

Abstract
Nuclear modifier genes have been proposed to modify the phenotypic expression of mitochondrial DNA mutations. Using a targeted exome-sequencing approach, here we found that the p.191Gly>Val mutation in mitochondrial tyrosyl-tRNA synthetase 2 (YARS2) interacts with the tRNASer(UCN) 7511A>G mutation in causing deafness. Strikingly, members of a Chinese family bearing both the YARS2 p.191Gly>Val and m.7511A>G mutations displayed much higher penetrance of deafness than those pedigrees carrying only the m.7511A>G mutation. The m.7511A>G mutation changed the A4:U69 base-pairing to G4:U69 pairing at the aminoacyl acceptor stem of tRNASer(UCN) and perturbed tRNASer(UCN) structure and function, including an increased melting temperature, altered conformation, instability, and aberrant aminoacylation of mutant tRNA. Using lymphoblastoid cell lines derived from symptomatic and asymptomatic members of these Chinese families and control subjects, we show that cell lines harboring only the m.7511A>G or p.191Gly>Val mutation revealed relatively mild defects in tRNASer(UCN) or tRNATyr metabolism, respectively. However, cell lines harboring both m.7511A>G and p.191Gly>Val mutations displayed more severe defective aminoacylations and lower tRNASer(UCN) and tRNATyr levels, aberrant aminoacylation, and lower levels of other tRNAs, including tRNAThr, tRNALys, tRNALeu(UUR), and tRNASer(AGY), than those in the cell lines carrying only the m.7511A>G or p.191Gly>Val mutation. Furthermore, mutant cell lines harboring both m.7511A>G and p.191Gly>Val mutations exhibited greater decreases in the levels of mitochondrial translation, respiration, and mitochondrial ATP and membrane potentials, along with increased production of reactive oxygen species. Our findings provide molecular-level insights into the pathophysiology of maternally transmitted deafness arising from the synergy between tRNASer(UCN) and mitochondrial YARS mutations.
Keywords: hearing, mitochondrial DNA (mtDNA), transfer RNA (tRNA), pathogenesis, mitochondrial respiratory chain complex, mitochondrial disease, RNA metabolism, reactive oxygen species (ROS), mitochondrial metabolism, translation, maternal inheritance, mitochondrial translation, mutation, synergy, hearing loss, mitochondrial tRNA, tyrosyl-tRNA synthetase 2 (YARS2), pathophysiology, oxidative stress, tRNASer(UCN), maternally transmitted deafness, genetic disorder
Introduction
Defects of mitochondrial tRNA metabolisms have been associated with both syndromic deafness (hearing loss with other medical problems, such as diabetes) and nonsyndromic deafness (where hearing loss is the only obvious medical problem) (1–5). In humans, mitochondrial genomes (mtDNA)3 encode 13 subunits of the oxidative phosphorylation system (OXPHOS), two rRNAs and 22 tRNAs required for translation (6, 7). The formation of functional tRNA molecules used for protein synthesis requires the transcription, nucleolytic processing, posttranscriptional nucleotide modifications, and aminoacylation (4–9). These proteins involved in the tRNA maturation processing, especially mitochondrial tRNA synthetases, encoded by nuclear genes, were synthesized in the cytosol and subsequently imported into mitochondria (7, 9–11). These deafness-associated tRNA mutations have structural and functional consequences for corresponding tRNAs (1, 12). These included the aberrant processing of 3′ end tRNASer(UCN) precursor, caused by m.7445A>G mutation (13, 14), instability of the folded secondary structure of tRNAGlu due to m.14692A>G mutation (15), deficient m1G37 modification of tRNAAsp caused by m.7551A>G mutation (16), and defective aminoacylation of tRNAHis resulting from m.12201T>C mutation (17). Furthermore, alterations in the LARS2, KARS, IARS2, and NARS2 encoding mitochondrial leucyl-tRNA, lysyl-tRNA, isoleucyl-tRNA, and asparaginyl-tRNA synthetases have been associated with syndromic deafness, respectively (18–21). Moreover, nonsyndromic deafness in some families was caused by the coexistence of the 12S rRNA m.1555A>G mutation and p.Ala10Ser mutation in TRMU responsible for the biosynthesis of τm5s2U at the wobble position of tRNAGln, tRNAGlu, and tRNALys (22, 23). However, the pathophysiology underlying deafness-linked aberrant tRNA metabolisms remains poorly understood.
As shown in Fig. 1A, the deafness-associated tRNASer(UCN) 7511A>G mutation converted the A4-U69 base-pairing into a G4-U69 base-pairing at the aminoacyl acceptor stem of this tRNA (24–27). This base-pairing may play an important role in the stability and identity of tRNA (24, 25). We therefore hypothesized that the m.7511A>G mutation perturbed both structure and function of tRNASer(UCN). The m.7511A>G mutation was identified in several families from different ethnic groups, with varying expressivity and penetrance of deafness (27–30). In particular, 9 of 10 matrilineal relatives in a three-generation Chinese pedigree carrying the m.7511A>G mutation exhibited hearing impairment, in contrast with only a small portion of hearing-impaired matrilineal relatives in two French pedigrees and one Japanese family carrying the same mtDNA mutation (27–30). These findings suggest that the nuclear modifier genes, especially those involved in mitochondrial tRNA metabolism, contributed to the phenotypic expression of m.7511A>G mutation. By target exome sequencing (genes encoding 20 mitochondrial tRNA synthetases and 25 tRNA modifying enzymes), we identified the known variant (c.572G>T, p.191Gly>Val) in the YARS2 gene encoding the mitochondrial tyrosyl-tRNA synthetase (31, 32) that interacted with the m.7511A>G mutation to cause hearing loss in a three-generation Chinese family with extremely high penetrance of hearing loss. In the present study, we further investigated the impact of the m.7511A>G mutation on the structure and function of tRNASer(UCN). The effects of YARS2 p.191Gly>Val and m.7511A>G mutations on mitochondrial functions were first assessed for the tRNA metabolism, including aminoacylation capacities and stability of tRNA, through the use of lymphoblastoid mutant cell lines derived from members of the Chinese family (individuals carrying only the m.7511A>G mutation, only the YARS2 p.191Gly>Val mutation or both m.7511A>G and heterozygous or homozygous p.191Gly>Val mutations), and genetically unrelated control subjects lacking these mutations. These cell lines were further evaluated for an effect on mitochondrial translation, respiration, production of ATP, mitochondrial membrane potential, and reactive oxygen species (ROS).
Figure 1.

The analysis of stability and conformation of tRNASer(UCN). A, cloverleaf structure of human mitochondrial tRNASer(UCN) (12). An arrow denotes the location of the m.7511A>G mutation. B, thermal stability of WT (A4) and mutant (G4) tRNASer(UCN). Absorbance (Abs) of WT and mutant (MT) was measured at 260 nm with a heating rate of 1 °C/min from 25 to 95 °C (red curves). First derivative, generated with the expression dA/dT, showed the rate of absorbance change (blue curves). The calculations were based on three independent experiments. C, in vitro analysis of the conformation of tRNASer(UCN). WT and mutant tRNASer(UCN) transcripts were electrophoresed through native polyacrylamide gel, electroblotted, and hybridized with the DIG-labeled oligonucleotide probes specific for tRNASer(UCN). D, Northern blot analysis of tRNA under native conditions. Two micrograms of total mitochondrial RNA from mutant and control cell lines were electrophoresed through native polyacrylamide gel, electroblotted, and hybridized with DIG-labeled oligonucleotide probes specific for the tRNASer(UCN) and tRNALeu(CUN), respectively.
Results
The m.7511A>G mutation altered the stability and conformation of tRNASer(UCN)
As shown in Fig. 1A, the m.7511A>G mutation changed the typical A4-U69 base-pairing into a noncanonical G4-U69 base-pairing at the acceptor stems. To experimentally test the effect of m.7511A>G mutation on the stability of tRNASer(UCN), we examined the melting temperatures (Tm) of WT (A4) and mutant (G4) tRNASer(UCN) transcripts. These Tm values were determined by calculating the derivatives of the absorbance against a temperature curve. As shown in Fig. 1B, the Tm values for WT (A4) and mutant (G4) tRNASer(UCN) transcripts were 41.7 and 51 °C, respectively. These data suggested that the tRNASer(UCN) with a G4:U69 bp may be more stable than the tRNASer(UCN) with an A4:U69 bp.
As shown in Fig. 1C, electrophoretic patterns showed that the mutant (G4) tRNASer(UCN) transcript migrated faster than the WT (A4) tRNASer(UCN) transcript under native conditions. However, there was no difference of migration pattern between WT (A4) and mutant (G4) tRNASer(UCN) transcripts under denaturing conditions. These data indicated that the m.7511A>G mutation resulted in the conformational change of tRNASer(UCN).
