

Abstract
The mitochondrial, or intrinsic, apoptosis pathway is regulated mainly by members of the B-cell lymphoma 2 (BCL-2) protein family. BCL-2–associated X apoptosis regulator (BAX) plays a pivotal role in the initiation of mitochondria-mediated apoptosis as one of the factors causing mitochondrial outer-membrane permeabilization (MOMP). Of current interest are endogenous BAX ligands that inhibit its MOMP activity. Mitochondrial-derived peptides (MDPs) are a recently identified class of mitochondrial retrograde signaling molecules and are reported to be potent apoptosis inhibitors. Among them, humanin (HN) has been shown to suppress apoptosis by inhibiting BAX translocation to the mitochondrial outer membrane, but the molecular mechanism of this interaction is unknown. Here, using recombinant protein expression, along with light-scattering, CD, and fluorescence spectroscopy, we report that HN and BAX can form fibers together in vitro. Results from negative stain EM experiments suggest that BAX undergoes secondary and tertiary structural rearrangements and incorporates into the fibers, and that its membrane-associating C-terminal helix is important for the fibrillation process. Additionally, HN mutations known to alter its anti-apoptotic activity affect fiber morphology. Our findings reveal for the first time a potential mechanism by which BAX can be sequestered by fibril formation, which can prevent it from initiating MOMP and committing the cell to apoptosis.
Keywords: apoptosis, mitochondrial apoptosis, B-cell lymphoma 2 (Bcl-2) family, conformational change, fibril, Bax, electron microscopy, fibers, humanin, BCL-2-associated X apoptosis regulator (BAX), β-sheet, protein aggregation, fibrillation
Introduction
Apoptosis is a programmed process by which cells can initiate their own death when prompted by some intracellular or extracellular stimuli. The mitochondrial pathway of apoptosis, or the intrinsic pathway, is regulated primarily by members of the B-cell lymphoma 2 (BCL-2)2 protein family. Some of these proteins stimulate the release of apoptotic factors into the cytosol from the mitochondria by causing mitochondrial outer-membrane permeabilization (MOMP) (1, 2). Expression levels and activities of BCL-2 proteins are tightly controlled to maintain cell homeostasis while also remaining primed to affect MOMP. Factors that can enhance or inhibit MOMP via interfering with BCL-2 pathologies are of interest for disease states where apoptosis is misregulated, e.g. cancers, neurological disorders, autoimmune diseases, and cardiovascular conditions (3).
BCL-2 family members are classified by their pro-apoptotic, anti-apoptotic, or signal-sensitizing functions and share regions of sequence homology called BCL-2 homology (BH) domains. The six anti-apoptotic and three pro-apoptotic BCL-2 proteins share up to four BH domains (BH1–4) and adopt similar globular folds: Seven amphipathic α-helices surrounding a single hydrophobic core helix and a ninth C-terminal helix with membrane-associating functions. This all-helical structure forms a characteristic hydrophobic groove along the surface of these proteins that can interact with BH3 domains from other members of the BCL-2 family (4). Eight BH3-only proteins are signaling effectors for initiating apoptosis and are all intrinsically disordered, with the exception of BH3 interacting-domain death agonist (BID) (5).
Subsequent to their activation, two pro-apoptotic BCL-2 proteins, BAX and Bak, congregate at the mitochondrial outer membrane (MOM) and oligomerize, which induces MOMP (6). Both proteins exist in equilibrium between the MOM and cytosol; however, BAX is primarily in the cytosol, whereas Bak is localized to membranes (7). It is well-established that translocation of BAX to the MOM is a prerequisite for apoptosis initiation, and it is considered to be the final, irreversible trigger for apoptosis via the mitochondrial pathway (8). This shift is modulated in several ways. Anti-apoptotic BCL-2 proteins can interact directly with BAX, preventing it from associating with the MOM (9), or they can retrotranslocate BAX from the MOM to the cytosol in an active process (10–12). Additionally, pro-apoptotic BH3-only proteins directly or indirectly regulate the recruitment of BAX to the MOM and membrane permeabilization activity (13–15). These can bind with BAX in the cytosol to affect translocation or they can bind to the MOM first and then recruit BAX to it (16).
Among the multi–BH domain BCL-2 proteins, BAX has some unique properties because of its membrane-associating C-terminal helix (α9). Mutating any of α9's residues affects membrane association and truncating it by as few as five residues abolishes MOM translocation (17). The NMR structure of BAX shows that α9 is sequestered and folds into the BH3-binding groove where it acts as an autoinhibitor (18). This is a major inactive conformation of BAX; however, FRET analysis in mouse embryonic fibroblast cells demonstrates that α9 can also be extended when monomeric BAX is still in the cytosol (19). Helix α9 is also directly implicated in BAX's oligomerization and membrane permeabilization activity. When BAX is translocated to the MOM, the entire protein adopts a completely extended conformation that facilitates intermolecular contacts between BAX proteins at two sites, α9-α9 and BH3-BH3 (19). These observations suggest that BAX's uniquely regulated BH3-binding groove and C-terminal helix are of great significance for its apoptotic function.
The present work was inspired by factors that are naturally produced by the mitochondria to regulate the effects of pro-apoptotic BCL-2 oligomerization on itself. Mitochondrial-derived peptides (MDP) are short proteins encoded by alternate small open reading frames within the region of the mitochondrial genome that is transcribed for the 12S and 16S rRNA subunits of the mitochondrial ribosome (20, 21). MDPs play an essential role in age-related diseases and have been described as cytoprotective, metaboloprotective, and apoptotic regulators (22–25). Humanin (HN), the first described MDP (26), has been reported to have specific interactions with cytosolic BAX, inhibiting MOM translocation and therefore acting as a potent apoptosis antagonist (27). Guo et al. (28) showed that expressing HN in neurons could attenuate apoptosis induced by staurosporine. Conversely, when they knocked down HN expression, cancer cells were sensitized to BAX-induced apoptosis (28). Furthermore, HN was shown to interfere with the oligomerization of BAX and BID, preventing their interaction and associated membrane localization (29–31).
A description of HN's specific mechanism of action against BAX currently does not exist, and the present work was conducted to elucidate the molecular interactions between HN and BAX. Here, using a combination of quantitative spectroscopic methods, negative staining EM, and proteolytic digestion we investigated fibril formation and the concomitant sequestration of structurally reorganized BAX. The fibrillation process was rapid and complex. Both molecules are required for significant aggregation to occur, and mature fibrils could be isolated from the reactions. We showed that BAX is structurally part of the fiber lattice after undergoing secondary and tertiary structure conformational changes and that a portion of the protein is protected from digestion by trypsin.
We sought to further elucidate the interactions of HN with BAX by observing the effects of BAX mutations on formation of the fibers. Two BAX variants, namely the helix α9 deletion (BAX ΔC) and the S184V mutants, were assessed for their HN fibril formation abilities. BAX with increased helix α9 exposure from the S184V mutation formed fiber more readily than WT. Counterintuitively, the α9 deletion mutation, exposing the major BH3-binding groove, also showed an increase in fiber formation propensity relative to WT BAX, albeit to a lesser degree than the S184V mutant. Taken together, those results allowed us to propose a novel BAX rearrangement mechanism involving both the helix α9 and the major BH3-binding groove upon HN binding, all while exposing the N terminus. Our finding reveals for the first time a novel mechanism for BAX inhibition by sequestration into a stable fibril structure.
