

Abstract
The protein quality control machinery of the endoplasmic reticulum (ERQC) ensures that client proteins are properly folded. ERQC substrates may be recognized as nonnative by the presence of exposed hydrophobic surfaces, free thiols, or processed N-glycans. How these features dictate which ERQC pathways engage a given substrate is poorly understood. Here, using metabolic labeling, immunoprecipitations, various biochemical assays, and the human serpin antithrombin III (ATIII) as a model, we explored the role of ERQC systems in mammalian cells. Although ATIII has N-glycans and a hydrophobic core, we found that its quality control depended solely on free thiol content. Mutagenesis of all six Cys residues in ATIII to Ala resulted in its efficient secretion even though the product was not natively folded. ATIII variants with free thiols were retained in the endoplasmic reticulum but not degraded. These results provide insight into the hierarchy of ERQC systems and reveal a fundamental vulnerability of ERQC in a case of reliance on the thiol-dependent quality control pathway.
Keywords: endoplasmic reticulum (ER), N-linked glycosylation, disulfide, protein folding, unfolded protein response (UPR), endoplasmic reticulum-associated protein degradation (ERAD), serpin, ER-mediated protein quality control (ERQC), protein homeostasis, protein maturation, UPR
Introduction
Protein maturation is an error-prone process, which, if allowed to proceed unchecked, would cause major cellular dysfunction. To prevent such calamities, quality control processes monitor the integrity of maturing polypeptide chains. For proteins that traverse the endoplasmic reticulum (ER),2 including secretory and plasma membrane proteins and proteins that reside in endomembrane compartments, proteins evaluated as native are allowed to exit to the Golgi. Proteins deemed nonnative are initially targeted for ER retention and potential repair. If the nonnative properties persist and the proteins accumulate in the ER, two processes can be initiated (1, 2). The unfolded protein response (UPR) is activated, causing a transcription-based remodeling of the ER contents in an attempt to maintain protein homeostasis (3, 4). If the interrogated proteins are determined to be irreparably or terminally misfolded, they are eventually targeted for destruction to recycle components and ensure that a potentially toxic misfolded substrate is not released from the ER or cell (5, 6).
A small number of quality control factors are responsible for evaluating the thousands of different client proteins that traverse the ER (7–9). Therefore, general protein hallmarks of foldedness must be queried to provide an efficient and plastic quality control process that supports the evaluation of a large number of substrates. Regardless of cellular location, exposed hydrophobic regions appear to be a defining signature of folding intermediates, misfolded proteins, or unassembled oligomers—all forms of protein that are frequently recognized by molecular chaperones and should be retained in the ER (10, 11). The ER is also an oxidizing environment that supports the formation of disulfide bonds assisted by a family of ER-resident oxidoreductases (12). These oxidoreductases are involved in a thiol-dependent quality control process (13–15). The vast majority of proteins that travel through the mammalian secretory pathway are also modified in the ER with multiple 14-residue N-linked oligosaccharides with the composition Glc3Man9GlcNAc2 (16). The maturation of the glycan provides a quality control code that reports on the fitness of the attached protein (17). Glycosidases and transferases orchestrate the glycan composition based on protein structural features, supporting binding and sorting of glycoproteins by quality control carbohydrate-binding factors. Among these are the lectins calnexin and calreticulin, which bind monoglucosylated glycans (18–20). Whereas the use of multiple quality control interrogation mechanisms that are based on the features of the specific protein is thought to maximize coverage and minimize mistakes, the interplay between these various quality control mechanisms is poorly understood. For instance, if there is overlap or redundancy in coverage, how is it determined which quality control process dominates? Or can multiple pathways query the same protein?
Serpins have been used extensively as model substrates to study protein quality control, as mutations in several inhibitory serpins are associated with degradation and accumulation of these proteins in the ER (21, 22). Functional inhibitory serpins are metastable; they fold to a kinetically trapped state that allows them to store potential energy, which is deployed to inhibit their cognate protease (22–25). This metastability and the need to avoid alternative conformations that might be more stable when folding likely make serpin maturation particularly problematic and may provide an explanation for why serpin misfolding is associated with a large number of pathologies, or serpinopathies (26). Functional, properly folded, inhibitory serpins form inactive covalent complexes, with their cognate proteases providing a simple activity assay to determine whether a secreted serpin has successfully folded.
The human serpin antithrombin III (ATIII) is an inhibitory serpin that is modified by multiple N-linked glycans. The three native ATIII disulfide bonds have been used to map its cellular folding pathway (27). ATIII inhibits thrombin, thus playing an essential role in the blood coagulation cascade. Mutations in ATIII are commonly associated with thrombosis, and some of these mutations also lead to accumulation of ATIII in the ER (25, 28). Therefore, ATIII provides an apt model substrate to explore various ER quality control processes. Here, we made use of ATIII variants with disrupted folding by mutating native Cys residues to Ala. We hypothesized that these variants would be targets for the thiol-based quality control pathway. We also predicted that these variants would be compromised in their folding in such a way that they would present nonnative hydrophobic surfaces and potentially glycan signals. Thus, these variants should reveal the interplay and hierarchy of ER quality control systems.
We found that quality control of ATIII relied solely on thiol-dependent quality control, and diversion from the thiol-dependent quality control pathway led to improper secretion of misfolded and inactive protein. These results demonstrate that ER quality control pathways do not necessarily act redundantly and the general features of a substrate do not automatically dictate which quality control pathways will be engaged.
Results
Misfolded and inactive Cys-less ATIII is efficiently secreted
As shown previously, the three intramolecular disulfide bonds of ATIII (Cys-8/Cys-128, Cys-21/Cys-95, and Cys-247/Cys-430) are required for folding to the functional, metastable conformation (27). We began to explore how the ERQC handles misfolded ATIII by determining the fate of ATIII with all of its Cys residues mutated to Ala (Cys-less ATIII). Chinese hamster ovary (CHO) cells transfected with WT or Cys-less ATIII were pulsed for 30 min with [35S]Met/Cys and chased for the designated times (Fig. 1A). ATIII from the lysates and media was then immunoisolated using antibodies directed toward the C-terminal Myc tag present on the ATIII constructs. Samples were analyzed by reducing SDS-PAGE and phosphorimaging. As expected, secreted ATIII shows two bands due to incomplete glycosylation at Asn-135 (29). Surprisingly, the level of secretion of WT and Cys-less ATIII was similar, reaching levels in the media after 3 h of chase of 48.0 and 43.6%, respectively (Fig. 1, A and B). Strikingly, the remaining in-cell ATIII fraction for both constructs remained stable, as degradation did not appear to occur even for Cys-less ATIII (Fig. 1A, lanes 1–4 and 9–12).
Figure 1.

Inactive and misfolded ATIII Cys-less is improperly but efficiently secreted. A, ATIII and ATIII Cys-less were expressed in CHO cells. Cells were radiolabeled with [35S]Cys/Met for 30 min and chased for the indicated times. At each time point, cell lysate and media were collected. Cells were lysed in MNT buffer. ATIII was immunoprecipitated using anti-Myc antibodies. Samples were resolved by reducing 9% SDS-PAGE. B, quantification of ATIII secretion from A. The lysate and media were quantified, and ATIII secretion is presented as a percentage of ATIII in the media to ATIII in the 0-h lysate. C, ATIII WT and ATIII Cys-less were expressed in CHO cells and radiolabeled with [35S]Cys/Met for 30 min and chased for 3 h. Cells were lysed in MNT buffer. Cell lysate and media were collected, and ATIII was immunoprecipitated using anti-Myc antibodies and washed with buffer containing 0.5% CHAPS. ATIII was then eluted from the immunoprecipitation beads by incubation with 0.5 mg/ml c-Myc peptide for 1 h at 37 °C. Sample was then evenly split between treated and nontreated samples. 0.648 mg (2 units) of thrombin were added to treated samples, whereas nontreated samples received an equal volume of water. Samples were then incubated at 37 °C for 1 h and resolved by reducing 9% SDS-PAGE. D, ATIII WT and Cys-less were expressed in CHO cells. Cells were radiolabeled with [35S]Cys/Met for 30 min and chased for 3 h, and cell lysate and media were collected. Cells were lysed in MNT buffer. ATIII was immunoprecipitated using anti-Myc antibodies. 0.05 μg of trypsin was added to treated samples, and an equal volume of water was added to nontreated samples. Samples were then incubated for the indicated times at 37 °C and resolved by reducing 9% SDS-PAGE. E, quantification of ATIII remaining post-trypsin degradation from D. F, ATIII WT and Cys-less were expressed in CHO cells. Cells were radiolabeled for 30 min with [35S]Cys/Met and chased for 2 h, and cell lysate and media were collected. Cells were lysed in MNT buffer. ATIII was immunoprecipitated using anti-Myc antibodies. Lysate and medium samples were treated with Endo H or PNGase F or left untreated. Samples were then resolved on a reducing 9% SDS-PAGE. All experiments are representative of three independent experiments. Error bars, S.D.
Normally, the ERQC machinery interrogates proteins as they traverse the secretory pathway, sensing whether their native structures have been reached (8, 30, 31), and only properly folded proteins are packaged into COPII vesicles for eventual secretion, whereas misfolded or nonnative structures are directed for ER retention (32). To assess the completeness of folding of Cys-less ATIII, the activity of the product secreted into the media was analyzed by gel shifts due to its ability to form a covalent inhibitory complex with its target protease, thrombin. When the protease thrombin cleaves the reactive center loop of the serpin ATIII, an acyl bond is formed between the loop of ATIII and thrombin. The formation of the covalent complex can be visualized by a gel shift on SDS-PAGE. Inactive ATIII does not demonstrate this gel shift, whereas partially active ATIII, although not functional as a thrombin inhibitor, can act as a substrate for thrombin while not forming a covalent bond. This leads to ATIII being cleaved and running at a lower molecular weight (Fig. 1C). Cells were pulsed for 30 min and chased for 3 h, and ATIII from lysate and media samples was immunoisolated. Protease inhibition by serpins requires large conformational changes (23, 24). Therefore, prior to performing the activity assay, ATIII was eluted from the beads and antibody using excess Myc-peptide to remove steric constraints. The eluted fraction was equally divided between thrombin-treated and nontreated samples and analyzed by reducing SDS-PAGE. Whereas WT ATIII in the media was active as shown by the formation of a thrombin and ATIII complex (Fig. 1C, lane 4), Cys-less ATIII was completely inactive (Fig. 1C, lane 8). Thrombin not only cannot form a complex with misfolded Cys-less ATIII, it also does not have an accessible reactive center loop, as there was no ATIII cleaved band from the media sample observed upon thrombin addition.
To further characterize the state of folding of the secreted Cys-less ATIII, we used protease sensitivity. A natively folded protein should be relatively resistant to protease digestion as compared with an unfolded protein as, generally, natively folded proteins adopt more compact conformations that expose fewer sites for cleavage. Cells were pulsed for 30 min and chased for 3 h, and ATIII was immunoprecipitated from media samples. Samples were then divided equally between untreated samples and trypsin-treated for either 0, 15, 30, or 60 min. Samples were then analyzed by SDS-PAGE. Secreted Cys-less ATIII showed significantly higher protease sensitivity compared with WT (Fig. 1, D and E). This result indicates that Cys-less ATIII is secreted like a natively folded protein, despite the fact that it is an inactive and significantly misfolded protein. Both the secretion of Cys-less ATIII and its stability in the cell suggest that it is evaluated as properly folded by the ERQC network even though it has not achieved its native, functional fold.
