accumulated in the outer segment. In contrast, localization of endomembrane proteins was not altered. Trafficking and confinement of most outer segment-resident proteins appeared to be unaffected or only minimally affected in Cep290 mutant mice. One notable exception was rhodopsin (RHO), which severely mislocalized to inner segments during the initial stage of degeneration. Similar mislocalization phenotypes were observed in Cep290rd16 mice. These results suggest that a failure of protein confinement at the connecting cilium and consequent accumulation of inner segment membrane proteins in the outer segment, along with insufficient RHO delivery, is part of the disease mechanisms that cause retinal degeneration in CEP290-associated ciliopathies. Our study provides insights into the pathomechanisms of retinal degenerations associated with compromised ciliary gates.
Keywords: cilia, retinal degeneration, photoreceptor, intracellular trafficking, genetic disease, ciliary gate, connecting cilium, diffusion barrier, gatekeeper, outer segment
Introduction
Photoreceptor cells in the retina are highly compartmentalized neurons. Although proteins involved in the phototransduction cascade are confined to a compartment called the outer segment, ones responsible for energy metabolism and protein/lipid synthesis are confined to another compartment, the inner segment. Proteins that constitute the outer segment are synthesized in the inner segment and transported to the outer segment through a narrow channel, called the connecting cilium. The outer segment is a primary cilium-related compartment, and the connecting cilium is equivalent to the transition zone in primary cilia. Because connecting cilia are the only conduit between the inner and the outer segments, they are situated at a critical place to control protein movement between the two compartments and maintain compartment-specific protein localization.
Although outer segments share many features with primary cilia, they have several unique features that are not observed in primary cilia (1). For example, outer segments are much larger than primary cilia, accounting for 25–60% of the total cell volume depending on cell types (rod versus cone) and species. Outer segments are also filled with membranous structures called discs. The large size and high membrane content of the outer segment provide a space to accommodate a large amount of transmembrane and lipidated proteins that constitute the phototransduction cascade. In addition, outer segments are constantly and rapidly regenerated. Old discs are shed from the distal end of the outer segment, and new discs are formed at the base (2). Constant and rapid regeneration of a large organelle necessitates a high volume of lipid and protein transport through the connecting cilium. At the same time, proteins that are not authorized to pass the connecting cilium (in either direction) should be kept in their designated compartments. Several proteins at the connecting cilium are thought to organize a gate to control protein movement between the inner and the outer segments (3).
CEP290 is one of such proteins. In the model organisms Chlamydomonas reinhardtii and Caenorhabditis elegans, CEP290 is found at the ciliary transition zone and is required to build Y-shaped linkers that extend from the axonemal microtubules to the ciliary membrane (4–6). Although cilia/flagella still form in the absence of CEP290 in these organisms, ciliary protein compositions are altered (4, 6). In mammalian primary cilia, CEP290 is localized to the transition zone (7–10), and loss of CEP290 reduces ARL13B and ADCY3 levels within cilia while increasing the rate of ciliary entry of smoothened (SMO) in fibroblasts (11). These studies establish the current model for CEP290 function: a ciliary gatekeeper that regulates protein trafficking in and out of the ciliary compartment at the transition zone (4–6, 8, 11–14). In photoreceptors, CEP290 is localized to the connecting cilium (15, 16) and is expected to control protein movement between the inner and the outer segments.
Mutations in human CEP290 cause various ciliopathies ranging from isolated retinal dystrophy (e.g. Leber congenital amaurosis) to syndromic diseases such as neonatal lethal Meckel-Gruber syndrome (MKS)2 with multiorgan malformations (17–23). Despite considerable variations in phenotypic severity, retinopathy is present in almost all cases regardless of the involvement of other organs. This suggests that photoreceptors are particularly susceptible to deficiencies in CEP290 function. Based on the ciliary gatekeeper model, anticipated functions of CEP290 in photoreceptors include the following: (i) permitting or facilitating entry of outer segment-bound proteins into the outer segment; (ii) blocking unauthorized entry of inner segment proteins into the outer segment; and (iii) preventing diffusion of outer segment proteins into the inner segment. In line with the first function listed above, Cep290 mutant mice fail to develop outer segments. The connecting cilium and the outer segment are entirely absent in Cep290 null mice (16). Partial loss of CEP290 function in rd16 mice, which have an in-frame deletion of exons 36–40 (based on reference sequence transcript NM_146009), allows formation of membrane-bound connecting cilia, but outer segments are rudimentary and severely malformed (15, 16, 24). These studies show that CEP290 is essential for outer segment biogenesis and, either directly or indirectly, is required for the trafficking of outer segment proteins.
However, precise roles of CEP290 in photoreceptors and disease mechanisms that induce retinal degeneration in CEP290-associated ciliopathies are not sufficiently understood. This is partly because of the critical requirement of CEP290 for the outer segment biogenesis in mouse models. As described above, Cep290 mutant mice have no or only rudimentary outer segments (15, 16, 24). Although these phenotypes demonstrate the requirement of CEP290 for the outer segment biogenesis, a complete lack or severe malformation of the outer segment precludes further investigation of CEP290's role in protein trafficking and confinement between the inner and the outer segments. For instance, although CEP290 is likely required for the trafficking of at least certain outer segment proteins, specifically which proteins require CEP290 is unclear. The requirement of CEP290 for protein confinement between the inner and the outer segments has not been demonstrated either. Contrary to the findings in mouse models, Cep290 (or the C-terminal half of Cep290) does not appear to be essential for outer segment biogenesis in zebrafish (25). In cep290fh297/fh297 mutants, which have a nonsense mutation (p.Q1217X) near the middle of the reading frame, retinas develop normally during embryogenesis. In addition, retinal degeneration is slow and limited to cones in this model. Interestingly, although RHO mislocalization is detected at 6 months post-fertilization, the degeneration of rods is not observed. Apart from RHO mislocalization, obvious signs of disrupted ciliary trafficking (e.g. accumulation of vesicular materials near the ciliary base in electron micrographs) are also not observed in this model. Therefore, precise roles of CEP290, including its gating functions, remain to be determined in photoreceptors.
In this work, we sought to test the current model of CEP290 function as a ciliary gatekeeper in photoreceptors and advance our understanding of the pathomechanisms underlying CEP290-associated retinopathies. We avoided the aforementioned limitations of the CEP290 mouse models with constitutive mutations by using a model with a conditional allele and disrupting CEP290 functions after the connecting cilium assembly. In addition, we reasoned that disruption of ciliary gates would have a profound impact on the localization of inner segment membrane proteins, because of the physical properties of the outer segment (i.e. large size, high membrane content, and continuous disc renewal) and its tendency to house membrane proteins (1, 26). Therefore, we examined (mis)trafficking of not only outer segment-resident proteins but also various inner segment membrane proteins.
Results
Characterization of the Cep290fl conditional allele
To avoid the outer segment biogenesis defects in constitutive Cep290 mutant mice, we employed a mouse line with a Cep290 conditional allele (Cep290fl) (27) and aimed to disrupt CEP290 functions after the connecting cilium was assembled. The Cep290fl allele has LoxP sites flanking exons 37 and 38 (Fig. 1A) (27). Hereafter, we use the term Cep290Δ to denote the Cep290fl allele lacking these two exons after CRE-mediated excision. Removal of these exons causes a frameshift followed by a premature termination codon. This change may induce nonsense-mediated decay of the mutant mRNAs, rendering Cep290Δ a null or a strong hypomorphic allele. If not, mutant mRNAs are expected to produce a C-terminally truncated protein (p.L1673HfsTer6) with a predicted molecular mass of ∼196 kDa. This truncated protein is likely to maintain some functions of full-length CEP290. rd1 (in Pde6b) and rd8 (in Crb1) mutations present in the cryo-recovered mice were removed by breeding with WT 129S6/SvEvTac mice. For robust and consistent excision in the vast majority of photoreceptors after the connecting cilium assembly, Cep290fl/fl mice were crossed to iCre75 (Cre hereafter) transgenic mice that express CRE recombinase under the control of mouse rhodopsin (Rho) promoter in rods (28).
Figure 1.

Cep290Δ is a hypomorphic allele encoding two species of CEP290 deletion mutants. A, schematics of Cep290 allele structures. Yellow boxes represent Cep290 exons. Exon numbers are described above the yellow boxes. Numbers below the yellow boxes indicate the sizes of respective exons (in bp). Locations of LoxP sites (open triangles) and a Neo cassette (white box) are marked. Dotted lines depict splicing events between exons 35, 36, and 39 in the Cep290Δ allele. Black arrows indicate the binding sites of the primers used for RT-PCR (F, forward; R, reverse). B, two mutant forms (black and red arrowheads) of CEP290 are detected by CEP290 immunoblotting in Cep290fl/fl;Cre+ retinas. Cep290fl/fl;Cre− littermates were used as a WT control. Open arrowhead indicates full-length CEP290. Numbers on the left mark the location of protein standards. C, Cep290Δ allele produces two species of mRNAs. Predicted sizes of RT-PCR products (F1, F2, and F3) are shown at the bottom. Numbers on the left indicate locations of DNA size markers (in kb). A no-reverse-transcriptase control (−RT) was used as a negative control. Each lane represents individual animals. D, chromatogram of RT-PCR product sequencing reactions around exon–exon junctions. Vertical dotted lines mark exon–exon junctions. E, schematic representation of CEP290 protein products encoded by WT, Cep290Δ, and Cep290rd16 alleles. Major structural domains are depicted (SMC, structural maintenance of chromosomes protein homology domain; MYO-TAIL, myosin-tail homology domain). The Cep290Δ allele produces two species of CEP290 deletion mutants, p.L1673HfsX6 and p.D1606_K1789del.
Because the protein product from the Cep290Δ allele has not been determined, we extracted proteins from Cep290fl/fl;Cre− (i.e. control) and Cep290fl/fl;Cre+ retinas at post-natal day (P) 21 and performed CEP290 immunoblotting (Fig. 1B). Immunoblotting results revealed the production of ∼210-kDa proteins in Cep290fl/fl;Cre+ retinas, which is close to the predicted 196-kDa C-terminally–truncated protein (Fig. 1B; black arrowhead). The presence of full-length CEP290 (open arrowhead) in Cep290fl/fl;Cre+ retinas is expected, because Cre is expressed only in rods, and other cells in the retina (including cones) produce full-length CEP290. Interestingly, we noticed that there was an additional protein band (red arrowhead) slightly smaller than full-length CEP290. Basal exon skipping and nonsense-associated altered splicing have been observed in human CEP290 (29–32). By these mechanisms, exons containing nonsense or frameshift mutations are skipped, and the resulting mutant mRNAs maintain the reading frame to produce near–full-length CEP290 proteins instead of being degraded or producing severely truncated proteins. In mouse Cep290, we noticed that omitting exon 36 in the Cep290Δ allele prevents frameshift (Fig. 1A). To test whether basal exon skipping or nonsense-associated altered splicing occurs, we analyzed Cep290 mRNAs from Cep290fl/fl;Cre− and Cep290fl/fl;Cre+ retinas. Reverse transcription PCR (RT-PCR) with primers specific to exons 34 and 41 (forward and reverse primers, respectively) produced a single fragment in controls but three fragments in Cep290fl/fl;Cre+ retinas (Fig. 1C). Sequence analyses of the PCR products revealed that the largest fragment (F1; 1224 bp) was derived from unexcised alleles and contained all exons between exons 34 and 41 (Fig. 1D). Fragment 2 (F2) (872 bp), which was the main PCR product from the Cep290fl/fl;Cre+ retinas, contained exons 35, 36, and 39 but not 37 and 38. This indicates that F2 is derived from the Cep290Δ allele. Fragment 3 (F3) (672 bp) was devoid of exon 36 in addition to exons 37 and 38, indicating that F3 was derived from the Cep290Δ allele, but exon 36 was skipped during splicing. Because no smaller PCR product was detected in control retinas, our data suggest that exon 36 skipping is a result of nonsense-associated altered splicing rather than basal exon skipping. Loss of exons 36–38 (i.e. 552 bp) causes an in-frame deletion of 184 amino acids (∼20 kDa), which are part of the region deleted in rd16 mutants (Fig. 1E). RT-PCR data also suggest that CRE-mediated excision of exons 37 and 38 occurs before P21 in the vast majority of rods. In addition, it is noteworthy that F2 is at least 12-fold more abundant than F3, but the quantity of protein products derived from these transcripts (black and red arrowheads in Fig. 1B) is comparable. Therefore, the near–full-length in-frame deletion mutant (p.D1606_K1789del) appears to be significantly more stable than the truncated mutant (p.L1673HfsX6). In summary, our data show that Cep290Δ is a hypomorphic allele encoding two species of mutant proteins, in which the myosin-tail homology domain is disrupted.
