

Abstract
Lytic polysaccharide monooxygenases (LPMOs) catalyze oxidative cleavage of recalcitrant polysaccharides such as cellulose and chitin and play an important role in the enzymatic degradation of biomass. Although it is clear that these monocopper enzymes have extended substrate-binding surfaces for interacting with their fibrous substrates, the structural determinants of LPMO substrate specificity remain largely unknown. To gain additional insight into substrate specificity in LPMOs, here we generated a mutant library of a cellulose-active family AA10 LPMO from Streptomyces coelicolor A3(2) (ScLPMO10C, also known as CelS2) having multiple substitutions at five positions on the substrate-binding surface that we identified by sequence comparisons. Screening of this library using a newly-developed MS-based high-throughput assay helped identify multiple enzyme variants that contained four substitutions and exhibited significant chitinolytic activity and a concomitant decrease in cellulolytic activity. The chitin-active variants became more rapidly inactivated during catalysis than a natural chitin-active AA10 LPMO, an observation likely indicative of suboptimal substrate binding leading to autocatalytic oxidative damage of these variants. These results reveal several structural determinants of LPMO substrate specificity and underpin the notion that productive substrate binding by these enzymes is complex, depending on a multitude of amino acids located on the substrate-binding surface.
Keywords: cellulose, chitin, substrate specificity, protein stability, high-throughput screening, auxiliary activity family 10 (AA10), carbohydrate-active enzyme, glycosidic bond cleavage, lytic polysaccharide monooxygenase (LPMO), renewable energy
Introduction
Lytic polysaccharide monooxygenases (LPMOs)3 constitute a class of enzymes that employ a powerful oxidative mechanism to cleave glycosidic bonds within crystalline regions of recalcitrant biomasses, such as cellulose and chitin. After the discovery of their catalytic function in 2010 (1), LPMOs have been classified as auxiliary activities (AA) in the carbohydrate-active enzyme database (cazy.org4 (2)) where they are categorized in families AA9–11 and AA13–16 on the basis of sequence similarity. LPMOs are abundant in Nature, where they are frequently produced by fungi and bacteria involved in the degradation of structural polysaccharides in plants, crustaceans, fungi, insects, and molluscs. Thus, LPMOs hold an important role in the Earth's carbon cycle. Some studies suggest alternative functions, including a role as virulence factors in microbial pathogenicity, that are not associated with biomass turnover (3–5).
LPMOs engage in a synergistic interplay with hydrolytic enzymes (e.g. cellulases and chitinases) during the conversion of recalcitrant polysaccharides, an important aspect of LPMO functionality that was observed prior to unraveling their mode of action (6, 7). The crystalline surface of densely packed polysaccharides can be disrupted by the oxidative action of LPMOs, thereby generating access points for hydrolytic enzymes in areas that otherwise would be less accessible (1, 8–10). The resulting boosting effect on biomass turnover makes LPMOs key components in the development of industrial bioprocessing technology (11, 12).
Although some LPMOs are expressed as single catalytic domains, others are associated with a carbohydrate-binding module (CBM) through a flexible linker region (13). CBMs are thought to increase the effective enzyme concentration on the substrate surface by anchoring the catalytic domain in the proximity of substrate-binding sites (14, 15). Two conserved histidines, one of which is the N-terminal amino acid of the mature LPMO, are located on the flat substrate-binding surface of the catalytic domain and constitute the catalytic center of the LPMO. Together with the α-amino group and a conserved tyrosine or phenylalanine in the axial position, these equatorial histidines coordinate a single copper ion, the reduction of which from Cu(II) to Cu(I) is crucial for catalysis (16). In fungal LPMOs, the N-terminal histidine tends to be methylated (16), but this post-translational modification has not been observed in LPMOs of bacterial origin.
LPMO catalysis has generally been believed to require molecular oxygen and a reductant that delivers two electrons for each catalytic cycle (1, 17, 18). However, a recent study has indicated that LPMOs may employ H2O2 as co-substrate in a reaction that does not require two reductant-delivered electrons per cycle (19). Although reduced LPMOs are prone to oxidative damage when exposed to high levels of O2 or H2O2 in the absence of a proper substrate (19–21), well-controlled H2O2-mediated reactions result in drastically improved catalytic rates (22) and stable reaction kinetics (12, 23). Notably, under the conditions normally used in LPMO reactions, H2O2 will be formed either by O2 reacting with reduced LPMO molecules that are not bound to the substrate (24, 25) or by reactions involving O2 and the reductant in the presence of trace amounts of transition metals.
LPMOs may be active on cellulose, hemicellulose, chitin, starch, and/or oligosaccharides (26). Most known LPMOs are active on cellulose or chitin, which are among the most abundant polysaccharides on Earth, both consisting of β-1,4-linked monomers (glucose in cellulose and N-acetylglucosamine (GlcNAc) in chitin) rotated 180° relative to each other. LPMO-mediated cleavage of cellulose and chitin occurs by hydroxylation of the C1 or C4 carbon of the sugar monomers adjacent to the scissile glycosidic bond (1, 16, 27), and different LPMOs can be limited to one of these mechanisms or be capable of both. Oxidation of the C1 carbon results in the formation of 1,5-δ-lactones that are spontaneously converted to their more stable aldonic acid forms, whereas oxidation of the C4 carbon produces 4-ketoaldoses that are hydrated to their corresponding gemdiol form (25). Of note, for chitin, only C1 oxidation has been observed.
NMR-based studies of LPMO–substrate interactions (28, 29), modeling (30), and mutational studies (20, 30–33) suggest that precise arrangements of multiple amino acid side chains covering a major fraction of the substrate-binding surface dictate enzyme properties such as oxidative regioselectivity and substrate preferences. Simultaneously, these multiple side-chain arrangements ensure stability for the LPMO by promoting precise substrate binding that protects the LPMO from oxidative self-inactivation (20). To create more insight into the structural determinants of LPMO substrate specificity, we have generated and screened a rationally-designed mutant library to convert the cellulose-active LPMO10C from Streptomyces coelicolor A3(2) (hereinafter called ScLPMO10C, previously known as CelS2 (34)) to a chitin-specific enzyme. The mutant library, containing multiple mutations at five positions on the substrate-binding surface (Fig. 1), was designed based on conservation patterns detected in 130 AA10 sequences (Table S1), including LPMOs acting on cellulose (C1-specific and mixed C1/C4-oxidizing), chitin (C1-specific), and LPMOs with as-yet unknown substrate specificity. The generated library, containing 4320 variants, was subsequently screened using an MS-based high-throughput assay for LPMO activity.
Figure 1.