There is a formal possibility that Cys-less ATIII is being improperly secreted and misevaluated by the ER quality control network because it is secreted via an unconventional secretory pathway, thereby escaping ERQC. In traversing the full secretory pathway, glycoproteins trafficked from the ER pass through the Golgi, where their glycans are extensively remodeled to receive complex glycans. However, proteins that pass through recently identified pathways for unconventional secretion, both including and excluding the ER, can bypass portions of the canonical secretory pathway such as the Golgi and thus lack remodeled complex glycans (33, 34). The differential susceptibility of secretory proteins to glycosidases PNGase F, which can cleave both complex and high-mannose glycans acquired in the Golgi, and Endo H, which can only cleave high-mannose glycans, can be used to ask whether a given protein has traversed the canonical secretory pathway. Proteins carrying glycans found in the ER or on secreted proteins that have bypassed the Golgi are sensitive to both PNGase F and Endo H, whereas proteins carrying glycans that have passed through the Golgi are sensitive to PNGase F and resistant to Endo H. Glycosidase sensitivity, analyzed by size shifts using SDS-PAGE, can therefore be used as an assay for unconventional secretion. As expected, both WT and Cys-less ATIII in the cell lysate were sensitive to PNGase F and Endo H, indicating that cellular ATIII mainly resides in the ER. In contrast, both WT and Cys-less ATIII from the cell media were sensitive to PNGase F and resistant to Endo H (Fig. 1E, lanes 5, 6, 11, and 12), showing that both had received complex glycans due to passage through the Golgi. Therefore, the Cys-less variant of ATIII utilizes the conventional secretory pathway.
Taken together, these results indicate that Cys-less ATIII, an inactive and misfolded protein, evades ER protein quality control and instead is secreted similarly to WT ATIII. These results support the hypothesis that free thiols are essential for ERQC recognition of misfolded ATIII and its consequent retention in the ER. Concomitantly, evasion of thiol-dependent quality control in this case allowed improper secretion rather than attempts by a complementary quality control branch to correct the misfolding or action of the ER-associated degradation pathway to eliminate the misfolded product.
Cellular retention and characterization of ATIII disulfide mutants
According to the recently discovered cellular pathway of disulfide bond formation for ATIII, the C-terminal disulfide, Cys-247/Cys-430, must form first in order for the two N-terminal disulfides, Cys-8/Cys-128 and Cys-21/Cys-95, to form properly (27). If the C-terminal disulfide is mutated, this model predicts that the remaining two disulfides would not form, leading to the generation of free thiols that may support ER retention. In contrast, this model predicts that mutating either pair of Cys residues that comprise the two N-terminal disulfides, C8A/C128A or C21A/C95A, allows the two remaining disulfides to form, reconciling all thiols into disulfides. This previous work, which elucidated the disulfide formation pathway of homogeneously glycosylated ATIII missing the partially recognized N-linked glycan at position Asn-135, led to the predictions that ATIII quality control is mediated, at least in part, by free thiols directing ER retention (27).
To characterize the disulfide requirements for proper ATIII quality control and secretion, and to test the predictions for ATIII disulfide mutants, the secretion rates of the three disulfide mutants (C8A/C128A, C21A/C95A, and C247A/C430A) and WT ATIII (all constructs possessing all four native glycosylation sites) were analyzed via pulse-chase in cells. Transfected cells were pulsed for 30 min with [35S]Met/Cys and chased for the designated times. Samples were analyzed by nonreducing and reducing SDS-PAGE and phosphorimaging (Fig. 2A).
Figure 2.

Cellularly retained ATIII C247A/C430A is inactive and contains a higher level of free thiols. A, WT ATIII and ATIII disulfide mutants were expressed in CHO cells. Cells were radiolabeled with [35S]Cys/Met for 30 min and chased for the indicated times. At each time point, cell lysate and media were collected and processed as described in the legend to Fig. 1A. B, quantification of ATIII secretion from A. The lysate and media were quantified, and ATIII secretion is presented as a percentage of ATIII in the media to ATIII in the 10-min lysate. C, ATIII and ATIII disulfide mutants were expressed in CHO cells, radiolabeled with [35S]Cys/Met for 30 min, and chased for 3 h. Cells were lysed in MNT buffer. Cell lysate and media were collected, and ATIII was immunoprecipitated using anti-Myc antibody and washed with 0.5% CHAPS buffer. ATIII was then eluted from the immunoprecipitation beads by incubation with 0.5 mg/ml c-Myc peptide for 1 h at 37 °C. Samples were then evenly split between treated and nontreated samples. 0.648 mg (2 units) of thrombin were added to treated samples, whereas nontreated samples received an equal volume of water. Samples were then incubated at 37 °C for 1 h and resolved by reducing 9% SDS-PAGE. D, WT ATIII and ATIII disulfide mutants were expressed in CHO cells, radiolabeled with [35S]Cys/Met for 30 min, and chased for 30 min. Cells were lysed in MNT buffer. To treated samples, PEG-maleimide was added to a final concentration of 1.4 mm, and to the nontreated samples, N-ethyl maleimide was added to a final concentration of 5 mm. Samples were incubated at room temperature for 30 min. Treated samples were then quenched with DTT at a final concentration of 100 mm. ATIII was immunoprecipitated using anti-Myc antibodies, and samples were resolved by reducing 9% SDS-PAGE. E, quantification of PEG-maleimide–unmodified ATIII from D. The percentage PEG-maleimide–unmodified ATIII was determined by quantifying the amount of unmodified ATIII in PEG-maleimide–treated and –nontreated samples and dividing the treated sample by the untreated sample. All experiments are representative of three independent experiments. Error bars, S.D.
Secretion of C247A/C430A ATIII was significantly reduced compared with WT, C8A/C128A, and C21A/C95A ATIII (Figs. 1B and 2A (lanes 13–15)). After a 2-h chase, WT secretion levels reached 50% compared with 8% for C247A/C430A ATIII. The secretion of the N-terminal disulfide mutants, ATIII C8A/C128A and C21A/C95A, was similar to WT secretion levels. Nonreducing SDS-PAGE displayed an increase in disulfide-linked adducts for all mutants compared with WT ATIII. Similar levels of aggregated ATIII were found in the Triton-insoluble fractions for all constructs (Fig. 2A, lanes 16–18).
Whereas secreted WT ATIII was active, as observed by the formation of the thrombin-ATIII complex (Fig. 2C, lane 4), the disulfide mutants displayed little activity and did not form an inhibitory complex (Fig. 2C, lanes 8, 12, and 16). This demonstrates that all of the ATIII disulfide mutants analyzed are inactive proteins, including C8A/C128A and C21A/C95A ATIII, which were efficiently secreted into the cell media. Thus, like Cys-less ATIII, the ATIII N-terminal disulfide mutants were mistakenly evaluated as natively folded by the ER quality control process. By contrast, the inactive C247A/C430A ATIII construct, which lacked the key early forming disulfide previously shown to be critical for initiation of proper folding, was properly retained.
A question that emerges from these data is whether secretion correlates with protection or inaccessibility of thiols. To address this question, a PEG-maleimide (PEG-Mal) modification gel-shift assay was performed on the ATIII mutants. PEG-Mal modifies exposed free thiols, and the bulky PEG group causes an increase in mass that can be visualized by a gel shift. Transfected cells were pulsed for 30 min and then chased for 30 min. Lysate and medium samples were split equally, and one half was treated with PEG-Mal and analyzed by reducing SDS-PAGE.
For all ATIII constructs analyzed, secreted ATIII found in the media had insignificant levels of PEG-Mal modification, which in turn indicates near absence of accessible free thiols (Fig. 2D, lanes 4, 8, 12, and 16; see Fig. 2E for quantification). This result suggests that in ATIII constructs that are successfully secreted to the media, any thiols had either formed disulfide bonds or were otherwise inaccessible, most likely through partial folding. The level of accessible thiols modifiable by PEG-MAL for WT, C8A/C128A, and C21A/C95A ATIII constructs in cell lysates was similar, ranging from 55 to 72% (Fig. 2E). In contrast, the level of C247A/C430A ATIII modified by PEG-Mal was 89%, arguing that in this construct, there is little sequestration of the Cys thiols. This result supports the model that the disulfide between Cys-247 and Cys-430 is critical for folding, and its absence leads to poor formation of the two N-terminal disulfides (27). Free thiols are present in all forms of ATIII in the cell lysates, including WT, because the intracellular pool of protein is sampled during formation and isomerization of intermediate disulfide bonds. These results suggest that C247A/C430A ATIII is retained in the ER because its folding is impaired so that its free thiols cannot form disulfide bonds, whereas C8A/C128A and C21A/C95A ATIII are secreted because they can initiate folding and hence partially form disulfides, which, together with partial folding, protect their free thiols. These findings support both the hypotheses that the C-terminal disulfide of ATIII must form before the N-terminal disulfides for ATIII to fold correctly and that free thiols lead to retention in the ER.
A single Cys is sufficient to retain ATIII in the ER
As the results in Figs. 1 and 2 suggest that free thiols play a significant role in the ER retention of ATIII, we next investigated whether the presence of a single Cys would be sufficient to retain ATIII in the ER. To this end, six single Cys mutants were created by adding the individual Cys residues back at their natural sites in the Cys-less ATIII background (A8C, A21C, A95C, A128C, A247C, A430C), and their secretion was analyzed by pulse-chase. Cells were pulsed for 30 min and chased for 10, 60, and 120 min. When secretion was analyzed by reducing SDS-PAGE, three single Cys mutants, A8C, A21C, and A247C, were poorly secreted as compared with Cys-less ATIII. A128C and A95C were secreted at a similar level to Cys-less ATIII. Although there was no significant difference between secretion of A430C and Cys-less, A430C secreted to a lower level than Cys-less (Fig. 3, A and B). When secretion was analyzed by nonreducing SDS-PAGE, a high-molecular weight band of secreted single-Cys ATIII was present, suggesting that a portion of secreted single-Cys ATIII mutants were in the form of a redox-dependent complex in which the Cys was no longer a free thiol but rather was in an intermolecular disulfide (Fig. 3A, top NR image). Altogether, these results demonstrate that thiol-dependent quality control is a robust retention mechanism, as a single free thiol led to the cellular retention of ATIII.
Figure 3.

ATIII with a single free thiol is retained in the ER. A, ATIII Cys-less and ATIII single-Cys mutants were expressed in CHO cells. Cells were radiolabeled with [35S]Cys/Met for 30 min and chased for the indicated times. At each time point, cell lysate and media were collected and processed as described in the legend to Fig. 1A. Samples were resolved by both reducing (R) and nonreducing (NR) 9% SDS-PAGE. B, quantification from A. Asterisks, statistical significance relative to Cys-less. All experiments are representative of three individual experiments. Error bars, S.D.
C247A/C430A ATIII is stably retained in the ER in a redox-dependent complex
We sought to obtain a better understanding of the cellular fate of the C247A/C430A ATIII, which was inefficiently secreted. CHO cells expressing WT, Cys-less, or C247A/C430A ATIII were imaged by immunofluorescence confocal microscopy, and co-localization with ER (KDEL) and Golgi (giantin) markers was monitored. All three variants were found throughout the ER, as observed by extensive co-localization with the KDEL ER marker (Fig. 4A). Some co-localization of all three constructs with the Golgi marker was also observed. These results indicated that regardless of their different fates and properties, after 16 h of expression, these three ATIII variants localize throughout the ER.
Figure 4.