Results
BAX together with WT humanin forms fibers
The interaction between BAX and HN was first interrogated using fluorescence spectroscopy. Scattering of the excitation beam reports on the aggregation propensity of the protein and peptide mixture, whereas the emission spectra provide information on potential conformational changes associated with their interaction. Addition of HN into a solution of BAX causes enhanced light scattering at a wavelength of 280 nm (Fig. 1a). Individual data points are available in Table S1. It is worth noting that this scattering could be observed in a matter of seconds after the peptide was introduced to BAX. Increased scattering was not observed when HN was added into buffer alone or buffer was titrated into BAX. The scattering plot as a function of HN concentration shows significant aggregation formation between HN and BAX that continues to form beyond the equal molar ratio of the two. This hints at an aggregation mechanism that involves a single molecule of BAX and multiple HN peptides. A shift from 340 nm to 332 nm was observed in the maximum fluorescence emission spectra of BAX upon addition of HN, indicating a conformational change in BAX (Fig. S1).
Figure 1.

Formation of HN aggregations, EM imaging of novel fibers, and secondary structure characterization. A, increase of scattered photons from a 280 nm laser caused by aggregations in solution as a function of HN concentration in the presence (circle) or absence (square) of 5 μm BAX. Error bars were calculated from the S.D. of three replicate titrations. Control titrations of buffer alone into BAX did not show increased light scattering. B, EM images of aggregations revealed multifibrous superstructures with a background of single fibers. The contrast in this image was enhanced with 0.5% pixel saturation by histogram stretching. C, EM of centrifuge-purified fibers. The expanded view shows fibers stacked against each other forming bundles with some individual fibers associating with multiple bundles. Fibers can also appear alone or associated in nonparallel configurations but are not branching. This image is unaltered from the original data collection. D, CD spectra of centrifuge purified fibers and control samples. Data are displayed with multiple y-axes as described in “Experimental procedures” and spectra are color-coordinated with their axes. Spectra were produced from three accumulated scans on a single sample. Fibers separated from a solution of 50 μm BAX and 500 μm HN (solid blue line) produced a spectrum with a single valley at 222 nm indicating the presence of predominantly anti-parallel β-sheets. A control spectrum of 25 μm BAX (solid red line) yielded two minima showing the globular α-helical BAX structure; the 50 μm HN spectrum (dashed blue line) reflected disordered conformation.
EM was used to probe the morphology of these aggregates directly. The aggregates were imaged as fibers and came in two major conformations: Single fibers and bundles (Fig. 1b). The latter contained multiple fibers organized in parallel and appeared to be highly ordered. These fibers were not observed on EM grids prepared from solutions containing BAX or HN alone. The fiber bundles could be separated from single fibers by centrifugation. Separation allowed imaging of the bundled fibers in much greater detail (Fig. 1c). A statistical analysis of single fibers was performed to describe the average fiber dimensions. Images of the fibers were isolated from the background using difference of Gaussian band-pass filtering (32), and their dimensions were measured automatically using the ridge detection plugin for ImageJ/FIJI (33) (Fig. S2). These fibers showed a relatively narrow distribution in width (Fig. S2e), and the length ranged from 10 to 250 nm with a maximum distribution around 40 nm (Fig. S2f). The average bundled fiber, however, is clearly much longer.
Conformational changes in BAX upon fibrillation with HN were further probed using circular dichroism (CD) (Fig. 1d). The CD spectrum of BAX alone is representative of α helices with minima at 209 nm and 222 nm. A resuspension of purified fibers produces a CD spectrum with one minimum at 222 nm, characteristic of β-sheet structure (34). These results demonstrate that HN induces structural rearrangements in BAX effecting a significant loss of its α-helical structure and formation of β-sheet upon fibrillation. Additionally, the CD spectrum of HN peptide alone is characteristic of disordered protein, demonstrating that the protein and peptide are together required for β-sheet fibrillation. Their interaction instigates mutual conformational changes that result in fibers.
To investigate this phenomenon under ionizing conditions, we undertook a salt concentration study. Light scattering titrations in the presence of NaCl revealed a significant increase in the amount of scattered light over the standard reaction conditions (Fig. S3a). Fibers were centrifuge purified as before; however, EM images of fibers purified from a no-salt reaction versus a salt-containing reaction were quite different. The same bundled structures were observed in the reaction with no salt (Fig. S3b). Fibers from the salt-containing reaction produced only short fibers. There was no sign of the bundled structures and some fibers could be seen only loosely associating with each other (Fig. S3c). Taken together, the observation that salt inhibits the formation of parallel bundles but also increases light scattering suggests that the BAX/HN interaction is enhanced by ionic conditions and that interactions between individual fibers might be mediated by electrostatic interactions.
Fibers structure is altered by functionally important humanin mutants
The fiber formation kinetics for two HN mutants were evaluated: C8A HN, showing reduced apoptotic inhibition; and S14G HN, exhibiting increased protective properties (35). Light scattering experiments were conducted for the mutants and compared with the WT (Fig. S4). A peptide derived from the BAX-binding domain of viral mitochondria-localized inhibitor of apoptosis (vMIA) was used as a control (36). There was no statistically relevant variation in the titration scattering profile among the three HN peptides at these concentrations, and vMIA does not cause any increase in light scattering beyond forming a complex with BAX.
Both mutants present variation in fiber structure versus the WT (Fig. 2a–f). The C8A mutant has a dense network with branching nodes of globular structures connecting fibers instead of the parallel bundles seen in the WT (Fig. 2c). In addition, the fibers are nonuniform in their diameter and can appear thicker or thinner than the WT (Fig. 2d). The S14G mutant produces similar structure to the WT, but there are fewer parallel bundles (Fig. 2e). The fibers are also generally thicker than WT fibers. Bundled S14G fibers have a smaller diameter than fibers that are unassociated with other fibers. Single fibers could be found with diameters ranging from 15–50 nm (Fig. 2f). No fibril structures were observed in samples containing vMIA (data not shown). It appears that sequence specific interactions between the peptide and BAX are required for fiber formation, and their modification can alter fiber morphologies.
Figure 2.

Fiber formation of mutant peptides. A and B, control samples of the WT network (A) and single WT fibers (B) are imaged as before. C, the C8A mutant produced shorter, more irregularly shaped fibers and showed clear evidence of branching. Their diameters are not uniform, and the fibers form into a dense network. D, the C8A mutant cannot form into single fibers. There is always some branching or fibers are attached by globular nodes. E, the S14G mutant has similar networking properties to the WT, except there are fewer fibers running in parallel. F, single S14G fibers can appear as seen in the WT, but their diameters are larger on average ranging from 15 to 50 nm.
BAX helix α9 propagates fibrillation
To further investigate the role of BAX in fiber formation we used two BAX mutants, namely, BAX ΔC and BAX S184V. These mutations are highlighted on the Bax structure (Fig. 3a). In the first case, helix α9 was deleted (after residue Gln-171) to expose the major BH3-binding groove (18). In the second case, a serine-to-valine substitution in position 184 (a phosphorylation site that regulates MOMP activity) enhances helix α9 hydrophobicity, hence increasing α9 flexibility and MOM association (17). Light scattering experiments were performed by titrations of WT HN peptide into WT BAX and each BAX mutant. Both BAX mutants featured enhanced fiber formation rates over WT BAX (Fig. 3b). The individual data points for this graph are available in Tables S2A–S2C. BAX ΔC only has a slight rate enhancement over WT BAX during the first part of the titration. After an excess amount of peptide has been titrated, this mixture results in comparable scattering to the WT BAX. Conversely, BAX S184V shows a significantly enhanced rate of fibrillation over the WT. It reaches an end point plateau early in the titrations with a scattering value close to the end points for the full WT and ΔC titrations. This indicates that exposure of helix α9 can accelerate fibrillation. Because BAX ΔC does not inhibit fiber formation, in fact, it is comparably better than the WT BAX, this indicates that the helix α9 is not a sole determinant for fiber formation. Exposing the BH3 groove or simply making the protein less stable overall may facilitate fiber formation.