Structural overview of targeted residues in ScLPMO10C. A displays a side view of the crystal structure of the catalytic domain of WT ScLPMO10C (PDB code 4OY7 (34)) with the substrate-binding surface facing downwards. The side chains of residues targeted for mutation are shown with green-colored carbons, and carbons in the side chains of the two histidines that coordinate the copper ion (orange sphere) are colored gray. B shows the substrate-binding surface with residue numbering according to the PDB structure, which is used throughout this study. Note that His-35 is the N-terminal residue of the mature protein and that the mutational work was carried out on the full-length enzyme that includes a family 2 CBM connected by a flexible Pro/Thr-rich linker (13). C shows amino acid frequencies encountered at each mutated position (1–5) in multiple sequence alignments of sequences belonging to various LPMO10 subgroups: ScLPMO10C-like (26 sequences, C1-oxidizing, cellulose); MaLPMO10B-like (28 sequences, C1/C4 oxidation of cellulose and C1 oxidation of chitin); and SmLPMO10A-like (49 sequences, C1-oxidizing, chitin). Position 4 (ScLPMO10C_Y111) is exclusively found in some ScLPMO10C-like sequences and is always present with the upstream motif Asn–Trp–Phe in which the first and the last make up positions 2 and 3 in the library. Mutations included in the library are shown in the top right corner of panel C. Panels A and B were made using PyMOL and the graphs in panel C were generated using WebLogo (36). A blue star in A–C marks the location of the second histidine (His-144) of the histidine brace. See Table S1 for the sequences used in the analyses and Fig. S1 for a structural comparison of natural LPMO10s with experimentally-verified different substrate specificities.
Results
Library design and mutant generation
Library design was based on multiple criteria, one of which was restricting library size. Thus, we focused on residues for which there were experimental data indicating a role in substrate binding. Furthermore, library design was limited to positions for which it was possible to compare multiple LPMOs structurally, i.e. positions in regions where sequence alignments were reliable and structures were similar. The selected positions all occur in the loop region that is referred to as the “L2 loop,” which covers a relatively large area on the flat substrate-binding surface of ScLPMO10C and that contains most of the sequence variation at the LPMO surface. Fig. 1 shows the location of residues Tyr-79, Asn-80, Phe-82, Tyr-111, and Trp-141 (positions 1–5) that were targeted for mutation. In terms of subsites, the mutations affect subsites −4 to −2, which are known to play an important role in substrate binding, both in cellulose-active (35) and chitin-active (30) LPMOs. These residues were postulated to dictate substrate specificity based on conservation patterns identified when comparing the sequences of cellulose- and chitin-active AA10 LPMOs, as outlined in Fig. 1C. Furthermore, a 2H/1H exchange experiment had previously shown that Tyr-54, Glu-55, Gln-57, and Thr-111 in SmLPMO10A (positions 1–3 and 5, see below) interact with crystalline chitin (29). The selected positions showed limited natural variation (Fig. 1), which allowed us to cover the naturally used sequence space in a library of acceptable size. Of course, residues in other locations, e.g. on the “other” side of the active site (+ subsites), could also play a role in determining substrate specificity, but it is much more difficult to structurally compare LPMOs in this region.
Tyr-79 (position 1) is the only aromatic residue on the AA10 substrate-binding surface that stacks with the carbohydrate substrate (−4 subsite in SmLPMO10A (30)). This position has been targeted in recent site-directed mutagenesis studies that revealed its importance for both substrate binding (20, 30) and enzyme activity and stability (20, 31). The most common natural residues at this position are Tyr and Trp, but nonaromatic residues also occur. Thus, the Tyr at this position was mutated to Trp/Thr/Asp/Glu and Ala. Alanine substitutions were included at all positions.
The second position, Asn-80 in ScLPMO10C, was selected on the basis of being highly conserved in C1 and mixed C1/C4-oxidizing cellulose-active LPMO10s, and its different and strictly-conserved nature in chitin-active LPMO10s, where it is a Glu. Mutation of this residue in chitin-oxidizing SmLPMO10A resulted in strongly-reduced substrate binding and abolished enzyme activity (6, 20). In the library, Asn-80 in ScLPMO10C was mutated to residues with similar side chain size and polarity (Gln/Asp/Glu), as well as to Ser and Ala, which are also, but rarely, found at this position.
The third position, Phe-82 in ScLPMO10C, was recently shown to be important for the regioselectivity of cellulose oxidation in an LPMO10 with mixed C1/C4 activity (31). LPMO10s with mixed C1/C4-cellulose oxidation activity tend to have an Asn at this position (see Fig. 1C), whereas those that are strict C1 oxidizers tend to have Phe (cellulose-oxidizers) or Gln (chitin-oxidizers) at this position. Of note, C1/C4 oxidizers active on cellulose tend to show activity on chitin, such as MaLPMO10B from Micromonospora aurantiaca. Mutation of Asn-85 in MaLPMO10B to Phe almost abolished the C4 activity but also the chitin-oxidizing activity (31), indicating an important role in substrate interactions. Position 3 was mutated to Ala/Gln/Asn/His/Tyr based on the natural variation in all types of LPMO10s.
Position 4, Tyr-111 in ScLPMO10C, is located in a less-conserved loop in ScLPMO10C and other cellulose C1-oxidizing LPMO10s. This extra loop (ScLPMO10C_P110_Y111_D112) always comes with the upstream motif Asn–Trp–Phe (ScLPMO10C-N80-W81-F82), which includes position 2 and 3. Position 4 was mutated to Ala/Phe/His to vary the side-chain properties at this position.
The fifth position, Trp-141 in ScLPMO10C, is highly conserved within each of the LPMO10 substrate-specificity groups. Although the typical chitin oxidizing LPMOs (e.g. SmLPMO10A-like) tend to have a polar Thr at this position, the majority of LPMO10s with mixed activities (C1/C4 on cellulose and C1 on chitin) have a Gln, and the strict C1-oxidizing cellulose-active LPMO10s (ScLPMO10C-like) have a Trp at this position. Here, Trp-141 was mutated to Ala/Gln/Thr/Phe. The final library contained 4320 variants.
High-throughput library screening
Chitin activity was measured using a high-throughput MS-based assay for detection of oxidized chito-oligomers. An Agilent RapidFire 365 system was applied, and the solid-phase extraction and triple-quadrupole MS detection methods used were established based on reaction products generated by the positive control enzyme SmLPMO10A (CBP21) acting on β-chitin as substrate. As there is no chromatographic separation between the analytes in the RapidFire system, the triple quadrupole mass spectrometer was run in MRM mode to obtain maximum selectivity and sensitivity. Testing with a mixture of oxidized chito-oligosaccharides led to the identification of fragments that selectively represented the various oxidized species. The precursor ions used to fragment and subsequently quantify daughter fragments in the triple quadrupole instrument were the [M + H2O − H+] species of the aldonic acid forms of oxidized chitin oligomers consisting of 2–6 monomers (Fig. 2 and Fig. S2). Protocols for LPMO enzyme reactions in 384-well microplates were established using robotic liquid handling methods optimized to evenly distribute suspensions of solid β-chitin.
Figure 2.

Screening for chitinolytic activity. Chitinolytic activity was assessed using 2 g/liter β-chitin and 1 mm ascorbic acid (reductant) at pH 6.0 and 40 °C. The figure shows RapidFire ESI triple quadrupole MRM chromatograms for M2 (four independent wells, light green) and WT ScLPMO10C (four independent wells, gray). The ESI MRM intensity is given on the y axis, while time (seconds) is on the x axis; note the different scales on the y axis. Chromatograms A–E show the following: A, DP2ox: ESI MRM m/z 439.2 → m/z 116.0; B, DP3ox: m/z 643.3 → m/z 625.3; C, DP4ox: 845.3 m/z → m/z 668.2; D, DP5ox: m/z 1048.2 → m/z 871.2; E, DP6ox: m/z 1251.5 → m/z 871.3. Underlying spectral data are shown in Fig. S2. A clone was defined as positive if, for at least one oxidized product, the difference between the average of the green peaks was higher than the average of the gray peaks by at least 10 times the standard deviation in the average for the gray peaks. The picture shows that the developed method failed for DP3ox, which shows similar signals for M2 and ScLPMO10C.