ATIII C247A/C430A is retained and diffuse throughout the ER. A, ATIII WT, ATIII Cys-less, and ATIII C247A/C430A were expressed in CHO cells. Cells were fixed in buffer containing 3.7% formaldehyde; permeabilized in buffer containing 0.1% Triton X-100; and stained using anti-Myc antibodies, KDEL, and giantin primary antibodies, as indicated, goat anti-mouse IgG secondary antibody, and DAPI. Cells were imaged using a confocal epifluorescence microscope at ×100 oil immersion. B, ATIII WT, ATIII Cys-less, and ATIII C247A/C430A were expressed in CHO cells. Cells were lysed in MNT. Media and lysate fractions were collected and split equally between DTT-treated and -nontreated fractions. Sucrose gradients were generated by solubilizing sucrose into MNT, and a 10–40% gradient was established. Samples were then laid on top of the sucrose gradient. Gradients were then centrifuged at 38,000 rpm for 18 h. Samples were taken from the gradient by pipetting 1 ml from the top of the gradient. Samples were TCA-precipitated and then resolved on a 9% reducing SDS-PAGE and imaged by Western blotting with Myc tag antibody.
Despite their similar cellular localization, we hypothesized that misfolded ATIII variants may not be monomeric within the ER. We therefore investigated whether ATIII variants were present in complexes within the ER. Cell lysate was layered on top of a 10–40% sucrose gradient and subjected to ultracentrifugation. Fractions were then analyzed by SDS-PAGE and immunoblotting. Both Cys-less and C247A/C430A ATIII are found in complexes that are larger than WT (Fig. 4B). The addition of the reducing agent DTT after cell lysis causes C247A/C430A ATIII to shift to lower-molecular weight fractions, whereas the addition of DTT had no effect on WT and Cys-less ATIII, suggesting that C247A/C430A is in a complex formed via accessible thiols and that retention of C247A/C430A is redox-dependent. Cys-less ATIII is also in a complex in the media, which may be due to secretion as an aggregate or secretion while bound to a partner. These data suggest that C247A/C430A ATIII is retained in a multimeric, disulfide-dependent complex in the ER, whereas ATIII Cys-less is secreted in a nonmonomeric state.
WT ATIII is more efficiently reglucosylated than Cys-less and C247A/C430A ATIII
The carbohydrate-binding chaperone calnexin and its soluble paralogue calreticulin play an important role in glycoprotein folding, quality control, and ER retention (1, 35–37). Within the calnexin/calreticulin cycle, the folding sensor UDP-glucose:glycoprotein glucosyltransferase 1 (UGGT1) acts as a gatekeeper of secretion by reglucosylating misfolded or incompletely folded proteins, which then allows calnexin/calreticulin to rebind the glycoprotein, leading to ER retention (1, 38, 39). Furthermore, recent X-ray crystal structures have shown that UGGT1 proteins from both Thermomyces dupontii and Chaetomium thermophilum have four thioredoxin-like domains (40, 41). Oxidoreductases are frequently comprised of multiple thioredoxin domains, contributing to their oxidizing, reducing, or isomerizing activities (42, 43). Although the active oxidoreductase motif, Cys-Xaa-Xaa-Cys, is not present in any of these domains, the multiple Cys residues present in UGGT1 and the thioredoxin-like folds likely aid UGGT1 in substrate recognition. We therefore investigated whether UGGT1 reglucosylation, and by extension the N-glycan quality control pathway, contributed to the ER retention of C247A/C430A ATIII.
We have previously developed a cell-based reglucosylation assay (39, 44, 45). Briefly, an Alg6-defective CHO cell line, MI8-5 CHO, generates glycans lacking glucoses on their A-branches (46) (Fig. S1). Therefore, in this cell line, a monoglucosylated glycan can only be generated by UGGT1. In contrast, in WT cells, the presence of a monoglucosylated glycan could indicate either trimming of two glucoses from the original Glc3Man9GlcNAc2 glycan or reglucosylation of an unglucosylated side chain by UGGT1. MI8-5 CHO cells were treated with N-butyl deoxynojirimycin (DNJ) for 30 min prior to the 30-min pulse and then throughout the indicated chase times. DNJ is a glucosidase inhibitor and therefore traps monoglucosylated proteins in their monoglucosylated state. Monoglucosylated proteins were first pulled down using recombinant GSH S-transferase (GST)-calreticulin, and from this pulldown, ATIII was immunoprecipitated using anti-Myc antibodies.
Whereas all three variants of ATIII, WT, Cys-less, and C247A/C430A, were reglucosylated by UGGT1, WT ATIII was found to be modified most efficiently, followed by C247A/C430A ATIII and then Cys-less ATIII (Fig. 5A). As WT ATIII was recognized at a higher level than the other two ATIII variants, UGGT1 modification does not explain the greatly increased ER retention of C247A/C430A.
Figure 5.

Retention of C247A/C430A in the ER does not require UGGT1. A, ATIII variants were expressed in MI8-5 CHO cells. 30 min prior to the pulse and throughout the chase, cells were treated with 0.5 mm DNJ. Cells were radiolabeled with [35S]Cys/Met for 30 min and chased for the indicated times. At each time point, cells were lysed in MNT buffer. 80% of the cell lysate was affinity-purified with GSH S-transferase–tagged calreticulin (GST-CRT), whereas 20% of the cell lysate was immunopurified with anti-Myc antibody. GST-tagged calreticulin affinity purifications were then eluted in buffer containing 1% SDS, diluted in MNT, and immunopurified using anti-Myc tag antibody. Samples were resolved by reducing 9% SDS-PAGE. B, quantification of reglucosylation in A. Percentage reglucosylation for each ATIII variant was calculated by quantifying the bands corresponding to ATIII, multiplying the lysate band by 4, and dividing the amount of ATIII in the sequential IP by the amount of ATIII in the nonsequential IP at each time point. All reglucosylation values are normalized to WT ATIII. C, same as A, except DNJ was added 15 min prior to each time point, not throughout the experiment. D, quantification of reglucosylation in C. Percentage reglucosylation was calculated as described in B. E, ATIII and ATIII C247A/C430A were expressed in both WT and UGGT1−/− MEF cells. Cells were radiolabeled with [35S]Cys/Met for 30 min and chased for the indicated times. At each time point, cell lysate and media were collected and processed as described in the legend to Fig. 1A. F, quantification of ATIII secretion from E. The lysate and media were quantified, and ATIII secretion is presented as a percentage of ATIII in the media to ATIII in the 0-h lysate. All experiments are representative of three independent experiments. Error bars, S.D.
To explore the timing of reglucosylation, DNJ was added for only 15 min prior to each time point, which allows for detecting reglucosylation at that specific window of time. Using this experimental scheme, C247A/C430A ATIII was found to be reglucosylated more than either WT or Cys-less ATIII after 2 h (Fig. 5, C and D). These results suggest that UGGT1 best recognizes WT ATIII, but once WT has folded and trafficked out of the ER, ATIII C247A/C430A becomes a better substrate for modification, as it is still present in the ER where UGGT1 has access to it.
To confirm that UGGT1 is not retaining the free thiol-carrying mutant, C247A/C430A ATIII, the secretion of WT and C247A/C430A ATIII was examined in WT and Uggt1−/− mouse embryonic fibroblast (MEF) cell lines using a pulse-chase approach. Cells were pulsed for 30 min and chased for 0 or 2 h before the media and lysate were collected. ATIII was immunoprecipitated from the media and lysate, and the percentage fraction of ATIII in each pool was analyzed as described previously. WT ATIII was secreted similarly in both WT and Uggt1−/− MEF cells, suggesting that repetitive rounds of calnexin/calreticulin binding were not needed for efficient ATIII secretion (Fig. 5, E and F). In contrast, ATIII C247A/C430A was poorly secreted in either WT or Uggt1−/− MEF cells, indicating that in the absence of UGGT1, the misfolded ATIII mutant C247A/C430A was still efficiently retained within the cell. Therefore, all together, UGGT1 does not appear to play the determinative role in C247A/C430A ATIII quality control.
C247A/C430A ATIII is a poor ER-associated degradation (ERAD) substrate
When a misfolded protein is persistently retained in the ER, the ERAD process generally degrades the protein to maintain proper secretory pathway flow and protein homeostasis (6, 47). Because C247A/C430A ATIII is retained in the ER, we would have expected it to be degraded. The stability of C247A/C430A ATIII was therefore compared with WT ATIII and the classical ERAD substrate α-1-antitrypsin null Hong Kong (A1AT NHK), which is also an inhibitory serpin and serves as a positive control for ERAD.
After an 8-h chase, both WT and C247A/C430A ATIII were stable, with 74 and 115% of the total protein remaining relative to zero time, respectively (Fig. 6, A and B). WT ATIII accumulated in the media, whereas C247A/C430A ATIII largely accumulated in the Triton-insoluble fraction. In contrast, A1AT NHK was turned over rapidly, with 48% total protein remaining after 8 h of chase and only a small fraction being secreted. Interestingly, although ATIII C247A/C430A was retained in the ER, it is a poor ERAD substrate as it was not efficiently degraded.
Figure 6.

ATIII C247A/C430A is poorly degraded. A, ATIII WT and disulfide mutants were expressed in CHO cells. Cells were radiolabeled with [35S]Cys/Met for 30 min and chased for the indicated times. At each time point, cell lysate, media, and Triton X-100–insoluble fractions were collected and processed as described in the legend to Fig. 1A. B, quantification of A. The percentage of each protein remaining was calculated by quantifying ATIII present in each fraction of the reducing gel and dividing by the amount of ATIII immediately after the chase (0 h). The amount of protein in the indicated fraction at each time point is represented as a fraction of the total amount of protein present at that time point. All experiments are representative of three independent experiments. Error bars, S.D.
Overexpression of ATIII constructs activates the IRE1α arm of the UPR pathway
One possible explanation for the lack of degradation of C247A/C430A ATIII retained in the ER is that expression of this impaired protein does not activate the UPR. Thus, we sought to examine whether UPR pathways were induced by overexpression of ATIII variants in CHO cells. The UPR consists of three branches controlled by the ER sensors IRE1α, ATF6, and PERK, and each sensor initiates a signaling cascade, which can increase the folding capacity of the ER, decrease the protein load in the ER, or increase the capacity of ERAD (48). Thus, we carried out experiments to test whether any of the three UPR branches were up-regulated upon expression of WT, C247A/C430A, or Cys-less ATIII or NHK A1AT.
Upon activation, IRE1α splices an unconventional intron from X-box protein 1 mRNA (Xbp1), creating a spliced form of Xbp1 (Xbp1s). A frameshift caused by the splicing supports the translation of an active transcription factor, XBP1s, which leads to the up-regulation of multiple genes involved in protein folding and ERAD (49, 50). Therefore, the production of Xbp1s is indicative of IRE1α activation. Tunicamycin (Tm), which inhibits N-linked glycosylation and leads to a strong activation of the UPR, was used as a positive control. CHO cells were either transfected with the serpin variants or treated with Tm for 24 h. RNA was then collected, and cDNA was generated. In all cases, Xbp1s was generated, suggesting that IRE1α is activated by ATIII/A1AT overexpression regardless of the construct (Fig. 7A). The level of activation during NHK ATIII and A1AT overexpression appears to be less than that of Tm, suggesting that overexpression of the proteins was not maximally activating IRE1α.
Figure 7.