Localization and interactions of CEP290 mutant proteins expressed from the Cep290Δ allele
We examined localization of CEP290 mutant proteins in Cep290fl/fl;Cre+ retinas. In both normal (Cep290fl/?;Cre−) and Cep290fl/fl;Cre+ retinas (at P25), CEP290 immunoreactivity was detected at the connecting cilium (Fig. 2A). One notable difference was although CEP290 was found throughout the connecting cilium in normal photoreceptors, it was enriched within the proximal half of the connecting cilium in Cep290fl/fl;Cre+ retinas. However, because our CEP290 antibody recognizes all three forms of CEP290, including the WT (Fig. 1B), it was difficult to unequivocally determine the localization of individual mutants in Cep290fl/fl;Cre+ retinas. To address this, we generated human CEP290 expression cassettes that mimic the deletion mutants with N-terminal FLAG tags, and we probed FLAG–CEP290 localization in transfected IMCD3 cells using anti-FLAG antibodies (Fig. 2B). To facilitate the identification of transfected cells, a small amount of GFP-tagged INPP5E, which localizes to primary cilia (33), was co-transfected and used as a ciliary marker. As described previously (7–10), full-length CEP290 localized to the base of primary cilia and centriolar satellites. Localization of both FLAG–CEP290(1–1671) and FLAG–CEP290Δ(1605–1788) was comparable with that of the full-length protein. These results indicate that both deletion mutants properly localize to the transition zone in cilia.
Figure 2.
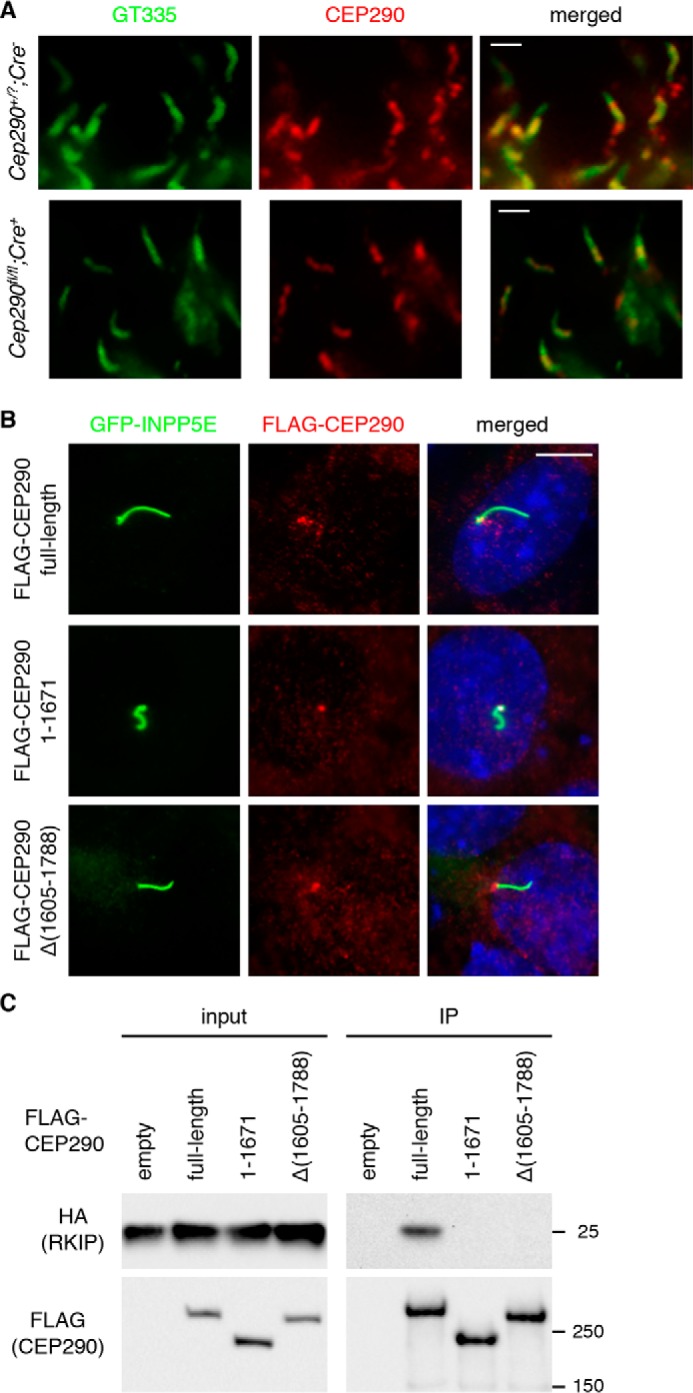
Localization and protein–protein interactions of CEP290 mutant proteins expressed from the Cep290Δ allele. A, localization of CEP290 mutant proteins (red) in normal and Cep290fl/fl;Cre+ mouse retinas at P25. Anti-polyglutamylation antibody GT335 was used to label connecting cilia (green). Merged images are shown on the right. Scale bar represents 1 μm. B, localization of FLAG-tagged CEP290 deletion mutants (red) in IMCD3 cells. GFP-INPP5E was co-transfected to mark transfected cells' primary cilia (green). Cells were counterstained with DAPI to show nuclei (blue). Representative images from five independent experiments are shown (n >75). Scale bar, 5 μm. C, CEP290 mutant proteins expressed from the Cep290Δ allele do not interact with RKIP. FLAG-tagged CEP290 mutant variants were transfected into HEK293T cells that stably expressed HA-RKIP, and cell lysates were subjected to immunoprecipitation (IP) with anti-FLAG antibodies. Empty vector was used as a negative control. Numbers on the right mark the location of protein standards.
We then tested protein–protein interactions of CEP290 deletion mutants. Residues commonly deleted in both p.L1673HfsX6 and p.D1606_K1789del mutants are part of the region deleted in CEP290rd16 mutant proteins (Fig. 1E). Raf-1 kinase inhibitory protein (RKIP) was previously shown to bind to this region (34). Therefore, we tested whether CEP290 deletion mutants expressed from the Cep290Δ allele maintained their ability to interact with RKIP. To this end, we generated a HEK293T cell line stably expressing HA-tagged mouse RKIP, transfected the aforementioned FLAG–CEP290 expression cassettes, and performed immunoprecipitation with anti-FLAG antibodies. As shown in Fig. 2C, HA–RKIP was co-precipitated with full-length CEP290 but not with either of the CEP290 deletion mutants. These data show that both mutant proteins expressed in our mouse model lost their ability to interact with RKIP.
Localization of outer segment resident proteins in Cep290fl/fl;Cre+ retinas
CEP290-associated ciliopathy patients with mutations in the myosin-tail homology domain present with more severe phenotypes than predicted by a model based on the total quantity of full-length or near–full-length CEP290 proteins (30). In addition, disruption of this domain in rd16 mice causes outer segment biogenesis defects and rapid degeneration of photoreceptors (15). These findings suggest that the myosin-tail homology domain is essential for CEP290's function and is possibly involved in the trafficking and confinement of outer segment proteins. Therefore, we examined localization of outer segment-resident proteins in Cep290fl/fl;Cre+ conditional mutant mice.
To this end, we probed localization of RHO, PRPH2, ROM1, ABCA4, PDE6B, GUCY2D, ATP8A2, and CNGA1 in Cep290+/fl;Cre+ (control) and Cep290fl/fl;Cre+ retinas. Although RHO localizes to both disc membranes and outer segment plasma membranes (35, 36), PRPH2, ROM1, ABCA4, GUCY2D, and ATP8A2 specifically localize to disc membranes (37–42). CNGA1 specifically localizes to the outer segment plasma membrane (43), which is contiguous with the inner segment plasma membrane (hereafter our use of the term inner segment encompasses all parts of the photoreceptors, including the ellipsoid, myoid, soma, axon, and synaptic terminals but excluding the outer segment). At P11, when the connecting cilium assembly is completed and the outer segment is rapidly growing (44), no significant differences were observed between control and Cep290fl/fl;Cre+ retinas with respect to the localization of outer segment-resident proteins, including RHO (Fig. S1). At P20, most outer segment-resident proteins examined still showed normal localization to the outer segment (Fig. 3). RHO was the only exception, and significant mislocalization was observed in the inner segments (Fig. 3A), indicating that RHO mislocalization is one of the earliest events that occur upon disruption of the myosin-tail homology domain.
Figure 3.

Localization of outer segment-resident proteins in Cep290fl/fl;Cre+ mice. Localization of RHO (A), PRPH2 (B), ROM1 (C), ABCA4 (D), PDE6B (E), GUCY2D (F), ATP8A2 (G), and CNGA1 (H) was probed in Cep290+/fl;Cre+ (at P20) and Cep290fl/fl;Cre+ (at P20 and P40) retinas. I, retinal sections of Ift88+/fl;Cre+ (at P22) and Ift88fl/fl;Cre+ (at P22 and P40) mice were immunostained with anti-RHO antibody and shown for comparisons with Cep290fl/fl;Cre+ mice. Red brackets indicate outer segments. Sections were counterstained with DAPI to show nuclei (blue). At least three animals, both male and female, were used per group, and representative images are shown. Scale bar represents 50 μm. J, length of the outer segment in Cep290fl/fl;Cre+ and Ift88fl/fl;Cre+ retinas. Lengths of the outer segment were measured in areas 0.5–1.0 mm away from the optic nerve. Data are from five animals per genotype (two sections per animal). Mean and standard deviation (S.D.; error bar) are shown. Asterisk indicates statistical significance (two-tailed, two-sample t test assuming unequal variances; p < 0.01).
To test whether outer segment protein localization deteriorates as photoreceptors degenerate, we examined the localization of the above eight proteins at P40 (Fig. 3). Twenty days (from P20 to P40) are sufficient for the outer segment to renew entirely in mouse retinas (2), and ∼30–40% of photoreceptors were lost by P40 in Cep290fl/fl;Cre+ retinas. However, no significant mislocalization of PRPH2, ROM1, ABCA4, PDE6B, GUCY2D, ATP8A2, and CNGA1 was observed. In addition, the length of the outer segment was not significantly reduced despite the progression of degeneration and cell loss. This observation sharply contrasts with the rapid shortening of the outer segment in Ift88fl/fl;Cre+ mice, in which intraflagellar transport (IFT) to maintain the outer segment is ablated, between P22 (when RHO mislocalization is first noticeable) and P40 (red bracket; Fig. 3, I and J). Therefore, most outer segment proteins except RHO appear to be properly transported and confined to the outer segment in Cep290fl/fl;Cre+ rods.
Accumulation of inner segment membrane proteins in the outer segment upon disruption of the CEP290 myosin-tail homology domain
We then examined whether disruption of the myosin-tail homology domain affected the confinement of inner segment membrane proteins. STX3 is a SNARE (Soluble N-ethymaleimide-sensitive factor Attachment protein REceptor) protein with a single transmembrane domain at its C terminus and facilitates membrane/vesicle fusion for exocytosis. In normal photoreceptors, STX3 is found in the plasma membrane throughout the inner segment but not in the outer segment (Fig. 4A, left) (45, 46). Because STX3 and its interacting partner STXBP1 mislocalize to the outer segment in Bardet-Biedl syndrome (BBS) mutant retinas (46–48), we speculated that these proteins might need an intact ciliary gate for their inner segment-restricted localization. Indeed, STX3 and STXBP1 were found mislocalized to outer segments in Cep290fl/fl;Cre+ retinas at P20 (Fig. 4, A and B; also see Fig. 4E for quantification).
Figure 4.