Figure 3.
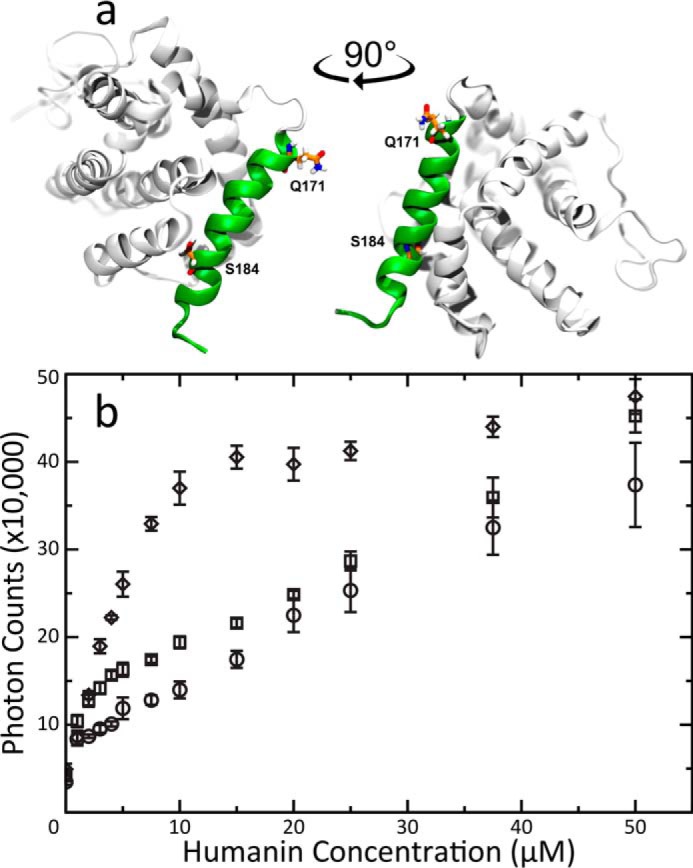
Solvent-exposed α9 propagates fiber formation. A, The NMR structure of BAX (PDB 1F16) highlighting the changes made by mutations. Residues Gln-171 and Ser-184 are rendered in sticks. Gln-171 is the final residue in the C-terminal truncated mutant. Helix 9 is rendered in green. BAX models were created using Visual Molecular Dynamics (68). B, Light scattering titrations of HN into 5 μM solutions of either WT (circle), BAX ΔC (square) or S184V (diamond) mutant BAX. Error bars were calculated from the standard deviation of three replicate titrations. BAX ΔC fibrillation has a slight rate enhancement during the sub-stoichiometric phase of the titration but eventually averages into the WT BAX curve. In contrast BAX S184V, with its flexible α9 helix, shows a greatly enhanced fiber formation rate.
BAX is incorporated into the fibers
To determine whether there is a segment of BAX being incorporated into the fiber core, we performed a trypsin fragmentation assay on the fibers and with BAX alone as a control. After fibrillation was allowed to proceed for 1 h, trypsin was added to the reactions and fragmentation products were analyzed after 2 h of digestion. The total ion chromatogram of a control digestion with BAX alone shows several peaks that could be assigned to six fragments in the N-terminal half of BAX (Fig. 4a and Fig. S5). Fiber samples lose three of those six BAX fragments, the three most C-terminal of them (Fig. 4d). A typical mass spectrum for an unprotected fragment in the BAX control sample is shown (Fig. 4b). An additional peak (no. 12) appears in the fiber digestion that correlates to a fragment spanning residues 10–190 of BAX (Fig. 4, c and d). This is nearly the entire protein minus nine N-terminal and two C-terminal residues. Deconvoluted mass spectra for each peak showing fragment ion masses are available in Fig. S5. A summary of these data relating the observed and theoretical ions to the expected molecular weight is available in Table S3. The full-length HN peptide and five fragments were also detected (Fig. S6). This digestion pattern implies that nearly all of BAX is protected by the fiber to some degree and that some portion of the N terminus remains exposed. However, it isn't clear from these early digestion experiments what the exact conformation of BAX is within the fibers.
Figure 4.

Fibers protect BAX from proteolytic digestion by trypsin. A, total ion chromatograms for BAX digested with trypsin (above) and for the fiber digested under the same conditions (below). The fibers formed protect some part of BAX from digestion as evidenced by several peaks missing with the fiber samples. B, the deconvoluted MS for Peak 4 showing a typical mass intensity for a fragment that only appears in the control sample. C, the deconvoluted MS for Peak 12 showing the correct mass for a segment of BAX that is incorporated into the fiber. D, fragments identified in BAX-only control digestions (green) and digestions with fibers (blue) overlaid with the sequence of WT BAX. Residues labeled red precede trypsin cleavage sites. In the control digestions the N-terminal half of BAX can be detected in six fragments. When a fiber sample is digested, the three fragments closer to the C-terminal end of the protein are no longer observed and a new peak appears corresponding to a fragment that is nearly the full length of the protein.
The differences in fiber morphology among HN mutants and tryptic digestion patterns of BAX mixed with HN, along with varying fibrillation kinetic profiles of BAX mutants, suggest that BAX plays an integral role in formation of the fiber structure. To confirm that BAX is indeed tightly incorporated into the fibers rather than just acting as a nucleation site for HN fibrillation, an mAb (2D2) specific to the N-terminal region (residues 3–16) of BAX and a gold-conjugated secondary antibody were used. BAX was preincubated with the primary antibody for 1 h before HN was added to induce fibrillation. After an additional hour, the secondary antibody was added, and samples were incubated for one more hour. After centrifugation, EM samples were taken from the supernatant. Isolated fibers could be observed that were decorated with gold nanoparticles (Fig. 5). These gold particles are distributed along the fibers rather than concentrated at the ends of them, indicating sequestration of BAX within the fibers rather than a nucleating event. No conclusion could be drawn based on the spatial arrangement of these gold particles, because only a fraction of BAX are labeled in this experiment. It is also interesting to note, because 2D2-bound BAX still reacts to form fiber, this confirms that the N-terminal region of BAX is exposed outside of the fiber.
Figure 5.

BAX-specific antibody labeling of fibers. Antibody labeling of Bax/HN fibers with a BAX-specific antibody (2D2) targeted to the protein's N terminus. Secondary antibodies conjugated to 12 nm gold particles indicate points along the fibers where Bax has been integrated into them. EM images illustrate examples of BAX sequestration in networked bundles (left) and single fibers (right). These pictures were unaltered from the original data collection.
Discussion
There is a growing body of work describing small molecules that can activate BAX. Some of them target hot spots within the protein's BH3-binding sites. A screen focused on the major BH3 groove identified three small molecule BAX activators that inhibit Ser-184 phosphorylation by Atk, a known BAX-inactivating posttranslational modification (37, 38). Other screens have focused on the N-terminal trigger site, a small BH3-interacting pocket formed by helices α1 and α6 that has been described as a site for allosteric BAX activation and stimulating MOM recruitment (39, 40). A lead compound was found to activate BAX-mediated MOMP and oligomerization activity (41). Other compounds have been discovered that target the multisite surface region proximal to the N-terminal trigger site that is ripe for allosterically regulating BAX (42, 43). Many of these compounds' proposed interaction sites overlap with the BAX-binding site for vMIA, a cytomegalovirus protein that ensures host cell survival and replication by specifically inhibiting a semi-activated form of BAX at the MOM (36, 44). The overlap of this site with ligands that induce opposing BAX functions suggests that this multisite surface area might be a region of structural susceptibility that is reinforced by vMIA but destabilized by BAX-activating compounds (43).