IRE-1 but not ATF6 or PERK is activated by ATIII overexpression. A, ATIII variants and A1AT NHK were expressed in CHO cells for 24 h. Control cells were treated with DMSO or Tm for 24 h. Cells were lysed, and RNA was collected. cDNA was generated and amplified via PCR using XPB-1–specific primers. B, ATIII variants and A1AT NHK were expressed in CHO cells for 24 h. Control cells were treated with either DMSO or Tm for 24 h. Cells were then lysed with MNT and TCA-precipitated. 5% whole-cell lysate was then resolved on a 9% SDS-PAGE and imaged by Western blotting using anti-Myc antibody (ATIII), anti-PERK antibody, and anti-A1AT antibody. C, ATIII variants and A1AT NHK were expressed in CHO cells for 24 h. Control cells were treated with either DMSO or Tm for 24 h. Cells were then lysed with MNT and TCA-precipitated. 5% whole-cell lysate was then resolved on a 9% SDS-PAGE and imaged by Western blotting using the indicated antibodies. D, quantification of B. Relative BiP expression was calculated by normalizing all BiP levels to DMSO, using GAPDH as a loading control. All experiments are representative of three independent experiments. Error bars, S.D.
Next, the second branch of the UPR, activation of the kinase PERK, was tested. During activation by ER stress, PERK undergoes trans-autophosphorylation, which activates PERK and leads to multiple downstream effects, including up-regulating chaperone expression, translational attenuation, and cell cycle arrest (51, 52). To test for the activation of PERK, CHO cells were either transfected or treated with Tm for 24 h. Cells were then lysed and TCA-precipitated before immunoblotting. Phosphorylated PERK (pPERK) can be distinguished from nonphosphorylated PERK by an increase in mass on a gel. Overexpression of ATIII variants and A1AT NHK did not generate pPERK (Fig. 7B). This suggested that PERK was not activated by the overexpression of these proteins.
The third branch of the UPR, activation of the transcription factor ATF6, was also examined. Activation of ATF6 leads to the up-regulation of multiple ER chaperones and ERAD factors (53, 54). One of the prime targets of ATF6 is the Hsp70 family member BiP (50). As such, the level of BiP expression was examined as an indicator of ATF6 activation. CHO cells were either transfected or treated with Tm for 24 h. Cells were then lysed and TCA-precipitated before immunoblotting. BiP expression was not found to be up-regulated by the overexpression of any of the constructs when compared with the positive control Tm (Fig. 7, C and D). Therefore, ATF6 was not activated by the overexpression of ATIII or A1AT variants. In total, these results indicate that the only branch of UPR activated by overexpression of ATIII variants and A1AT NHK was the IRE1α pathway.
C247A/C430A ATIII interacts poorly with ERAD factors
IRE1α activation leads to up-regulation of numerous ER-resident proteins, including the ERAD factors ER degradation–enhancing α-mannosidase-like (EDEM) proteins EDEM1, EDEM2, and EDEM3 (50, 55, 56). Up-regulation of ERAD factors is expected to lead to robust degradation of misfolded and ER-retained proteins, as they are thought to be responsible for directing glycosylated misfolded proteins like C247A/C430A ATIII to the ERAD pathway (57–62). However, C247A/C430A ATIII was degraded significantly less than the ERAD substrate A1AT NHK (Fig. 6B). A possible explanation is that C247A/C430A ATIII does not bind well to EDEM1, EDEM2, and EDEM3. To test this, we co-transfected CHO cells with either WT or C247A/C430A ATIII or A1AT NHK, and EDEM1, EDEM2, and EDEM3. Cells were pulsed for 30 min and chased for the indicated times. Cells were lysed with buffer containing Triton X-100 (MNT). Interactions were queried by performing co-immunoprecipitations. EDEM1, EDEM2, and EDEM3 all associated efficiently with NHK A1AT, as demonstrated by their co-immunoprecipitation with the A1AT pulldowns (Fig. 8, A–F, lanes 21 and 22). In contrast, neither WT nor C247A/C430A ATIII showed significant association with any of the EDEMs. We conclude that the misfolded and ER-retained C247A/C430A ATIII is a poor substrate for recognition by these ERAD sorting factors, thus providing a possible explanation for the stability of C247A/C430A ATIII retained in the ER.
Figure 8.

ATIII C247A/C430A binds poorly to EDEM1, EDEM2, and EDEM3. A, FLAG-tagged EDEM1 was co-expressed with Myc-tagged ATIII WT, Myc-tagged ATIII C247A/C430A, and A1AT NHK in CHO cells, as indicated. Cells were radiolabeled with [35S]Cys/Met for 30 min and chased for the indicated times. Cells were lysed in MNT buffer. Lysates were then split equally and immunoprecipitated with either anti-Myc, FLAG, or A1AT antibodies, as indicated. Samples were resolved by reducing 9% SDS-PAGE and imaged by phosphorimaging. EDEM1 is denoted by an asterisk, whereas ATIII and A1AT NHK are denoted by a filled circle. B, quantification of EDEM1 co-immunoprecipitation from A. Percentage co-immunoprecipitation was determined by dividing the amount of EDEM1 immunoprecipitated by ATIII or NHK by total EDEM1. C–F, either FLAG-tagged EDEM2 or HA-tagged EDEM3 was co-expressed with Myc-tagged ATIII WT, Myc-tagged ATIII C247A/C430A, and A1AT NHK in CHO cells. Lysates were treated the same as in previous panels and immunoprecipitated using the indicated antibodies. Quantifications were conducted as described in B. All experiments are representative of three independent experiments. Error bars, S.D.
Discussion
A central question regarding protein quality control in the ER is how thousands of proteins that pass through the secretory pathway are evaluated by a small number of quality control factors. The ERQC must efficiently and accurately enable natively folded proteins to exit to their final destination and identify incompletely folded or misfolded proteins so that they are retained in the ER to allow fresh starts on proper folding or degradation. ERQC must rely on features of secretory pathway clients that indicate those with nonnative characteristics, such as exposed hydrophobic residues, mispaired or free Cys residues, or processed N-glycans (15, 18, 63, 64) (Fig. 9). Exposed hydrophobic residues are most notably monitored by the BiP/ERdj network in the ER (65–67), whereas disulfides are formed and monitored for their integrity by a group of some 20 oxidoreductases or protein-disulfide isomerase family members (8, 68, 69). Glycosidases, transferases, lectin chaperones, and sorting factors comprise an N-linked carbohydrate-dependent glycoprotein quality control system (1, 63). How these different quality control pathways work together to ensure that only native proteins are passed along the secretory pathway is poorly understood.
Figure 9.
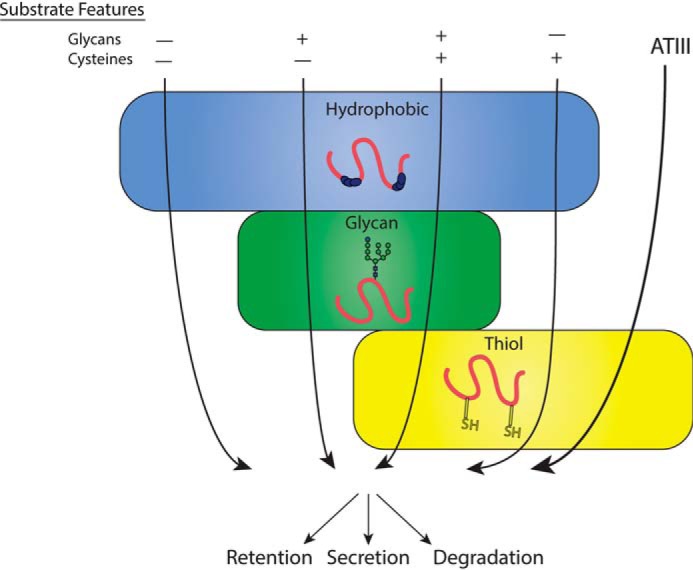
ATIII quality control model. Substrate features are generally understood to dictate the ER quality control pathways substrates engage, as depicted by arrows corresponding to different combinations of substrate features traversing quality control pathways. ATIII does not follow expected quality control pathways, but rather engages only the thiol-dependent quality control.
In this work, we have focused on a member of the serpin family of secreted proteins to gain insight into ERQC. The serpin native fold is complex and challenged by the need to adopt a metastable state that is required for inhibitory activity. Many examples of mutant serpins have been characterized to have secretion defects; often these are associated with diseases arising from loss of function and/or toxicity caused by retention and aggregation in the ER (22, 27, 70, 71). Whereas the fold is conserved in the serpin family, the number and locations of Cys and disulfide bonds, as well as N-linked glycans, vary among serpins. Therefore, we anticipated that different serpins would engage different quality control systems. We selected ATIII for this study as it is a glycoprotein with six Cys that are paired into three intramolecular disulfide bonds in its properly folded native, functional state (27, 72). Therefore, it is expected to engage the three quality control pathways: hydrophobic-, thiol-, and glycan-dependent.
Our previous work provided insight into the cellular folding pathway of ATIII: the C-terminal disulfide between Cys-247 and Cys-430 must form first for the protein to achieve its native fold with the next two disulfides in place (27). Cys residues in proteins that traverse the ER are generally either buried or reside in disulfide bonds once a protein is natively folded and assembled (15, 73). Exposed, unpaired Cys present in unpaired oligomers can be recognized by the thiol quality control pathway and retrieved from the ER-Golgi intermediate compartment and retained in the ER via a pH-dependent interaction with ERp44, as in the case of IgM (15, 74), acetylcholinesterase (75), SUMF1 (76), and adiponectin (14), whereas IgG CH1 domain mutants are free thiol quality control substrates retained by an unclear mechanism (77). However, little is known about the role of the thiol-dependent quality control process for monomeric, secreted proteins. Previous work has also been limited by a lack of a clear method to detect the folded status of secreted protein, thereby leaving the possibility that Cys mutants are properly folded and secreted rather than evading quality control as inactive and misfolded mutants. Therefore, we sought to delineate the role of oligomerization from thiol-dependent quality control while monitoring folded status by examining the quality control of a monomeric protein, ATIII.
Strikingly, mutation of all six Cys residues of ATIII to Ala resulted in the efficient secretion of a significantly misfolded and inactive protein; adding back a single native Cys to the Cys-less construct resulted in efficient ER retention in all but one case; and all disulfide mutants with free thiols were retained, whereas disulfide mutants without free thiols were not. These compelling results indicate that ATIII relies almost completely on thiol-dependent quality control for its interrogation within the ER. Our data therefore expand the clientele of thiol-dependent quality control by demonstrating that ATIII, a monomeric and secreted protein, is a substrate of thiol-dependent quality control. Furthermore, it is notable that no other features of ATIII that signal incomplete folding or misfolding were exploited by ER quality control to retain the nonnative species, demonstrating a lack of redundancy between ER quality control pathways.
Interestingly, there are other proteins that possess unpaired Cys residues in their native sequences and nonetheless are efficiently secreted, including the serpin A1AT, which possesses a single Cys (71). Additionally, when Cys 128 was added back to Cys-less ATIII, the resulting protein was secreted at a level near that of the Cys-less variant. The observation of a higher-molecular weight species in the nonreducing gel for this variant suggests that a population formed a disulfide bond either in an ATIII dimer or with another luminal species, cloaking the free thiol and allowing secretion of this ATIII variant (Fig. 3). Thus, not all free thiols designate ER retention, and there must be additional determinants that contribute to a retention outcome. The most obvious factor is the solvent accessibility of a Cys, but even for ATIII, an active mutant with an extra solvent-accessible Cys (R57C) is properly secreted, whereas other mutants with a seventh Cys (Y63C, F402C, and Y2166C) are retained (27, 78), and some data suggest that Cys solvent accessibility does not impede secretion of functional WT A1AT (79, 80). How accessible single Cys residues are to thiol-reactive proteins in the cell is not well-understood. Additional factors involved in recognition may include the pKa of a given Cys, as Cys displays a wide variety of reactivity, which is dictated by interactions with residues in the local environment (81), the presence of local hydrophobic domains, or oxidative modifications of a Cys. Future work should address these questions to determine the specific requirements of a thiol-dependent quality control substrate.