Localization of STX3 and its interacting proteins in Cep290fl/fl;Cre+ mice. Control (Cep290+/fl;Cre+ at P20) and Cep290fl/fl;Cre+ (at P20 and P40) mouse retinal sections were immunostained with STX3 (A), STXBP1 (B), SNAP25 (C), and VAMP2 (D) antibodies (green). To delineate outer segments, sections were co-stained with PRPH2, RHO, ROM1, and GRK1 antibodies (red). DAPI was used to label nuclei (blue). Merged images are shown on the right. Five animals, both male and female, were used per group, and representative images are shown. Scale bar represents 50 μm. E, quantification of inner segment protein mislocalization to the outer segment. Depicted are ratios of integrated fluorescence intensities in the outer segment relative to the photoreceptor cell layer. Data are from five mice (two sections per mouse and two areas per section). Mean and standard deviation (S.D.; error bar) are shown. Asterisks indicate statistical significance (two-tailed, two-sample t test assuming unequal variances; p < 0.01).
These results prompted us to examine the localization of other inner segment-specific membrane proteins. SNAP25 and VAMP2 are SNARE proteins that interact with STX3. Although both SNAP25 and VAMP2 are restricted to the inner segment, SNAP25 is a t-SNARE protein localizing to the plasma membrane, whereas VAMP2 is a v-SNARE protein present on secretory vesicles. Also, although SNAP25 is almost evenly distributed throughout the inner segment, VAMP2 is highly enriched at synaptic terminals (Fig. 4, C and D, left). In 20-day-old Cep290fl/fl;Cre+ mice, SNAP25 was found dispersed throughout the cell, including the outer segment (Fig. 4, C and E). In contrast, VAMP2 localization was not altered in Cep290fl/fl;Cre+ rods (Fig. 4, D and E).
IMPG2 (interphotoreceptor matrix proteoglycan 2) is a single transmembrane domain-containing proteoglycan found in the interphotoreceptor matrix, an extracellular matrix between the retinal pigmented epithelium and the outer limiting membrane of the retina (49, 50). Mutations in IMPG2 are associated with retinitis pigmentosa and vitelliform macular dystrophy (51–53). We previously identified IMPG2 as one of the proteins enriched in Lztfl1 mutant outer segments (46). When probed with an antibody raised to its C-terminal intracellular domain, IMPG2 immunoreactivity was mostly found within the ellipsoid and myoid zones of the inner segment in normal photoreceptors (Fig. 5A; also see Fig. S2 for antibody characterization and discussion). In Cep290fl/fl;Cre+ retinas, however, a significant amount of IMPG2 immunoreactivity was detected in outer segments in addition to inner segments (Fig. 5, A and F).
Figure 5.

Localization of inner segment membrane proteins in Cep290fl/fl;Cre+ mice. Retinal sections from Cep290+/fl;Cre+ (at P20) and Cep290fl/fl;Cre+ (at P20 and P40) mice were labeled with IMPG2 (A), HCN1 (B), and ATP1A3 (C) antibodies (green). Outer segments were delineated with ABCA4 or RHO (A), ATP8A2 (B), and GUCY2D (C) antibodies (red). Nuclei were counterstained with DAPI. D and E, localization of endomembrane proteins SYP (D) and LAMP1 (E) was examined using respective antibodies. At least three animals were used per group, and representative images are shown. Scale bar represents 50 μm. F, quantification of inner segment membrane protein mislocalization to the outer segment. Others are the same as in Fig. 4.
HCN1 (K+/Na+ hyperpolarization-activated cyclic nucleotide-gated channel 1) and ATP1A3 (Na+/K+-transporting ATPase1 subunit α3) are transmembrane proteins localizing to the inner segment plasma membrane (54–56). Within the inner segment, HCN1 and ATP1A3 are enriched within the myoid and ellipsoid zones (Fig. 5, B, C, and F). Mild mislocalization of HCN1 was observed throughout the outer segment in Cep290fl/fl;Cre+ retinas. ATP1A3 also exhibited mild mislocalization to outer segments, but there was a considerable cell–to–cell variation with respect to the severity of mislocalization. Synaptophysin (SYP) and lysosome-associated membrane glycoprotein 1 (LAMP1) are integral membrane proteins that specifically localize to synaptic vesicles and lysosomes, respectively. As expected, SYP was found at synaptic terminals and LAMP1 was in the ellipsoid zone in normal photoreceptors. Localization of these endomembrane proteins was not altered in Cep290fl/fl;Cre+ retinas (Fig. 5, D and E).
To test whether mislocalization of inner segment proteins becomes more prevalent or severe as retinal degeneration progresses, we examined the localization of the aforementioned proteins at P40 (Figs. 4 and 5, right panels). By this age, the majority (∼60%) of STX3 and STXBP1 localized to the outer segment (see Fig. 4E for quantification). It should be noted that this is an underestimation, because Stx3 and Stxbp1 are expressed in not only rods but also cones, and residual signals in the synaptic terminals (which are included in quantification as a part of the inner segment) are mostly from cones, in which Cre is not expressed (see Fig. S3). SNAP25 showed a moderate increase in mislocalization at P40, but the signal intensity in the outer segment did not exceed that in the inner segment. Mislocalization of other inner segment proteins was not appreciably exacerbated from what was observed at P20. These data show that the myosin-tail homology domain of CEP290 is essential for confinement of inner segment membrane proteins. Our data also suggest that proteins on the plasma membranes are primarily subject to diffusion, and that STX3 and STXBP1 are particularly susceptible to accumulation in the outer segment.
Disc morphogenesis defects in Cep290fl/fl;Cre+ retinas
We next examined the impact of loss of CEP290 myosin-tail homology domain on disc morphogenesis and the connecting cilium ultrastructure by transmission EM (TEM). Under our fixation condition using half-strength Karnovsky's fixative, “closed” discs are more intensely stained compared with “open” discs, which exist only at the proximal end of the outer segment in normal retinas (Fig. 6, A and B and A′ and B′). In 25-day-old Cep290fl/fl;Cre+ mouse retinas, more intensely stained disc membranes were intermixed with less dense disc membranes (Fig. 6, C–E and C′ and D′). In addition, longitudinally aligned discs were observed in a subset of photoreceptors (Fig. 6, red arrowheads). These data suggest that disc morphogenesis process is disturbed in Cep290fl/fl;Cre+ retinas. In contrast, but consistent with the previous findings in rd16 mice (15, 57), we were not able to find obvious structural changes in the connecting cilium (Fig. 6, E–H). Therefore, loss of CEP290 myosin-tail homology domain does not appear to cause overt structural defects within the connecting cilium.
Figure 6.

Altered disc morphogenesis in Cep290fl/fl;Cre+ retinas. Transmission electron micrographs from normal (A, B, and F) and Cep290fl/fl;Cre+ (C–E, G, and H) mouse retinas are shown. A′–D′ are enlarged images of the red dotted boxes in A–D, respectively. Red arrowheads indicate longitudinally aligned disc membranes. Scale bars in A–E and A′–D′ denote 1 μm and in F–H denote 0.2 μm.
Mislocalization of inner segment membrane proteins in Cep290fl/fl;Cre+ retinas is not due to loss of BBSome functions
Accumulation of STX3 and STXBP1 in the outer segment is a phenotype commonly observed in BBS mutant photoreceptors (46–48). While CEP290 is a stationary protein constituting the ciliary gate, BBSomes (a protein complex composed of eight BBS proteins (58, 59)) are adapter complexes that link ciliary cargos to IFT particles and transport them in and out of the ciliary compartment (60–67). To test whether STX3 and STXBP1 mislocalization in Cep290fl/fl;Cre+ retinas is mediated by loss or dysfunction of the BBSome, we examined the quantity and localization of BBSome components as well as a BBSome regulator LZTFL1 (68). Immunoblot analyses of retinal extracts from 25-day-old Cep290+/fl;Cre? (control) and Cep290fl/fl;Cre+ mice indicated that there was a slight to moderate (12–25%) reduction of photoreceptor-specific proteins (RHO, GRK1, PRPH2, and PDE6A) in Cep290fl/fl;Cre+ retinas (Fig. 7A). This is consistent with some loss of photoreceptors by this age. BBSome components (BBS2 and BBS7) and LZTFL1 showed similar levels of reduction. These data suggest that reduction of BBS proteins is proportionate to the loss of photoreceptors and that BBS protein levels are not affected by impaired CEP290 functions in Cep290fl/fl;Cre+ retinas. We then examined BBSome assembly by immunoprecipitation. As shown in Fig. 7B, all BBSome components tested were similarly pulled down by BBS7 antibodies in control and Cep290fl/fl;Cre+ retinas. These data indicate that BBSome assembly is not altered in Cep290fl/fl;Cre+ retinas.
Figure 7.
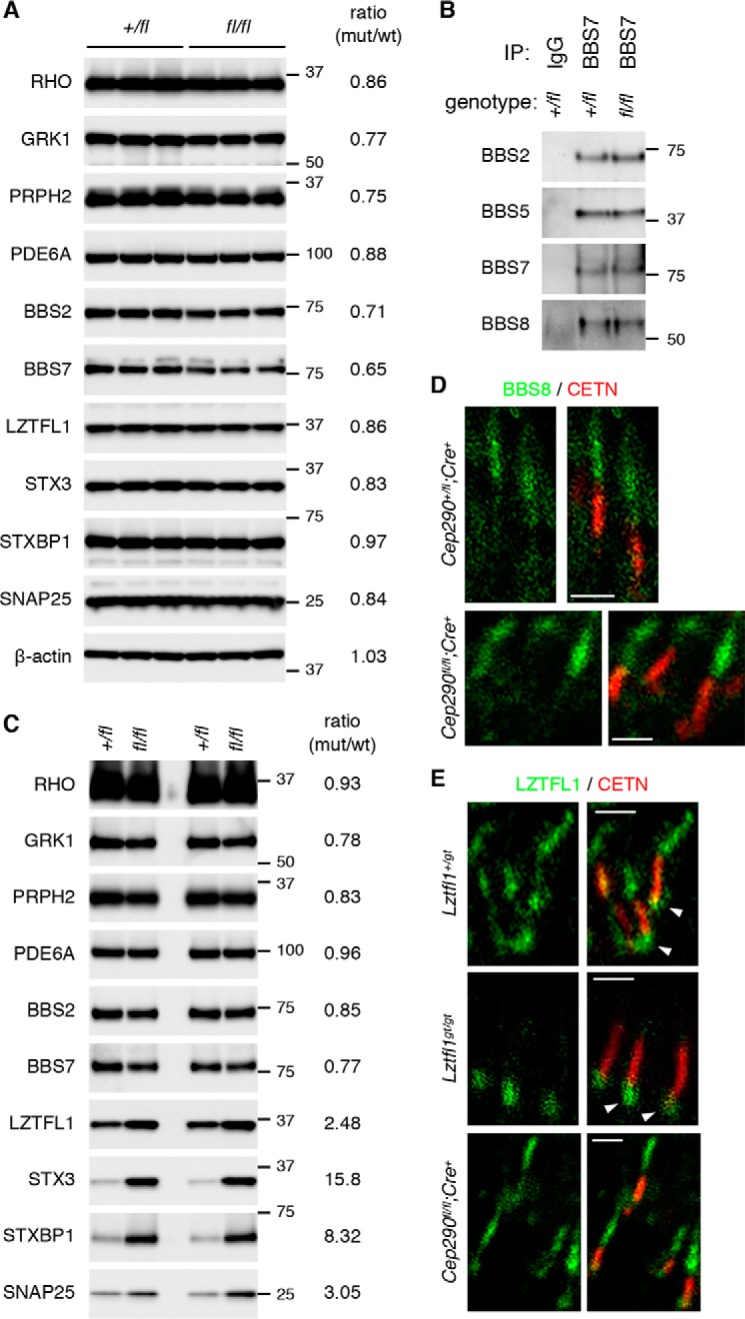
Expression and localization of BBS proteins are not altered in Cep290fl/fl;Cre+ retinas. A, immunoblot analysis of Cep290+/fl;Cre? and Cep290fl/fl;Cre+ retinal protein extracts. Each lane represents individual animals, and three animals were analyzed per genotype. Thirty μg of proteins were loaded per lane. Locations of protein standards are shown on the right. Numbers in ratio column are relative band intensities of the indicated proteins in Cep290fl/fl;Cre+ retinas compared with those in Cep290+/fl;Cre? retinas. B, BBSome assembly is not altered in Cep290fl/fl;Cre+ retinas. Retinal protein extracts from Cep290+/fl;Cre? and Cep290fl/fl;Cre+ retinas were subjected to immunoprecipitation (IP) with anti-BBS7 antibody. Normal rabbit IgG was used as a negative control. Co-precipitated BBS proteins were detected by immunoblotting. C, immunoblot analysis of outer segment fractions isolated from Cep290+/fl;Cre? and Cep290fl/fl;Cre+ retinas. Each lane represents individual outer segment preparations, and the results from two independent preparations are shown. Five μg of proteins were loaded per lane. Others are same as in A. D, localization of BBS8 (green) in Cep290+/fl;Cre+ and Cep290fl/fl;Cre+ photoreceptors. Connecting cilia were labeled with anti-CETN antibody (red). Scale bar represents 1 μm. E, localization of LZTFL1 (green) in control (Lztfl1+lgt), Lztfl1gt/gt, and Cep290fl/fl;Cre+ photoreceptors. White arrowheads indicate binding of anti-LZTFL1 antibody to cross-reacting protein(s) around the basal body. Others are same as in D.