Clinical presentation of a hearing-impaired Han Chinese pedigree
One Han Chinese hearing-impaired proband carrying the m.7511A>G mutation was identified among 2651 Chinese hearing-impaired probands but absent in 574 Chinese hearing-normal controls (28). As shown in Fig. S1A, the Chinese family exhibited extremely high penetrance of hearing loss. As shown in Fig. S2 and Table S1, 9 of 10 matrilineal relatives exhibited the variable degree of hearing impairment (two with mild hearing loss, six with moderate hearing loss, and one with severe hearing loss), whereas none of other members in this family had hearing loss. The age-at-onset of hearing loss ranged from 5 to 55 years old, with an average of 25 years old. There was no evidence that any of the other members of this family had any other causes to account for hearing loss. These matrilineal relatives showed no other clinical abnormalities, including cardiac failure, muscular diseases, visual failure, and neurological disorders. Further analysis showed that the m.7511A>G mutation was present in homoplasmy in all matrilineal relatives but not in other members of this family (Fig. S1B).
Targeting exome sequence analysis
The higher penetrance of hearing loss in this Chinese family implied that nuclear modifier genes, especially for genes involved in mitochondrial tRNA metabolism, influence the phenotypic manifestation of m.7511A>G mutation. To test this hypothesis, we performed targeting exome-sequencing analyses of 45 genes encoding 20 mitochondrial tRNA synthetases and 25 tRNA-modifying enzymes (Table S2) among seven matrilineal relatives (II-5, II-7, III-3, III-4, III-5, III-6, and III-7) and two married-in controls (II-4 and II-5) of WZD200 pedigree carrying the m.7511A>G mutation. As a result, we identified the known (c.572G>T, p.191Gly>Val) mutation in the YARS2 gene encoding the mitochondrial tyrosyl-tRNA synthetase in six hearing-impaired matrilineal relatives but not in the hearing-normal matrilineal relative (III-7). We further analyzed the presence of the c.572G>T mutation in three symptomatic members and six asymptomatic subjects of this Chinese family and 13 symptomatic members and five asymptomatic subjects of a Japanese family (30), by restriction fragment length polymorphism analysis, because the c.572G>T mutation disrupted a Tsp45I site (32). In the Chinese family, the symptomatic subjects (II-1 and II-5) and married-in control (I-1) carried the homozygous c.572G>T mutation, the symptomatic subjects (I-2, II-2, II-7, III-3, III-4, III-5 and III-6) harbored the heterozygous c.572G>T mutation, and the asymptomatic individual (III-7) and three married controls lacked the c.572G>T mutation (Fig. S1A and Table S1). However, this mutation was absent in the members of the Japanese family (30). These suggested that the c.572G>T mutation may increase the penetrance of hearing loss in the Chinese family.
Reductions in the steady-state levels of mitochondrial tRNAs
To test if the m.7511A>G mutation affected the conformation of tRNASer(UCN) ex vivo, total RNAs from mitochondria isolated from various cell lines were electrophoresed through 15% polyacrylamide gel (native condition) and then electroblotted onto a positively charged nylon membrane (Roche Applied Science) for hybridization analysis with digoxigenin (DIG)-labeled oligodeoxynucleotide probes for tRNASer(UCN) and tRNALeu(CUN), respectively. As shown in Fig. 1D, the electrophoretic patterns showed that the tRNASer(UCN) in the mutant cell lines (III-7) carrying the m.7511A>G mutation migrated faster than those of one cell line (A61) lacking this mutation. However, there were no differences in the migration of tRNALeu(CUN) between WT and mutant cell lines.
To further examine the effect of m.7511A>G and c.572G>T mutations on the stability of tRNA, mitochondrial RNAs from various cell lines were subjected to Northern blotting and hybridized with DIG-labeled oligodeoxynucleotide probes specific for tRNASer(UCN), tRNATyr, tRNAGlu, tRNAAsp, tRNAMet, tRNALys, tRNALeu(UUR), and 5S rRNA, respectively. For comparison, the average levels of each tRNA in various cell lines were normalized according to the level of the 5S rRNA. As shown in Fig. 2, the steady-state levels of tRNASer(UCN) in the cell line III-7, bearing only the m.7511A>G mutation, and tRNATyr in the cell line I-1, carrying the homozygous c.572G>T mutation, were decreased 49.5 and 33.7%, respectively, as compared with those in the control cell line (A61) lacking these mutations. Strikingly, cells harboring both m.7511A>G and c.572G>T mutations exhibited drastic decreases in levels of tRNASer(UCN) and tRNATyr as well as various reductions in the other tRNAs (Fig. 2B). In particular, the average steady-state levels of tRNASer(UCN), tRNATyr, tRNAGlu, tRNAAsp, tRNAMet, tRNALys, and tRNALeu(UUR) in mutant cell lines carrying both m.7511A>G and homozygous c.572G>T mutations were decreased by 72.2, 58.9, 33.5, 69.6, 86.1, 13.5, and 21.2%, as compared with the average values in the control cell line (A61), respectively. Furthermore, the average steady-state levels of tRNASer(UCN), tRNATyr, tRNAGlu, tRNAAsp, tRNAMet, tRNALys, and tRNALeu(UUR) in mutant cell lines carrying both m.7511A>G and heterozygous c.572G>T mutations were decreased by 75.8, 51.1, 35.5, 64.4, 71.1, 11.2, and 26.1%, as compared with the average values in the control cell line (A61), respectively.
Figure 2.

Northern blot analysis of tRNA under denaturing conditions. A, Northern blot analysis of tRNA under denaturing conditions. Two micrograms of total mitochondrial RNA from various cell lines were electrophoresed through a denaturing polyacrylamide gel, electroblotted, and hybridized with DIG-labeled oligonucleotide probes for the tRNASer(UCN), tRNATyr, tRNAGlu, tRNAAsp, tRNAMet, tRNALys, and tRNALeu(UUR), respectively. B, quantification of tRNA levels. Shown is average relative content of each tRNA per cell, normalized to the average content per cell of 5S rRNA in mutant cell lines harboring both the m.7511A>G and heterozygous YARS2 p.Gly191Val mutations, both the m.7511A>G and homozygous YARS2 p.Gly191Val mutations, only the homozygous YARS2 p.Gly191Val mutation, or only the m.7511A>G mutation and control cell lines lacking these mutations. The values for the mutant cell lines are expressed as percentages of the average values for the control cell lines. The calculations were based on three independent experiments. Error bars, S.D. p, significance, according to the t test, of the differences between mutant and control cell lines.
Defects in tRNA aminoacylation
The aminoacylation capacities of tRNASer(UCN), tRNATyr, tRNAThr, tRNALeu(UUR), tRNALys, and tRNASer(AGY) in various control and mutant cell lines were examined by using electrophoresis in an acid polyacrylamide/urea gel system to separate uncharged tRNA species from the corresponding charged tRNA, electroblotting and hybridizing with the above tRNA probes. As shown in Fig. 3A, the slower-migrating band (top band) represents the charged tRNA, and the faster-migrating band (bottom band) represents uncharged tRNA. The electrophoretic patterns revealed two stacked bands present for the WT tRNASer(UCN) and two well-separated bands for the mutant tRNASer(UCN). Furthermore, either charged or uncharged tRNASer(UCN) migrated faster in all mutant cell lines carrying the m.7511A>G mutation than those in other cell lines lacking the mutation. To further distinguish nonaminoacylated tRNA from aminoacylated tRNA, samples of tRNAs were deacylated after heating for 10 min at 60 °C (pH 9.0) and then run in parallel. As shown in Fig. 3, the deacylated samples gave only one band (uncharged tRNA) in both mutant and control cell lines.
Figure 3.

In vivo aminoacylation assays. A, 4–8 μg of total mitochondrial RNA purified from various cell lines under acid conditions were electrophoresed at 4 °C through an acid (pH 5.0) 10% polyacrylamide, 8 m urea gel, electroblotted, and hybridized with DIG-labeled oligonucleotide probes specific for the tRNASer(UCN), tRNATyr, tRNAThr, tRNALeu(UUR), tRNALys, and tRNASer(AGY), respectively. The samples from control and mutant cell lines were also deacylated (DA) by heating for 10 min at 60 °C at pH 9.0, electrophoresed, and hybridized with DIG-labeled oligonucleotide probes as above. B, quantification of aminoacylated proportions of tRNASer(UCN), tRNATyr, tRNAThr, tRNALeu(UUR), tRNALys, and tRNASer(AGY) in the mutant and controls. The calculations were based on three independent experiments. Graph details and symbols are explained in the legend to Fig. 2.
As shown in Fig. 3, ∼10% decreases in the aminoacylated efficiency of tRNASer(UCN) in the cell line III-7, bearing only the m.7511A>G mutation, and 25.4% reductions in the aminoacylated efficiency of tRNATyr in the cell line I-1, carrying only the homozygous c.572G>T mutation, were observed, as compared with those in the control cell line (A61). Strikingly, cells harboring both m.7511A>G and c.572G>T mutations exhibited greater reductions in aminoacylated efficiencies of tRNASer(UCN) and tRNATyr as well as various reductions in those of other tRNAs (Fig. 3A). In particular, the aminoacylated efficiencies of tRNASer(UCN), tRNATyr, tRNAThr, tRNALeu(UUR), tRNALys, and tRNASer(AGY) in mutant cell lines carrying both m.7511A>G and homozygous c.572G>T mutations were 37.9, 62.5, 88.6, 40.7, 65.1, and 74.3% of the average values in the control cell line (A61), respectively. Furthermore, the aminoacylated efficiencies of tRNASer(UCN), tRNATyr, tRNAThr, tRNALeu(UUR), tRNALys, and tRNASer(AGY) in mutant cell lines carrying both m.7511A>G and heterozygous c.572G>T mutations were 44, 62.8, 111.2, 51.6, 99, and 88.6% of the average values in the control cell line (A61), respectively.