Why did the other quality control pathways that monitor structural features associated with protein misfolding or incomplete folding not recognize the Cys-less ATIII variant? It is clear from our results that this construct is not natively folded, as it is inactive and protease-labile. A major cellular strategy to recognize “unfoldedness” in a substrate is the exposure of hydrophobic surfaces or sequences. In the ER lumen, the most well-studied quality control mediators that utilize hydrophobic surface belong to the BiP-ERdj network. Serpins have an ellipsoidal, watermelon-shaped fold that increases their surface/volume ratio relative to other globular proteins that are more spherical (82, 83), suggesting that the serpins are likely to be more hydrophilic than more common, more spherical protein folds. This higher hydrophilicity and lower hydrophobicity could interfere with BiP binding. Future work may explore the possibility of a poor interaction of Cys-less ATIII with BiP directly.
The carbohydrate chaperone system of the ER is responsible for directing the folding and retention of aberrant glycoproteins (31, 63). Whereas retention is mediated by binding to the ER-resident lectin chaperones, calnexin and calreticulin, the decision for chaperone rebinding or ER retention is made by UGGT1. Although UGGT1 is understood to reglucosylate proteins that it determines to be nonnative, the parameters by which UGGT1 selects substrates remain incompletely understood. One general possibility is that UGGT1 recognizes generally misfolded proteins, as in the case of misfolded mutants of A1AT (45). A second hypothesis suggests that UGGT1 specifically recognizes on-pathway folding intermediates, so as to promote productive folding rather than futile rounds of reglucosylation of irreparable substrates (84). Although it is hard to envision distinguishing features of on-pathway proteins from off-pathway targets, our cellular results are consistent with this model, as WT ATIII was reglucosylated at a higher level than the misfolded off-pathway mutants of ATIII (Fig. 5, A and B). Studies using purified components have found that UGGT1 favors modifying glycopeptides that have hydrophobic patches C-terminal to the glycan (85). Analyzing ATIII with this pattern in mind did not reveal such hydrophobic sequences C-terminal to the N-linked glycans; by comparison, A1AT, which is a better substrate of UGGT1 (45), displayed two strong hydrophobic patches following glycans at Asn-70 and -271 (Fig. S2). This observation would account for the apparent poor recognition of ATIII by the glycan-dependent pathway of ERQC and underline its reliance on the thiol-dependent quality control pathway. The presence of the robust thiol-dependent pathway for monitoring ATIII may have alleviated any evolutionary pressure to maintain motifs suitable for reglucosylation and the reliance on the UGGT1-directed carbohydrate-dependent quality control pathway. Further work examining the substrate preferences of UGGT1 in a cellular context is required to elucidate the selection process for this intriguing folding sensor.
The remaining puzzle in our findings is why persistent retention of misfolded ATIII variants does not correlate with degradation. We postulate that stability of retained ATIII mutants is promoted by weak interactions with the ERAD-promoting proteins EDEM1/2/3 (Fig. 8). Why C247A/C430A ATIII is not recognized by the EDEMs is unclear. However, the presence of higher molecular weights of C247A/C430A ATIII, as demonstrated by sucrose gradients (Fig. 4), may indicate that C247A/C430A ATIII is present in small, disulfide-linked aggregates with which the EDEMs are unable to interact. Despite weak degradation, cells expressing C247A/C430A ATIII did not show higher UPR activation as compared with cells expressing other ATIII variants (Fig. 7). As such, it is unclear how cells are capable of maintaining proteostasis during retention of this substrate, although it is possible that long-term retention may more strongly activate UPR or potentially other mechanisms such as autophagy.
It is notable that even other members of the serpin family, which fold to very similar structures, do not use the same quality control pathway as ATIII. Neuroserpin, although trafficked through the secretory pathway, lacks Cys residues and as such does not engage thiol-dependent quality control. Multiple serpins, such as serpin B3 (squamous cell carcinoma antigen 1 (SCCA1)), the human serpin with the highest sequence identity (39%) to ATIII, are cytoplasmic and therefore also must use unique quality control pathways as compared with secretory serpins. This suggests that universal protein quality control mechanisms are not generally applicable, as even highly related proteins do not use the same quality control pathways. Rather, slight differences in specific substrate features dictate which quality control pathways are engaged.
Together, these results point to hierarchies in ERQC that are tailored to particular proteins through the process of evolution. We found that the serpin ATIII, a protein containing multiple glycans and a hydrophobic core and characterized in its native state by three disulfide bonds, is solely reliant on the thiol-dependent quality control pathway. In the case of ATIII, this heavy reliance on the thiol-dependent quality control pathway introduced a vulnerability, which was revealed by removing all six Cys residues from ATIII. The resulting protein was completely incapable of folding to a native, functional state but nonetheless bypassed ERQC and was secreted efficiently. These results highlight the potential lack of redundancy between ER quality control pathways and demonstrate that general substrate features do not necessarily predict the quality control pathways of a substrate. Future studies should explore the reasons that ATIII variants were poorly recognized by key players in the other quality control systems as well as conduct a more detailed analysis of other serpins to elucidate the nuances of protein quality control and accurately predict the quality control pathways a substrate will engage. This predictive understanding will open the door to therapeutic approaches to improve ERQC and thus address defects in protein maturation and secretion.
Experimental procedures
Cell culture
MI8-5 CHO cells were a gift from S. Krag (Johns Hopkins University, Baltimore, MD). CHO-K1 (lot 62960170) cells were purchased from ATCC. Cells were authenticated by a universal mycoplasma detection kit (catalog no. 30-012K, ATCC). WT MEF and Uggt1−/− MEF cells were a gift from R. J. Kaufman and were generated as described previously (86, 87). CHO and MI8-5 CHO cells were grown in α-minimum essential medium supplemented with 10% FBS and 1% penicillin/streptomycin at 37 °C and 34 °C, respectively, and 5% CO2. MEF cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% FBS and 1% penicillin/streptomycin at 37 °C and 5% CO2. All cell culture reagents were purchased from Thermo Fisher Scientific.
Reagents
The plasmid for pGEX-3X GST-calreticulin was from M. Michalak (University of Alberta, Edmonton, Canada). Antibodies used were as follows: monoclonal mouse Myc tag 9B11 antibody, rabbit monoclonal C33E10 PERK (Cell Signaling), monoclonal mouse HA probe 12CA5 (Santa Cruz Biotechnology, Inc.), monoclonal mouse KDEL 10C3 DyLight 488 (Enzo Life Sciences), goat anti-mouse secondary antibody Alexa Fluor 594 (Thermo Fisher Scientific), monoclonal mouse anti-FLAG M2 F1804 (Millipore–Sigma), rabbit polyclonal anti-A1AT (Dako), and mouse monoclonal GAPDH 374 (Millipore–Sigma). Rabbit polyclonal affinity-purified anti-BiP was from L. Hendershot (St. Jude Children's Research Hospital). S-tag agarose was purchased from Millipore–Sigma. All ATIII constructs, EDEM1, and EDEM2 were cloned into a pcDNA3.1(+) vector. pcDNA3.1(+)EDEM3-HA was from N. Hosokawa (Kyoto University). All chemicals were purchased from Millipore–Sigma, except where indicated.
Metabolic labeling
Cells were pulse-labeled for 30 min with 60 μCi of EasyTag Express35S Protein Labeling Mix [35S]Cys/Met (PerkinElmer Life Sciences) in 3-cm plates and 120 μCi of [35S]Cys/Met in 6-cm plates. Immediately after pulse, cells were washed with PBS and either lysed in lysis buffer with protease inhibitors (MNT; 20 mm MES, 100 mm NaCl, 30 mm 0.5% Triton X-100, 50 μm calpain inhibitor I, 1 μm pepstatin, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 400 μm phenylmethylsulfonyl fluoride, and 20 mm N-ethylmaleimide) or chased for the indicated time using regular growth media. Media and Triton X-100–insoluble fractions were collected where indicated.
Immunoprecipitations and SDS-PAGE
After lysis, samples were vortexed at high speed at 4 °C for 5 min and then centrifuged at high speed at 4 °C for 5 min. The supernatant was then precleared using protein A–Sepharose beads (GE Healthcare) by end-over-end rotation for 1 h at 4 °C. Samples were then centrifuged at 3,000 rpm for 5 min at 4 °C, and the beads were discarded. Samples were then incubated with protein A–Sepharose beads and the indicated antibody overnight at 4 °C under end-over-end rotation. Samples were then washed with Triton's wash (100 mm Tris-HCl, pH 8.6, 300 mm NaCl, 0.1% SDS, 0.05% Triton X-100) or lysis buffer without protease inhibitors, where indicated. The Triton X-100–insoluble pellet was either discarded or solubilized in 1% SDS in 100 mm Tris-HCl, pH 8, by trituration followed by high-speed vortexing at room temperature, heating for 10 min at 95 °C, dilution in lysis buffer, and sonication. Samples were eluted from beads using Werner's sample buffer (30 mm Tris-HCl, pH 6.8, 9% SDS, 15% glycerol, 0.05% bromphenol blue), and SDS-PAGE was performed. Radiolabeled samples were imaged by phosphorimaging using a GE Typhoon FLA 9500 phosphorimager (GE Healthcare) and quantified using ImageQuant (Fujifilm).
Secretion assay
Cells were seeded onto 3-cm plates, transfected, and metabolically labeled using [35S]Cys/Met, as described previously. After 30 min of pulse and the indicated time of chase, media and lysate portions were collected, and immunoprecipitations were performed as previously described. After washing with Triton wash, sample buffer was added, and samples were analyzed by SDS-PAGE and imaged by phosphorimaging.
Activity assay
Cells were seeded into 6-cm plates, transfected, and metabolically labeled using [35S]Cys/Met, as described previously. After 30 min of pulse and 3 h of chase, the media and lysate portions were collected, and immunoprecipitations were performed as described previously. After washing with 0.5% CHAPS HBS (0.5% CHAPS, 50 mm HEPES, pH 7.5, 200 mm NaCl), samples were eluted from beads with 10 μl of 0.5 mg/ml c-Myc peptide (Millipore–Sigma) for 1 h at 37 °C. The beads were then discarded, and the sample was split equally between treated and nontreated; 2 units of thrombin was added to treated samples, and an equal volume of water was added to nontreated samples. Samples were then incubated for 1 h at 37 °C before the addition of sample buffer. All samples were then analyzed by SDS-PAGE and imaged by phosphorimaging.
Protease sensitivity assay
Cells were seeded into 6-cm plates, transfected, and metabolically labeled using [35S]Cys/Met, as described previously. After 30 min of pulse and 3 h of chase, the media and lysate portions were collected and split equally into treated and nontreated samples. Samples were immunoprecipitated as described previously. After washing with 0.5% CHAPS HBS, 0.05 μg of trypsin was added to treated samples, and an equal volume of water was added to nontreated samples. Samples were then incubated for 15, 30, or 60 min at 37 °C before the addition of sample buffer. All samples were then analyzed by SDS-PAGE and imaged by phosphorimaging.