To assess the quantity of BBS proteins within the outer segment, we isolated outer segments from Cep290+/fl;Cre? and Cep290fl/fl;Cre+ retinas and conducted immunoblot analyses (Fig. 7C). After normalization to total protein quantities, no significant differences were observed between normal and CEP290-deficient outer segments with respect to the quantity of outer segment-resident proteins. BBSome components, BBS2 and BBS7, also showed no significant differences. Interestingly, however, there was a more than 2-fold increase in LZTFL1 quantity in outer segments from Cep290fl/fl;Cre+ retinas. Consistent with the mislocalization of STX3 and STXBP1 in the outer segment, there was a large increase of these proteins in Cep290fl/fl;Cre+ outer segments. SNAP25 showed an ∼3-fold increase in Cep290fl/fl;Cre+ outer segments.
We then examined the localization of BBS proteins in normal and Cep290fl/fl;Cre+ photoreceptors. While CEP290 is localized to the connecting cilium, BBS5 was previously shown to localize along the axoneme in the outer segment (69). BBS5 was also detected within the connecting cilium at a lower intensity. Very similar localization patterns were observed with BBS8 and LZTFL1 (Fig. 7, D and E). LZTFL1 immunoreactivity was also detected around the basal body, but this staining persisted in Lztfl1 mutant (46) retinas, indicating that signals around the basal bodies are from cross-reacting protein(s) (Fig. 7E, arrowheads). Localization of BBS8 and LZTFL1 in Cep290fl/fl;Cre+ retinas was comparable with that of normal photoreceptors (Fig. 7, D and E). These data indicate that, despite some quantitative changes in LZTFL1 in the outer segment, overall localization patterns of the BBSome and LZTFL1 are not altered in Cep290fl/fl;Cre+ retinas. Therefore, we conclude that the accumulation of STX3 and STXBP1 in the outer segment in Cep290fl/fl;Cre+ retinas is unlikely due to alterations in BBSome functions.
Accumulation of inner segment membrane proteins in the outer segment in rd16 mice
We next examined whether similar protein mislocalization was observed in rd16 mice (Fig. 8). As described previously (15), retinal degeneration in rd16 mice was already evident at P15, and outer segments were rudimentary, suggesting that outer segment biogenesis is severely impaired in rd16 mice. RHO mislocalization was also evident in the inner segment (Fig. 8B). In addition, presumably due to the outer segment biogenesis defect, low level mislocalization of outer segment proteins was detected in general. However, the highest immunoreactivity of all outer segment-resident proteins examined was detected in the outer segment, suggesting that these proteins are still delivered to the outer segment in rd16 mice. Mislocalization of CNGA1 was relatively obvious compared with other outer segment proteins and predominantly restricted to the distal (ellipsoid) portion of the inner segment as opposed to being dispersed throughout the inner segment like RHO (Fig. 8H).
Figure 8.
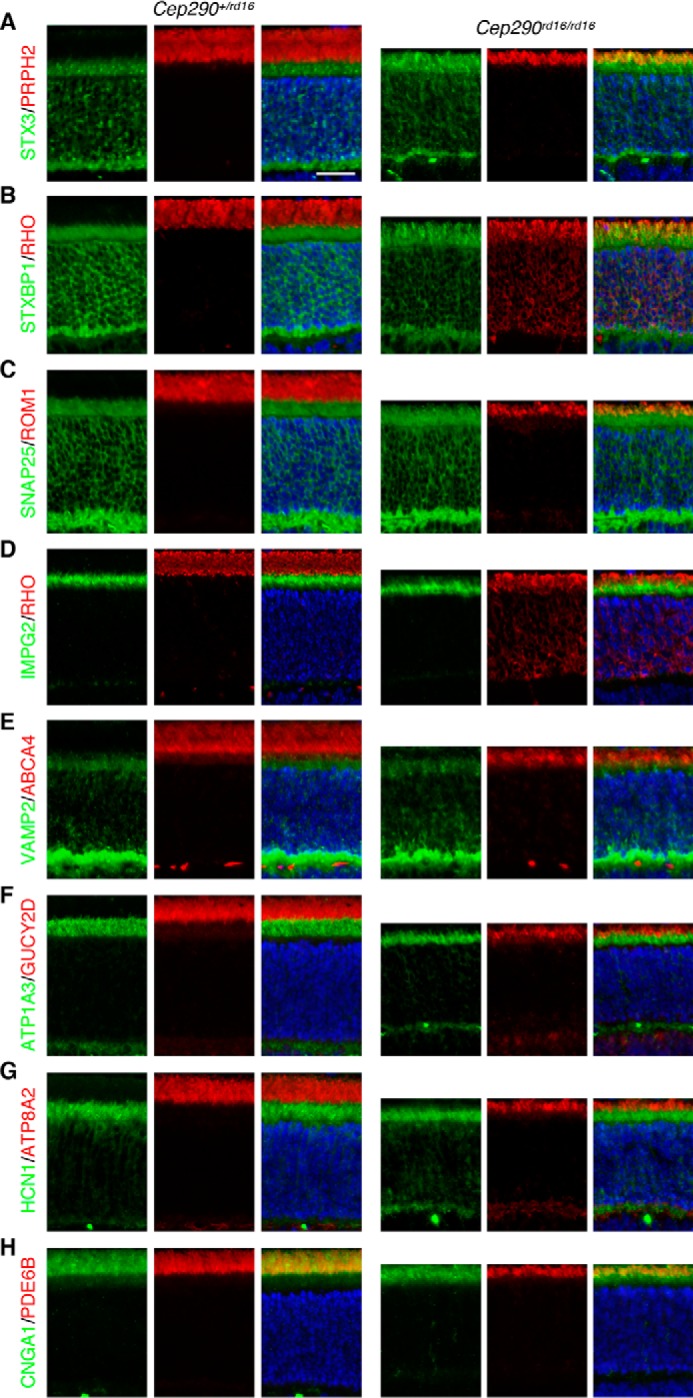
Disruption of compartmentalized protein localization in Cep290rd16/rd16 mice. Localization of outer segment-resident proteins (PRPH2 (A), RHO (B and D), ROM1 (C), ABCA4 (E), GUCY2D (F), ATP8A2 (G), PDE6B (H), and CNGA1 (H); all in red except CNGA1, which is in green) and inner segment membrane proteins (STX3 (A), STXBP1 (B), SNAP25 (C), IMPG2 (D), VAMP2 (E), ATP1A3 (F), and HCN1 (G); all in green) was examined by indirect immunofluorescence microscopy. Scale bar represents 25 μm.
We then examined the localization of inner segment membrane proteins in rd16 mice. Proteins that exhibited significant mislocalization in Cep290fl/fl;Cre+ retinas (STX3, STXBP1, SNAP25, and IMPG2) all showed obvious mislocalization to the outer segment in rd16 retinas (Fig. 8). In contrast, localization of VAMP2, ATP1A3, HCN1, SYP, and LAMP1 was not noticeably altered in rd16 retinas (Fig. 8 and data not shown). These results indicate that protein mislocalization phenotypes are comparable in Cep290fl/fl;Cre+ and rd16 retinas and that similar pathomechanisms underlie retinal degeneration in rd16 mice as in Cep290fl/fl;Cre+ mice.
Discussion
Mutations that cause CEP290-associated ciliopathies are found throughout the gene. The vast majority of them are truncating mutations (i.e. nonsense or frameshift mutations) (23). This means that most mutant alleles are destined to produce either no proteins (if mutant mRNAs are degraded by nonsense-mediated decay) or more likely C-terminally truncated proteins. Certain truncating mutations avoid these detrimental consequences by skipping exons and producing near–full-length proteins, resulting in unexpectedly mild phenotypes (29–32). The myosin-tail homology domain is within the C-terminal third of CEP290 and therefore is not produced in a large proportion of CEP290 mutant alleles with truncating mutations. The mouse models used in our study represent CEP290-associated retinopathies in which the myosin-tail homology domain is disrupted. rd16 mice have an in-frame deletion of 299 amino acids (amino acids 1606–1904) within this domain. Rods in Cep290fl/fl;Cre+ mice are effectively compound heterozygotes expressing two deletion mutants, p.L1673HfsX6 and p.D1606_K1789del. Our study demonstrates that CEP290, more specifically the myosin-tail homology domain of CEP290, is essential for protein confinement between the inner and the outer segments and thus maintains compartmentalized protein localization in photoreceptors. Disruption of this domain and consequent deficits in CEP290 function cause encroachment of select inner segment plasma membrane proteins on the outer segment and mislocalization of RHO to the inner segment. Therefore, our study supports the current model of CEP290 function as a ciliary gatekeeper and identifies disruption of compartmentalized protein localization as part of the disease mechanisms underlying CEP290-associated retinopathies.
Our study shows that the myosin-tail homology domain is essential for CEP290's function as a ciliary gatekeeper, particularly to prevent diffusion of inner segment membrane proteins into the outer segment. A subset of inner segment membrane proteins rapidly accumulates in the outer segment upon disruption of the myosin-tail homology domain. STX3 and STXBP1, in particular, show striking accumulation in outer segments as degeneration progresses. Other inner segment membrane proteins tested show various degrees of accumulation in the outer segment. SNAP25 and IMPG2 exhibit significant accumulation in the outer segment, but their density in the outer segment does not exceed that in the inner segment. It is tantalizing to speculate that there are additional factor(s) that induce STX3 and STXBP1 enrichment in the outer segment. Mislocalization of ATP1A3 and HCN1 is mild, and localization of VAMP2, LAMP1, and SYP is not affected by the loss of CEP290 myosin-tail homology domain.
In contrast, the myosin-tail homology domain appears to be dispensable for the trafficking and confinement of most outer segment-resident proteins except RHO. In rd16 mice, the myosin-tail homology domain is constitutively disrupted, and the outer segment biogenesis is severely impaired. Most outer segment proteins, however, manage to be delivered to the rudimentary outer segment. The low-level mislocalization of outer segment proteins is likely secondary to the outer segment biogenesis defects, including the small size and disorganized disc structures. In Cep290fl/fl;Cre+ retinas, in which the myosin-tail homology domain is disrupted after the connecting cilium is formed, mislocalization of outer segment-resident proteins is not observed except for RHO. Furthermore, the length of the outer segment does not change significantly for 20 days while degeneration is progressing. Zebrafish cep290fh297/fh297 mutants, which have a nonsense mutation (p.Gln-1217*) before the myosin-tail homology domain, exhibit partial mislocalization of RHO but not of rhodopsin kinase GRK1 or a transducin subunit GNB1 at 6 months post-fertilization (25). Although degradation of mis-trafficked proteins in the inner segment could contribute to the apparent lack of mislocalized proteins, these data suggest that most outer segment proteins do not require the myosin-tail homology domain for their outer segment-specific localization. It remains to be determined whether the residual part of CEP290, which is expressed in Cep290fl/fl;Cre+ and rd16 retinas, is sufficient for the trafficking and confinement of outer segment proteins and, if so, specifically which proteins require CEP290 for their outer segment localization. Among the outer segment proteins examined, RHO was the only protein that showed consistent mislocalization in both Cep290fl/fl;Cre+ and rd16 retinas. It is currently unclear whether mislocalized RHOs originate from the outer segment (i.e. by leakage), whether they are newly synthesized proteins that failed to be delivered, or both. More sophisticated approaches such as a pulse–chase experiment are needed to address this question.