The designed LPMO library was expressed in Escherichia coli XL10 Gold and screened for production of LPMOs with activity toward β-chitin as described under “Experimental procedures.” A primary screen of 11,520 transformants led to selection of 42 transformants. After a second screening of these mutants for verification of activity and after discarding two mutants that contained unintended mutations discovered during sequencing, 37 transformants remained, representing 27 unique mutants, as detailed below.
Sequence analysis of putative chitin-active mutants
Table 1 provides sequence information for the 37 mutants that remained after the screening process. The collection contained 27 unique mutants, including two that were detected four times, one that was detected three times, and two that were detected twice. The combined frequencies of residues among all of the 37 variants (Table 1A) and among the variants that were detected multiple times (Table 1B) suggested a consensus sequence for chitinolytic activity of Tyr, Asp, Xaa, Phe, and Gln for positions 79, 80, 82, 111, and 141, respectively. At position 79, 95% of the selected variants had kept the WT amino acid. At the other four positions, mutation frequencies were higher, and all were mutated, relative to WT ScLPMO10C, in the deduced consensus sequence. Both Asn-80 and Tyr-111 were substituted in ∼75% of the 37 mutants, with the most common substitutions being N80D and Y111F. Residue Phe-82 was replaced by a variety of different amino acids in 95% of the putative chitin-active variants, and almost 50% of these carried the mutation F82Q. Trp-141 was the only residue that was mutated in all selected variants, with W141Q being the most frequent substitution.
Table 1.
Sequence analysis and selection of mutants
Part A shows the frequency of different substitutions of the targeted residues (Tyr-79, Asn-80, Phe-82, Tyr-111, and Trp-141) in the 37 selected putatively chitin-active mutants. Mutations included in the library are shown below the boxes. Note that for most variants the chitin activity was not verified experimentally beyond the screening procedure, so the presence of false-positives cannot be excluded. Part B shows the frequency of different substitutions in variants that occurred multiple times. Part C shows the three mutants (M2, M5, and M18) that were selected for further analysis, as well as four additional mutants that were generated by site-directed mutagenesis or selected from the mutant collection to verify the contribution of individual residues to the chitin activity of M18. Darker shading of certain residues indicates identity with the wildtype residue.
Based on these observations, a subset of the mutants that were detected multiple times and carried the mutation pattern Tyr-79/Asp-80/Xaa-82/Phe-111/Gln-141 (M2, M5, and M18; Table 1C) were selected for production and further investigation. Of note, this mutation pattern was present in 11 of the 37 selected mutants. The contribution of individual mutations to chitinolytic activity was investigated by studying the effect of mutating single residues in M18, the most active variant (see below), back to the WT sequence. This required construction of two variants of M18, whereas the effect of the other reverse mutations could be assessed by characterization of variants that were among the 37 selected mutants (Table 1C).
Production of putative chitin-active mutants
The selected ScLPMO10C variants and ScLPMO10C WT were expressed and purified to electrophoretic homogeneity (Fig. S3) using anion-exchange purification followed by size-exclusion chromatography. The yields of purified protein were typically 10–20 mg per liter of culture.
Verification of chitinolytic activity
Analysis of reaction products after incubation of β-chitin with purified ScLPMO10C variants containing the mutation pattern Tyr-79/Asp-80/Xaa-82/Phe-111/Gln-141 showed that the selected enzymes indeed were active toward chitin (Fig. 3). MALDI-ToF mass spectra of reaction products generated by the three mutants (Fig. 3, D–F) were very similar to spectra of products generated by the well-known chitin-active LPMO from Serratia marcescens (SmLPMO10A or CBP21) after degradation of β-chitin (Fig. 3, A and B). Comparison of the zoom-in views of the hexamer cluster (GlcNAc5GlcNAc1A) for SmLPMO10A (Fig. 3B) and the three mutants (Fig. 3, D–F) shows identical dominating signals representing the aldonic acid sodium adduct (m/z = 1276) and the disodium adduct of the aldonate (m/z = 1298). There are, however, some minor differences: next to the peak for the lactone form (m/z 1258), the spectra for the mutants show signals at m/z = 1256 and m/z = 1260 (see arrows in Fig. 3, D–F), which could indicate LPMO side activities, as discussed below. Using the same reaction setup and analytical procedures, no oxidized chito-oligomers were detected for reactions with WT ScLPMO10C (Fig. 3C).
Figure 3.

Oxidized products generated from chitin. A shows the results of MALDI-ToF MS analysis after a 24-h incubation of the positive control LPMO (SmLPMO10A) with 10 g/liter β-chitin at 40 °C in sodium phosphate (pH 6.0) in the presence of 1 mm ascorbic acid (reducing agent) and displays the C1-oxidized products (aldonic acids and lactones) that can be expected for a chitin-active LPMO. Peaks representing oxidized chito-oligomers are marked with their m/z value (see A for details). B shows a zoom-in view of the hexamer cluster displaying SmLPMO10A-generated products within the 1250 to 1320 m/z range. The peaks that are marked with an asterisk represent potassium adducts, which are absent in the spectra for the ScLPMO10C mutants due to different storage buffers (SmLPMO10A was stored in potassium phosphate (pH 6.0), and the ScLPMO10C mutants in sodium phosphate (pH 6.0)). C shows a zoom-in view of the hexamer cluster area for WT ScLPMO10C and shows no oxidized products. D–F display zoom-in views of the hexamer cluster area for mutants M2, M5, and M18, respectively. Arrows in B, D–F indicate minor signals at m/z = 1256 and m/z = 1260 that are more prominent in the mutants (relative to the signals for regular oxidized products) compared with SmLPMO10A.