Glycosylation assay
Cells were seeded onto 3-cm plates, transfected, and metabolically labeled using [35S]Cys/Met, as described previously. After 30 min of pulse and 2 h of chase, the media and lysate portions were collected, and immunoprecipitations were performed as described previously. After washing, samples were treated with either Endo H or PNGase F (New England Biolabs), according to the manufacturer's instructions. Samples were then analyzed by SDS-PAGE and imaged by phosphorimaging.
Assay for the presence of free thiols
Cells were seeded onto 3-cm plates, transfected, and metabolically labeled using [35S]Cys/Met, as described previously. After 30 min of pulse and 30 min of chase, the media and lysate portions were collected and split equally into treated and nontreated samples. To treated samples, 100 μl of PEG maleimide (Millipore–Sigma) was added to a final concentration of 1.4 mm, and to the nontreated samples, 100 μl of N-ethyl maleimide was added to a final concentration of 5 mm. Samples were then incubated at room temperature for 30 min. Treated samples were then quenched by adding DTT (Thermo Fisher Scientific) to a final concentration of 100 mm. Immunoprecipitations were then performed as previously described. All samples were then analyzed by SDS-PAGE, imaged by phosphorimaging, and quantified using ImageQuant (Fujifilm).
Immunofluorescence
CHO cells were seeded onto glass coverslips in α-minimum essential medium supplemented with 10% FBS. 24 h later, cells were transfected with the indicated plasmid using polyethyleneimine (Polysciences Inc., Warrington, PA) according to the manufacturer's instructions. After 16 h, cells were washed with PBS (2.7 mm KCl, 1.5 mm KH2PHO4, 136.9 mm NaCl, 8 mm Na2PHO4, pH 7.4) and fixed using 3.7% formaldehyde for 15 min, permeabilized using 0.1% Triton X-100 in PBS for 15 min, and then washed with PBS and blocked with 10% FBS in PBS for 30 min. Cells were incubated in Myc tag antibody in 10% FBS in PBS (1:500) for 1 h, washed with PBS, blocked with 10% FBS, and incubated with highly cross-adsorbed goat-anti mouse IgG secondary antibody conjugated to Alexa Fluor 594 (1:500) for 1 h. Cells were then incubated with KDEL antibody conjugated to DyLight 488 (Enzo Life Sciences) (1:200) for 1 h. Cells were then washed with PBS and mounted to glass slides using Vectashield with DAPI (Vector Laboratories, Inc., Burlingame, CA) and sealed with nail polish. All images were acquired using an Olympus Fluoview FV1000 confocal microscope. Images were taken at ×100 oil immersion. Images were processed with Adobe Photoshop.
Sucrose gradients
Cells were seeded onto 10-cm plates and transfected. Media and lysate fractions were collected. Lysate fractions were then split equally between DTT-treated and nontreated samples. Treated samples contained 100 mm DTT. Sucrose gradients were made by solubilizing sucrose into MNT to a concentration of 10 or 40%. 6 ml of 10% sucrose was then laid on top of an equal volume of 40% sucrose, and the gradient was established using a BioComp model 153 gradient station (BioComp Instruments, Fredericton, Canada). Samples were laid on top of the 10–40% sucrose gradient. Gradients were centrifuged at 38,000 rpm for 18 h in a Beckman Optima L-90K Ultracentrifuge. Samples were taken from the gradient by pipetting 1 ml from the top of the gradient using a wide-bore pipette tip, treated with TCA to a final concentration of 10%, and incubated for 30 min. Samples were then centrifuged at 14,000 rpm in a bench-top centrifuge for 10 min at 4 °C, washed with acetone, and centrifuged at 14,000 rpm for 10 min at 4 °C. Reducing sample buffer was added to all samples, followed by analysis by SDS-PAGE and Western blotting.
Western blotting
After SDS-PAGE, gels were washed with ultrapure water, and transfer membrane (Immobilon-FL, Millipore) was pretreated according to the manufacturer's instructions. Transfer was then conducted using a wet-transfer apparatus (Invitrogen Novex mini-cell). Blots were blocked in 5% milk, 2% BSA in PBS solution for 1 h under gentle shaking at room temperature. Primary antibodies at the indicated concentration in a 5% milk, 2% BSA, PBST (2.7 mm KCl, 1.5 mm KH2PO4, 136.9 mm NaCl, 8 mm Na2PHO4, 0.5% Tween 20, pH 7.4) solution were added to blots and incubated overnight at 4 °C under gentle rotation. Blots were then washed with PBST and incubated with IRDye 800CW-conjugated goat secondary antibody against the appropriate species (LI-COR Biosciences, Lincoln, NE) in a 5% milk, 2% BSA, 0.02% SDS, PBST solution for 1 h at 25 °C under gentle shaking. Blots were then washed with PBST and imaged using a LI-COR Odyssey CLx imaging system.
GST-calreticulin pulldown
Recombinant GST-calreticulin was expressed in Escherichia coli and purified as described previously (39, 88). MI8-5 cells were seeded into 3-cm plates, transfected, and metabolically labeled using [35S]Cys/Met, as described previously. Cells were either treated with N-butyl deoxynojirimycin (Toronto Research Chemicals) for 30 min prior to the experiment and then continuously throughout or for 15 min prior to each time point, as indicated. 80% of the cell lysate was incubated with 8 μg of GST-calreticulin prebound to GSH beads (GE Life Sciences) overnight at 4 °C, whereas 20% of the lysate was immunoprecipitated with Myc antibody, as described previously. All samples were washed with Triton wash, and reducing sample buffer was added to the immunoprecipitations. GST-calreticulin samples were treated with 20 μl of elution buffer (1% SDS in 10 mm Tris, pH 7.5, 150 mm NaCl) at 95 °C for 10 min and centrifuged for 2 min at 2000 rpm. The supernatant was collected and quenched with MNT buffer, followed by immunoprecipitation with protein A–Sepharose beads with Myc antibody, as described previously. Beads were then washed with Triton wash, and the sample was treated with reducing sample buffer. Sample was then analyzed by SDS-PAGE, imaged with phosphorimaging, and quantified using ImageQuant (Fujifilm).
XBP1 splicing
CHO cells were seeded onto 10-cm plates and transfected for 24 h. Untransfected cells were either treated with tunicamycin in DMSO at a concentration of 5 μg/ml or with DMSO alone for 24 h. Total RNA was then collected using an RNeasy kit (Qiagen). cDNA was then generated using avian myeloblastosis virus reverse transcriptase (New England Biolabs) and mouse Xbp1 primers (89), according to the manufacturer's instructions. The resulting cDNA product was then separated using a 2.5% agarose gel and visualized using a G:BOX (Syngene).
BIP expression
Cells were seeded onto 10-cm plates and transfected for 24 h. Untransfected cells were treated either with tunicamycin (5 μg/ml) in DMSO or with DMSO alone for 24 h. Cells were then lysed using MNT and TCA-precipitated. TCA precipitation was conducted by adding TCA to cell lysate to a final concentration of 10%. Cell lysate was then briefly rotated and allowed to incubate on ice for 15 min before spinning at 14,000 rpm for 10 min at 4 °C. Supernatant was then aspirated and washed with acetone and centrifuged for 10 min at 4 °C. Supernatant was then aspirated, and the remaining precipitant was allowed to dry for 5 min at room temperature and briefly at 65 °C. Reducing sample buffer was then added, and 5% of total lysates were resolved on a 9% reducing SDS-PAGE. Gels were then imaged by Western blotting, as described previously, using the following antibodies: anti-BiP (1:1000), Myc (1:2500), A1AT (1:500), and GAPDH (1:1000).
PERK phosphorylation
Cells were seeded onto 10-cm plates and transfected for 24 h. Untransfected cells were treated either with tunicamycin (5 μg/ml) in DMSO or with DMSO alone for 24 h. Cells were then lysed using MNT and TCA-precipitated as described previously. 5% of total lysates were resolved on a 9% reducing SDS-PAGE. Gels were then imaged by Western blotting, as described previously, using the following antibodies: PERK (1:500), Myc (1:2500), and A1AT (1:500).
Co-immunoprecipitations
Cells were seeded onto 3-cm plates, transfected, and metabolically labeled using [35S]Cys/Met, as described previously. After 30 min of pulse and the indicated chase times, cells were lysed, and the lysate was collected. For EDEM co-IPs, half of the plates were lysed in MNT. For OS9.2 and XTP3B, half of the plates were lysed in buffer containing 150 mm NaCl, 5 mm EDTA, pH 7.4, 50 mm Tris, pH 7.4, 1% Triton X-100, 50 μm calpain inhibitor I, 1 μm pepstatin, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 400 μm phenylmethylsulfonyl fluoride, and 20 mm N-ethylmaleimide. In all cases, the other half of the plates were lysed with 2% CHAPS HBS buffer. Cell lysates were then split equally and immunoprecipitated for either substrate or ERAD factor. ATIII was immunoprecipitated as described previously. AlAT NHK was immunoprecipitated using A1AT antibody, and protein A–agarose beads EDEM1/2 were immunoprecipitated using FLAG tag and protein A–agarose beads, whereas EDEM3 was immunoprecipitated using HA tag and protein A–agarose beads. IPs were then washed with Triton wash buffer if lysed using 1% triton X-100, or 0.5% CHAPS HBS buffer if lysed using 2% CHAPS HBS. After washes, sample buffer was added, and samples were analyzed by 9% reducing SDS-PAGE and imaged by phosphorimaging.
Statistics
Percentages of reglucosylation were calculated by dividing the number obtained from quantification of bands in the GST-calreticulin lanes by the number obtained from quantification of bands in the single immunoprecipitation lanes multiplied by 4 to correct for the amount of cell lysate used in each pulldown. The comparative percentage of reglucosylation was then obtained by dividing the amount of reglucosylation of mutant protein over that of WT. For all quantifications, error bars were calculated by determining the S.D. of three independent samples. Statistical significance was determined by using an unpaired t test with a confidence interval of 95%.
Author contributions
B. M. A. and D. N. H. conceptualization; B. M. A. data curation; B. M. A. validation; B. M. A., H. K., and D. N. H. investigation; B. M. A. and H. K. methodology; B. M. A., A. G., and D. N. H. writing-original draft; B. M. A., L. M. G., A. G., and D. N. H. writing-review and editing; D. N. H. formal analysis; D. N. H. funding acquisition; D. N. H. project administration.
Supplementary Material
This work was supported by NIGMS, National Institutes of Health, Grant GM086874 (to D. N. H.) and Chemistry-Biology Interface Program Training Grant T32GM008515 (to B. M. A.). This work was also supported by GM1118161 (to L. M. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2.
- ER
- endoplasmic reticulum
- UPR
- unfolded protein response
- ATIII
- antithrombin III
- ERQC
- ER quality control
- CHO
- Chinese hamster ovary
- PNGase F
- peptide:N-glycosidase F
- Endo H
- endoglycosidase H
- Mal
- maleimide
- UGGT1
- UDP-glucose:glycoprotein glucosyltransferase 1
- DNJ
- N-butyl deoxynojirimycin
- GST
- glutathione S-transferase
- MEF
- mouse embryonic fibroblast
- ERAD
- endoplasmic reticulum–associated protein degradation
- A1AT NHK
- α-1-antitrypsin null Hong Kong
- Tm
- tunicamycin
- pPERK
- phosphorylated PERK
- FBS
- fetal bovine serum
- DAPI
- 4′,6-diamidino-2-phenylindole
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- EDEM
- ER degradation–enhancing α-mannosidase-like
- IP
- immunoprecipitation.