Although the number of proteins examined in this study is limited, it is noteworthy that all proteins that mislocalize upon disruption of the myosin-tail homology domain are plasma membrane–localizing proteins. For instance, STX3 and SNAP25 are transmembrane proteins known to localize to the inner segment plasma membrane (45, 54–56). STXBP1 does not have a transmembrane domain or a lipid anchor. However, STXBP1 localization tightly correlates with that of STX3 in both BBS and Cep290 mutant retinas at all ages tested (this study and see Ref. 46). In addition, STXBP1–STX3 protein complexes are readily detectable by immunoprecipitation followed by silver staining in retinal extracts (Fig. S4). These observations suggest that most STXBP1 proteins are likely associated with STX3 in photoreceptors. Therefore, STXBP1 can be considered as a peripheral membrane protein associated with the plasma membrane through STX3. IMPG2 is a single transmembrane protein identified as a constituent of the interphotoreceptor matrix (50). Because the procedures used in the prior study to isolate IMPG2 from the interphotoreceptor matrix were expected not to extract integral membrane proteins, Acharya et al. (50) predicted that IMPG2 might be cleaved before the transmembrane domain to release the N-terminal fragment into the extracellular matrix. Indeed, we found that recombinant IMPG2 expressed in HEK293T cells was efficiently cleaved (Fig. S2A). The C-terminal fragment containing the transmembrane domain and the intracellular domain, to which our anti-IMPG2 antibody was raised, localized to the plasma membrane (Fig. S2B). These data suggest that IMPG2 immunoreactivity detected by our IMPG2 antibody in the retina is from full-length IMPG2 in the secretory pathway (including endoplasmic reticulum and Golgi) or the C-terminal cleavage product that localizes to the plasma membrane. In contrast, localization of endomembrane proteins (VAMP2, SYP, and LAMP1) is not affected by the loss of the myosin-tail homology domain. These proteins are actively sorted and targeted to their destinations upon synthesis and during recycling and therefore are not subject to diffusion along the plasma membrane. ATP1A3 and HCN1 localize to the inner segment plasma membrane but exhibit only mild mislocalization. We speculate that these proteins may be targeted and retained in the inner segment plasma membrane by unknown mechanisms. Indeed, contrary to STX3, STXBP1, and SNAP25, which are evenly distributed throughout the inner segment in normal photoreceptors, ATP1A3 and HCN1 are significantly enriched in the myoid and ellipsoid zones, implying that they do not freely diffuse within the inner segment. Taken together, our data suggest that a subset of inner segment plasma membrane proteins is susceptible to diffusion to the outer segment upon disruption of the myosin-tail homology domain.
Based on our study, we propose that failure of protein confinement at the connecting cilium and the consequent accumulation of inner segment plasma membrane proteins in the outer segment combined with insufficient RHO delivery underlie retinal degeneration in CEP290-associated ciliopathies (Fig. 9). In normal photoreceptors, CEP290 is a part of the ciliary gate that confines inner segment plasma membrane proteins to the inner segment. The myosin-tail homology domain is crucial for this function. CEP290 is also required for the trafficking and/or confinement of RHO to the outer segment. In photoreceptors with compromised CEP290 functions, ciliary gates are impaired, allowing diffusion of select inner segment plasma membrane proteins into the outer segment. Aberrant accumulation of inner segment membrane proteins in nascent discs combined with insufficient delivery of RHO disturbs the disc morphogenesis process. The abundance of membranes in the outer segment is likely to contribute to the accumulation of inner segment membrane proteins. Inner segment mislocalization of RHO is likely another key pathomechanism that triggers photoreceptor death, as mutations disrupting RHO trafficking commonly cause retinal degeneration (Ref. 70 and references therein). The precise roles of CEP290 for outer segment protein trafficking and confinement as well as the mechanisms by which the myosin-tail homology domain blocks inner segment membrane proteins remain to be determined. It also should be noted that both CEP290 mouse models used in this study possess hypomorphic alleles and not a null. Cep290 null mutants completely lack the connecting cilium and the outer segment (16). Therefore, although not fully understood, one should bear in mind that CEP290 has additional roles in the connecting cilium biogenesis and that the N-terminal two-thirds of CEP290 might be necessary for outer segment protein trafficking and confinement. Finally, ciliary gates are composed of multiple proteins of the “NPHP” and “MKS modules” (5, 6, 8, 9, 71). Loss–of–function mutations in these genes/proteins commonly cause retinal degeneration. Similar pathomechanisms may underlie retinal degenerations in these diseases.
Figure 9.

Model for pathomechanisms of CEP290-associated retinopathies. CEP290 is a component of the Y-link that functions as a ciliary gatekeeper at the connecting cilium. In normal photoreceptors (left), CEP290 allows entry of outer segment-bound proteins while blocking that of inner segment proteins such as STX3, STXBP1, SNAP25, and IMPG2. In photoreceptors with compromised CEP290 function (right), defective ciliary gates allow entry of not only outer segment-bound proteins but also certain inner segment plasma membrane proteins that do not have specific targeting signals. Accumulation of inner segment membrane proteins in nascent discs interferes with normal disc closure. Although precise roles of CEP290 in RHO trafficking are currently uncertain, a subset of RHO mislocalizes to the inner segment and RHO mislocalization is likely to contribute to retinal degeneration in CEP290-associated ciliopathies.
Experimental procedures
Animal study approval
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Iowa and were conducted in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Mice
Cep290fl (Cep290tm1Jgg/J; stock number 013701) (27) and Cep290rd16 (BXD24/TyJ-Cep290rd16/J, stock number 000031) mice were obtained from The Jackson Laboratory. iCre75 transgenic mice (28) were obtained from Dr. Ching-Kang Chen (Baylor College of Medicine). Ift88fl and Lztfl1 gene-trap mice were from our colonies and were previously described (46, 72). The original Cep290fl mice that we obtained had both rd1 (in Pde6b) and rd8 (in Crb1) mutations. These mutations were eliminated by breeding with WT 129S6/SvEvTac mice (Taconic Biosciences), and all animals used in this study were Pde6b+/+;Crb1+/+ in B6;129S6 mixed backgrounds. Cep290rd16 mice were maintained in a BXD;B6 mixed background to obtain heterozygous littermates (i.e. Cep290+/rd16)as a control. All primers for genotyping were from Integrated DNA Technologies, and their sequences are described in Table S1. All animals were maintained in 12-h light/dark cycles and fed ad libitum standard mouse chow. PCR protocols are available upon request.
RNA extraction and reverse transcription (RT)-PCR
Mice were euthanized by CO2 asphyxiation followed by cervical dislocation. Eyes were enucleated, and the anterior segment was removed using micro-dissecting scissors. The neural retina was separated from the rest of the ocular tissues with forceps and snap-frozen in liquid nitrogen. Upon completion of retina collection, frozen retinas were placed on ice, immersed in 1 ml of TRI Reagent (Sigma), and homogenized with a PT1200E Polytron homogenizer (Kinematica). Total RNAs were extracted following the manufacturer's instruction. One μg of total RNA was used for cDNA synthesis using SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific) and random hexamers (Thermo Fisher Scientific) following the manufacturer's instruction. Cep290 cDNA fragments between exons 34 and 41 were PCR-amplified with Universe High-Fidelity Hot Start DNA polymerase (Bimake) and two primers (F-Cep290-e34, 5′-GCCGAAATCTCATCACACAATG-3′, and R-Cep290-e41, 5′-GCTTCTCCTTCCTTCTCCTTTAG-3′). PCR products were separated in a 1.2% agarose gel and purified with a Gel/PCR DNA fragment extraction kit (IBI Scientific). Purified DNAs were sequenced by Sanger sequencing using the aforementioned two primers (F-Cep290-e34 and R-Cep290-e41).
Cell culture, plasmids, and transfection
HEK293T and IMCD3 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific) and DMEM/F-12 (Thermo Fisher Scientific), respectively, supplemented with 10% (v/v) fetal bovine serum (Sigma) and 100 units/ml penicillin/streptomycin (Thermo Fisher Scientific) at 37 °C in a humidified 5% CO2 incubator. Plasmids containing full-length human CEP290 ORF (NM_025114.4) and mouse Rkip (also known as Pebp1; BC083063) were obtained from Dr. Val Sheffield (University of Iowa) and Transomic Technologies, respectively. To generate FLAG-tagged CEP290 expression vectors, CEP290 fragments encoding residues 1–2479, 1–1671, 1–1604, and 1789–2479 were PCR-amplified using Q5 High-Fidelity DNA Polymerase (New England Biolabs) and cloned into the EcoRI and XhoI sites of the pSS-FS vector (33) using a GenBuilder cloning kit (GenScript). HA-tagged RKIP expression vectors were generated by PCR amplification of the full-length Rkip fragment (residues 1–187) and insertion at the EcoRI and XhoI sites of the pCS2HA vector, which was produced by replacing the 6×Myc tag with an HA tag in the pCS2+MT vector (73). For GFP-tagged INPP5E fusion proteins, the full-length human INPP5E coding sequence (BC028032; Open Biosystems) was inserted into the XhoI and BamHI sites of pEGFP-C3 (Clontech). All constructs were sequence-verified by Sanger sequencing. HEK293T cells that stably express HA–RKIP were generated by co-transfection of pCS2HA-Rkip with pUS2puro using FuGENE HD Transfection Reagent (Promega) followed by puromycin selection (1.2 μg/ml; Thermo Fisher Scientific). Transient transfection of CEP290 expression cassettes was conducted using FuGENE HD following the manufacturer's instruction.
Immunofluorescence microscopy
Immunofluorescence microscopy in IMCD3 cells was conducted as described previously (33).
For immunohistochemistry of connecting cilium-localizing proteins, mice were euthanized by CO2 asphyxiation followed by cervical dislocation. Mouse eyes were enucleated and the anterior segment was carefully removed using micro-dissecting scissors. The posterior chamber was placed in a cryo-mold, embedded in Neg-50 Frozen Section Medium (Thermo Fisher Scientific), and frozen in a 2-methylbutane bath chilled with liquid nitrogen. Ten-μm cryosections were collected on Superfrost Plus Microscope Slides (Thermo Fisher Scientific) using a CryoStar NX70 Cryostat (Thermo Fisher Scientific). Sections were fixed in 4% (w/v) paraformaldehyde (PFA)/PBS fixative for 8 min at 4 °C and washed in PBS three times at room temperature, prior to permeabilization and primary antibody binding as described below.
For immunofluorescence microscopy in retinal sections, mouse eyes were collected as above and immersed in 4% PFA/PBS fixative. A small puncture was created between the lens and the sclera using a 26-gauge needle. After 5 min of fixation, the lens and the anterior chamber were removed with micro-dissecting scissors. The eye cups were further fixed in 4% PFA/PBS for 3 h at 4 °C. After washing with PBS (three times for 10 min each), eye cups were infiltrated and embedded in acrylamide as described previously (74). After solidification, extra acrylamide was carefully removed, and eye cups were placed in Neg-50 Frozen Section Medium, frozen in a dry-ice/ethanol bath, and stored at −80 °C. Five- to 8-μm cryosections were collected on Superfrost Plus microscope slides using a CryoStar NX70 cryostat. Retinal sections were permeabilized with PBS-T (PBS with 0.1% Triton X-100), blocked with 5% BSA, 5% normal goat serum (Vector Laboratories) in PBS-T, and incubated with the indicated primary antibodies for 2–3 h at room temperature. After washing, sections were incubated with secondary antibodies conjugated to Alexa Fluor 488 or 568 (Thermo Fisher Scientific) for 2 h at room temperature in the dark. After rinsing, Vectashield Mounting Medium with 4′,6-diamidino-2′-phenylindole dihydrochloride (DAPI) (Vector Laboratories) was added, and images were taken using an Olympus IX71 microscope or a Zeiss LSM 710 confocal microscope. Intensity range of images was adjusted by linear level adjustments using Adobe Photoshop CC 2018 (Adobe Systems). For fluorescence intensity quantification, 20–25-μm width areas were randomly selected, and integrated fluorescence intensities were measured within the outer segment and the entire photoreceptor cells using ImageJ to calculate the ratio of fluorescence intensities in the outer segment. Two-tailed, two-sample t-tests assuming unequal variances were used for statistical analyses. p values smaller than 0.01 were regarded as statistically significant.