Quantitative analysis of chitin degradation
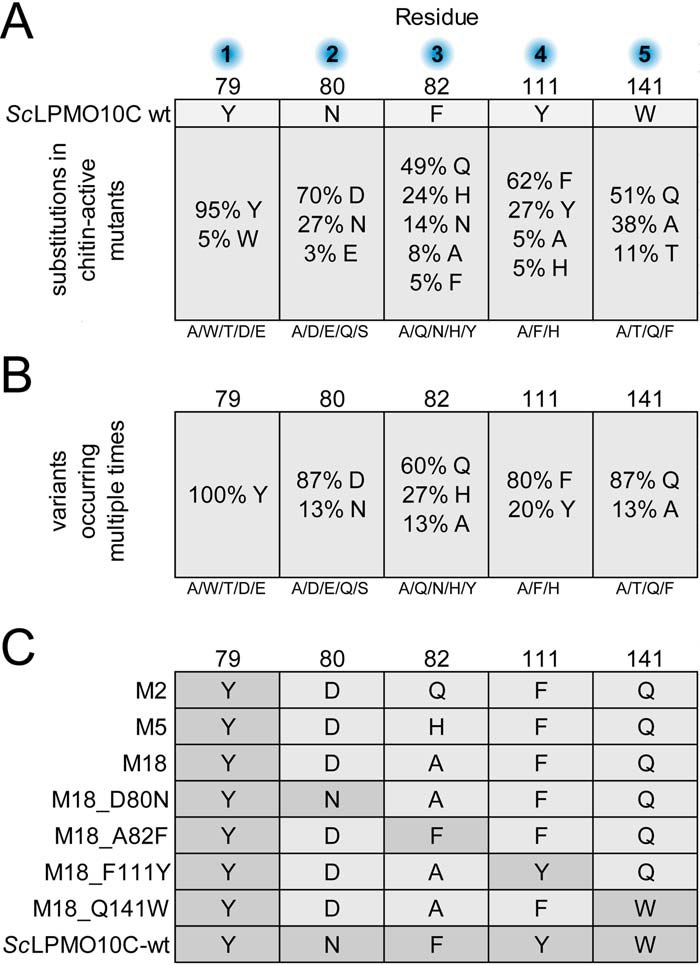
Monitoring of chitin degradation over time showed that the mutants generated oxidized products at an initial rate close to that of SmLPMO10A and that the product yield obtained in reactions with the best mutant, M18, after 4 h was about half of the yield obtained with SmLPMO10A (Fig. 4). Interestingly, a hardly detectable but still significant amount of oxidized product was detected in the reaction with ScLPMO10C WT, suggesting that the ScLPMO10C WT has a hitherto undetected very weak intrinsic capability of cleaving chitin. Fig. 4 shows that, although the mutants had good initial rates, product formation stopped earlier, compared with SmLPMO10A. M18 was able to generate products over a longer period compared with M2 and M5.
Figure 4.

Time course of chitin degradation. SmLPMO10A, ScLPMO10C WT, and the three chitin-active ScLPMO10C variants M2, M5, and M18 were incubated at 1 μm with 10 g/liter β-chitin at 40 °C and pH 6.0 in the presence of 1 mm ascorbic acid (reducing agent) for 4 h. A chitinase (SmChiC) was also added at 0.25 μm to obtain more rapid solubilization of LPMO-generated chain ends from the insoluble substrate. The reaction was stopped by vacuum filtering, and the soluble oxidized products were converted to oxidized dimers by treatment with chitobiase prior to analysis. All reactions were performed in triplicate. The error bars show ± S.D. (n = 3).
The stagnation of the product formation could be caused by reductant-, substrate-, or enzyme-related issues and was investigated by adding fresh reductant, fresh substrate, or fresh enzyme to the reaction at a point where the product generation had stopped (Fig. S4A). This experiment showed that enzyme inactivation is the primary reason for the cessation of product formation, as only the addition of fresh enzyme led to recovery of product generation (Fig. S4B).
Thermostability of the mutants
The apparent melting temperatures (Tm) of the mutants were determined by monitoring the effect of temperature on the binding of a fluorescent dye (SYPRO Orange), where increased binding of the dye (monitored in a real-time PCR machine) indicates exposure of the hydrophobic core of the protein upon unfolding. The melting curves in Fig. 5 show that SmLPMO10A (Tm = 71.2 °C) and ScLPMO10C WT (64.1 °C) are more stable than M2 (52.5 °C), M5 (53.5 °C), and M18 (54.8 °C). The decreased thermal stability of the mutants is somewhat surprising as the mutants contain only four mutations relative to the WT that are all located on the protein surface. Still, the apparent melting temperatures of the mutants are well above the reaction temperature used in the experiments (i.e. 40 °C).
Figure 5.

Mutational effects on thermostability. The plots show the apparent Tm of copper-saturated SmLPMO10A, ScLPMO10C WT, M2, M5, and M18. The derivative of the fluorescence signal (−dRFU/dT, where RFU is relative fluorescence units) is plotted as a function of the temperature (39). The reactions contained 0.1 g/liter LPMO and were heated from 25 to 99 °C, at a rate of 1.5 °C/min, in the presence of a fluorescent dye (SYPRO Orange). The scans were performed four times for each protein, and the figures show a typical scan. All apparent Tm values had standard deviations below ±0.1 °C.
Oxidative stability in H2O2-driven reactions
Based on the recent finding that H2O2 is a relevant co-substrate of LPMOs (19), it is conceivable that LPMO products observed in reactions without added H2O2 are in fact generated through H2O2-mediated catalysis after H2O2 has been formed from O2 (24, 25). This would mean that the apparent initial reaction rates reflect the rate-limiting step of the H2O2 generation from O2.
H2O2-mediated catalysis was investigated in reactions with M18 by adding 15 μm H2O2 at 15-min intervals (Fig. 6A). Reaction samples taken in between the addition of fresh H2O2 (after 7.5, 22.5, 37.5, and 52.5 min) indicated that the reactions must be fast as both M18 and the positive control, SmLPMO10A, had depleted the H2O2 in less than 7.5 min, meaning that the product yields did not increase any further until fresh H2O2 was added. Overall, the progress curves of Fig. 6A indicate that, initially, the SmLPMO10A and M18 show similar catalytic rates but that their stabilities differ, similar to what was concluded from Fig. 4. Thus, SmLPMO10A and M18 differ in terms of how much of the added H2O2 is consumed in productive LPMO reactions rather than in nonproductive reactions with either ascorbic acid or reduced LPMOs, where the latter may generate oxidative damage on the enzyme leading to enzyme inactivation (19, 20). This difference was even more prominent in reactions where the amount of added H2O2 was increased to 100 μm; in this case SmLPMO10A still performed well, whereas M18 was almost immediately inactivated (Fig. S5; note that SmLPMO10A generated many more products when fed with this higher amount of H2O2). Control reactions, displayed in Fig. 6B, showed that SmLPMO10A, ScLPMO10C WT, and M18 were hardly active in reactions where H2O2 was replaced by water, emphasizing the importance of H2O2 to drive the reaction at these low initial ascorbic acid concentrations. In these control experiments, the two chitin-active LPMOs, SmLPMO10A and M18, did show some product formation over time due to the repetitive addition of small amounts of ascorbic acid (Fig. 6D).
Figure 6.

H2O2 as co-substrate. A shows the amount of solubilized oxidized sites produced by SmLPMO10A (black line), M18 (dashed black line), and ScLPMO10C WT (dotted gray line) over 60 min in a reaction with 10 g/liter β-chitin in 50 mm sodium phosphate (pH 6.0), which were supplied with 100 μm AscA and 15 μm H2O2 every 15 min (indicated by arrows). The enzyme concentration was 1 μm in all cases. Aliquots were withdrawn between H2O2 additions (at 7.5, 22.5, 37.5, and 52.5 min) and immediately prior to addition of fresh H2O2 (at 0, 15, 30, and 45 min). The apparent plateaus in product levels between the intermediate sampling points (e.g. at 7.5 min) and the subsequent point of addition of fresh H2O2 and AscA (e.g. at 15 min) are because all the H2O2 added at the preceding addition (in this example, 0 min) was consumed at the intermediate sampling point (7.5 min). The lines connecting the points are drawn for illustration purposes only, and actual reaction rates were higher than suggested by the slopes of the ascending lines. B shows control reactions related to A. In the reactions labeled “without H2O2,” water was added to the reactions instead of H2O2. The graph also shows H2O2-driven product formation by WT ScLPMO10C (same curve as in A). Note the difference in the scale of the y axis between A and B; the product levels shown in B are very low, making quantification difficult. C shows the total amounts of oxidized sites at the end of the reactions shown in A (i.e. after 60 min). D shows the effect of varying the amount of added ascorbic acid in the reactions displayed in A (addition of 15 μm H2O2 + varying amounts of AscA every 15 min) for M18 and shows that essentially similar product yields were obtained when using a 10 times lower AscA concentrations compared with the reactions shown in A. The control reaction to the left shows that the LPMO is essentially inactive in the absence of added reductant. All reactions were performed in triplicates, and the error bars show ± S.D. (n = 3). Prior to analysis, soluble oxidized products were converted to oxidized dimers by treatment with a chitobiase. The total amount of oxidized products was determined by treating the complete reaction mixture with a mixture of chitinases and chitobiase, which leads to complete solubilization of the chitin and to conversion of all oxidized products, soluble or insoluble (i.e. still part of the chitin), to chitobionic acid.