Decreases in the levels of mitochondrial proteins
To assess whether the c.572G>T mutation enhanced the defects in mitochondrial translation associated with m.7511A>G mutation, a Western blot analysis was carried out to examine the levels of seven mtDNA encoding polypeptides (of respiratory complex) in various cell lines with VDAC as a loading control. As shown in Fig. 4A, the levels of ND1, ND4, ND5, and ND6 (subunits 1, 4, 5, and 6 of NADH dehydrogenase); CYTB (apocytochrome b); CO1 (subunit 1 of cytochrome c oxidase); and ATP6 (subunit 6 of the H+-ATPase) exhibited variable reductions in mutant cell lines, as compared with those of the control cell line. As shown in Fig. 4B, the average levels of ND1, ND4, ND5, ND6, CO1, CYTB, and ATP6 in mutant cell lines carrying only the homozygous c.572G>T mutation, the m.7511A>G mutation, or both the m.7511A>G and heterozygous or homozygous c.572G>T mutations were 84.9, 77.7, 61.7, and 49.8% of those in the control cell line (A61), respectively. In particular, the levels of ND1, ND4, ND5, ND6, CO1, CYTB, and ATP6 in the cell line carrying only m.7511A>G mutation were 35.4, 97.8, 70.2, 83.7, 52.8, 102.6, and 101.3% of those in control cell line (A61) (p < 0.05), respectively.
Figure 4.

Western blot analysis of mitochondrial proteins. A, 5 μg of total mitochondrial proteins from various cell lines were electrophoresed through a denaturing polyacrylamide gel, electroblotted, and hybridized with antibodies specific for ND1, ND4, ND5, ND6, CO1, CYTB, and ATP6 and with VDAC as a loading control, respectively. B, quantification of mitochondrial proteins. The levels of mitochondrial proteins in mutant and control cell lines were determined as described elsewhere (17, 32). The average of three determinations for each cell line is shown. Graph details and symbols are explained in the legend to Fig. 2.
We then examined the levels of seven subunits (mtDNA-encoding CO2 and six nucleus-encoding proteins) of the phosphorylation system (OXPHOS) in control and mutant cell lines by Western blot analysis. As shown in Fig. 5A, the levels of NDUFS3, NDUFB8 (subunits of NADH:ubiquinone oxidoreductase), CO2, and COX10 (subunits of cytochrome c oxidase) were decreased in the mutant cell lines. By contrast, the levels of other mitochondrial proteins (ATP5A, UQCRC2, and SDHB) in mutant cell lines were comparable with those in the control cell line. As illustrated in Fig. 5B, the average levels of NDUFS3, NDUFB8, CO2, and COX10 in mutant cell lines carrying both m.7511A>G and heterozygous c.572G>T mutations were 42.2, 71.8, 50.3, and 70.5% of those in the control cell line (A61). In particular, the levels of NDUFS3, NDUFB8, CO2, and COX10 in mutant cell line carrying both m.7511A>G and homozygous c.572G>T mutations were 19.4, 52.7, 47.1, and 70% of those in control cell line (A61), respectively (Fig. 5B).
Figure 5.

Western blot analysis of 7 OXPHOS subunits. A, 5 μg of total mitochondrial proteins from various cell lines were electrophoresed through a denaturing polyacrylamide gel, electroblotted, and hybridized with an antibody mixture specific for subunits of each OXPHOS complex and with GAPDH as a loading control. B, quantification of the levels of ATP5A, UQCRC2, SDHB, CO2, COX10, NDUFS3, and NDUFB8 in mutant and control cell lines as described elsewhere (32, 47). Graph details and symbols are explained in the legend to Fig. 2.
Reduced activities of respiratory complexes I, III, and IV
To examine whether the c.572G>T mutation worsened the respiratory deficiency caused by m.7511A>G mutation, we measured the activities of respiratory complexes by isolating mitochondria from mutant and control cell lines (33, 34). As shown in Fig. 6, the activity of complex I in mutant cell lines carrying only the c.572G>T mutation, the m.7511A>G mutation, or both the m.7511A>G and heterozygous or homozygous c.572G>T mutations were 84.8, 68.3, 42.6, and 33.2% of the control cell line (A61), respectively. The activities of complex III in mutant cell lines carrying only the c.572G>T mutation, the m.7511A>G mutation, or both the m.7511A>G and heterozygous or homozygous c.572G>T mutations were 105.5, 97.5, 89.0, and 67.9% of the control cell line (A61), respectively. Furthermore, the activities of complex IV in mutant cell lines carrying only the c.572G>T mutation, the m.7511A>G mutation, or both the m.7511A>G and heterozygous or homozygous c.572G>T mutations were 89.1, 73.9, 47.3, and 49.1% of the control cell line (A61), respectively. However, the activities of complex II in the mutant cell lines were comparable with those in the control cell line (A61).
Figure 6.

Respiration assays. A, enzymatic activities of respiratory chain complexes. The activities of respiratory complexes were investigated by enzymatic assay on complexes I, II, III, and IV in mitochondria isolated from various cell lines. B, an analysis of O2 consumption in the various cell lines using different inhibitors. The rates of O2 (OCR) were first measured on 2 × 104 cells of each cell line under basal conditions and then sequentially added to oligomycin (1.5 μm), FCCP (0.5 μm), rotenone (1 μm), and antimycin A (1 μm) at the indicated times to determine different parameters of mitochondrial functions. C, graphs presented the ATP-linked OCR, proton leak OCR, maximal OCR, reserve capacity, and nonmitochondrial OCR in mutant and control cell lines. Nonmitochondrial OCR was determined as the OCR after rotenone/antimycin A treatment. Basal OCR was determined as OCR before oligomycin minus OCR after rotenone/antimycin A. ATP-linked OCR was determined as OCR before oligomycin minus OCR after oligomycin. Proton leak was determined as basal OCR minus ATP-linked OCR. Maximal OCR was determined as the OCR after FCCP minus nonmitochondrial OCR. Reserve capacity was defined as the difference between maximal OCR after FCCP minus basal OCR. OCR values were expressed in pmol of oxygen/min/μg of protein. The average values of three determinations for each cell line were shown. Graph details and symbols are explained in the legend to Fig. 2.
Respiration defects in mutant cells
To further assess whether the m.7511A>G and c.572G>T mutations altered cellular bioenergetics, we examined the oxygen consumption rates (OCR) of various mutant and control cell lines using a Seahorse Bioscience XF-96 extracellular flux analyzer (35, 36). In this system, a single experiment can measure all major aspects of mitochondrial coupling and respiratory control, including basal respiration, O2 consumption attributed to ATP production, proton leak, maximum respiratory rate, reserve capacity, and nonmitochondrial respiration (Fig. 6B). As shown in Fig. 6C, the basal OCR in the mutant cell lines carrying only the c.572G>T mutation, the m.7511A>G mutation, or both the m.7511A>G and heterozygous or homozygous c.572G>T mutations were 92.5, 68.6, 47.0, and 36.9% of the mean values measured in the control cell lines (p < 0.05), respectively. To investigate which of the enzyme complexes of the respiratory chain was affected in the mutant cell lines, OCR was measured after the sequential addition of oligomycin (to inhibit the ATP synthase), carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) (to uncouple the mitochondrial inner membrane and allow for maximum electron flux through the electron transfer chain), rotenone (to inhibit complex I), and antimycin A (to inhibit complex III) (56). The differences between the basal OCR and the drug-insensitive OCR yielded the amount of ATP-linked OCR, proton leak OCR, maximal OCR, reserve capacity, and nonmitochondrial OCR. As illustrated in Fig. 6C, the ATP-linked OCR, proton leak OCR, maximal OCR, reserve capacity, and nonmitochondrial OCR in mutant cell line carrying only homozygous c.572G>T mutation were 94.7, 82.1, 76.3, 63.2, and 106.4% of those in the control cell lines. The values in mutant cell line carrying only the m.7511A>G mutation were 72.5, 50.3, 73.9, 78.2, and 77.5% of the control cell line. Notably, those in mutant cell lines carrying both m.7511A>G and heterozygous c.572G>T mutations were 43.3, 64.4, 41.1, 36.3, and 94.8%, and those in mutant cell lines carrying both m.7511A>G and homozygous c.572G>T mutations were 34.1, 50.3, 37.3, 37.7, and 46.9%, relative to the mean values measured in the control cell lines, respectively.
Reduced levels in mitochondrial ATP production
To examine the capacity of oxidative phosphorylation, we measured the levels of cellular and mitochondrial ATP production using a luciferin/luciferase assay. Populations of cells from various mutant and control cell lines were incubated in the medium in the presence of glucose (total cellular ATP production) or 2-deoxy-d-glucose with pyruvate (mitochondrial ATP production). As shown in Fig. 7, the levels of mitochondrial ATP production in mutant cell lines carrying only the c.572G>T mutation, the m.7511A>G mutation, or both the m.7511A>G and heterozygous or homozygous c.572G>T mutations were 77.6, 58.9, 62.9, and 44.8% of the control cell lines. Moreover, the levels of total cellular ATP production in the above mutant cell lines were 97.5, 84.3, 79.5, and 67.1%, relative to the mean value measured in the control cell lines, respectively.