Inhibitors of BAX MOMP activity are arguably just as important as activators but are identified less frequently. A screen using a vesicle release assay found small molecule BAX inhibitors that are believed to stabilize hydrophobic interactions of the core helix which inhibits BH3 triggering of the N-terminal site (45). Other compounds have been identified that disrupt BAX oligomerization at the MOM and were found to protect neurons from BAX-induced apoptosis (46). Despite confirmation of the existence of these BAX-interacting small molecules, no structures of BAX ligated with any of them have been published, presumably because of the protein's tendency to aggregate. Therefore, the molecular mechanisms of these compounds are poorly understood. Another promising avenue to study mechanisms for BAX inhibition is using peptides. The structure of BAX complexed with a peptide derived from the cytomegalovirus protein vMIA has been determined by NMR (36). Additionally, peptides derived from Ku70, a protein involved in DNA double-strand break repair, were found to suppress BAX by inhibiting MOM translocation in a process independent of its repair functions (47, 48).
Here we have described an unexpected and alternative regulation of BAX by fiber formation with one of its known endogenous peptide inhibitors, HN. The fibrillation process is rapid and complex and involves a significant structural rearrangement of BAX. BAX with increased helix α9 exposure from the S184V mutation forms fibers more readily than WT. Counterintuitively, our mutation that removes α9, thus exposing the major BH3-binding groove, also shows an increase in fiber formation propensity relative to WT BAX, albeit less than the S184V mutant. The differential protease digestion profiles between BAX and BAX in the fiber with HN confirms the BAX conformational change associated with fiber formation as indicated by the shift in its fluorescence emission and CD spectra. We showed by EM that the morphology of the fibers formed between BAX and different HN variants is distinct. These results clearly show that the conformations of BAX as it is incorporated into these fibers are not alike.
HN is also known to be conformationally dynamic, and these changes have important implications for the fibrillation process with BAX. CD and NMR spectroscopy have been used to investigate HN secondary structure propensity in various conditions. Multiple reports describe HN as generally disordered in aqueous solutions but gaining α-helical structure in lipophilic environments. Conformational dynamics between the disordered and α-helical states can change with HN mutations (49–58). Some β-strand and β-sheet forming activity has also been described for HN with the C8A mutation disrupting this process, but C8A HN still forms disordered aggregations that are reversible with the monomer state (59). WT HN and some mutant peptides substituted with d-serine at position 14 were shown to form β-sheet fibers as characterized by CD- and thioflavin T–binding assays (60). Additionally, anti-parallel β-sheet structures have been reported when the peptide is interacting with membrane models and vesicles (61–63), and some structures have been proposed for the interactions between HN and β-amyloid fibers (64–66). HN could be modified to take advantage of its intrinsic β-sheet activity in a variety of ways. Mutations or incorporation of nonnatural amino acids might increase fibrillation propensity with BAX, increase the peptide's half-life in the cytosol, or localize it to a specific cellular compartment, bearing in mind that these changes must not alter the intercellular and intracellular trafficking of HN.
HN has been described interacting with BID in an α-helical state by NMR. This was achieved using a truncated mutant of the peptide that does not form aggregations when titrated with BID (30). The mutant HN was reported to be interacting with BID with an α-helical conformation in the hydrophobic groove analogous to the BH3-binding groove in BAX. This was proposed to be a potential mechanism by which HN disrupts BID-BH3 interactions with other BCL-2 proteins. Given our data concerning the kinetics of fiber formation related to α9 flexibility and BH3 groove exposure, it is likely that WT HN engages BAX in a similar manner to BID at the major BH3 groove. We further propose a hypothesis that the full-length WT HN induces a mutual conformational change between the two molecules after first engaging BAX in its BH3-binding groove, which results in the fibers described herein.
Considering these observations together, we propose a fibrillation mechanism where BAX and HN initially interact in a way that is mediated by α9 and the major BH3 groove. This interaction disrupts the hydrophobic core of BAX as it unfolds into an extended conformation that is incorporated into the growing fiber with the N terminus of BAX exposed outside of it. We cannot rule out the possibility of a molten globule transition state. Because of the rapid rate of fiber formation, however, studying these intermediate forms of the BAX and HN aggregates is quite challenging. Taking into account the CD spectrum, kinetics of aggregation, and protease protection, it appears that some portion of the BAX C-terminal region switches into β-strand structure and interdigitates with HN peptides in anti-parallel β-sheets to form the mature fibers. We further hypothesize that modulation of the anti-apoptotic activities of HN variants is related to the consequences of different BAX conformations within the fibers. One possible consequence is increasing the overall stability of BAX sequestration within the fiber which might be enhanced depending on its conformation in the fiber. The other consequence is the rate of incorporation of BAX into the fiber. For instance, the S14G mutant of HN, which has higher protective effect, either can hold BAX with much higher stability in the fiber or can incorporate a lot more BAX into the fiber, whereas the C8A mutant of HN, with its attenuated protective abilities, cannot sequester BAX at the same rates or as stably as the WT peptide.
In conclusion, here we show that BAX and HN can interact to form fibers. There are some hints of molecular details of this interaction such as the involvement of helix α9, BH3 groove, or the exposed N-terminal of BAX, and we propose that BAX is fully incorporated into the fiber in an extended conformation. This process represents a novel structural mechanism for BAX inhibition by sequestration into the fibril superstructure which effectively prevents BAX from being activated to initiate MOMP and apoptosis. This alternative inhibition mechanism offers a new concept for therapeutic strategies that target BCL-2 proteins and regulate apoptosis in a diverse range of diseases.
Experimental procedures
BAX expression and purification
Protein expression was performed as described previously (67). Escherichia coli cell pellets were resuspended and homogenized in TEN buffer (0.1 m Tris-Cl, pH 8.0, 10 mm EDTA, and 1.0 mm NaCl) with Benzonase® Nuclease (Merck KGaA, Darmstadt, Germany) and cOmpleteTM Protease Inhibitor mixture (Roche). Insoluble material was pelleted by ultracentrifugation for 40 min at 185,500 × g and 4 °C. A 20-ml chitin column was prepared by washing the resin (New England Biolabs, Ipswich, MA) with 10 column volumes (CV) of water and then equilibrating with 15 CV of TEN buffer. The supernatant was passed over the column, immobilizing BAX. Self-cleavage of the intein inserted between BAX and its chitin binding protein fusion partner was induced by percolating a solution of TEN buffer with 50 mm DTT into the resin and allowing the resin to incubate in this solution for about 40 h at 4 °C. After cleavage, the BAX/DTT-containing eluate was collected and buffer exchanged into 20 mm Tris, pH 8.0, using a PD-10 column (GE Healthcare). The protein was further purified by ion-exchange chromatography using a 5 ml HiTrap® 5 Q HP column (GE Healthcare) with a linear anion salt gradient up to 800 mm NaCl over 20 CV. The BAX-containing fractions were pooled, concentrated, and purified by size exclusion chromatography (Sephadex G-75, GE Healthcare) using 20 mm sodium acetate pH 6.3, 150 mm NaCl, 1 mm DTT, and 0.1 mm EDTA buffer. BAX-containing fractions were pooled, concentrated, and exchanged into storage buffer (20 mm sodium acetate, pH 6.3, and 0.1% sodium azide) by filter centrifugation. Purified protein was stored at −20 or −80 °C.