Fig. 6A shows that SmLPMO10A stays active for the full hour of the reaction. In fact, product formation seems to increase over time, which may be due to the accumulation of ascorbic acid, leading to in situ H2O2 production and/or may reflect that soluble products are more easily released as the reaction progresses (37). To gain further insight in the utilization of H2O2, we determined the total amount of oxidized sites in the reaction mixtures. Fig. 6C shows that SmLPMO10A indeed generated slightly more than 60 μm oxidized products (of which 34.6% was soluble), in line with the total amount of added H2O2 being 60 μm. In contrast, M18 only generated ∼35 μm product (of which 41.7% was soluble), indicating that almost half of the added H2O2 was not converted to products. Of note, a control experiment showed that product formation after 1 h by M18 was similar in H2O2-driven reactions with 10 times less ascorbic acid (Fig. 6D), showing that both product formation and enzyme inactivation are driven by added H2O2 and that reduction of the LPMO is not rate-limiting.
It is conceivable that the higher frequency of enzyme inactivation in M18 relative to SmLPMO10A could be due to weaker substrate binding of the former, because nonsubstrate-bound reduced LPMOs are susceptible to damaging reactions with H2O2. However, due to the presence of the CBM2, WT ScLPMO10C binds well to chitin (Fig. S6) (38) and so does M18 (which also has an intact CBM2). It would, of course, be interesting to assess binding of the catalytic domain of M18 to chitin and compare this with binding of the catalytic domain of the WT enzyme. However, despite many attempts, we have not been able to produce the catalytic domain of M18 alone (i.e. M18 without the linker and the CBM2).
Effect of individual mutations on chitinolytic activity
To investigate the contribution of each mutated residue to chitinolytic activity, M18 (Tyr-79/Asp-80/Ala-82/Phe-111/Gln-141) was used as a template to reverse single mutations back to the WT sequence. Two of these combinations (M18_A82F and M18_F111Y) were already present among the mutants that had passed the screening process and could be produced directly from the library. The two other residue combinations (M18_D80N and M18_Q141W) were generated by site-directed mutagenesis. Progress curves (Fig. 7) showed that none of these single mutations reduced the chitinolytic activity of M18 to the (very low) level of WT ScLPMO10C, but all variants performed significantly worse than M18. This suggests a cumulative effect of the mutations. Similarly to the observations made when comparing M2 and M5 with M18 (Fig. 4), the mutational effects seem to primarily relate to stability. Three of the mutants exhibited an initial rate very similar to M18, but became inactivated at earlier time points compared with M18, whereas M18_Q141W showed a slightly reduced initial rate and slightly retarded inactivation.
Figure 7.
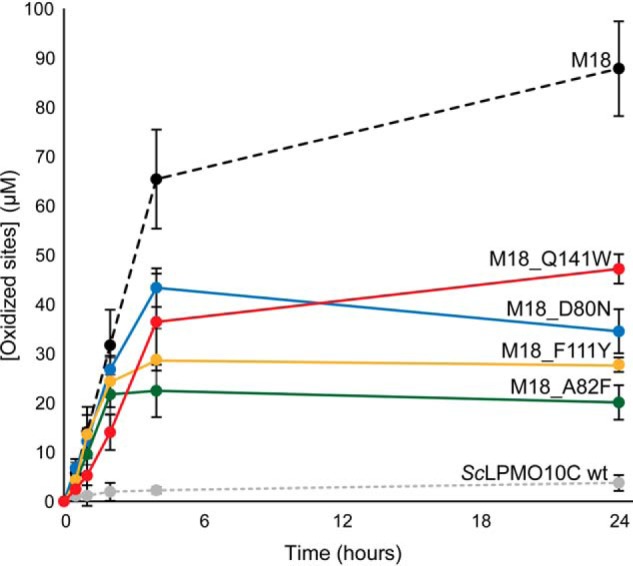
Effect of single mutations on chitin degradation by M18. ScLPMO10C WT, M18, and the four M18 mutants were incubated at 1 μm with 10 g/liter β-chitin at 40 °C in 50 mm sodium phosphate buffer (pH 6.0) in the presence of 1 mm ascorbic acid for 24 h. SmChiC was also added, at 0.25 μm, to obtain more rapid solubilization of LPMO-generated oxidized chain ends from the insoluble substrate. The reaction was stopped by filtration, and soluble oxidized products were degraded to oxidized dimers with chitobiase prior to analysis. All reactions were performed in triplicates. The error bars show ± S.D. (n = 3).
Residual cellulolytic activity
All variants generated C1-oxidized cello-oligomers upon degradation of phosphoric acid-swollen cellulose (PASC) (Fig. 8). C4 oxidation did not occur, because there were no detectable levels of C4-oxidized products and because the levels of native products, which may emerge from C4-oxidized products as a result of the high pH used during HPAEC (40), were low (Fig. 8A). Even though cellulose activity was not completely lost, the mutants exhibited a drastic decrease in cellulolytic activity compared with WT ScLPMO10C (Fig. 8B), with product yields after 4 h amounting to 3–7% of the yields obtained with the WT enzyme (set to 100% product yield). M18_Q141W stood out in that product yields were high (52.5% of WT levels) and because this variant showed stable kinetics, in contrast to all the other variants, which did not generate products after the 1st h of incubation. Fig. 8B shows that all mutants, except M18_Q141W were as good as inactive after 1 h of incubation, whereas WT ScLPMO10C maintained activity also after 1 h. This indicates that the low activity on cellulose, resulting from the mutations needed to convert ScLPMO10C into a chitin-active LPMO, is associated with reduced enzyme stability in reactions with cellulosic substrates.
Figure 8.