Figure 7.

Measurement of cellular and mitochondrial ATP levels. Mutant and control cell lines were incubated with 10 mm glucose or 5 mm 2-deoxy-d-glucose plus 5 mm pyruvate to determine ATP generation under mitochondrial ATP synthesis. Average rates of ATP level per cell line are shown. A, ATP level in mitochondria; B, ATP level in total cells. Three determinations were made for each cell line. Graph details and symbols are explained in the legend to Fig. 2.
Decreases in mitochondrial membrane potentials
The mitochondrial membrane potential (ΔΨm) generated by proton pumps (complexes I, III, and IV) is an essential component in the process of energy storage during oxidative phosphorylation (37). We examined the levels of ΔΨm in the mutant and control cell lines using a fluorescence probe JC-10 assay system. The ratios of fluorescence intensity excitation/emission = 490/590 and 490/530 nm (FL590/FL530) were recorded to reflect the ΔΨm level of each sample. As shown in Fig. 8, the ΔΨm levels of mutant cell lines harboring only the c.572G>T mutation, the m.7511A>G mutation, or both the m.7511A>G and heterozygous or homozygous c.572G>T mutations were 97.5, 71.1, 67.7, and 64.1% of the mean values measured in the control cell lines, respectively. In contrast, the ΔΨm levels in mutant cell lines in the presence of FCCP were comparable with those measured in the control cell lines.
Figure 8.

Mitochondrial membrane potential analysis. ΔΨm was measured in various cell lines using a fluorescence probe JC-10 assay system. The ratios of fluorescence intensity excitation/emission = 490/590 and 490/530 nm (FL590/FL530) were recorded to delineate the ΔΨm level of each sample. The relative ratios of FL590/FL530 geometric mean between mutant and control cell lines were calculated to reflect the level of ΔΨm. Shown are relative ratios of JC-10 fluorescence intensity at excitation/emission = 490/530 nm and 490/590 nm in the absence (A) and presence (B) of 10 μm FCCP. The average of three determinations for each cell line is shown. Graph details and symbols are explained in the legend to Fig. 2.
Increase of ROS production
Respiratory deficiency can increase the production of ROS (38, 39). In this study, we measured the levels of ROS generation in mutant and control cell lines with flow cytometry under normal and H2O2-stimulated conditions. To detect the capacity of reaction upon increasing levels of ROS under oxidative stress, we calculated the ratio of geometric mean intensity between unstimulated and stimulated with H2O2 in each cell line. As shown in Fig. 9, the levels of ROS generation in the mutant cell lines harboring only the c.572G>T mutation, the m.7511A>G mutation, or both the m.7511A>G and heterozygous or homozygous c.572G>T mutations were 119.3, 109.6, 127.3, and 140.6% of the control cell lines.
Figure 9.
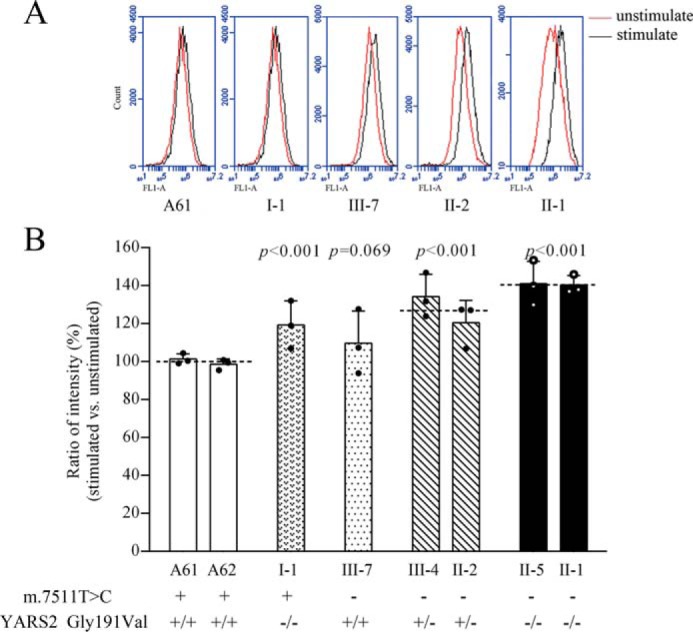
Measurement of ROS. Shown is the ratio of geometric mean intensity between the levels of ROS generation in the vital cells with or without H2O2 stimulation. The rates of total ROS production in various cell lines were analyzed by measurement of fluorescence using a BD Accuri C6 flow cytometer system. A, flow cytometry histogram showing fluorescence of cell lines without (red) or with (black) H2O2 stimulation. B, relative ratios of fluorescence intensity were calculated in the absence and presence of H2O2. The average of three determinations for each cell line is shown. Graph details and symbols are explained in the legend to Fig. 2.
Discussion
The pathogenicity of tRNASer(UCN) 7511A>G mutation
In the present study, we further investigated the molecular mechanism of the deafness-associated m.7511A>G mutation. Indeed, the occurrence of the m.7511A>G mutation in several hearing-impaired families from different ethic backgrounds strongly indicated that this mutation is involved in the pathogenesis of deafness (26–30). The m.7511A>G mutation caused the substitution of the A4:U69 base-pairing with G4:U69 base-pairing at the aminoacyl acceptor stem of tRNASer(UCN) (12, 26, 27). In fact, this A4:U69 base-pairing may play an important role in the stability and identity of tRNA (12, 24, 25, 39–42). Therefore, it was hypothesized that m.7511A>G mutation led to structural and functional consequences for tRNASer(UCN), including the processing of RNA precursors, stability, and aminoacylation of tRNASer(UCN). In particular, the substitution A4:U69 base-pairing with G4:U69 base-pairing caused by the m.7511A>G mutation may restrict the accessible conformation space of tRNASer(UCN) (43–45). Here, the altered structure of tRNASer(UCN) caused by the m.7511A>G mutation was evidenced by the increased melting temperature and electrophoretic mobility of mutated tRNA with respect to the WT molecule in vitro or ex vivo. The instability of mutant tRNA was further supported by marked reductions in the steady-state level of tRNASer(UCN) in the cybrid mutant cell lines (26) and lymphoblastoid cell lines carrying the m.7511A>G mutation in the present study.
Furthermore, the substitution A4:U69 base-pairing with G4:U69 base-pairing induced by the m.7511A>G mutation may result in the faulty interaction of tRNASer(UCN) with mitochondrial seryl-tRNA synthetase, thereby altering the aminoacylation properties of tRNASer(UCN) (39, 43–46). Indeed, all human AlaRS mischarged to noncognate tRNAs, such as tRNACys and tRNAAsp, with the G4:U69 bp (45, 46). Therefore, mutant tRNASer(UCN) with G4:U69 bp can be mischarged with other amino acids. In this study, the possible mischarging to noncognate tRNAs of mutant tRNASer(UCN) may account for the improperly aminoacylated tRNASer(UCN), as suggested by the aberrantly aminoacylated tRNASer(UCN) in the mutant cell lines and faster electrophoretic mobility of mutated tRNA with respect to the WT molecules. Alternatively, the mutant tRNASer(UCN) may be charged to a lesser extent by the mitochondrial seryl-synthetase. In this study, only mildly reduced efficiencies of aminoacylated tRNASer(UCN) were observed in a mutant cell line carrying only the m.7511A>G mutation, in contrast to marked decreases of aminoacylation in the tRNALeu(UUR) with the 14A>G substitution or tRNALys with the 55A>G mutation (47–49). Improper aminoacylation and instability of tRNASer(UCN) were responsible for marked reductions in the level of tRNASer(UCN) observed in a cell line carrying the m.7511A>G mutation, as in the cases of other pathogenic tRNA mutations (47–51). The aberrant tRNASer(UCN) metabolism resulted in the impairment of mitochondrial translation, defective oxidative phosphorylation, and increasing production of oxidative reactive species (15, 16, 17, 26). The resultant mitochondrial dysfunctions would lead to the dysfunction or death of cochlear cells, thereby contributing to the development of hearing loss.