Engineering expression plasmids for BAX mutants
The serine in position 184 of hBAX was mutated to a valine using a QuikChange® II XL Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) and two custom primers: 5′-GGA GTG CTC ACC GCC GTG CTC ACC ATC TGG AA-3′ and its reverse complement strand (Eurofins Genomics, Louisville, KY). The cDNA for hBAX in a pTYB1 vector (New England Biolabs, Ipswich, MA) was used as a template. The entire coding region of the fusion protein was verified by sequencing (Macrogen USA, Rockville, MD). The 1–171 hBAX mutant was produced by deletion using a Q5 Site-Directed Mutagenesis Kit (New England Biolabs) and two custom primers 5′-CTG CCA CGT GGG CGT CCC-3′ and 5′-TGC TTT GCC AAG GGT ACC AAT GTT TTA ATG G-3′ (Eurofins Genomics), hybridizing to the redesigned hBAX C terminus DNA sequence and the N terminus sequence of the intein tag, respectively. The reliability of the entire coding sequence was verified by sequencing (Macrogen USA).
Fiber formation and purification
Solid HN peptides (Anaspec, Fremont, CA) were predissolved in reaction buffer (20 mm sodium acetate, pH 6.3) to 1 mm stock. Fibers were produced by preparing solutions of BAX and HN in the reaction buffer. Fibers to be purified for EM were made by combining 5 μm BAX with 50 μm HN in the reaction buffer in a volume of 100 μl and allowing the reaction to proceed at room temperature for 1 h. After the incubation period, fibers were pelleted by centrifugation for 15 min at 21,000 × g and room temperature. The pellets were further purified by successive washes in 1 ml reaction buffer and then 1 ml of ultrapure water. Pelleted fibers were resuspended a final time in 100 μl of water, and this final solution was used to prepare samples for EM.
Light scattering and fluorescence spectroscopy
A QuantaMasterTM 8000 Series spectrofluorometer (PTI, Birmingham, NJ) with an excitation wavelength of 280 nm and a vertical polarizer was used. Emission wavelengths were recorded for 340 nm at 54.70 ° and 280 nm with a vertical polarizer. All slit widths were set to 1 mm. For emission scanning experiments, the emission wavelengths were recorded from 290 to 470 nm. For titration experiments, the number of photons at each titration point was averaged from continuous measurement over 1 min at a rate of one measurement per second using the PTI FelixGX software (Horiba, Kyoto, Japan). All titrations were carried out, in triplicate, at 24 °C in quartz cells with a reaction volume of 200 μl. Solutions containing 5 μm BAX were titrated with 1 mm HN up to a final HN concentration of 50 μm in a buffer of 20 mm sodium acetate, pH 6.3, at 24 °C, a 5% volume increase. For the titrations with mutant proteins in Fig. 3b, each BAX stock solution was ultracentrifuged at 69,500 × g for 45 min to remove any potential aggregates immediately before performing titrations.
Circular dichroism
CD spectra were recorded from 200 nm to 260 nm on a Jasco J-715 spectropolarimeter (Jasco Analytical Instruments, Easton, MD) equipped with a Jasco PTC-348WI temperature controller. Samples were measured in volumes of 200 μl using a 1 mm cuvette at 24 °C. Each experiment is reported as an average of three accumulated scans and spectra were baseline corrected by subtraction of a spectrum collected on the reaction buffer. Scans were collected in continuous mode at a rate of 50 nm per min with 1 nm bandwidth. For this experiment a highly concentrated solution of the fibers was required to measure a spectrum with reasonable signal to noise. To achieve this, fibers were purified as described in “Fiber formation and purification,” but a fibrillation reaction with 10 times the amount of starting material was used containing 50 μm of BAX and 500 μm of HN. The concentrations of control experiments with 25 μm BAX or 50 μm HN only were chosen to accurately report on their CD spectra with good signal to noise. Data were plotted concurrently with two y-axes to accommodate the ellipticity differences between the two control samples.
Negative stain electron microscopy
Images were collected on a JEOL JEM-1200EX Transmission Electron Microscope (accelerating voltage 80 keV) (JEOL Ltd., Akishima, Tokyo, Japan) equipped with an AMT XR-60 digital camera (Advanced Microscopy Techniques, Woburn, MA). Uranyl formate (UF) applied as a 1% solution at pH 4.5 was used as a contrasting agent. The UF solution was prepared by dissolving UF salt to 1% w/v in boiling water and then titrating in 5 m NaOH until the yellow color deepened and the final pH measured about 4.5 by litmus test. Holey Formvar EM grids (Electron Microscopy Sciences, Hatfield, PA) were ionized by glow discharge (Pelco easiGlowTM, Ted Pella Inc., Redding, CA) for 30 s and 5 μl of fiber solution, prepared as described in “Fiber formation and purification,” was allowed to adhere to the grid surface for 2 min. Grids were prepared with 5 μl of 1:50 or 1:100 dilutions of the purified fiber solutions. Excess liquid was removed using the side blot method, and the grids were washed by passing them through three drops of water and reblotted. 5 μl of 1% UF was applied for 2 min and blotted away; the staining was then repeated a second time. After the final blot, the grids were allowed to dry overnight resting on filter paper in the dark.
For the secondary antibody gold-labeling experiments, BAX was diluted into the 2D2 primary antibody solution (ab77566, Abcam, Cambridge, MA) to a final concentration of 5 μm with a volume of 100 μl. The BAX/antibody interaction was then allowed to preincubate at room temperature for 2 h with gentle agitation. 50 μm HN was added and fibrillation was allowed to proceed for 1 h. The reaction was centrifuged to remove large aggregations and gelatin from the antibody solution. The 12 nm gold-conjugated secondary antibody (ab105286, Abcam) was diluted 1:5 in water and 100 μl of this was added to the primary antibody-labeled fibers. This secondary antibody labeling was allowed to proceed at room temperature for 1 h with gentle agitation. The reaction was centrifuged a final time and dilutions of the supernatant were applied to EM grids as described previously.
Analysis of fiber dimensions
EM images were first processed using the GDSC difference of Gaussian band-pass filter plugin for ImageJ with σ1 and σ2 values of 20 and 10 pixels, respectively. This produced a black background with the fibers highlighted in white. Following this, the ridge detection plugin was used to automatically detect and measure the fibers using a σ value of 7 pixels. The data were manually trimmed to remove data points from fibers that were too overlapped to resolve their path or if they intersected with the edge of a frame. 281 fibers in total were measured to produce the distribution histograms. Sample images from the dataset are provided in Fig. S2.
It is important to note that ridge detection algorithm used in the analysis consistently identified the extreme inside edge of the fibers, ignoring the dark-stained area. This resulted in underestimation of the measurements by roughly 4 nm compared with measurements taken manually.
Trypsin digestions
Fibrillation reactions were prepared in a 20-mm sodium acetate buffer at pH 6.3 with 20 μl reaction volumes using 20 μm of BAX and 200 μm HN. Fibrillation was allowed to proceed for 1 h at room temperature with gentle agitation. After this, 1 μl of trypsin (Trypsin Gold MS Grade, Promega, Madison, WI) was added to a final concentration of 50 ng/μl. Digestions were incubated for 2 h at 37 °C in a 300-rpm shaking incubator and were quenched by addition of 2.2 μl of 10% TFA.