References
- 1. Lamriben L., Graham J. B., Adams B. M., and Hebert D. N. (2016) N-Glycan-based ER molecular chaperone and protein quality control system: the calnexin binding cycle. Traffic 17, 308–326 10.1111/tra.12358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lederkremer G. Z. (2009) Glycoprotein folding, quality control and ER-associated degradation. Curr. Opin. Struct. Biol. 19, 515–523 10.1016/j.sbi.2009.06.004 [DOI] [PubMed] [Google Scholar]
- 3. Kozutsumi Y., Segal M., Normington K., Gething M.-J., and Sambrook J. (1988) The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332, 462–464 10.1038/332462a0 [DOI] [PubMed] [Google Scholar]
- 4. Hwang J., and Qi L. (2018) Quality control in the endoplasmic reticulum: crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 43, 593–605 10.1016/j.tibs.2018.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McCracken A. A., and Brodsky J. L. (1996) Assembly of ER-associated protein degradation in vivo: dependence on cytosol, calnexin and ATP. J. Cell Biol. 132, 291–298 10.1083/jcb.132.3.291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Olzmann J. A., Kopito R. R., and Christianson J. C. (2013) The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb. Perspect. Biol. 5, a013185 10.1101/cshperspect.a013185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huh W.-K., Falvo J. V., Gerke L. C., Carroll A. S., Howson R. W., Weissman J. S., and O'Shea E. K. (2003) Global analysis of protein localization in budding yeast. Nature 425, 686–691 10.1038/nature02026 [DOI] [PubMed] [Google Scholar]
- 8. Määttänen P., Gehring K., Bergeron J. J., and Thomas D. Y. (2010) Protein quality control in the ER: the recognition of misfolded proteins. Semin. Cell Dev. Biol. 21, 500–511 10.1016/j.semcdb.2010.03.006 [DOI] [PubMed] [Google Scholar]
- 9. Tamura T., Sunryd J. C., and Hebert D. N. (2010) Sorting things out through endoplasmic reticulum quality control. Mol. Membr. Biol. 27, 412–427 10.3109/09687688.2010.495354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ellgaard L., and Helenius A. (2003) Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell. Biol. 4, 181–191 10.1038/nrm1052 [DOI] [PubMed] [Google Scholar]
- 11. Kim Y. E., Hipp M. S., Bracher A., Hayer-Hartl M., and Hartl F. U. (2013) Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 82, 323–355 10.1146/annurev-biochem-060208-092442 [DOI] [PubMed] [Google Scholar]
- 12. Bulleid N. J., and Ellgaard L. (2011) Multiple ways to make disulfides. Trends Biochem. Sci. 36, 485–492 10.1016/j.tibs.2011.05.004 [DOI] [PubMed] [Google Scholar]
- 13. Isidoro C., Maggioni C., Demoz M., Pizzagalli A., Fra A. M., and Sitia R. (1996) Exposed thiols confer localization in the endoplasmic reticulum by retention rather than retrieval. J. Biol. Chem. 271, 26138–26142 10.1074/jbc.271.42.26138 [DOI] [PubMed] [Google Scholar]
- 14. Wang Z. V., Schraw T. D., Kim J.-Y., Khan T., Rajala M. W., Follenzi A., and Scherer P. E. (2007) Secretion of the adipocyte-specific secretory protein adiponectin critically depends on thiol-mediated protein retention. Mol. Cell. Biol. 27, 3716–3731 10.1128/MCB.00931-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anelli T., Sannino S., and Sitia R. (2015) Proteostasis and “redoxtasis” in the secretory pathway: tales of tails from ERp44 and immunoglobulins. Free Radic. Biol. Med. 83, 323–330 10.1016/j.freeradbiomed.2015.02.020 [DOI] [PubMed] [Google Scholar]
- 16. Helenius A., and Aebi M. (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049 10.1146/annurev.biochem.73.011303.073752 [DOI] [PubMed] [Google Scholar]
- 17. Hebert D. N., and Molinari M. (2012) Flagging and docking: dual roles for N-glycans in protein quality control and cellular proteostasis. Trends Biochem. Sci. 37, 404–410 10.1016/j.tibs.2012.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hammond C., Braakman I., and Helenius A. (1994) Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc. Natl. Acad. Sci. U.S.A. 91, 913–917 10.1073/pnas.91.3.913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hebert D. N., Foellmer B., and Helenius A. (1995) Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell 81, 425–433 10.1016/0092-8674(95)90395-X [DOI] [PubMed] [Google Scholar]
- 20. Peterson J. R., Ora A., Van P. N., and Helenius A. (1995) Transient, lectin-like association of calreticulin with folding intermediates of cellular and viral glycoproteins. Mol. Biol. Cell 6, 1173–1184 10.1091/mbc.6.9.1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu Y., Choudhury P., Cabral C. M., and Sifers R. N. (1997) Intracellular disposal of incompletely folded human α1-antitrypsin involves release from calnexin and post-translational trimming of asparagine-linked oligosaccharides. J. Biol. Chem. 272, 7946–7951 10.1074/jbc.272.12.7946 [DOI] [PubMed] [Google Scholar]
- 22. Gooptu B., and Lomas D. A. (2009) Conformational pathology of the serpins: themes, variations, and therapeutic strategies. Annu. Rev. Biochem. 78, 147–176 10.1146/annurev.biochem.78.082107.133320 [DOI] [PubMed] [Google Scholar]
- 23. Huntington J. A., Read R. J., and Carrell R. W. (2000) Structure of a serpin-protease complex shows inhibition by deformation. Nature 407, 923–926 10.1038/35038119 [DOI] [PubMed] [Google Scholar]
- 24. Dementiev A., Dobó J., and Gettins P. G. (2006) Active site distortion is sufficient for proteinase inhibition by serpins: structure of the covalent complex of α1-proteinase inhibitor with porcine pancreatic elastase. J. Biol. Chem. 281, 3452–3457 10.1074/jbc.M510564200 [DOI] [PubMed] [Google Scholar]
- 25. Corral J., de la Morena-Barrio M. E., and Vicente V. (2018) The genetics of antithrombin. Thromb. Res. 169, 23–29 10.1016/j.thromres.2018.07.008 [DOI] [PubMed] [Google Scholar]
- 26. Davies M. J., and Lomas D. A. (2008) The molecular aetiology of the serpinopathies. Int. J. Biochem. Cell Biol. 40, 1273–1286 10.1016/j.biocel.2007.12.017 [DOI] [PubMed] [Google Scholar]
- 27. Chandrasekhar K., Ke H., Wang N., Goodwin T., Gierasch L. M., Gershenson A., and Hebert D. N. (2016) Cellular folding pathway of a metastable serpin. Proc. Natl. Acad. Sci. U.S.A. 113, 6484–6489 10.1073/pnas.1603386113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Perry D. J., and Carrell R. W. (1996) Molecular genetics of human antithrombin deficiency. Hum. Mutat. 7, 7–22 [DOI] [PubMed] [Google Scholar]
- 29. Picard V., Ersdal-Badju E., and Bock S. (1995) Partial glycosylation of antithrombin III asparagine-135 is caused by the serine in the third position of its N-glycosylation consensus sequence and is responsible for production of the β-antithrombin III isoform with enhanced heparin affinity. Biochemistry 34, 8433–8440 10.1021/bi00026a026 [DOI] [PubMed] [Google Scholar]
- 30. Hurtley S. M., and Helenius A. (1989) Protein oligomerization in the endoplasmic reticulum. Annu. Rev. Cell Biol. 5, 277–307 10.1146/annurev.cb.05.110189.001425 [DOI] [PubMed] [Google Scholar]
- 31. Adams B. M., Oster M. E., and Hebert D. N. (2019) Protein quality control in the endoplasmic reticulum. Protein J. 38, 317–329 10.1007/s10930-019-09831-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barlowe C. K., and Miller E. A. (2013) Secretory protein biogenesis and traffic in the early secretory pathway. Genetics 193, 383–410 10.1534/genetics.112.142810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fatal N., Suntio T., and Makarow M. (2002) Selective protein exit from yeast endoplasmic reticulum in absence of functional COPII coat component sec13p. Mol. Biol. Cell. 13, 4130–4140 10.1091/mbc.02-05-0082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rabouille C. (2017) Pathways of unconventional protein secretion. Trends Cell Biol. 27, 230–240 10.1016/j.tcb.2016.11.007 [DOI] [PubMed] [Google Scholar]
- 35. Hebert D. N., Foellmer B., and Helenius A. (1996) Calnexin and calreticulin promote folding, delay oligomerization and suppress degradation of influenza hemagglutinin in microsomes. EMBO J. 15, 2961–2968 10.1002/j.1460-2075.1996.tb00659.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vassilakos A., Cohen-Doyle M. F., Peterson P. A., Jackson M. R., and Williams D. B. (1996) The molecular chaperone calnexin facilitates folding and assembly of class I histocompatibility molecules. EMBO J. 15, 1495–1506 10.1002/j.1460-2075.1996.tb00493.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kozlov G., Muñoz-Escobar J., Castro K., and Gehring K. (2017) Mapping the ER interactome: the P domains of calnexin and calreticulin as plurivalent adapters for foldases and chaperones. Structure 25, 1415–1422.e3 10.1016/j.str.2017.07.010 [DOI] [PubMed] [Google Scholar]
- 38. Sousa M., and Parodi A. J. (1995) The molecular basis for the recognition of misfolded glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. EMBO J. 14, 4196–4203 10.1002/j.1460-2075.1995.tb00093.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pearse B. R., Gabriel L., Wang N., and Hebert D. N. (2008) A cell-based reglucosylation assay demonstrates the role of GT1 in the quality control of a maturing glycoprotein. J. Cell Biol. 181, 309–320 10.1083/jcb.200712068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Satoh T., Song C., Zhu T., Toshimori T., Murata K., Hayashi Y., Kamikubo H., Uchihashi T., and Kato K. (2017) Visualization of a flexible modular structure of the ER folding-sensor enzyme UGGT. Sci. Rep. 7, 12142 10.1038/s41598-017-12283-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roversi P., Marti L., Caputo A. T., Alonzi D. S., Hill J. C., Dent K. C., Kumar A., Levasseur M. D., Lia A., Waksman T., Basu S., Soto Albrecht Y., Qian K., McIvor J. P., Lipp C. B., et al. (2017) Interdomain conformational flexibility underpins the activity of UGGT, the eukaryotic glycoprotein secretion checkpoint. Proc. Natl. Acad. Sci. U.S.A. 114, 8544–8549 10.1073/pnas.1703682114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Holmgren A., Söderberg B.-O., Eklund H., and Brändén C.-I. (1975) Three-dimensional structure of Escherichia coli thioredoxin-S2 to 2.8 Å resolution. Proc. Natl. Acad. Sci. U.S.A. 72, 2305–2309 10.1073/pnas.72.6.2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Martin J. L. (1995) Thioredoxin: a fold for all reasons. Structure 3, 245–250 10.1016/S0969-2126(01)00154-X [DOI] [PubMed] [Google Scholar]