The YARS2 p.191Gly>Val mutation enhanced the phenotypic manifestation of the m.7511A>G mutation
Genetic modifiers involved in mitochondrial tRNA metabolism modulate the phenotypic manifestation of the deafness-associated 12S rRNA mutations (22, 23, 52). In this study, the penetrances of hearing loss in this Chinese family harboring both the m.7511A>G and YARS2 p.191Gly>Val mutations were significantly higher than those in the French and Japanese families carrying only the m.7511A>G mutation (29, 30). Furthermore, cell lines bearing both p.191Gly>Val and m.7511A>G mutations exhibited greater mitochondrial dysfunctions than those carrying only p.191Gly>Val or m.7511A>G mutation. Strikingly, mutant cell lines harboring both m.7511A>G and p.191Gly>Val mutations exhibited not only more decreases in the aminoacylation efficiencies of tRNASer(UCN) and tRNATyr but also deficient aminoacylation of tRNAThr, tRNALys, tRNALeu(UUR), and tRNASer(AGY), as compared with those in the cell lines carrying only the p.191Gly>Val or m.7511A>G mutation. The aberrantly aminoacylated tRNA makes the mutant tRNA metabolically less stable and more subject to degradation, thereby lowering the level of the tRNA in mutant cell lines (17, 26, 40). In the present study, mutant cell lines bearing only the m.7511A>G mutation exhibited 48% reductions in the level of tRNASer(UCN), and mutant cell lines harboring only the p.191Gly>Val mutation displayed 33.7% decreases in the level of tRNATyr., respectively. By contrast, mutant cell lines harboring both m.7511A>G and homozygous p.191Gly>Val mutations reveled 70% decreases in the level of tRNASer(UCN) and 59% reductions in the level of tRNATyr as well as various decreases in the levels of other tRNAs, including tRNAGlu, tRNAAsp, tRNAMet, tRNALys, and tRNALeu(UUR). Notably, ∼70% reductions in the steady-state levels of tRNASer(UCN) in the cells carrying both p.191Gly>Val and m.7511A>G mutations were in good agreement with the 75% decrease in the levels of tRNASer(UCN) in those in the mutant cybrid cells bearing the m.7511A>G, tRNAAla 5655A>G, and ND1 3308T>C mutations (26). These data strongly suggested that the synergic interaction between the YARS2 p.191Gly>Val and m.7511A>G mutations mediated mitochondrial tRNA metabolisms, especially exacertbating the defects of tRNASer(UCN) and tRNATyr metabolisms. Notably, mutations in the TRMU involved in biosynthesis of τm5s2U at the wobble position of tRNAGln, tRNAGlu, and tRNALys affected the metabolism of not only tRNALys, tRNAGlu, tRNAGln, but also other mitochondrial tRNA (22, 53).
Both shortage of and aberrant aminoacylation of tRNAs led to impairments of mitochondrial translation. In this investigation, 50% decreases in the levels of mtDNA encoding proteins observed in the mutant cells carrying both the m.7511A>G and p.191Gly>Val mutations are below the proposed threshold level (50%) to produce a clinical phenotype associated with a mtDNA mutation (23, 47, 48, 54). The defects of mitochondrial translation were responsible for the respiratory deficiency, uncoupling of the oxidative pathway for ATP synthesis, diminished mitochondrial membrane potentials, and overproduction of ROS (7, 48, 52, 55). In particular, more drastic decreases of oxygen consumption rates, mitochondrial ATP production, and mitochondrial membrane potentials and increases of ROS production were observed in the cell lines carrying both the p.191Gly>Val and m.7511A>G mutations than those in cell lines carrying only the p.191Gly>Val or m.7511A>G mutation. These mitochondrial dysfunctions yielded a preferential effect on the hair cells and neurons in the cochlea, because cochlear functions depend on a very high rate of ATP production (56–58). This would result in the dysfunction or death of hair cells and neurons in the cochlea carrying both the p.191Gly>Val and m.7511A>G mutations, thereby producing a phenotype of hearing loss.
In summary, we demonstrated that the pathophysiology of maternally inherited deafness was manifested by aberrant tRNA metabolisms due to the combination of YARS2 p.191Gly>Val with tRNASer(UCN) 7511A>G mutations. The m.7511A>G mutation altered both the structure and function of tRNASer(UCN). The p.191Gly>Val mutation deteriorated the aberrant tRNA metabolisms associated with the m.7511A>G mutation. The aberrant tRNA metabolisms resulted in defective mitochondrial translation, respiratory deficiency, decreasing ATP production, and increasing ROS production. These biochemical defects led to the high penetrance and occurrence of deafness in the Chinese family carrying both the m.7511A>G and p.191Gly>Val mutations. Our findings provide new insights into the pathophysiology of maternally inherited deafness, manifested by the synergetic interaction between mitochondrial and nuclear gene products underlying aberrant tRNA metabolism.
Experimental procedures
Subjects
One Han Chinese family (WZD200), as shown in Fig. S1A, was recruited from the Otology Clinics of Wenzhou Medical University (Zhejiang, China), as described previously (28). Comprehensive history-taking, physical examination, and audiological examination were performed to identify any syndromic findings, history of exposure to aminoglycosides, and genetic factors related to hearing impairment in all available members of this Chinese pedigree, as detailed previously (59, 60). The 574 control subjects were from a panel of unaffected subjects of Han Chinese ancestry from the same region. This study followed the principles of the Declaration of Helsinki. Informed consent was obtained from the participants prior to their participation in the study, under protocols approved by the Ethics Committees of Zhejiang University and the Wenzhou Medical University.
Mitochondrial DNA-sequencing analysis
Genomic DNA was isolated from whole blood of participants using the QIAamp DNA Blood Mini Kit (Qiagen, catalog no. 51104). The entire mtDNAs of the family members of WZD200 (I-1, II-1, II-2, III-5, and III-7) and one Chinese control subject (A61) were PCR-amplified in 24 overlapping fragments using sets of the light (L) and heavy (H) strand oligonucleotide primers, as described previously (61). These sequence results were compared with the updated consensus Cambridge sequence (GenBankTM accession number NC_012920) (6). For the analysis for the presence and level of the m.7511A>G mutation, the PCR DNA fragments (117 bp) spanning the tRNASer(UCN) gene were amplified using genomic DNA as the template and the oligodeoxynucleotides 5′-CCCCATGGCCTCCATGACTTTTTAAA-3′ and 5′-CTACTTGCGCTGCATGTGCCATTAAGAT-3′. The resultant 117-bp segments were digested with the restriction enzyme DraI and analyzed by electrophoresis through a 14% polyacrylamide gel. After ethidium bromide staining, the ImageQuant program was used to determine the proportions of digested and undigested PCR product to ascertain whether the m.7511A>G mutation was present in homoplasmy in these subjects (Fig. S1B).
Target exome sequencing
A panel of exome sequencings (genes encoding 20 mitochondrial tRNA synthetases and 25 tRNA-modifying enzymes, Table S1) of seven matrilineal relatives (II-5, II-7, III-3, III-4, III-5, III-6, and III-7) carrying the m.7511A>G mutation and two married-in controls (II-4 and II-6) of WZD200 pedigree were performed by BGI (Shenzhen, China). High-quality genomic DNA (3 μg) was captured by hybridization using the SureSelect XT Human All Exon 50Mb kit (Agilent Technologies). Samples were prepared according to the manufacturer's instructions. Each captured library was run on a HiSeq 2000 instrument, and sequences were generated as 90-bp pair-end reads. An average of 82 million paired reads were generated per sample, the mean duplication rate was 6.37%, and 98% of the targeted region was covered by at least 50 × mean depth. All sequencing reads were mapped to the human reference genome (GRCh37) at UCSC. The software SOAPsnp was used to assemble the consensus sequence and call genotypes in target regions. GATK (Indel Genotyper version 1.0) was used for indel detection. The threshold for filtering SNPs included the following criteria. SNP quality score should be ≧20; sequencing depth should be between 4 and 200; estimated copy number should be no more than 2; and the distance between two SNPs should be larger than 5.
Mutation analysis of YARS2 gene
Five pairs of primers for PCR-amplifying exons and their flanking sequences, including splicing-donor and acceptor-consensus sequences of YARS2, were used for this analysis, as described previously (32). Fragments spanning five exons and flanking sequences from seven matrilineal relatives (II-5, II-7, III-3, III-4, III-5, III-6, and III-7) and three married-in controls (I-1, II-4, and II-6) carrying the m.7511A>G mutation in the Chinese family and two genetically unrelated Chinese controls were PCR-amplified, purified, and subsequently analyzed by Sanger sequencing. These sequence results were compared with the YARS2 genomic sequence (RefSeq NC_000012.12). Genotyping for the c.572G>T mutation in other subjects was PCR-amplified for exon 1 and followed by digestion of the 626-bp segment with the restriction enzyme Tsp45I. The forward and reverse primers for exon 1 are 5′-GACTCGCTTCATGTGGGTCAT-3′ and 5′-CGAAGGGCAGCAACTACAATC-3′, respectively. The Tsp45I-digested products were analyzed on 10% polyacrylamide gel (Fig. S1C).
Cell lines and culture conditions
Lymphoblastoid cell lines were immortalized by transformation with the Epstein–Barr virus, as described elsewhere (62). Cell lines derived from five members of the Chinese family (hearing-impaired subjects II-2 and III-4 harboring both m.7511A>G and heterozygous c.572G>T mutations, II-1 and II-5 carrying both m.7511A>G and homozygous c.572G>T mutations, a hearing-normal individual (I-1) bearing only the homozygous c.572G>T mutation, and one hearing-normal subject (III-7) carrying only the m.7511A>G mutation) and two genetically unrelated control individuals (A61 and A62) lacking these mutations (Table S3) were grown in the RPMI 1640 medium (Invitrogen) supplemented with 10% fetal bovine serum.