HPLC-mass spectrometry
To create samples for peptide separation and mass determinations, 10 μl of the quenched digestion was combined with 40 μl of a mixture of 5% acetonitrile and 0.05% TFA in water. 5 μl of the sample was injected into an Agilent 1260 series HPLC system equipped with an autosampler maintained to 4 °C and a capillary pump system. Peptides were eluted from a reverse phase C18 HPLC column (Zorbax 300SB-C18, 1.0 × 50 mm, 3.5 μm particle size, Agilent Technologies, Wilmington, DE) with a flow rate of 20 μl per minute at a gradient profile of 0 to 50% acetonitrile at 1% per minute. Electrospray MS was performed on an Agilent 6530C accurate mass quadrupole-TOF system (Agilent Technologies) equipped with double electrospray ionization (ESI) source and Q-TOF analyzer. Mass spectra were obtained at positive polarity in the range of 100 to 2500 m/z. Mass spectra were analyzed using the Agilent software MassHunter version B.06 and matched to fragment digestions predicted using GPMAW (version 12, Lighthouse Data, Odense, Denmark).
Author contributions
D. L. M., D. W. K., and S. J. formal analysis; D. L. M. validation; D. L. M., D. W. K., S. J., M.-P. S., Y. H., C. K. E. B., and D.-Y. L. investigation; D. L. M., D. W. K., and S. J. visualization; D. L. M. and D. W. K. methodology; D. L. M. and M.-P. S. writing-original draft; N. T. conceptualization; N. T. resources; N. T. supervision; N. T. project administration; N. T. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Drs. Grzegorz Piszczek and Di Wu of the Biophysics Core facility at NHLBI for advice and expertise regarding light scattering, fluorescence, and CD. We appreciate the advice and help of Drs. Jennifer Lee and Ryan McGlinchey throughout the project for fiber expertise. Dr. Jiansen Jiang provided invaluable advice on preparation of samples for EM. Finally, we thank Dr. Christian Evenhuis for advice setting up ridge detection analysis of the fibers.
This work was supported by NHLBI Intramural Research Programs, National Institutes of Health (to N. T.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S6 and Tables S1–S3.
- BCL-2
- B-cell lymphoma 2
- BAX
- BCL-2–associated X apoptosis regulator
- BH
- BCL-2 homology
- BID
- BH3 interacting-domain death agonist
- CV
- column volume
- HN
- humanin
- MDP
- mitochondrial-derived peptide
- MOM
- mitochondrial outer membrane
- MOMP
- MOM permeabilization
- UF
- uranyl formate
- vMIA
- viral mitochondria-localized inhibitor of apoptosis.
References
- 1. Edlich F. (2018) BCL-2 proteins and apoptosis: Recent insights and unknowns. Biochem. Biophys. Res. Commun. 500, 26–34 10.1016/j.bbrc.2017.06.190 [DOI] [PubMed] [Google Scholar]
- 2. Bilinovich S. M., Davis C. M., Morris D. L., Ray L. A., Prokop J. W., Buchan G. J., and Leeper T. C. (2014) The C-terminal domain of SRA1p has a fold more similar to PRP18 than to an RRM and does not directly bind to the SRA1 RNA STR7 region. J. Mol. Biol. 426, 1753–1765 10.1016/j.jmb.2014.01.007 [DOI] [PubMed] [Google Scholar]
- 3. Czabotar P. E., Lessene G., Strasser A., and Adams J. M. (2014) Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 15, 49–63 10.1038/nrm3722 [DOI] [PubMed] [Google Scholar]
- 4. Youle R. J., and Strasser A. (2008) The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59 10.1038/nrm2308 [DOI] [PubMed] [Google Scholar]
- 5. Hinds M. G., Smits C., Fredericks-Short R., Risk J. M., Bailey M., Huang D. C. S., and Day C. L. (2007) Bim, Bad and Bmf: Intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Differ. 14, 128–136 10.1038/sj.cdd.4401934 [DOI] [PubMed] [Google Scholar]
- 6. Nechushtan A., Smith C. L., Lamensdorf I., Yoon S. H., and Youle R. J. (2001) Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J. Cell Biol. 153, 1265–1276 10.1083/jcb.153.6.1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schellenberg B., Wang P., Keeble J. A., Rodriguez-Enriquez R., Walker S., Owens T. W., Foster F., Tanianis-Hughes J., Brennan K., Streuli C. H., and Gilmore A. P. (2013) Bax exists in a dynamic equilibrium between the cytosol and mitochondria to control apoptotic priming. Mol. Cell. 49, 959–971 10.1016/j.molcel.2012.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wei M. C., Zong W. X., Cheng E. H., Lindsten T., Panoutsakopoulou V., Ross A. J., Roth K. A., MacGregor G. R., Thompson C. B., and Korsmeyer S. J. (2001) Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730 10.1126/science.1059108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fletcher J. I., Meusburger S., Hawkins C. J., Riglar D. T., Lee E. F., Fairlie W. D., Huang D. C. S., and Adams J. M. (2008) Apoptosis is triggered when prosurvival Bcl-2 proteins cannot restrain Bax. Proc. Natl. Acad. Sci. 105, 18081–18087 10.1073/pnas.0808691105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Edlich F., Banerjee S., Suzuki M., Cleland M. M., Arnoult D., Wang C., Neutzner A., Tjandra N., and Youle R. J. (2011) Bcl-xL retrotranslocates Bax from the mitochondria into the cytosol. Cell 145, 104–116 10.1016/j.cell.2011.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Todt F., Cakir Z., Reichenbach F., Youle R. J., and Edlich F. (2013) The C-terminal helix of Bcl-xL mediates Bax retrotranslocation from the mitochondria. Cell Death Differ. 20, 333–342 10.1038/cdd.2012.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Todt F., Cakir Z., Reichenbach F., Emschermann F., Lauterwasser J., Kaiser A., Ichim G., Tait S. W., Frank S., Langer H. F., and Edlich F. (2015) Differential retrotranslocation of mitochondrial Bax and Bak. EMBO J. 34, 67–80 10.15252/embj.201488806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ren D., Tu H. C., Kim H., Wang G. X., Bean G. R., Takeuchi O., Jeffers J. R., Zambetti G. P., Hsieh J. J. D., and Cheng E. H. Y. (2010) BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science 330, 1390–1393 10.1126/science.1190217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim H., Tu H. C., Ren D., Takeuchi O., Jeffers J. R., Zambetti G. P., Hsieh J. J. D., and Cheng E. H. Y. (2009) Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol. Cell. 36, 487–499 10.1016/j.molcel.2009.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuwana T., Bouchier-Hayes L., Chipuk J. E., Bonzon C., Sullivan B. A., Green D. R., and Newmeyer D. D. (2005) BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell. 17, 525–535 10.1016/j.molcel.2005.02.003 [DOI] [PubMed] [Google Scholar]
- 16. Lovell J. F., Billen L. P., Bindner S., Shamas-Din A., Fradin C., Leber B., and Andrews D. W. (2008) Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135, 1074–1084 10.1016/j.cell.2008.11.010 [DOI] [PubMed] [Google Scholar]