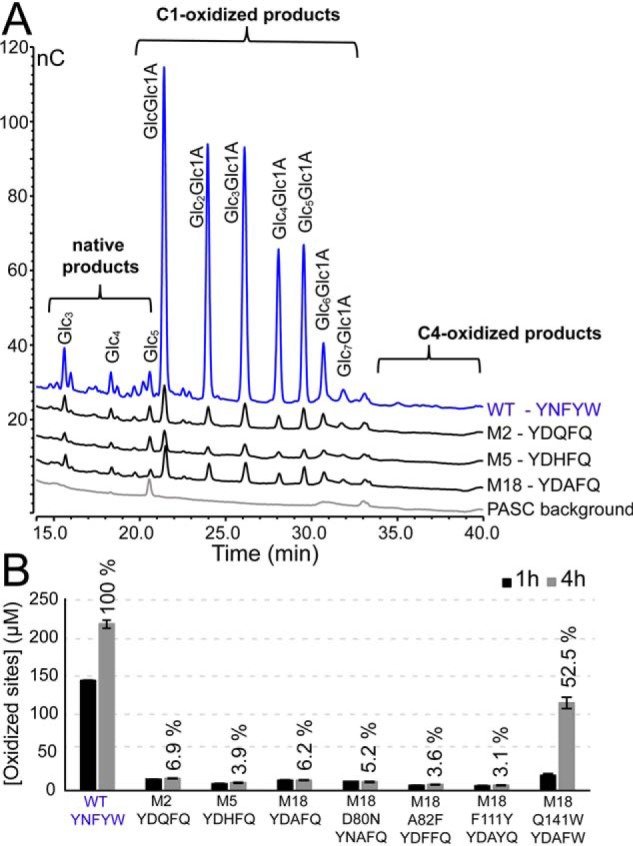
Residual cellulolytic activity. ScLPMO10C WT and mutants were incubated at 1 μm with 5 g/liter PASC and 1 mm ascorbic acid at 40 °C and in 50 mm sodium phosphate (pH 6.0). A shows oxidized products after ∼24 h of incubation, varying from the dimer (GlcGlc1A) to the octamer (Glc7Glc1A), generated by ScLPMO10C WT, M2, M5, and M18. The background signal of the substrate (PASC) incubated with LPMO in the absence of reductant is also shown. Note that the size of the peaks of the WT chromatogram was reduced by 25% before making the plot. Regions where C4-oxidized products would elute are marked. B shows the amount of soluble oxidized sites in aliquots that were withdrawn after 1 and 4 h of incubation; in this case oxidized products were quantified after degrading them to a mixture of oxidized dimers (GlcGlc1A) and trimers (Glc2Glc1A) only, by treatment with the endoglucanase TfCel5A. Relative activities were calculated relative to the amount of product generated by the WT enzyme after 4 h (100%). Testing of the cellulolytic activity of M18 with Avicel gave similar results. All reactions were performed in triplicate, and the error bars show ± S.D. (n = 3).
When comparing the activity of M18 and ScLPMO10C WT on β-chitin and PASC, it is clear that the substrate specificity has been dramatically shifted toward a preference for chitin. Although M18 only gave a relative yield of 6.2% compared with the WT enzyme (100%) on PASC, M18 produced a relative yield of 100% compared with the WT enzyme (1.4%) on β-chitin (Fig. 9). This represents approximately a 16-fold decrease in cellulosic activity and approximately a 70-fold increase in chitinolytic activity (note that the estimated increase in chitinolytic activity is inaccurate as the product generation by the WT on chitin is close to the lower detection limit).
Figure 9.

Comparison of chitinolytic and cellulolytic activity. Comparison of solubilized oxidized products generated in 4 h by 1 μm M18 or 1 μm ScLPMO10C WT from PASC (5 g/liter) or β-chitin (10 g/liter). 100% activity for ScLPMO10C WT on PASC is equal to 218.6 μm (sum of GlcGlc1A and Glc2Glc1A, obtained after cellulase treatment of soluble products as described under “Experimental procedures”). 100% activity for M18 on β-chitin is equal to 20.6 μm (GlcNAcGlcNAc1A, obtained after chitobiase treatment of soluble products as described under “Experimental procedures”). The solubilized oxidized products were converted to oxidized dimers before quantification. Note that quantification of products generated from β-chitin by the WT enzyme is inaccurate as the product levels are close to the lower detection limit. All reactions were performed in triplicates, and the error bars show ± S.D. (n = 3).
Discussion
By screening of a rationally designed mutant library, we were able to convert ScLPMO10C from an almost exclusively cellulose-active LPMO into a chitin-active LPMO with only weak remaining cellulose-activity. None of the chitin-active variants showed C4-oxidizing activity on cellulose, as is the case for naturally occurring AA10s that act on both cellulose and chitin. As such, M18 and some of the M18 derivatives are AA10 LPMOs that have never been encountered in Nature so far: exclusively C1-oxidizing and with activity on chitin and cellulose. In this respect, M18_Q141W stands out, because it retains much of the chitin-degrading ability of M18 while regaining much of the cellulose-degrading ability of ScLPMO10C. Of note, while strictly C1-oxidizing enzymes with activity on both chitin and cellulose have not been encountered among the characterized naturally occurring AA10 LPMOs, a recently described LPMO of eukaryotic origin that is the founding member of the AA15 family (41) has these properties.
Despite the success of our approach, the selected mutants are not perfect chitin-active LPMOs in the sense that they become inactivated faster during catalysis, compared with e.g. natural chitin-active SmLPMO10A. Recent mutagenesis studies on chitin-active (20) and cellulose-active (13, 31) AA10s have shown that a considerable fraction of mutations with a negative effect on LPMO performance primarily affect LPMO stability under turnover conditions. These studies showed clear correlations between decreased substrate binding and increased inactivation. Bissaro et al. (19) have shown that reduced LPMOs are prone to oxidative damage as a consequence of the formation of reactive oxygen species through reaction of the reduced copper with O2 or H2O2 in the absence of substrate. Accordingly, in their mutational study, Loose et al. (20) not only showed a correlation between decreased substrate binding and an increased rate of inactivation, but also that increased inactivation is correlated to increased auto-catalytic damage (i.e. chemical modification) of residues in the catalytic center.
Accumulating data indicate that the stability of LPMOs likely depends on the ability of the enzyme to precisely bind to the substrate in a way that confines the emerging highly powerful reactive oxygen species to only act productively, i.e. abstracting a hydrogen atom from the C1 in the scissile glycosidic bond. Modeling studies have shown that, indeed, binding of the correct substrate strongly confines the spatial orientation of the reactive oxygen species (30, 42), whereas structural studies of the binding of oligomeric substrates to a fungal LPMO (43) have shown that binding of a preferred substrate leads to a more confined catalytic center compared with binding of a less-preferred substrate. Thus, we propose, that binding of chitin to M18 is not yet “perfect.” This will lead to imprecise coordination of reactive oxygen species in the space between the active-site copper and the chemical bond that is to be cleaved and may lead to oxidation of the active-site residues, which results in irreversible inactivation of the LPMO.
On the latter note, the MALDI-ToF analyses of Fig. 3 show that the three tested mutants, and in particular the two less stable mutants, M2 and M5, generated minor amounts of unexpected products (at m/z 1256 and m/z 1260). Both these species, which could be a double-oxidized and a native product, respectively, are almost or completely absent in the reaction with SmLPMO10A. Although the origin and nature of these species remain unknown, it is conceivable that imprecise binding of the substrate, leading to less confinement of the reactive oxygen species, may lead to nonspecific reactions giving unusual products. It is also possible that auto-catalytic damage to the catalytic center reduces enzyme specificity as the reaction progresses.