UV melting assays
UV melting assays were carried out as described previously (50, 63). The WT and mutant tRNASer(UCN) transcripts were generated as detailed elsewhere (64). The transcripts were diluted in buffer including 50 mm sodium phosphate (pH 7.0), 50 mm NaCl, 5 mm MgCl2, and 0.1 mm EDTA. Absorbance against temperature melting curves were measured at 260 nm with a heating rate of 0.5 °C/min from 25 to 95 °C through an Agilent Cary 100 UV spectrophotometer.
Mitochondrial tRNA analysis
Total mitochondrial RNAs were obtained from mitochondria isolated from lymphoblastoid cell lines (∼2.0 × 108 cells), as described previously (65). The tRNA Northern blot analysis was performed as detailed elsewhere (63). Oligodeoxynucleotide for tRNASer(UCN), tRNATyr, tRNALys, tRNAMet, tRNALeu(UUR), tRNALeu(CUN), tRNAAsp, tRNAGlu, and 5S rRNA were as detailed elsewhere (55). The hybridization and quantification of density in each band were performed as detailed previously (63).
The aminoacylation assays were carried out as detailed previously (63, 66). To further distinguish nonaminoacylated tRNA from aminoacylated tRNA, total RNAs were treated by heat shock for 10 min at 60 °C at pH 9.0 and then run in parallel (63, 66). DIG-labeled oligodeoxynucleotide probes for tRNASer(UCN), tRNATyr, tRNASer(AGY), tRNALeu(UUR), tRNALys, and tRNAThr were as described above. Quantification of density in each band was performed as detailed previously (63, 66).
For the tRNA mobility shift assay, 2 μg of total mitochondrial RNAs were electrophoresed through a 10% polyacrylamide native gel at room temperature in 50 mm Tris-glycine buffer. After electrophoresis, the gels were treated according to the procedure for the tRNA Northern blot analysis described above.
Western blot analysis
Western blot analysis was performed as detailed previously (17, 32). The antibodies used for this investigation were from Abcam (GAPDH (ab8245), ND1 (ab74257), ND5 (ab92624), ND6 (ab81212), CO1 (ab14705), ATP6 (ab101908), NDUFS3 (ab14711), and total OXPHOS human WB antibody mixture (ab110411)), Novus (ND4 (NBP2-47365)), and Proteintech (VDAC (10866-1-AP), CYTB (55090-1-AP), and COX10 (10611-2-AP)). Peroxidase Affini Pure goat anti-mouse IgG and goat anti-rabbit IgG (Jackson) were used as a secondary antibody, and protein signals were detected using the ECL system (CWBIO). Quantification of density in each band was performed as detailed previously (17, 32).
Assays of activities of respiratory complexes
The enzymatic activities of complex I, II, III, and IV were assayed as detailed elsewhere (33, 67, 68). Briefly, complex I (NADH ubiquinone oxidoreductase) activity was determined by following the oxidation of NADH with ubiquinone as the electron acceptor. complex III (ubiquinone cytochrome c oxidoreductase) activity was measured as the reduction of cytochrome c (III) using d-ubiquinol-2 as the electron donor. The activity of complex IV (cytochrome c oxidase) was monitored by following the oxidation of cytochrome c (II).
Measurements of oxygen consumption
OCR in lymphoblastoid cell lines were measured with a Seahorse Bioscience XF-96 extracellular flux analyzer (Seahorse Bioscience), as detailed previously (17, 35, 36).
ATP measurements
The Cell Titer-Glo® luminescent cell viability assay kit (Promega) was used for the measurement of cellular and mitochondrial ATP levels, according to the modified manufacturer's instructions (17, 68).
Assessment of mitochondrial membrane potential
The JC-10 Assay Kit-Microplate (Abcam) was used to assess the mitochondrial membrane potential, according to a modification of the manufacturer's instructions (37).
Measurement of ROS production
ROS measurements were performed following the procedures detailed previously (40, 50, 69).
Computer analysis
Statistical analysis was performed using the unpaired, two-tailed Student's t test contained in the Microsoft Excel program (version 2017). Differences were considered significant at p < 0.05.
Author contributions
W. F., J. Z., W. K., L. C., M. A., Q. Y., M. W., X. T., and M.-X. G. data curation; W. F., J. Z., Q. Y., X. C., J. Q. M., and M.-X. G. formal analysis; W. F., M. A., M. W., and M.-X. G. investigation; W. F., J. Z., L. C., and N. S. methodology; W. F. writing-original draft; J. Z., W. K., Y. C., J. Q. M., W. G., and M.-X. G. resources; J. Z., Y. C., and M.-X. G. funding acquisition; N. S. validation; W. G. and M.-X. G. supervision; M.-X. G. conceptualization; M.-X. G. project administration; M.-X. G. writing-review and editing.
Supplementary Material
Acknowledgments
We are grateful to the patients and their family members for participating.
This work was supported by Ministry of Science and Technology of Zhejiang Province Grant 2018C03026; National Key Technologies R&D Program Grant 2014CB541704 from the Ministry of Science and Technology of China (to M.-X. G.); and National Natural Science Foundation of China Grants 81330024, 31671305, and 81600817 (to M.-X. G., Y. C., and J. Z., respectively). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1–S3 and Figs. S1 and S2.
- mtDNA
- mitochondrial DNA
- OXPHOS
- oxidative phosphorylation system
- ROS
- reactive oxygen species
- DIG
- digoxigenin
- OCR
- oxygen consumption rate(s)
- FCCP
- carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase.
References
- 1. Zheng J., Ji Y., and Guan M. X. (2012) Mitochondrial tRNA mutations associated with deafness. Mitochondrion 12, 406–413 10.1016/j.mito.2012.04.001 [DOI] [PubMed] [Google Scholar]
- 2. Abbott J. A., Francklyn C. S., and Robey-Bond S. M. (2014) Transfer RNA and human disease. Front. Genet. 5, 158 10.3389/fgene.2014.00158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sissler M., González-Serrano L. E., and Westhof E. (2017) Recent advances in mitochondrial aminoacyl-tRNA synthetases and disease. Trends Mol. Med. 23, 693–708 10.1016/j.molmed.2017.06.002 [DOI] [PubMed] [Google Scholar]
- 4. Boczonadi V., Ricci G., and Horvath R. (2018) Mitochondrial DNA transcription and translation: clinical syndromes. Essays Biochem. 62, 321–340 10.1042/EBC20170103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Suzuki T., Nagao A., and Suzuki T. (2011) Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu. Rev. Genet. 45, 299–329 10.1146/annurev-genet-110410-132531 [DOI] [PubMed] [Google Scholar]
- 6. Andrews R. M., Kubacka I., Chinnery P. F., Lightowlers R. N., Turnbull D. M., and Howell N. (1999) Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 23, 147 10.1038/13779 [DOI] [PubMed] [Google Scholar]
- 7. Wallace D. C. (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 39, 359–407 10.1146/annurev.genet.39.110304.095751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ojala D., Montoya J., and Attardi G. (1981) tRNA punctuation model of RNA processing in human mitochondria. Nature 290, 470–474 10.1038/290470a0 [DOI] [PubMed] [Google Scholar]
- 9. Mercer T. R., Neph S., Dinger M. E., Crawford J., Smith M. A., Shearwood A. M., Haugen E., Bracken C. P., Rackham O., Stamatoyannopoulos J. A., Filipovska A., and Mattick J. S. (2011) The human mitochondrial transcriptome. Cell 146, 645–658 10.1016/j.cell.2011.06.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Attardi G., and Schatz G. (1988) Biogenesis of mitochondria. Annu. Rev. Cell Biol. 4, 289–333 10.1146/annurev.cb.04.110188.001445 [DOI] [PubMed] [Google Scholar]
- 11. Hällberg B. M., and Larsson N. G. (2014) Making proteins in the powerhouse. Cell Metab. 20, 226–240 10.1016/j.cmet.2014.07.001 [DOI] [PubMed] [Google Scholar]
- 12. Florentz C., Sohm B., Tryoen-Tóth P., Pütz J., and Sissler M. (2003) Human mitochondrial tRNAs in health and disease. Cell Mol. Life Sci. 60, 1356–1375 10.1007/s00018-003-2343-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guan M. X., Enriquez J. A., Fischel-Ghodsian N., Puranam R. S., Lin C. P., Maw M. A., and Attardi G. (1998) The deafness-associated mitochondrial DNA mutation at position 7445, which affects tRNASer(UCN) precursor processing, has long-range effects on NADH dehydrogenase subunit ND6 gene expression. Mol. Cell Biol. 18, 5868–5879 10.1128/MCB.18.10.5868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yan H., Zareen N., and Levinger L. (2006) Naturally occurring mutations in human mitochondrial pre-tRNASer(UCN) can affect the transfer ribonuclease Z cleavage site, processing kinetics, and substrate secondary structure. J. Biol. Chem. 281, 3926–3935 10.1074/jbc.M509822200 [DOI] [PubMed] [Google Scholar]