- 17. Nechushtan A., Smith C. L., Hsu Y. T., and Youle R. J. (1999) Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J. 18, 2330–2341 10.1093/emboj/18.9.2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suzuki M., Youle R. J., and Tjandra N. (2000) Structure of Bax: Coregulation of dimer formation and intracellular localization. Cell 103, 645–654 10.1016/S0092-8674(00)00167-7 [DOI] [PubMed] [Google Scholar]
- 19. Gahl R. F., He Y., Yu S., and Tjandra N. (2014) Conformational rearrangements in the pro-apoptotic protein, bax, as it inserts into mitochondria: A cellular death switch. J. Biol. Chem. 289, 32871–32882 10.1074/jbc.M114.593897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chu Q., Ma J., and Saghatelian A. (2015) Identification and characterization of sORF-encoded polypeptides. Crit. Rev. Biochem. Mol. Biol. 50, 134–141 10.3109/10409238.2015.1016215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ma J., Diedrich J. K., Jungreis I., Donaldson C., Vaughan J., Kellis M., Yates J. R. 3rd, and Saghatelian A. (2016) Improved identification and analysis of small open reading frame encoded polypeptides. Anal. Chem. 88, 3967–3975 10.1021/acs.analchem.6b00191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee C., Kim K. H., and Cohen P. (2016) MOTS-c: A novel mitochondrial-derived peptide regulating muscle and fat metabolism. Free Radic. Biol. Med. 100, 182–187 10.1016/j.freeradbiomed.2016.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zarse K., and Ristow M. (2015) A mitochondrially encoded hormone ameliorates obesity and insulin resistance. Cell Metab. 21, 355–356 10.1016/j.cmet.2015.02.013 [DOI] [PubMed] [Google Scholar]
- 24. Zapała B., Kaczyñski Ł., Kieć-Wilk B., Staszel T., Knapp A., Thoresen G. H., Wybrañska I., and Dembiñska-Kieć A. (2010) Humanins, the neuroprotective and cytoprotective peptides with antiapoptotic and anti-inflammatory properties. Pharmacol. Rep. 62, 767–777 10.1016/S1734-1140(10)70337-6 [DOI] [PubMed] [Google Scholar]
- 25. Gong Z., Tas E., and Muzumdar R. (2014) Humanin and age-related diseases: A new link? Front. Endocrinol. (Lausanne) 5, 210 10.3389/fendo.2014.00210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hashimoto Y., Ito Y., Niikura T., Shao Z., Hata M., Oyama F., and Nishimoto I. (2001) Mechanisms of neuroprotection by a novel rescue factor humanin from Swedish mutant amyloid precursor protein. Biochem. Biophys. Res. Commun. 283, 460–468 10.1006/bbrc.2001.4765 [DOI] [PubMed] [Google Scholar]
- 27. Jia Y., Lue Y. H., Swerdloff R., Lee K. W., Cobb L. J., Cohen P., and Wang C. (2013) The cytoprotective peptide humanin is induced and neutralizes Bax after pro-apoptotic stress in the rat testis. Andrology 1, 651–659 10.1111/j.2047-2927.2013.00091.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guo B., Zhai D., Cabezas E., Welsh K., Nouraini S., Satterthwait A. C., and Reed J. C. (2003) Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature 423, 456–461 10.1038/nature01627 [DOI] [PubMed] [Google Scholar]
- 29. Zhai D., Luciano F., Zhu X., Guo B., Satterthwait A. C., and Reed J. C. (2005) Humanin binds and nullifies bid activity by blocking its activation of Bax and Bak. J. Biol. Chem. 280, 15815–15824 10.1074/jbc.M411902200 [DOI] [PubMed] [Google Scholar]
- 30. Choi J., Zhai D., Zhou X., Satterthwait A., Reed J. C., and Marassi F. M. (2007) Mapping the specific cytoprotective interaction of humanin with the pro-apoptotic protein bid. Chem. Biol. Drug Des. 70, 383–392 10.1111/j.1747-0285.2007.00576.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ma Z. W., and Liu D. X. (2018) Humanin decreases mitochondrial membrane permeability by inhibiting the membrane association and oligomerization of Bax and Bid proteins. Acta Pharmacol. Sin. 39, 1012–1021 10.1038/aps.2017.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., and Cardona A. (2012) Fiji: An open-source platform for biological-image analysis. Nat. Methods. 9, 676–682 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Steger G. (1998) An unbiased detector of curvilinear structures. IEEE Trans. Pattern Anal. Mach. Intell. 20, 113–125 10.1109/34.659930 [DOI] [Google Scholar]
- 34. Micsonai A., Wien F., Kernya L., Lee Y.-H., Goto Y., Réfrégiers M., and Kardos J. (2015) Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 112, E3095–E3103 10.1073/pnas.1500851112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hashimoto Y., Niikura T., Tajima H., Yasukawa T., Sudo H., Ito Y., Kita Y., Kawasumi M., Kouyama K., Doyu M., Sobue G., Koide T., Tsuji S., Lang J., Kurokawa K., and Nishimoto I. (2002) A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer's disease genes and Aβ. Proc. Natl. Acad. Sci. U.S.A. 98, 6336–6341 10.1073/pnas.101133498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ma J., Edlich F., Bermejo G. A., Norris K. L., Youle R. J., and Tjandra N. (2012) Structural mechanism of Bax inhibition by cytomegalovirus protein vMIA. Proc. Natl. Acad. Sci. U.S.A. 109, 20901–20906 10.1073/pnas.1217094110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kale J., Kutuk O., Brito G. C., Andrews T. S., Leber B., Letai A., and Andrews D. W. (2018) Phosphorylation switches Bax from promoting to inhibiting apoptosis thereby increasing drug resistance. EMBO Rep. 19, e45235 10.15252/embr.201745235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xin M., Li R., Xie M., Park D., Owonikoko T. K., Sica G. L., Corsino P. E., Zhou J., Ding C., White M. A., Magis A. T., Ramalingam S. S., Curran W. J., Khuri F. R., and Deng X. (2014) Small-molecule Bax agonists for cancer therapy. Nat. Commun. 5, 4935 10.1038/ncomms5935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gavathiotis E., Suzuki M., Davis M. L., Pitter K., Bird G. H., Katz S. G., Tu H.-C., Kim H., Cheng E. H.-Y., Tjandra N., and Walensky L. D. (2008) BAX activation is initiated at a novel interaction site. Nature 455, 1076–1081 10.1038/nature07396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gavathiotis E., Reyna D. E., Davis M. L., Bird G. H., and Walensky L. D. (2010) BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol. Cell. 40, 481–492 10.1016/j.molcel.2010.