Outer segment isolation and immunoblotting
Photoreceptor outer segments were isolated as described previously (46). Briefly, eyes were enucleated from Cep290+/fl;Cre? and Cep290fl/fl;Cre+ mice at P25–28, and the anterior segment and the lens were removed using micro-dissecting scissors. The neural retina was separated from the pigmented retinal epithelium with forceps, collected in 63% sucrose/PBS, snap-frozen in liquid nitrogen, and stored in −80 °C until needed. Fifteen to 20 animals were used per genotype per preparation. Retinas with the same genotypes were pooled in 1.5-ml tubes on ice, and 63% sucrose/PBS was added to 1 ml. Retinal suspensions were gently pipetted up and down 10 times using a P1000 tip with a 1.5–2-mm orifice, and further vortexed for 30 s. Homogenates were centrifuged at 100 × g for 3 min, and supernatants were transferred to fresh 1.5-ml tubes and spun at 2,350 × g for 10 min at 4 °C. Supernatants were transferred to ultracentrifuge tubes, and layers of 42, 37, and 32% sucrose/PBS solutions (1.1 ml each) were overlaid. Homogenates were centrifuged at 116,000 × gave for 1 h at 4 °C using a Sorvall TH-660 rotor. Outer segments were collected at the 32–37% sucrose interface, and an equal volume of ice-cold PBS was added to the collected outer segment fractions. Outer segments were pelleted by spinning at 10,000 × g for 8 min at 4 °C and resuspended in a lysis buffer (PBS with 1% Triton X-100). Protein concentrations were measured with a DC Protein Assay kit (Bio-Rad) following the manufacturer's instruction. Five μg of proteins were loaded and separated on a 4–12% (w/v) NuPAGE Bis-Tris gel (Thermo Fisher Scientific) and transferred to a nitrocellulose membrane (Bio-Rad). Proteins were detected by indicated primary and secondary antibodies (see below) and SuperSignal West Dura Extended Duration Substrate (Thermo Fisher Scientific). Images were taken with a ChemiDoc system (Bio-Rad) and quantified with the Image Lab software (Bio-Rad).
Transmission EM (TEM)
Mice were anesthetized with a lethal dose of a ketamine/xylazine mixture (ketamine, 175 mg/kg body weight; xylazine, 25 mg/kg body weight) and perfused transcardially with half-strength Karnovsky's fixative (1.5% PFA with 1.5% glutaraldehyde in 0.1 m sodium cacodylate buffer (Electron Microscopy Sciences)). Eyes were enucleated, and the anterior segment and the lens were removed with micro-dissecting scissors. Eye cups were further fixed in half-strength Karnovsky's fixative overnight at 4 °C. After fixation, eye cups were rinsed three times in 0.1 m sodium cacodylate buffer for a total of 30 min and treated with 1% OsO4 (Electron Microscopy Sciences) containing 1.5% (w/v) potassium ferrocyanide for 1.5 h. After three washes in 0.1 m sodium cacodylate buffer, eye cups were rinsed in distilled water, treated with a solution of 2.5% (w/v) uranyl acetate (Electron Microscopy Sciences), dehydrated in graded ethanol concentrations up to 100% (v/v), treated with propylene oxide, and infiltrated and embedded with Eponate 12 resin (Ted Pella) before overnight polymerization in a 70 °C oven. Ultrathin sections (80-nm thickness) were prepared with a Leica EM UC6 Ultramicrotome, collected on copper grids, and contrasted with 5% uranyl acetate and lead citrate. Micrographs were obtained with a Jeol JEM 1230 transmission electron microscope equipped with a Gatan UltraScan 1000 2k × 2k CCD camera.
Immunoprecipitation
For CEP290–RKIP interaction studies, HEK293T cells expressing HA–RKIP were transfected with FLAG–CEP290 expression cassettes or empty vectors using FuGENE HD and harvested 30 h post-transfection. Cells were lysed in a lysis buffer (50 mm HEPES, pH 7.0, 150 mm NaCl, 1% (v/v) Nonidet P-40, 2 mm EDTA, 5% (v/v) glycerol, protease inhibitor mixture (Bimake)), and insoluble materials were removed by centrifugation at 15,000 × g for 10 min at 4 °C. Anti-FLAG M2 magnetic beads (Sigma) were added to protein extracts and incubated at 4 °C for 3 h with rotation. After washing four times with the lysis buffer, 1× NuPAGE LDS sample buffer (Thermo Fisher Scientific) was added to the beads to elute bound proteins. Eluted proteins were loaded and separated on a 4–12% (w/v) NuPAGE Bis-Tris gel, transferred to a nitrocellulose membrane, and detected by peroxidase-conjugated anti-FLAG and anti-HA antibodies (Sigma, A8592 and 12013819001).
For immunoprecipitation of the BBSome in retinal extracts, neural retinas from Cep290+/fl;Cre? and Cep290fl/fl;Cre+ mice were isolated as described above and lysed in 400 μl of a lysis buffer (50 mm HEPES, pH 7.0, 150 mm NaCl, 0.5% (v/v) Nonidet P-40, 2 mm EGTA, 2 mm MgCl2, 10% (v/v) glycerol, protease inhibitor mixture (Bimake)) by pipetting and vortexing. After removing insoluble materials by centrifugation at 15,000 × g for 10 min, 1.5–2 μg of BBS7 (Proteintech Group, 18961-1-AP), STX3 (Proteintech Group, 15556-1-AP), or rabbit polyclonal control IgG (Proteintech Group, 30000-0-AP) antibodies were added to the supernatants and incubated at 4 °C overnight with rotation. To precipitate antibodies and associated proteins, 10 μl of DynabeadsTM protein G magnetic beads (Thermo Fisher Scientific) were added and further incubated at 4 °C for 2 h with rotation. Beads were washed four times with 1 ml of a wash buffer (50 mm HEPES, pH 7.0, 150 mm NaCl, 0.1% Nonidet P-40, 2 mm EGTA, 2 mm MgCl2, 10% glycerol). Bound proteins were eluted by adding 25 μl of 1× NuPAGE LDS sample buffer. SDS-PAGE, immunoblotting, and imaging were conducted as described above except that rabbit primary antibodies were detected by peroxidase-conjugated protein A (Proteintech Group, SA00001-18).
Antibodies
The following primary antibodies were used for immunofluorescence microscopy and immunoblotting: ABCA4 (EMD Millipore, MABN2439); β-actin (Sigma, A1978); ATP1A3 (abcam, ab2826); BBS2 (Proteintech Group, 66246-1-AP); BBS5 (Santa Cruz Biotechnology, sc-515331); BBS7 (Proteintech Group, 18961-1-AP); BBS8 (Sigma, HPA003310); CEP290 (EMD Millipore, ABN1710, for immunohistochemistry); CEP290 (Proteintech Group, 22490-1-AP, for immunoblotting); CETN (EMD Millipore, 04-1624); CNGA1 (EMD Millipore, MABN468); FLAG (Sigma, F1804 and A8592 (peroxidase-conjugated)); GRK1 (abcam, ab2776); GUCY2D (Proteintech Group, 55127-1-AP); HA (peroxidase-conjugated; Sigma, 12013819001); HCN1 (NeuroMab, 75-110); IMPG2 (Thermo Fisher Scientific, PA5-64926); LAMP1 (Sigma, L1418); LZTFL1 (custom-made (68)); PDE6A (Proteintech Group, 21200-1-AP); PDE6B (Proteintech Group, 22063-1-AP); polyglutamylation modification/GT335 (AdipoGen, AG-20B-0020-C100); PRPH2 (Proteintech Group, 18109-1-AP); RHO (EMD Millipore, MAB5356); ROM1 (Proteintech Group, 21984-1-AP); SNAP25 (abcam, ab24737); STX3 (EMD Millipore, MAB2258), STXBP1 (Proteintech Group, 11459-1-AP), SYP (Cell Signaling, 5461); and VAMP2 (Cell Signaling, 13508). Rabbit polyclonal ATP8A2 antibody was custom-made using GST-tagged mouse ATP8A2 (amino acids 369–644) as an immunogen, followed by affinity-purification with MBP-tagged mouse ATP8A2 (amino acids 369–644) (Proteintech Group).
Secondary antibodies used are as follows: anti-mouse IgG conjugated to HRP (Cell Signaling Technology, 707; anti-rabbit IgG conjugated to HRP (Cell Signaling Technology, 7074); goat anti-mouse IgG conjugated to Alexa Fluor 488 (Thermo Fisher Scientific, A-11029); goat anti-mouse IgG conjugated to Alexa Fluor 568 (Thermo Fisher Scientific, A-11031); goat anti-rabbit IgG conjugated to Alexa Fluor 488 (Thermo Fisher Scientific, A32731); and goat anti-rabbit IgG conjugated to Alexa Fluor 568 (Thermo Fisher Scientific, A-11036).
Author contributions
P. D., B. H., and S. S. validation; P. D., B. H., S. B., A. R., and S. S. investigation; P. D., B. H., and S. S. visualization; P. D. and B. H. methodology; P. D., B. H., and S. S. writing-review and editing; S. S. conceptualization; S. S. data curation; S. S. formal analysis; S. S. supervision; S. S. funding acquisition; S. S. writing-original draft; S. S. project administration.
Supplementary Material
Acknowledgments
We thank Drs. Ching-Kang Chen (Baylor College of Medicine) and Val C. Sheffield (University of Iowa) for generously providing iCre75 transgenic mice and a plasmid with human CEP290 cDNA, respectively. We also thank Dr. Chantal Allamargot and J. Thomas Cribbs for excellent assistance in EM and Rkip subcloning, respectively. The University of Iowa was the recipient of National Institutes of Health Center Support Grant P30-EY025580.
This work was supported by National Institutes of Health Grants R01-EY022616 and R21-EY027431 (to S. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4 and Table S1.
- MKS
- Meckel-Gruber syndrome
- RHO
- rhodopsin
- DAPI
- 4′,6-diamidino-2′-phenylindole
- DMEM
- Dulbecco's modified Eagle's medium
- HRP
- horseradish peroxidase
- TEM
- transmission electron microscopy
- P
- post-natal
- PFA
- paraformaldehyde
- RKIP
- Raf-1 kinase inhibitory protein
- BBS
- Bardet-Biedl syndrome
- IFT
- intraflagellar transport
- SYP
- synaptophysin.