- 15. Wang M., Liu H., Zheng J., Chen B., Zhou M., Fan W., Wang H., Liang X., Zhou X., Eriani G., Jiang P., and Guan M. X. (2016) A deafness- and diabetes-associated tRNA mutation causes deficient pseudouridinylation at position 55 in tRNAGlu and mitochondrial dysfunction. J. Biol. Chem. 291, 21029–21041 10.1074/jbc.M116.739482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang M., Peng Y., Zheng J., Zheng B., Jin X., Liu H., Wang Y., Tang X., Huang T., Jiang P., and Guan M. X. (2016) A deafness-associated tRNAAsp mutation alters the m1G37 modification, aminoacylation and stability of tRNAAsp and mitochondrial function. Nucleic Acids Res. 44, 10974–10985 10.1093/nar/gkw726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gong S., Peng Y., Jiang P., Wang M., Fan M., Wang X., Zhou H., Li H., Yan Q., Huang T., and Guan M. X. (2014) A deafness-associated tRNAHis mutation alters the mitochondrial function, ROS production and membrane potential. Nucleic Acids Res. 42, 8039–8048 10.1093/nar/gku466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pierce S. B., Gersak K., Michaelson-Cohen R., Walsh T., Lee M. K., Malach D., Klevit R. E., King M. C., and Levy-Lahad E. (2013) Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am. J. Hum. Genet. 92, 614–620 10.1016/j.ajhg.2013.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou X. L., He L. X., Yu L. J., Wang Y., Wang X. J., Wang E. D., and Yang T. (2017) Mutations in KARS cause early-onset hearing loss and leukoencephalopathy: potential pathogenic mechanism. Hum. Mutat. 38, 1740–1750 10.1002/humu.23335 [DOI] [PubMed] [Google Scholar]
- 20. Schwartzentruber J., Buhas D., Majewski J., Sasarman F., Papillon-Cavanagh S., Thiffaut I., Sheldon K. M., Massicotte C., Patry L., Simon M., Zare A. S., McKernan K. J., FORGE Canada Consortium, Michaud J., Boles R. G., et al. (2014) Mutation in the nuclear-encoded mitochondrial isoleucyl-tRNA synthetase IARS2 in patients with cataracts, growth hormone deficiency with short stature, partial sensorineural deafness, and peripheral neuropathy or with Leigh syndrome. Hum. Mutat. 35, 1285–1289 10.1002/humu.22629 [DOI] [PubMed] [Google Scholar]
- 21. Simon M., Richard E. M., Wang X., Shahzad M., Huang V. H., Qaiser T. A., Potluri P., Mahl S. E., Davila A., Nazli S., Hancock S., Yu M., Gargus J., Chang R., Al-Sheqaih N., et al. (2015) Mutations of human NARS2, encoding the mitochondrial asparaginyl-tRNA synthetase, cause nonsyndromic deafness and Leigh syndrome. PLoS Genet. 11, e1005097 10.1371/journal.pgen.1005097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guan M. X., Yan Q., Li X., Bykhovskaya Y., Gallo-Teran J., Hajek P., Umeda N., Zhao H., Garrido G., Mengesha E., Suzuki T., del Castillo I., Peters J. L., Li R., Qian Y., et al. (2006) Mutation in TRMU related to transfer RNA modification modulates the phenotypic expression of the deafness-associated mitochondrial 12S ribosomal RNA mutations. Am. J. Hum. Genet. 79, 291–302 10.1086/506389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meng F., Cang X., Peng Y., Li R., Zhang Z., Li F., Fan Q., Guan A. S., Fischel-Ghosian N., Zhao X., and Guan M. X. (2017) Biochemical evidence for a nuclear modifier allele (A10S) in TRMU (methylaminomethyl-2-thiouridylate-methyltransferase) related to mitochondrial tRNA modification in the phenotypic manifestation of deafness-associated 12S rRNA mutation. J. Biol. Chem. 292, 2881–2892 10.1074/jbc.M116.749374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lovato M. A., Chihade J. W., and Schimmel P. (2001) Translocation within the acceptor helix of a major tRNA identity determinant. EMBO J. 20, 4846–4853 10.1093/emboj/20.17.4846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Giegé R., Sissler M., and Florentz C. (1998) Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 26, 5017–5035 10.1093/nar/26.22.5017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li X., Fischel-Ghodsian N., Schwartz F., Yan Q., Friedman R. A., and Guan M. X. (2004) Biochemical characterization of the mitochondrial tRNASer(UCN) T7511C mutation associated with nonsyndromic deafness. Nucleic Acids Res. 32, 867–877 10.1093/nar/gkh226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sue C. M., Tanji K., Hadjigeorgiou G., Andreu A. L., Nishino I., Krishna S., Bruno C., Hirano M., Shanske S., Bonilla E., Fischel-Ghodsian N., DiMauro S., and Friedman R. (1999) Maternally inherited hearing loss in a large kindred with a novel T7511C mutation in the mitochondrial DNA tRNASer(UCN) gene. Neurology 52, 1905–1908 10.1212/WNL.52.9.1905 [DOI] [PubMed] [Google Scholar]
- 28. Tang X., Zheng J., Ying Z., Cai Z., Gao Y., He Z., Yu H., Yao J., Yang Y., Wang H., Chen Y., and Guan M. X. (2015) Mitochondrial tRNASer(UCN) variants in 2651 Han Chinese subjects with hearing loss. Mitochondrion 23, 17–24 10.1016/j.mito.2015.05.001 [DOI] [PubMed] [Google Scholar]
- 29. Chapiro E., Feldmann D., Denoyelle F., Sternberg D., Jardel C., Eliot M. M., Bouccara D., Weil D., Garabédian E. N., Couderc R., Petit C., and Marlin S. (2002) Two large French pedigrees with non syndromic sensorineural deafness and the mitochondrial DNA T7511C mutation: evidence for a modulatory factor. Eur. J. Hum. Genet. 10, 851–856 10.1038/sj.ejhg.5200894 [DOI] [PubMed] [Google Scholar]
- 30. Li R., Ishikawa K., Deng J. H., Heman-Ackah S., Tamagawa Y., Yang L., Bai Y., Ichimura K., and Guan M. X. (2005) Maternally inherited nonsyndromic hearing loss is associated with the T7511C mutation in the mitochondrial tRNASer(UCN) gene in a Japanese family. Biochem. Biophys. Res. Commun. 328, 32–37 10.1016/j.bbrc.2004.12.140 [DOI] [PubMed] [Google Scholar]
- 31. Bonnefond L., Fender A., Rudinger-Thirion J., Giegé R., Florentz C., and Sissler M. (2005) Toward the full set of human mitochondrial aminoacyl-tRNA synthetases: characterization of AspRS and TyrRS. Biochemistry 44, 4805–4816 10.1021/bi047527z [DOI] [PubMed] [Google Scholar]
- 32. Jiang P., Jin X., Peng Y., Wang M., Liu H., Liu X., Zhang Z., Ji Y., Zhang J., Liang M., Zhao F., Sun Y. H., Zhang M., Zhou X., Chen Y., et al. (2016) The exome sequencing identified the mutation in YARS2 encoding the mitochondrial tyrosyl-tRNA synthetase as a nuclear modifier for the phenotypic manifestation of Leber's hereditary optic neuropathy-associated mitochondrial DNA mutation. Hum. Mol. Genet. 25, 584–596 10.1093/hmg/ddv498 [DOI] [PubMed] [Google Scholar]
- 33. Thorburn D. R., Chow C. W., and Kirby D. M. (2004) Respiratory chain enzyme analysis in muscle and liver. Mitochondrion 4, 363–375 10.1016/j.mito.2004.07.003 [DOI] [PubMed] [Google Scholar]
- 34. Scheffler I. E. (2015) Mitochondrial disease associated with complex I (NADH-CoQ oxidoreductase) deficiency. J. Inherit. Metab. Dis. 38, 405–415 10.1007/s10545-014-9768-6 [DOI] [PubMed] [Google Scholar]
- 35. Dranka B. P., Benavides G. A., Diers A. R., Giordano S., Zelickson B. R., Reily C., Zou L., Chatham J. C., Hill B. G., Zhang J., Landar A., and Darley-Usmar V. M. (2011) Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic. Biol. Med. 51, 1621–1635 10.1016/j.freeradbiomed.2011.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brand M. D., and Nicholls D. G. (2011) Assessing mitochondrial dysfunction in cells. Biochem. J. 435, 297–312 10.1042/BJ20110162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reers M., Smiley S. T., Mottola-Hartshorn C., Chen A., Lin M., and Chen L. B. (1995) Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 260, 406–417 10.1016/0076-6879(95)60154-6 [DOI] [PubMed] [Google Scholar]
- 38. Hayashi G., and Cortopassi G. (2015) Oxidative stress in inherited mitochondrial diseases. Free Radic. Biol. Med. 88, 10–17 10.1016/j.freeradbiomed.2015.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lenhard B., Orellana O., Ibba M., and Weygand-Durasević I. (1999) RNA recognition and evolution of determinants in seryl-tRNA synthesis. Nucleic Acids Res. 27, 721–729 10.1093/nar/27.3.721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jia Z., Zhang Y., Li Q., Ye Z., Liu Y., Fu C., Cang X., Wang M., and Guan M. X. (2019) A coronary artery disease-associated tRNAThr mutation altered mitochondrial function, apoptosis and angiogenesis. Nucleic Acids Res. 47, 2056–2074 10.1093/nar/gky1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hou Y. M., and Schimmel P. (1988) A simple structural feature is a major determinant of the identity of a transfer RNA. Nature 333, 140–145 10.1038/333140a0 [DOI] [PubMed] [Google Scholar]