- 44. Pearse B. R., Tamura T., Sunryd J. C., Grabowski G. A., Kaufman R. J., and Hebert D. N. (2010) The role of UDP-Glc:glycoprotein glucosyltransferase 1 in the maturation of an obligate substrate prosaposin. J. Cell Biol. 189, 829–841 10.1083/jcb.200912105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tannous A., Patel N., Tamura T., and Hebert D. N. (2015) Reglucosylation by UDP-glucose:glycoprotein glucosyltransferase 1 delays glycoprotein secretion but not degradation. Mol. Biol. Cell. 26, 390–405 10.1091/mbc.E14-08-1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Quellhorst G. J. Jr., O'Rear J. L., Cacan R., Verbert A., and Krag S. S. (1999) Nonglucosylated oligosaccharides are transferred to protein in MI8–5 Chinese hamster ovary cells. Glycobiology 9, 65–72 10.1093/glycob/9.1.65 [DOI] [PubMed] [Google Scholar]
- 47. Brodsky J. L. (2012) Cleaning up: endoplasmic reticulum associated degradation to the rescue. Cell 151, 1163–1167 10.1016/j.cell.2012.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Walter P., and Ron D. (2011) The unfolded protein response: from stress pathway to homeostatic response. Science 334, 1081–1086 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- 49. Calfon M., Zeng H., Urano F., Till J. H., Hubbard S. R., Harding H. P., Clark S. G., and Ron D. (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP1 mRNA. Nature 415, 92–96 10.1038/415092a [DOI] [PubMed] [Google Scholar]
- 50. Plate L., Cooley C. B., Chen J. J., Paxman R. J., Gallagher C. M., Madoux F., Genereux J. C., Dobbs W., Garza D., Spicer T. P., Scampavia L., Brown S. J., Rosen H., Powers E. T., Walter P., et al. (2016) Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. eLife 5, e15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shi Y., Vattem K. M., Sood R., An J., Liang J., Stramm L., and Wek R. C. (1998) Identification and characterization of pancreatic eukaryotic initiation factor 2 α-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol. 18, 7499–7509 10.1128/MCB.18.12.7499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hetz C., and Papa F. R. (2018) The unfolded protein response and cell fate control. Mol. Cell. 69, 169–181 10.1016/j.molcel.2017.06.017 [DOI] [PubMed] [Google Scholar]
- 53. Haze K., Yoshida H., Yanagi H., Yura T., and Mori K. (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell. 10, 3787–3799 10.1091/mbc.10.11.3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yoshida H., Matsui T., Yamamoto A., Okada T., and Mori K. (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 10.1016/S0092-8674(01)00611-0 [DOI] [PubMed] [Google Scholar]
- 55. Yoshida H., Matsui T., Hosokawa N., Kaufman R. J., Nagata K., and Mori K. (2003) A time-dependent phase shift in the mammalian unfolded protein response. Dev Cell. 4, 265–271 10.1016/S1534-5807(03)00022-4 [DOI] [PubMed] [Google Scholar]
- 56. Bernasconi R., Pertel T., Luban J., and Molinari M. (2008) A dual task for the XBP1-responsive OS-9 variants in the mammalian endoplasmic reticulum. J. Biol. Chem. 283, 16446–16454 10.1074/jbc.M802272200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Molinari M., Calanca V., Galli C., Lucca P., and Paganetti P. (2003) Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 299, 1397–1400 10.1126/science.1079474 [DOI] [PubMed] [Google Scholar]
- 58. Olivari S., Galli C., Alanen H., Ruddock L., and Molinari M. (2005) A novel stress-induced EDEM variant regulating endoplasmic reticulum associated glycoprotein degradation. J. Biol. Chem. 280, 2424–2428 10.1074/jbc.C400534200 [DOI] [PubMed] [Google Scholar]
- 59. Hirao K., Natsuka Y., Tamura T., Wada I., Morito D., Natsuka S., Romero P., Sleno B., Tremblay L. O., Herscovics A., Nagata K., and Hosokawa N. (2006) EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J. Biol. Chem. 281, 9650–9658 10.1074/jbc.M512191200 [DOI] [PubMed] [Google Scholar]
- 60. Cormier J. H., Tamura T., Sunryd J. C., and Hebert D. N. (2009) EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol. Cell 34, 627–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Araki K., and Nagata K. (2011) Protein folding and quality control in the ER. Cold Spring Harb. Perspect. Biol. 3, a007526 10.1101/cshperspect.a007526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lamriben L., Oster M. E., Tamura T., Tian W., Yang Z., Clausen H., and Hebert D. N. (2018) EDEM1's mannosidase-like domain binds ERAD client proteins in a redox-sensitive manner and possesses catalytic activity. J. Biol. Chem. 293, 13932–13945 10.1074/jbc.RA118.004183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Caramelo J. J., and Parodi A. J. (2015) A sweet code for glycoprotein folding. FEBS Lett. 589, 3379–3387 10.1016/j.febslet.2015.07.021 [DOI] [PubMed] [Google Scholar]
- 64. Behnke J., Mann M. J., Scruggs F.-L., Feige M. J., and Hendershot L. M. (2016) Members of the Hsp70 family recognize distinct types of sequences to execute ER quality control. Mol Cell. 63, 739–752 10.1016/j.molcel.2016.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hendershot L. M. (2004) The ER function BiP is a master regulator of ER function. Mt. Sinai J. Med. 71, 289–297 [PubMed] [Google Scholar]
- 66. Pobre K. F. R., Poet G. J., and Hendershot L. M. (2019) The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: getting by with a little help from ERdj friends. J. Biol. Chem. 294, 2098–2108 10.1074/jbc.REV118.002804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Preissler S., and Ron D. (2019) Early events in the endoplasmic reticulum unfolded protein response. Cold Spring Harb. Perspect. Biol. 11, a033894 10.1101/cshperspect.a033894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hatahet F., and Ruddock L. W. (2007) Substrate recognition by the protein disulfide isomerases. FEBS J. 274, 5223–5234 10.1111/j.1742-4658.2007.06058.x [DOI] [PubMed] [Google Scholar]
- 69. Tsunoda S., Avezov E., Zyryanova A., Konno T., Mendes-Silva L., Pinho Melo E., Harding H. P., and Ron D. (2014) Intact protein folding in the glutathione-depleted endoplasmic reticulum implicates alternative protein thiol reductants. eLife 3, e03421 10.7554/eLife.03421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Stein P. E., and Carrell R. W. (1995) What do dysfunctional serpins tell us about molecular mobility and disease? Nat. Struct. Biol. 2, 96–113 10.1038/nsb0295-96 [DOI] [PubMed] [Google Scholar]
- 71. Ronzoni R., Berardelli R., Medicina D., Sitia R., Gooptu B., and Fra A. M. (2016) Aberrant disulphide bonding contributes to the ER retention of α1-antitrypsin deficiency variants. Hum. Mol. Genet. 25, 642–650 10.1093/hmg/ddv501 [DOI] [PubMed] [Google Scholar]
- 72. Zhou Z. R., and Smith D. L. (1990) Location of disulfide bonds in antithrombin III. Biomed. Environ. Mass Spectrom. 19, 782–786 10.1002/bms.1200191206 [DOI] [PubMed] [Google Scholar]
- 73. Oka O. B., and Bulleid N. J. (2013) Forming disulfides in the endoplasmic reticulum. Biochim. Biophys. Acta 1833, 2425–2429 10.1016/j.bbamcr.2013.02.007 [DOI] [PubMed] [Google Scholar]
- 74. Sitia R., Neuberger M., Alberini C., Bet P., Fra A., Valetti C., Williams G., and Milstein C. (1990) Development regulation of IgM secretion: the role of the carboxy-terminal cysteine. Cell 60, 781–790 10.1016/0092-8674(90)90092-S [DOI] [PubMed] [Google Scholar]
- 75. Kerem A., Kronman C., Bar-Nun S., Shafferman A., and Velan B. (1993) Interrelations between assembly and secretion of recombinant human acetylcholinesterase. J. Biol. Chem. 268, 180–184 [PubMed] [Google Scholar]
- 76. Fraldi A., Zito E., Annunziata F., Lombardi A., Cozzolino M., Monti M., Spampanato C., Ballabio A., Pucci P., Sitia R., and Cosma M. P. (2008) Multistep, sequential control of the trafficking and function of the multiple sulfatase deficiency gene product, SUMF1 by PDI, ERGIC-53 and ERp44. Hum. Mol. Genet. 17, 2610–2621 10.1093/hmg/ddn161 [DOI] [PubMed] [Google Scholar]
- 77. Elkabetz Y., Argon Y., and Bar-Nun S. (2005) Cysteines in CH1 underlie retention of unassembled Ig heavy chains. J. Biol. Chem. 280, 14402–14412 10.1074/jbc.M500161200 [DOI] [PubMed] [Google Scholar]
- 78. Perry D. J., Daly M. E., Colvin B. T., Brown K., and Carrell R. W. (1995) Two antithrombin mutations in a compound heterozygote: Met20Thr and Tyr166Cys. Am. J. Hematol. 50, 215–216 10.1002/ajh.2830500310 [DOI] [PubMed] [Google Scholar]
- 79. Griffiths S. W., King J., and Cooney C. L. (2002) The reactivity and oxidation pathway of cysteine 232 in recombinant human α1-antitrypsin. J. Biol. Chem. 277, 25486–25492 10.1074/jbc.M203089200 [DOI] [PubMed] [Google Scholar]
- 80. Patschull A. O., Segu L., Nyon M. P., Lomas D. A., Nobeli I., Barrett T. E., and Gooptu B. (2011) Therapeutic target-site variability in α1-antitrypsin characterized at high resolution. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 67, 1492–1497 10.1107/S1744309111040267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Weerapana E., Wang C., Simon G. M., Richter F., Khare S., Dillon M. B., Bachovchin D. A., Mowen K., Baker D., and Cravatt B. F. (2010) Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795 10.1038/nature09472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Dima R. I., and Thirumalai D. (2004) Asymmetry in the shapes of folded and denatured states of proteins. J. Phys. Chem. B 108, 6564–6570 10.1021/jp037128y [DOI] [Google Scholar]
- 83. Liu L., Werner M., and Gershenson A. (2014) Collapse of a long axis: single-molecule Förster resonance energy transfer and serpin equilibrium unfolding. Biochemistry 53, 2903–2914 10.1021/bi401622n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Caramelo J. J., Castro O. A., de Prat-Gay G., and Parodi A. J. (2004) The endoplasmic reticulum glucosyltransferase recognizes nearly native glycoprotein folding intermediates. J. Biol. Chem. 279, 46280–46285 10.1074/jbc.M408404200 [DOI] [PubMed] [Google Scholar]
- 85. Taylor S. C., Thibault P., Tessier D. C., Bergeron J. J., and Thomas D. Y. (2003) Glycopeptide specificity of the secretory protein folding sensor UDP-glucose glycoprotein:glucosyltransferase. EMBO Rep. 4, 405–411 10.1038/sj.embor.embor797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Molinari M., Galli C., Vanoni O., Arnold S. M., and Kaufman R. J. (2005) Persistent glycoprotein misfolding activates the glucosidase II/UGT1-driven calnexin cycle to delay aggregation and loss of folding competence. Mol. Cell. 20, 503–512 10.1016/j.molcel.2005.09.027 [DOI] [PubMed] [Google Scholar]
- 87. Soldà T., Galli C., Kaufman R. J., and Molinari M. (2007) Substrate-specific requirements for UGT1-dependent release from calnexin. Mol. Cell. 27, 238–249 10.1016/j.molcel.2007.05.032 [DOI] [PubMed] [Google Scholar]
- 88. Baksh S., and Michalak M. (1991) Expression of calreticulin in Escherichia coli and identification of its Ca2+ binding domains. J. Biol. Chem. 266, 21458–21465 [PubMed] [Google Scholar]
- 89. Lee K., Tirasophon W., Shen X., Michalak M., Prywes R., Okada T., Yoshida H., Mori K., and Kaufman R. J. (2002) IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 16, 452–466 10.1101/gad.964702 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.