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gavathiotis E., Reyna D. E., Bellairs J. A., Leshchiner E. S., and Walensky L. D. (2012) Direct and selective small-molecule activation of proapoptotic BAX. Nat. Chem. Biol. 8, 639–645 10.1038/nchembio.995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brahmbhatt H., Uehling D., Al-awar R., Leber B., and Andrews D. (2016) Small molecules reveal an alternative mechanism of Bax activation. Biochem. J. 473, 1073–1083 10.1042/BCJ20160118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pritz J. R., Wachter F., Lee S., Luccarelli J., Wales T. E., Cohen D. T., Coote P., Heffron G. J., Engen J. R., Massefski W., and Walensky L. D. (2017) Allosteric sensitization of proapoptotic BAX. Nat. Chem. Biol. 13, 961–967 10.1038/nchembio.2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Arnoult D., Bartle L. M., Skaletskaya A., Poncet D., Zamzami N., Park P. U., Sharpe J., Youle R. J., and Goldmacher V. S. (2004) Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc. Natl. Acad. Sci. U.S.A. 101, 7988–7993 10.1073/pnas.0401897101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Garner T. P., Amgalan D., Reyna D. E., Li S., Kitsis R. N., and Gavathiotis E. (2019) Small-molecule allosteric inhibitors of BAX. Nat. Chem. Biol. 15, 322–330 10.1038/s41589-018-0223-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Niu X., Brahmbhatt H., Mergenthaler P., Zhang Z., Sang J., Daude M., Ehlert F. G. R., Diederich W. E., Wong E., Zhu W., Pogmore J., Nandy J. P., Satyanarayana M., Jimmidi R. K., Arya P., Leber B., Lin J., Culmsee C., Yi J., and Andrews D. W. (2017) A small-molecule inhibitor of Bax and Bak oligomerization prevents genotoxic cell death and promotes neuroprotection. Cell Chem. Biol. 24, 493–506 10.1016/j.chembiol.2017.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sawada M., Hayes P., and Matsuyama S. (2003) Cytoprotective membrane-permeable peptides designed from the Bax-binding domain of Ku70. Nat. Cell Biol. 5, 352–357 10.1038/ncb955 [DOI] [PubMed] [Google Scholar]
- 48. Sawada M., Sun W., Hayes P., Leskov K., Boothman D. A., and Matsuyama S. (2003) Ku70 suppresses the apoptotic translocation of bax to mitochondria. Nat. Cell Biol. 5, 320–329 10.1038/ncb950 [DOI] [PubMed] [Google Scholar]
- 49. Ramirez-Galicia G., Gutierrez-Gonzalez L. H., and Havel J. (2005) Structure of neuroprotective [G-14]humanin peptide by quantum chemistry, NMR and circular dichroism. Collect. Symp. Ser. 8, 62–64 10.1135/css200508062 [DOI] [Google Scholar]
- 50. Benaki D., Zikos C., Evangelou A., Livaniou E., Vlassi M., Mikros E., and Pelecanou M. (2005) Solution structure of humanin, a peptide against Alzheimer's disease-related neurotoxicity. Biochem. Biophys. Res. Commun. 329, 152–160 10.1016/j.bbrc.2005.01.100 [DOI] [PubMed] [Google Scholar]
- 51. Arakawa T., Niikura T., Tajima H., and Kita Y. (2006) The secondary structure analysis of a potent Ser14Gly analog of antiAlzheimer peptide, humanin, by circular dichroism. J. Pept. Sci. 12, 639–642 10.1002/psc.773 [DOI] [PubMed] [Google Scholar]
- 52. Benaki D., Zikos C., Evangelou A., Livaniou E., Vlassi M., Mikros E., and Pelecanou M. (2006) Solution structure of Ser14Gly-humanin, a potent rescue factor against neuronal cell death in Alzheimer's disease. Biochem. Biophys. Res. Commun. 349, 634–642 10.1016/j.bbrc.2006.08.087 [DOI] [PubMed] [Google Scholar]
- 53. Rojo-Dominguez A., Ramirez-Galicia G., Havel J., and Gutierrez-Gonzalez L. H. (2007) Structural preferences of neuroprotective S14G-humanin peptide analyzed by molecular modeling and circular dichroism. Protein Pept. Lett. 14, 618–624 10.2174/092986607780989903 [DOI] [PubMed] [Google Scholar]
- 54. Benaki D., Zikos C., Evangelou A., Vlassi M., Slaninova J., Livaniou E., Mikros E., and Pelecanou M. (2007) Structural studies of humanin and its derivatives in the investigation of their neuroprotective role against Alzheimer's disease. Collect. Symp. Ser. 9, 5–10 10.1135/css200709005 [DOI] [Google Scholar]
- 55. Kita Y., Niikura T., Arisaka F., and Arakawa T. (2008) The complex structure transition of humanin peptides by sodium dodecylsulfate and trifluoroethanol. Protein Pept. Lett. 15, 510–515 10.2174/092986608784567555 [DOI] [PubMed] [Google Scholar]
- 56. Yagisawa R., Kamiya N., Ikebe J., Umezawa K., and Higo J. (2008) Structure dependency of a 24-residue peptide humanin on solvent and preferential solvation by trifluoroethanol studied by multicanonical sampling. Chem. Phys. Lett. 455, 293–296 10.1016/j.cplett.2008.02.109 [DOI] [Google Scholar]
- 57. Arisaka F., Arakawa T., Niikura T., and Kita Y. (2009) Active form of neuroprotective humanin, HN, and inactive analog, S7A-HN, are monomeric and disordered in aqueous phosphate solution at pH 6.0; no correlation of solution structure with activity. Protein Pept. Lett. 16, 132–137 10.2174/092986609787316225 [DOI] [PubMed] [Google Scholar]
- 58. Arakawa T., Niikura T., and Kita Y. (2011) The biological activity of humanin analogs correlates with structure stabilities in solution. Int. J. Biol. Macromol. 49, 93–97 10.1016/j.ijbiomac.2011.04.003 [DOI] [PubMed] [Google Scholar]
- 59. Arakawa T., Niikura T., and Kita Y. (2014) Inactive C8A-humanin analog is as stable as a potent S14G-humanin analog. Mol. Med. Rep. 9, 375–379 10.3892/mmr.2013.1797 [DOI] [PubMed] [Google Scholar]
- 60. Hayashi K., Sasabe J., Chiba T., Aiso S., and Utsunomiya-Tate N. (2012) D-Ser-containing humanin shows promotion of fibril formation. Amino Acids. 42, 2293–2297 10.1007/s00726-011-0971-6 [DOI] [PubMed] [Google Scholar]
- 61. Pistolesi S., Rossini L., Ferro E., Basosi R., Trabalzini L., and Pogni R. (2009) Humanin structural versatility and interaction with model cerebral cortex membranes. Biochemistry. 48, 5026–5033 10.1021/bi900187s [DOI] [PubMed] [Google Scholar]
- 62. Hirano A., Shiraki K., Niikura T., Arakawa T., and Kita Y. (2010) Structure changes of natively disordered humanin in the presence of lipid. Int. J. Biol. Macromol. 46, 375–379 10.1016/j.ijbiomac.2010.01.012 [DOI] [PubMed] [Google Scholar]
- 63. Hirano A., Shiraki K., Niikura T., Arakawa T., and Kita Y. (2011) Structure of three humanin peptides with different activities upon interaction with liposome. Int. J. Biol. Macromol. 48, 360–363 10.1016/j.ijbiomac.2010.12.017 [DOI] [PubMed] [Google Scholar]
- 64. Maftei M., Tian X., Manea M., Exner T. E., Schwanzar D., von Arnim C. A. F., and Przybylski M. (2012) Interaction structure of the complex between neuroprotective factor humanin and Alzheimer's β-amyloid peptide revealed by affinity mass spectrometry and molecular modeling. J. Pept. Sci. 18, 373–382 10.1002/psc.2404 [DOI] [PubMed] [Google Scholar]
- 65. Esckilsen D., Evans H., Milletti M.-C., Guthrie J., Holmes H., and Heyl-Clegg D. (2017) Investigating the binding affinity of the peptide humanin and its analogs to amyloid beta. FASEB J. 31, 601–610 [Google Scholar]
- 66. Alsanousi N., Sugiki T., Furuita K., So M., Lee Y.-H., Fujiwara T., and Kojima C. (2016) Solution NMR structure and inhibitory effect against amyloid-β fibrillation of humanin containing a D-isomerized serine residue. Biochem. Biophys. Res. Commun. 477, 647–653 10.1016/j.bbrc.2016.06.114 [DOI] [PubMed] [Google Scholar]
- 67. He Y., Chen Y., Morris D. L., Lee D.-Y., and Tjandra N. (2020) Bax expression is optimal at low oxygen tension and constant agitation. Protein Expr. Purif. 165, 105501 10.1016/j.pep.2019.105501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Humphrey W., Dalke A., and Schulten K. (1996) VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.