- 42. Steiner R. E., and Ibba M. (2019) Regulation of tRNA-dependent translational quality control. IUBMB Life 71, 1150–1157 10.1002/iub.2080 [DOI] [PubMed] [Google Scholar]
- 43. Naganuma M., Sekine S., Chong Y. E., Guo M., Yang X. L., Gamper H., Hou Y. M., Schimmel P., and Yokoyama S. (2014) The selective tRNA aminoacylation mechanism based on a single G·U pair. Nature 510, 507–511 10.1038/nature13440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chong Y. E., Guo M., Yang X. L., Kuhle B., Naganuma M., Sekine S. I., Yokoyama S., and Schimmel P. (2018) Distinct ways of G:U recognition by conserved tRNA binding motifs. Proc. Natl. Acad. Sci. U.S.A. 115, 7527–7532 10.1073/pnas.1807109115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sun L., Gomes A. C., He W., Zhou H., Wang X., Pan D. W., Schimmel P., Pan T., and Yang X. L. (2016) Evolutionary gain of alanine mischarging to noncognate tRNAs with a G4:U69 base pair. J. Am. Chem. Soc. 138, 12948–12955 10.1021/jacs.6b07121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kuncha S. K., Mazeed M., Singh R., Kattula B., Routh S. B., and Sankaranarayanan R. (2018) A chiral selectivity relaxed paralog of DTD for proofreading tRNA mischarging in Animalia. Nat. Commun. 9, 511 10.1038/s41467-017-02204-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li R., and Guan M. X. (2010) Human mitochondrial leucyl-tRNA synthetase corrects mitochondrial dysfunctions due to the tRNALeu(UUR) A3243G mutation, associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms and diabetes. Mol. Cell Biol. 30, 2147–2154 10.1128/MCB.01614-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chomyn A., Enriquez J. A., Micol V., Fernandez-Silva P., and Attardi G. (2000) The mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episode syndrome-associated human mitochondrial tRNALeu(UUR) mutation causes aminoacylation deficiency and concomitant reduced association of mRNA with ribosomes. J. Biol. Chem. 275, 19198–19209 10.1074/jbc.M908734199 [DOI] [PubMed] [Google Scholar]
- 49. Enriquez J. A., Chomyn A., and Attardi G. (1995) MtDNA mutation in MERRF syndrome causes defective aminoacylation of tRNALys and premature translation termination. Nat. Genet. 10, 47–55 10.1038/ng0595-47 [DOI] [PubMed] [Google Scholar]
- 50. Zhou M., Wang M., Xue L., Lin Z., He Q., Shi W., Chen Y., Jin X., Li H., Jiang P., and Guan M. X. (2017) A hypertension-associated mitochondrial DNA mutation alters the tertiary interaction and function of tRNALeu(UUR). J. Biol. Chem. 292, 13934–13946 10.1074/jbc.M117.787028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jiang P., Wang M., Xue L., Xiao Y., Yu J., Wang H., Yao J., Liu H., Peng Y., Liu H., Li H., Chen Y., Guan M. X. (2016) A hypertension-associated tRNAAla mutation alters tRNA metabolism and mitochondrial function. Mol. Cell Biol. 36, 1920–1930 10.1128/MCB.00199-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Meng F., He Z., Tang X., Zheng J., Jin X., Zhu Y., Ren X., Zhou M., Wang M., Gong S., Mo J. Q., Shu Q., and Guan M. X. (2018) Contribution of the tRNAIle 4317A→G mutation to the phenotypic manifestation of the deafness-associated mitochondrial 12S rRNA 1555A→G mutation. J. Biol. Chem. 293, 3321–3334 10.1074/jbc.RA117.000530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang Q., Zhang L., Chen D., He X., Yao S., Zhang Z., Chen Y., and Guan M. X. (2018) Deletion of Mtu1 (Trmu) in zebrafish revealed the essential role of tRNA modification in mitochondrial biogenesis and hearing function. Nucleic Acids Res. 46, 10930–10945 10.1093/nar/gky758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guan M. X., Fischel-Ghodsian N., and Attardi G. (1996) Biochemical evidence for nuclear gene involvement in phenotype of non-syndromic deafness associated with mitochondrial 12S rRNA mutation. Hum. Mol. Genet. 5, 963–971 10.1093/hmg/5.7.963 [DOI] [PubMed] [Google Scholar]
- 55. Zhao X., Cui L., Xiao Y., Mao Q., Aishanjiang M., Kong W., Liu Y., Chen H., Hong F., Jia Z., Wang M., Jiang P., and Guan M. X. (2019) Hypertension-associated mitochondrial DNA 4401A>G mutation caused the aberrant processing of tRNAMet, all 8 tRNAs and ND6 mRNA in the light-strand transcript. Nucleic Acids Res. 47, 10340–10356 10.1093/nar/gkz742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ceriani F., Pozzan T., and Mammano F. (2016) Critical role of ATP-induced ATP release for Ca2+ signaling in nonsensory cell networks of the developing cochlea. Proc. Natl. Acad. Sci. U.S.A. 113, E7194–E7201 10.1073/pnas.1616061113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fischel-Ghodsian N. (1999) Mitochondrial deafness mutations reviewed. Hum. Mutat. 13, 261–270 [DOI] [PubMed] [Google Scholar]
- 58. Guan M. X. (2004) Molecular pathogenetic mechanism of maternally inherited deafness. Ann. N.Y. Acad. Sci. 1011, 259–271 10.1196/annals.1293.025 [DOI] [PubMed] [Google Scholar]
- 59. Zhao H., Li R., Wang Q., Yan Q., Deng J. H., Han D., Bai Y., Young W. Y., and Guan M. X. (2004) Maternally inherited aminoglycoside-induced and nonsyndromic deafness is associated with the novel C1494T mutation in the mitochondrial 12S rRNA gene in a large Chinese family. Am. J. Hum. Genet. 74, 139–152 10.1086/381133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yan X., Wang X., Wang Z., Sun S., Chen G., He Y., Mo J. Q., Li R., Jiang P., Lin Q., Sun M., Li W., Bai Y., Zhang J., Zhu Y. et al. (2011) Maternally transmitted late-onset non-syndromic deafness is associated with the novel heteroplasmic T12201C mutation in the mitochondrial tRNAHis gene. J. Med. Genet. 48, 682–690 10.1136/jmedgenet-2011-100219 [DOI] [PubMed] [Google Scholar]
- 61. Rieder M. J., Taylor S. L., Tobe V. O., and Nickerson D. A. (1998) Automating the identification of DNA variations using quality-based fluorescence re-sequencing: analysis of the human mitochondrial genome. Nucleic Acids Res. 26, 967–973 10.1093/nar/26.4.967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Miller G., and Lipman M. (1973) Release of infectious Epstein-Barr virus by transformed marmoset leukocytes. Proc. Natl. Acad. Sci. U.S.A. 70, 190–194 10.1073/pnas.70.1.190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhou M., Xue L., Chen Y., Li H., He Q., Wang B., Meng F., Wang M., and Guan M. X. (2018) A hypertension-associated mitochondrial DNA mutation introduces an m1G37 modification into tRNAMet, altering its structure and function. J. Biol. Chem. 293, 1425–1438 10.1074/jbc.RA117.000317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li Y., Chen J., Wang E., and Wang Y. (1999) T7 RNA polymerase transcription of Escherichia coli isoacceptors tRNALeu. Sci. China C Life Sci. 42, 185–190 10.1007/BF02880055 [DOI] [PubMed] [Google Scholar]
- 65. King M. P., and Attardi G. (1993) Post-transcriptional regulation of the steady-state levels of mitochondrial tRNAs in HeLa cells. J. Biol. Chem. 268, 10228–10237 [PubMed] [Google Scholar]
- 66. Enríquez J. A., and Attardi G. (1996) Analysis of aminoacylation of human mitochondrial tRNAs. Methods Enzymol. 264, 183–196 10.1016/S0076-6879(96)64019-1 [DOI] [PubMed] [Google Scholar]
- 67. Bourgeron T., Rustin P., Chretien D., Birch-Machin M., Bourgeois M., Viegas-Péquignot E., Munnich A., and Rötig A. (1995) Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat. Genet. 11, 144–149 10.1038/ng1095-144 [DOI] [PubMed] [Google Scholar]
- 68. Zhang J., Ji Y., Lu Y., Fu R., Xu M., Liu X., and Guan M. X. (2018) Leber's hereditary optic neuropathy (LHON)-associated ND5 12338T>C mutation altered the assembly and function of complex I, apoptosis and mitophagy. Hum. Mol. Genet. 27, 1999–2011 10.1093/hmg/ddy107 [DOI] [PubMed] [Google Scholar]
- 69. Mahfouz R., Sharma R., Lackner J., Aziz N., and Agarwal A. (2009) Evaluation of chemiluminescence and flow cytometry as tools in assessing production of hydrogen peroxide and superoxide anion in human spermatozoa. Fertil. Steril. 92, 819–827 10.1016/j.fertnstert.2008.05.087 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.