Accumulating data (this study and Refs. 20, 31, 32, 44) suggest that productive interactions between LPMOs and their polymeric substrates are governed by a multitude of interactions involving many of the residues in the substrate-binding surface. The four mutations in M18, relative to ScLPMO10C WT, seem to have a cumulative effect on the chitinolytic activity as none of the single mutations back to WT abolished chitinolytic activity (Fig. 7). Additionally, all the tested ScLPMO10C variants, except M18_Q141W, showed low relative cellulose activities of 3–7% compared with the WT (Fig. 8). Residue 141 stood out in that it alone seemed to be a major determinant of cellulolytic activity, as shown by the high cellulose activity of M18_Q141W. This is discussed further below.
Fig. 10 provides structural models of LPMO variants interacting with chitin. Although such models need to be used with care, they do shed some light on the mutational effects observed in this study. Tyr-111 (position 4) does not have an obvious analogue in the well-studied chitin-active SmLPMO10A. Fig. 10C illustrates that the protruding hydroxyl group of the Tyr side chain may interfere with substrate binding, and it is thus not surprising that this Tyr was mutated, mostly to Phe, in the large majority of selected chitin-active mutants. Of note, Fig. 10, B–D, shows that, next to the Ser-109–Tyr-111 loop that includes Tyr-111, ScLPMO10C contains another surface loop, Ser-182–Arg-185, that may interfere with chitin binding. It is possible that these two loops, which are absent or shorter in naturally-occurring chitin-active AA10 LPMOs, need further modifications to improve the properties of the chitin-active M18 mutant.
Figure 10.

Models of substrate binding by LPMOs. A shows a model (extracted from MD simulations performed on β-chitin (30)) of SmLPMO10A interacting with GlcNAc7. The side chains of the copper-binding histidines and four of the five residues that were targeted for mutagenesis in cellulose-active ScLPMO10C are shown as sticks (the shown residues correspond to Tyr-79, Asn-80, Phe-82, and Trp-141 in ScLPMO10C; Tyr-111 in ScLPMO10C does not have an analogue in SmLPMO10A). B shows a model of M18 interacting with GlcNAc7 that was generated by superposing the crystal structure of the catalytic domain of ScLPMO10C onto the existing SmLPMO10A–chitin model. The mutations found in M18 were introduced using the mutagenesis tool in PyMOL, and rotamers with the highest probabilities were selected. C shows the same model for WT ScLPMO10C, showing that two aromatic residues (Tyr-111 and Trp-141), which are both mutated in M18, may restrict contact with the chitin substrate. B–D highlight two loop regions, Ser-109–Pro-110–Tyr-111 and Ser-182–Pro-183–Gly-184–Thr-185 (red in B–D) that are extended in ScLPMO10C, relative to natural chitin-active LPMOs, and that may interfere with chitin binding. D shows a superposition of WT ScLPMO10C (green and red) and SmLPMO10A (gray) bound to β-chitin (yellow). The structures shown in D are rotated 90° clockwise relative to the structures shown in A–C.
The other four targeted residues all have similarly-positioned analogues in natural chitin-active LPMOs. Both MD simulations (30) and NMR studies (29) have shown that residues Tyr-54, Glu-55, Gln-57, and Thr-111 in SmLPMO10A (equivalent to Tyr-79, Asn-80, Phe-82, and Trp-141 in ScLPMO10C) interact with chitin during catalysis (mainly in the −5 to −2 subsites; Fig. 10). Most LPMOs, including LPMOs belonging to other families (28, 35), have an exposed aromatic residue near the −4 subsite that is important for substrate binding. The seemingly general importance of this interaction is supported by this study, because the corresponding residue in ScLPMO10C, Tyr-79 (position 1), was unchanged in 95% of the variants, whereas the other 5% carried a Trp. The preference for Tyr or Trp seems context-dependent, because a previous study has shown that the Y79W mutation has hardly any effect on the cellulolytic activity of ScLPMO10C, while mutating the Trp present at this position in the chitinolytic and cellulolytic C1/C4-oxidizing MaLPMO10B to Tyr (Y82W) reduced activity on both chitin and cellulose (31).
Fig. 10 does not reveal a possible explanation for the observation that the targeted asparagine residue (Asn-80, position 2) of ScLPMO10C was mutated to aspartate in 70% of the chitin-active mutants and in almost 90% of the mutants selected multiple times. Possibly, the resulting change in charge somehow affects the catalytic competence of the catalytic center, which, notably, is affected by substrate binding (35) and thus may vary for cellulose and chitin substrates. It is worth noting the Glu, which occurs at this position in SmLPMO10A, was present in the library but was only found in one of the 37 selected variants.
The mutants did not show any clear consensus for position 3 (Phe-82), but among the variants that were detected multiple times the most common mutation was F82Q (M2), followed by F82H (M5) and F82A (M18). It seems clear that reduction of the size of the side chain at this position is beneficial for chitinolytic activity, as the original Phe was replaced by a smaller residue in 95% of the selected mutants. Accordingly, a previous study had shown a negative effect of increasing the size of this residue in MaLPMO10B (31). Phe-82 is slightly buried in the surface and does not seem to interfere directly with substrate binding (Fig. 10). However, the side chain of Phe-82 is close to the side chain of Trp-141 (closest distance 3.4 Å), and this latter residue is of major importance for substrate binding and specificity (Fig. 10).
Trp-141 was the only residue that was mutated in all selected chitin-active variants, and nearly 50% of the mutants and 90% of the mutants occurring multiple times had the W141Q mutation in this position. Although this substitution does not correspond well with threonine being present in the corresponding position of strictly chitin-active SmLPMO10A-like LPMOs (Fig. 1C), it fits well with glutamine frequently being present in MaLPMO10B-like LPMOs. Of note, Loose et al. (20) showed that mutating the corresponding residue in SmLPMO10A (T111A) did not have a major impact on the activity, which could indicate that the primary reason for Trp not being favorable is its large size. Indeed, Fig. 10 shows that the large protruding side chain of Trp-141 could interfere with chitin binding. The W141Q mutation alone drastically reduced the cellulolytic activity of ScLPMO10C (31), but did not yield chitinolytic activity. Likewise, introduction of the Q141W mutation in M18 led to a minor reduction in chitinolytic activity but a large increase in cellulolytic activity.
The discovery of the peroxygenase activity of LPMOs (19) has created discussions in the field as to the nature of the natural co-substrate of the LPMOs (45–47). It has been claimed that H2O2-driven reactions are less specific and lead to more enzyme damage than O2-driven reactions (47), but this is disputed (21, 46). Although an in-depth discussion or experimental assessment of these issues is beyond the scope of this study, it is important to note that Fig. 6 shows that both SmLPMO10A and M18 exert peroxygenase activity. Importantly, also in this case, the primary difference between M18 and the natural chitin-active SmLPMO10A lies in the lower stability of the former.
In conclusion, the results presented in this study reveal structural determinants of the substrate specificity of LPMOs. However, first and foremost, these results underpin the complexity of substrate specificity in these fascinating enzymes. Apparently, substrate specificity depends on a multitude of correctly positioned collaborating residues and, despite the progress such as described here, rational design of substrate specificity does not yet seem feasible. Intriguingly, the multitude of mutations needed to confer chitinolytic activity upon ScLPMO10C and the rather poor properties of intermediate stages of evolution raise questions as to how natural evolution of LPMOs takes place. Most importantly, the present results add to the notion that stability, provided by an optimized LPMO–substrate combination that protects the LPMO from entering off-pathway processes, really is a key determinant of LPMO performance.