References
- 1. Seo S., and Datta P. (2017) Photoreceptor outer segment as a sink for membrane proteins: hypothesis and implications in retinal ciliopathies. Hum. Mol. Genet. 26, R75–R82 10.1093/hmg/ddx163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Young R. W. (1967) The renewal of photoreceptor cell outer segments. J. Cell Biol. 33, 61–72 10.1083/jcb.33.1.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baehr W., Hanke-Gogokhia C., Sharif A., Reed M., Dahl T., Frederick J. M., and Ying G. (2019) Insights into photoreceptor ciliogenesis revealed by animal models. Prog. Retin. Eye Res. 71, 26–56 10.1016/j.preteyeres.2018.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Craige B., Tsao C. C., Diener D. R., Hou Y., Lechtreck K. F., Rosenbaum J. L., and Witman G. B. (2010) CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J. Cell Biol. 190, 927–940 10.1083/jcb.201006105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Williams C. L., Li C., Kida K., Inglis P. N., Mohan S., Semenec L., Bialas N. J., Stupay R. M., Chen N., Blacque O. E., Yoder B. K., and Leroux M. R. (2011) MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J. Cell Biol. 192, 1023–1041 10.1083/jcb.201012116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li C., Jensen V. L., Park K., Kennedy J., Garcia-Gonzalo F. R., Romani M., De Mori R., Bruel A. L., Gaillard D., Doray B., Lopez E., Rivière J. B., Faivre L., Thauvin-Robinet C., Reiter J. F., et al. (2016) MKS5 and CEP290 dependent assembly pathway of the ciliary transition zone. PLos Biol. 14, e1002416 10.1371/journal.pbio.1002416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim J., Krishnaswami S. R., and Gleeson J. G. (2008) CEP290 interacts with the centriolar satellite component PCM-1 and is required for Rab8 localization to the primary cilium. Hum. Mol. Genet. 17, 3796–3805 10.1093/hmg/ddn277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Garcia-Gonzalo F. R., Corbit K. C., Sirerol-Piquer M. S., Ramaswami G., Otto E. A., Noriega T. R., Seol A. D., Robinson J. F., Bennett C. L., Josifova D. J., García-Verdugo J. M., Katsanis N., Hildebrandt F., and Reiter J. F. (2011) A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet. 43, 776–784 10.1038/ng.891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang T. T., Su J., Wang W. J., Craige B., Witman G. B., Tsou M. F., and Liao J. C. (2015) Super-resolution pattern recognition reveals the architectural map of the ciliary transition zone. Sci. Rep. 5, 14096 10.1038/srep14096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wiegering A., Dildrop R., Kalfhues L., Spychala A., Kuschel S., Lier J. M., Zobel T., Dahmen S., Leu T., Struchtrup A., Legendre F., Vesque C., Schneider-Maunoury S., Saunier S., Rüther U., and Gerhardt C. (2018) Cell type-specific regulation of ciliary transition zone assembly in vertebrates. EMBO J. 37, e97791 10.15252/embj.201797791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shimada H., Lu Q., Insinna-Kettenhofen C., Nagashima K., English M. A., Semler E. M., Mahgerefteh J., Cideciyan A. V., Li T., Brooks B. P., Gunay-Aygun M., Jacobson S. G., Cogliati T., Westlake C. J., and Swaroop A. (2017) In vitro modeling using ciliopathy-patient–derived cells reveals distinct cilia dysfunctions caused by CEP290 mutations. Cell Rep. 20, 384–396 10.1016/j.celrep.2017.06.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reiter J. F., Blacque O. E., and Leroux M. R. (2012) The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep. 13, 608–618 10.1038/embor.2012.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Betleja E., and Cole D. G. (2010) Ciliary trafficking: CEP290 guards a gated community. Curr. Biol. 20, R928–R931 10.1016/j.cub.2010.09.058 [DOI] [PubMed] [Google Scholar]
- 14. Garcia-Gonzalo F. R., and Reiter J. F. (2017) Open sesame: how transition fibers and the transition zone control ciliary composition. Cold Spring Harb. Perspect. Biol. 9, a028134 10.1101/cshperspect.a028134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chang B., Khanna H., Hawes N., Jimeno D., He S., Lillo C., Parapuram S. K., Cheng H., Scott A., Hurd R. E., Sayer J. A., Otto E. A., Attanasio M., O'Toole J. F., Jin G., et al. (2006) In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum. Mol. Genet. 15, 1847–1857 10.1093/hmg/ddl107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rachel R. A., Yamamoto E. A., Dewanjee M. K., May-Simera H. L., Sergeev Y. V., Hackett A. N., Pohida K., Munasinghe J., Gotoh N., Wickstead B., Fariss R. N., Dong L., Li T., and Swaroop A. (2015) CEP290 alleles in mice disrupt tissue-specific cilia biogenesis and recapitulate features of syndromic ciliopathies. Hum. Mol. Genet. 24, 3775–3791 10.1093/hmg/ddv123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. den Hollander A. I., Koenekoop R. K., Yzer S., Lopez I., Arends M. L., Voesenek K. E., Zonneveld M. N., Strom T. M., Meitinger T., Brunner H. G., Hoyng C. B., van den Born L. I., Rohrschneider K., and Cremers F. P. (2006) Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 79, 556–561 10.1086/507318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sayer J. A., Otto E. A., O'Toole J. F., Nurnberg G., Kennedy M. A., Becker C., Hennies H. C., Helou J., Attanasio M., Fausett B. V., Utsch B., Khanna H., Liu Y., Drummond I., Kawakami I., et al. (2006) The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat. Genet. 38, 674–681 10.1038/ng1786 [DOI] [PubMed] [Google Scholar]
- 19. Baala L., Audollent S., Martinovic J., Ozilou C., Babron M. C., Sivanandamoorthy S., Saunier S., Salomon R., Gonzales M., Rattenberry E., Esculpavit C., Toutain A., Moraine C., Parent P., Marcorelles P., et al. (2007) Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am. J. Hum. Genet. 81, 170–179 10.1086/519494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cideciyan A. V., Aleman T. S., Jacobson S. G., Khanna H., Sumaroka A., Aguirre G. K., Schwartz S. B., Windsor E. A., He S., Chang B., Stone E. M., and Swaroop A. (2007) Centrosomal-ciliary gene CEP290/NPHP6 mutations result in blindness with unexpected sparing of photoreceptors and visual brain: implications for therapy of Leber congenital amaurosis. Hum. Mutat. 28, 1074–1083 10.1002/humu.20565 [DOI] [PubMed] [Google Scholar]
- 21. Frank V., den Hollander A. I., Brüchle N. O., Zonneveld M. N., Nürnberg G., Becker C., Du Bois G., Kendziorra H., Roosing S., Senderek J., Nürnberg P., Cremers F. P., Zerres K., and Bergmann C. (2008) Mutations of the CEP290 gene encoding a centrosomal protein cause Meckel-Gruber syndrome. Hum. Mutat. 29, 45–52 10.1002/humu.20614 [DOI] [PubMed] [Google Scholar]
- 22. den Hollander A. I., Roepman R., Koenekoop R. K., and Cremers F. P. (2008) Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 27, 391–419 10.1016/j.preteyeres.2008.05.003 [DOI] [PubMed] [Google Scholar]
- 23. Coppieters F., Lefever S., Leroy B. P., and De Baere E. (2010) CEP290, a gene with many faces: mutation overview and presentation of CEP290 base. Hum. Mutat. 31, 1097–1108 10.1002/humu.21337 [DOI] [PubMed] [Google Scholar]
- 24. Rachel R. A., May-Simera H. L., Veleri S., Gotoh N., Choi B. Y., Murga-Zamalloa C., McIntyre J. C., Marek J., Lopez I., Hackett A. N., Zhang J., Brooks M., den Hollander A. I., Beales P. L., Li T., et al. (2012) Combining Cep290 and Mkks ciliopathy alleles in mice rescues sensory defects and restores ciliogenesis. J. Clin. Invest. 122, 1233–1245 10.1172/JCI60981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lessieur E. M., Song P., Nivar G. C., Piccillo E. M., Fogerty J., Rozic R., and Perkins B. D. (2019) Ciliary genes arl13b, ahi1 and cc2d2a differentially modify expression of visual acuity phenotypes but do not enhance retinal degeneration due to mutation of cep290 in zebrafish. PLoS ONE 14, e0213960 10.1371/journal.pone.0213960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baker S. A., Haeri M., Yoo P., Gospe S. M. 3rd, Skiba N. P., Knox B. E., and Arshavsky V. Y. (2008) The outer segment serves as a default destination for the trafficking of membrane proteins in photoreceptors. J. Cell Biol. 183, 485–498 10.1083/jcb.200806009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lancaster M. A., Gopal D. J., Kim J., Saleem S. N., Silhavy J. L., Louie C. M., Thacker B. E., Williams Y., Zaki M. S., and Gleeson J. G. (2011) Defective Wnt-dependent cerebellar midline fusion in a mouse model of Joubert syndrome. Nat. Med. 17, 726–731 10.1038/nm.2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li S., Chen D., Sauvé Y., McCandless J., Chen Y. J., and Chen C. K. (2005) Rhodopsin-iCre transgenic mouse line for Cre-mediated rod-specific gene targeting. Genesis 41, 73–80 10.1002/gene.20097 [DOI] [PubMed] [Google Scholar]
- 29. Littink K. W., Pott J. W., Collin R. W., Kroes H. Y., Verheij J. B., Blokland E. A., de Castro Miró M., Hoyng C. B., Klaver C. C., Koenekoop R. K., Rohrschneider K., Cremers F. P., van den Born L. I., and den Hollander A. I. (2010) A novel nonsense mutation in CEP290 induces exon skipping and leads to a relatively mild retinal phenotype. Invest. Ophthalmol. Vis. Sci. 51, 3646–3652 10.1167/iovs.09-5074 [DOI] [PubMed] [Google Scholar]
- 30. Drivas T. G., Wojno A. P., Tucker B. A., Stone E. M., and Bennett J. (2015) Basal exon skipping and genetic pleiotropy: a predictive model of disease pathogenesis. Sci. Transl. Med. 7, 291ra97 10.1126/scitranslmed.aaa5370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roosing S., Cremers F. P. M., Riemslag F. C. C., Zonneveld-Vrieling M. N., Talsma H. E., Klessens-Godfroy F. J. M., den Hollander A. I., and van den Born L. I. (2017) A rare form of retinal dystrophy caused by hypomorphic nonsense mutations in CEP290. Genes 8, E208 10.3390/genes8080208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barny I., Perrault I., Michel C., Soussan M., Goudin N., Rio M., Thomas S., Attié-Bitach T., Hamel C., Dollfus H., Kaplan J., Rozet J. M., and Gerard X. (2018) Basal exon skipping and nonsense-associated altered splicing allows bypassing complete CEP290 loss-of-function in individuals with unusually mild retinal disease. Hum. Mol. Genet. 27, 2689–2702 10.1093/hmg/ddy179 [DOI] [PubMed] [Google Scholar]
- 33. Humbert M. C., Weihbrecht K., Searby C. C., Li Y., Pope R. M., Sheffield V. C., and Seo S. (2012) ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc. Natl. Acad. Sci. U.S.A. 109, 19691–19696 10.1073/pnas.1210916109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Murga-Zamalloa C. A., Ghosh A. K., Patil S. B., Reed N. A., Chan L. S., Davuluri S., Peränen J., Hurd T. W., Rachel R. A., and Khanna H. (2011) Accumulation of the Raf-1 kinase inhibitory protein (Rkip) is associated with Cep290-mediated photoreceptor degeneration in ciliopathies. J. Biol. Chem. 286, 28276–28286 10.1074/jbc.M111.237560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kamps K. M., De Grip W. J., and Daemen F. J. (1982) Use of a density modification technique for isolation of the plasma membrane of rod outer segments. Biochim. Biophys. Acta 687, 296–302 10.1016/0005-2736(82)90558-2 [DOI] [PubMed] [Google Scholar]
- 36. Molday R. S., and Molday L. L. (1987) Differences in the protein composition of bovine retinal rod outer segment disk and plasma membranes isolated by a ricin-gold-dextran density perturbation method. J. Cell Biol. 105, 2589–2601 10.1083/jcb.105.6.2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Molday R. S., Hicks D., and Molday L. (1987) Peripherin. A rim-specific membrane protein of rod outer segment discs. Invest. Ophthalmol. Vis. Sci. 28, 50–61 [PubMed] [Google Scholar]
- 38. Moritz O. L., and Molday R. S. (1996) Molecular cloning, membrane topology, and localization of bovine rom-1 in rod and cone photoreceptor cells. Invest. Ophthalmol. Vis. Sci. 37, 352–362 [PubMed] [Google Scholar]
- 39. Illing M., Molday L. L., and Molday R. S. (1997) The 220-kDa rim protein of retinal rod outer segments is a member of the ABC transporter superfamily. J. Biol. Chem. 272, 10303–10310 10.1074/jbc.272.15.10303 [DOI] [PubMed] [Google Scholar]
- 40. Hallett M. A., Delaat J. L., Arikawa K., Schlamp C. L., Kong F., and Williams D. S. (1996) Distribution of guanylate cyclase within photoreceptor outer segments. J. Cell Sci. 109, 1803–1812 [DOI] [PubMed] [Google Scholar]
- 41. Kwok M. C., Holopainen J. M., Molday L. L., Foster L. J., and Molday R. S. (2008) Proteomics of photoreceptor outer segments identifies a subset of SNARE and Rab proteins implicated in membrane vesicle trafficking and fusion. Mol. Cell. Proteomics 7, 1053–1066 10.1074/mcp.M700571-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Coleman J. A., Kwok M. C., and Molday R. S. (2009) Localization, purification, and functional reconstitution of the P4-ATPase Atp8a2, a phosphatidylserine flippase in photoreceptor disc membranes. J. Biol. Chem. 284, 32670–32679 10.1074/jbc.M109.047415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cook N. J., Molday L. L., Reid D., Kaupp U. B., and Molday R. S. (1989) The cGMP-gated channel of bovine rod photoreceptors is localized exclusively in the plasma membrane. J. Biol. Chem. 264, 6996–6999 [PubMed] [Google Scholar]
- 44. Sedmak T., and Wolfrum U. (2011) Intraflagellar transport proteins in ciliogenesis of photoreceptor cells. Biol. Cell 103, 449–466 10.1042/BC20110034 [DOI] [PubMed] [Google Scholar]
- 45. Mazelova J., Ransom N., Astuto-Gribble L., Wilson M. C., and Deretic D. (2009) Syntaxin 3 and SNAP-25 pairing, regulated by omega-3 docosahexaenoic acid, controls the delivery of rhodopsin for the biogenesis of cilia-derived sensory organelles, the rod outer segments. J. Cell Sci. 122, 2003–2013 10.1242/jcs.039982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Datta P., Allamargot C., Hudson J. S., Andersen E. K., Bhattarai S., Drack A. V., Sheffield V. C., and Seo S. (2015) Accumulation of non-outer segment proteins in the outer segment underlies photoreceptor degeneration in Bardet-Biedl syndrome. Proc. Natl. Acad. Sci. U.S.A. 112, E4400–E4409 10.1073/pnas.1510111112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hsu Y., Garrison J. E., Kim G., Schmitz A. R., Searby C. C., Zhang Q., Datta P., Nishimura D. Y., Seo S., and Sheffield V. C. (2017) BBSome function is required for both the morphogenesis and maintenance of the photoreceptor outer segment. PLoS Genet. 13, e1007057 10.1371/journal.pgen.1007057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dilan T. L., Singh R. K., Saravanan T., Moye A., Goldberg A. F. X., Stoilov P., and Ramamurthy V. (2018) Bardet-Biedl syndrome-8 (BBS8) protein is crucial for the development of outer segments in photoreceptor neurons. Hum. Mol. Genet. 27, 283–294 10.1093/hmg/ddx399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hollyfield J. G. (1999) Hyaluronan and the functional organization of the interphotoreceptor matrix. Invest. Ophthalmol. Vis. Sci. 40, 2767–2769 [PubMed] [Google Scholar]
- 50. Acharya S., Foletta V. C., Lee J. W., Rayborn M. E., Rodriguez I. R., Young W. S. 3rd., and Hollyfield J. G. (2000) SPACRCAN, a novel human interphotoreceptor matrix hyaluronan-binding proteoglycan synthesized by photoreceptors and pinealocytes. J. Biol. Chem. 275, 6945–6955 10.1074/jbc.275.10.6945 [DOI] [PubMed] [Google Scholar]
- 51. Bandah-Rozenfeld D., Collin R. W., Banin E., van den Born L. I., Coene K. L., Siemiatkowska A. M., Zelinger L., Khan M. I., Lefeber D. J., Erdinest I., Testa F., Simonelli F., Voesenek K., Blokland E. A., Strom T. M., et al. (2010) Mutations in IMPG2, encoding interphotoreceptor matrix proteoglycan 2, cause autosomal-recessive retinitis pigmentosa. Am. J. Hum. Genet. 87, 199–208 10.1016/j.ajhg.2010.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. van Huet R. A., Collin R. W., Siemiatkowska A. M., Klaver C. C., Hoyng C. B., Simonelli F., Khan M. I., Qamar R., Banin E., Cremers F. P., Theelen T., den Hollander A. I., van den Born L. I., and Klevering B. J. (2014) IMPG2-associated retinitis pigmentosa displays relatively early macular involvement. Invest. Ophthalmol. Vis. Sci. 55, 3939–3953 10.1167/iovs.14-14129 [DOI] [PubMed] [Google Scholar]
- 53. Meunier I., Manes G., Bocquet B., Marquette V., Baudoin C., Puech B., Defoort-Dhellemmes S., Audo I., Verdet R., Arndt C., Zanlonghi X., Le Meur G., Dhaenens C. M., and Hamel C. P. (2014) Frequency and clinical pattern of vitelliform macular dystrophy caused by mutations of interphotoreceptor matrix IMPG1 and IMPG2 genes. Ophthalmology 121, 2406–2414 10.1016/j.ophtha.2014.06.028 [DOI] [PubMed] [Google Scholar]
- 54. Demontis G. C., Moroni A., Gravante B., Altomare C., Longoni B., Cervetto L., and DiFrancesco D. (2002) Functional characterisation and subcellular localisation of HCN1 channels in rabbit retinal rod photoreceptors. J. Physiol. 542, 89–97 10.1113/jphysiol.2002.017640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pan Y., Laird J. G., Yamaguchi D. M., and Baker S. A. (2015) A di-arginine ER retention signal regulates trafficking of HCN1 channels from the early secretory pathway to the plasma membrane. Cell. Mol. Life Sci. 72, 833–843 10.1007/s00018-014-1705-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Geering K. (2008) Functional roles of Na,K-ATPase subunits. Curr. Opin. Nephrol. Hypertens. 17, 526–532 10.1097/MNH.0b013e3283036cbf [DOI] [PubMed] [Google Scholar]
- 57. Cideciyan A. V., Rachel R. A., Aleman T. S., Swider M., Schwartz S. B., Sumaroka A., Roman A. J., Stone E. M., Jacobson S. G., and Swaroop A. (2011) Cone photoreceptors are the main targets for gene therapy of NPHP5 (IQCB1) or NPHP6 (CEP290) blindness: generation of an all-cone Nphp6 hypomorph mouse that mimics the human retinal ciliopathy. Hum. Mol. Genet. 20, 1411–1423 10.1093/hmg/ddr022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nachury M. V., Loktev A. V., Zhang Q., Westlake C. J., Peränen J., Merdes A., Slusarski D. C., Scheller R. H., Bazan J. F., Sheffield V. C., and Jackson P. K. (2007) A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129, 1201–1213 10.1016/j.cell.2007.03.053 [DOI] [PubMed] [Google Scholar]
- 59. Loktev A. V., Zhang Q., Beck J. S., Searby C. C., Scheetz T. E., Bazan J. F., Slusarski D. C., Sheffield V. C., Jackson P. K., and Nachury M. V. (2008) A BBSome subunit links ciliogenesis, microtubule stability, and acetylation. Dev. Cell 15, 854–865 10.1016/j.devcel.2008.11.001 [DOI] [PubMed] [Google Scholar]
- 60. Blacque O. E., Reardon M. J., Li C., McCarthy J., Mahjoub M. R., Ansley S. J., Badano J. L., Mah A. K., Beales P. L., Davidson W. S., Johnsen R. C., Audeh M., Plasterk R. H., Baillie D. L., Katsanis N., et al. (2004) Loss of C. elegans BBS-7 and BBS-8 protein function results in cilia defects and compromised intraflagellar transport. Genes Dev. 18, 1630–1642 10.1101/gad.1194004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lechtreck K. F., Johnson E. C., Sakai T., Cochran D., Ballif B. A., Rush J., Pazour G. J., Ikebe M., and Witman G. B. (2009) The Chlamydomonas reinhardtii BBSome is an IFT cargo required for export of specific signaling proteins from flagella. J. Cell Biol. 187, 1117–1132 10.1083/jcb.200909183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jin H., White S. R., Shida T., Schulz S., Aguiar M., Gygi S. P., Bazan J. F., and Nachury M. V. (2010) The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 141, 1208–1219 10.1016/j.cell.2010.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liew G. M., Ye F., Nager A. R., Murphy J. P., Lee J. S., Aguiar M., Breslow D. K., Gygi S. P., and Nachury M. V. (2014) The intraflagellar transport protein IFT27 promotes BBSome exit from cilia through the GTPase ARL6/BBS3. Dev. Cell 31, 265–278 10.1016/j.devcel.2014.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Eguether T., San Agustin J. T., Keady B. T., Jonassen J. A., Liang Y., Francis R., Tobita K., Johnson C. A., Abdelhamed Z. A., Lo C. W., and Pazour G. J. (2014) IFT27 links the BBSome to IFT for maintenance of the ciliary signaling compartment. Dev. Cell 31, 279–290 10.1016/j.devcel.2014.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Williams C. L., McIntyre J. C., Norris S. R., Jenkins P. M., Zhang L., Pei Q., Verhey K., and Martens J. R. (2014) Direct evidence for BBSome-associated intraflagellar transport reveals distinct properties of native mammalian cilia. Nat. Commun. 5, 5813 10.1038/ncomms6813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ye F., Nager A. R., and Nachury M. V. (2018) BBSome trains remove activated GPCRs from cilia by enabling passage through the transition zone. J. Cell Biol. 217, 1847–1868 10.1083/jcb.201709041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liu P., and Lechtreck K. F. (2018) The Bardet-Biedl syndrome protein complex is an adapter expanding the cargo range of intraflagellar transport trains for ciliary export. Proc. Natl. Acad. Sci. U.S.A. 115, E934–E943 10.1073/pnas.1713226115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Seo S., Zhang Q., Bugge K., Breslow D. K., Searby C. C., Nachury M. V., and Sheffield V. C. (2011) A novel protein LZTFL1 regulates ciliary trafficking of the BBSome and smoothened. PLoS Genet. 7, e1002358 10.1371/journal.pgen.1002358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Smith T. S., Spitzbarth B., Li J., Dugger D. R., Stern-Schneider G., Sehn E., Bolch S. N., McDowell J. H., Tipton J., Wolfrum U., and Smith W. C. (2013) Light-dependent phosphorylation of Bardet-Biedl syndrome 5 in photoreceptor cells modulates its interaction with arrestin1. Cell. Mol. Life Sci. 70, 4603–4616 10.1007/s00018-013-1403-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nemet I., Ropelewski P., and Imanishi Y. (2015) Rhodopsin trafficking and mistrafficking: signals, molecular components, and mechanisms. Prog. Mol. Biol. Transl. Sci. 132, 39–71 10.1016/bs.pmbts.2015.02.007 [DOI] [PubMed] [Google Scholar]
- 71. Sang L., Miller J. J., Corbit K. C., Giles R. H., Brauer M. J., Otto E. A., Baye L. M., Wen X., Scales S. J., Kwong M., Huntzicker E. G., Sfakianos M. K., Sandoval W., Bazan J. F., Kulkarni P., et al. (2011) Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell 145, 513–528 10.1016/j.cell.2011.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Haycraft C. J., Zhang Q., Song B., Jackson W. S., Detloff P. J., Serra R., and Yoder B. K. (2007) Intraflagellar transport is essential for endochondral bone formation. Development 134, 307–316 10.1242/dev.02732 [DOI] [PubMed] [Google Scholar]
- 73. Turner D. L., and Weintraub H. (1994) Expression of achaete-scute homolog 3 in Xenopus embryos converts ectodermal cells to a neural fate. Genes Dev. 8, 1434–1447 10.1101/gad.8.12.1434 [DOI] [PubMed] [Google Scholar]
- 74. Johnson L. V., and Blanks J. C. (1984) Application of acrylamide as an embedding medium in studies of lectin and antibody binding in the vertebrate retina. Curr. Eye Res. 3, 969–974 10.3109/02713688409167215 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.