Experimental procedures
Library design and generation of ScLPMO10C mutants
Based on conservation patterns detected in 130 AA10 sequences (Table S1), residues Tyr-79, Asn-80, Phe-82, Tyr-111, and Trp-141 in the N-terminal AA10 domain of ScLPMO10C (Uniprot ID Q9RJY2) were selected as targets in the design of the library (see under “Results” and Fig. 1). To limit the library size, amino acid substitutions in these positions were restricted on the basis of a naturally-observed sequence variation. Mutations to alanine were also included. The substitutions were as follows: Ala/Trp/Thr/Asp/Glu for Tyr-79; Ala/Asp/Glu/Gln/Ser for Asn-80; Ala/Gln/Asn/His/Tyr for Phe-82; Ala/Phe/His for Tyr-111; and Ala/Thr/Gln/Phe for Trp-141 (see Fig. 1C). This implies a library size of 4320 variants. The library was synthesized by GeneArt® Combinatorial Libraries (Life Technologies, Inc.), and the amplified library was cloned into plasmid pWW0 for controlled expression with the XylS/Pm system (48, 49). The plasmids harboring the ScLPMO10C encoding mutant genes were subsequently transformed into the E. coli XL10 gold expression host.
High-throughput screening of the mutant library
Cultivation of the mutant library
The library, in E. coli XL10 gold, was spread on LA agar supplemented with 100 μg/ml ampicillin to ensure single colonies. The agar plates were incubated at 37 °C overnight. Colonies were picked and inoculated to clear, flat, and sterile polystyrene 384 microwell plates (Nunc 242757) containing 30 μl of LB medium supplemented with 100 μg/ml ampicillin using a Genetix QPixII robot. 11,520 colonies were picked and stored as glycerol stocks at −80 °C.
The 11,520 transformants were inoculated from the glycerol stocks using a Tecan Freedom EVO-2 200 robot system equipped with an MCA384 well pipetting tool and cultivated in 96-well flat polystyrene microwell plates (Greiner 655163) with Costar lids (Corning 3931). Each well contained 80 μl of a defined basic medium (Red Hi+YE basic medium; see supporting Methods) with 100 μg/ml ampicillin and 2 μl of inoculum. The microwell plates with cultures were incubated at 37 °C, at 90% relative humidity, 900 rpm, and 3 mm amplitude in an Infors HT Multitron Pro shaker incubator for 20 h. To minimize evaporation, the microwell plates were incubated in stacks of five and wrapped in autoclave paper.
After incubation, 40 μl of defined induction medium (Red Hi+YE induction medium; see supporting Methods) supplemented with 1.5 mm m-toluate and 100 μg/ml ampicillin was added to the cultures using a Beckman Coulter Biomek NXP and SCARA robotic system. The cultures were then incubated at 16 °C, 900 rpm, for 24 h, wrapped in autoclave paper. Upon this incubation, the cells were pelleted by centrifugation of the microwell plates at 4700 rpm (3828 × g) for 20 min in a Thermo Fisher Scientific SL 40R laboratory centrifuge and supernatants were discarded. Release of proteins from the periplasmic space was promoted by addition of 120 μl of ion-free water and a subsequent vortex treatment using a Quantifoil Instruments GmbH BioShake 3000 Elm, at 1200 rpm for 60 s. Prior to screening, the cells were stored at −20 °C and were thus subjected to one freeze–thaw cycle. In the preceding method optimization, this procedure (treatment with ion-free water and one freeze–thaw cycle) had been found to liberate sufficient amounts of correctly processed LPMO molecules from the periplasmic space.
Primary screen for enzyme activity
96-well microwell plates containing water-treated cells were thawed at room temperature. Enzyme assays were performed in 384-well deep microwell plates (CorningTM 3965 CostarTM 384-Well Storage Block). The Tecan robot system equipped with an MCA384 well pipetting tool was used to add 20 μl of substrate solution (4 mg/ml β-chitin in 100 mm ammonium acetate buffer supplemented with 2 mm ascorbic acid and 15.8 μm CuCl2 (pH 6.0)) to each well of the 384-well plates. The Beckman and SCARA robot system was then used to add 20 μl of thawed cell suspension to each well containing substrate solution (one well per transformant). The microwell plates were then sealed and given a short spin in the centrifuge to collect all samples and substrate solution at the bottom of the wells. Next, the plates were vortexed on a BioShake at 2000 rpm for 20 s to ensure proper mixing. The plates were then incubated overnight at 40 °C, 1000 rpm, 3 mm amplitude in an Infors HT Multitron Pro shaker incubator. The enzyme reaction was stopped by addition of 160 μl of 100% acetonitrile. Plates were re-sealed, vortexed at 2000 rpm for 20 s, and centrifuged at 4700 rpm (3828 × g) for 15 min to pellet insoluble material. 80 μl of precipitate-free supernatant was transferred to new 384 deep microwell plates and stored at −20 °C awaiting analysis.
The presence of soluble oxidized chitin oligomers was assessed using an Agilent RapidFire 365 instrument (Agilent G9530A) coupled to an Agilent triple quadrupole MS detector (Agilent G6490A). An Agilent Hilic Rapidfire cartridge (HILIC Type H1) was used as stationary phase for solid-phase extraction, whereas the mobile phases used to retain and elute the oxidized chitin oligomers were a 90:10 (v/v) mixture of acetonitrile/water and a 10:90 (v/v) mixture of acetonitrile/water, each with 0.1% (v/v) formic acid and applied using a flow rate of 1 ml/min, respectively. The mass spectrometer was equipped with an ESI ion source run in negative mode (gas temperature: 220 °C; gas flow: 20 liter/min; nebulizer: 40 p.s.i.; sheath gas temperature: 400 °C; sheath gas flow: 11 liters/min; capillary: 3300 V; nozzle voltage: 500 V; iFunnel high: 90 V; iFunnel low: 60 V), and the chitin oligomers were quantified in MRM mode using the following mass transitions: DP2ox, m/z 439.2 → m/z 116.0; DP3ox, m/z 643.3 → m/z 625.3; DP4ox, 845.3 m/z → m/z 668.2; DP5ox, m/z 1048.2 → m/z 871.2; DP6ox, m/z 1251.5 → m/z 871.3; where DPnox indicates an oligomer of GlcNAc comprising n sugar residues of which one is oxidized to GlcNAc1A (e.g. DP3ox indicates (GlcNAc)n − 1GlcNAc1A), and where the first m/z value given for each oligomer represents [M − H+], where M is the mass of the aldonic acid.
Cultivation and screening of candidates selected for validation screening
42 of the 11,520 transformants were selected for further screening. The location of each selected candidate in the library was identified, and selected clones were spread on Petri dishes containing LA agar supplemented with 100 μg/ml ampicillin to obtain single colonies. The agar plates were incubated at 37 °C overnight. Four single colonies were selected for each candidate and inoculated to 120 μl of medium in 96-well flat polystyrene microwell plates (Greiner 655163) with Costar lids (Corning 3931). Two parallels were made: one microwell plate with LB medium supplemented with 100 μg/ml ampicillin for use as a glycerol stock, and one microwell plate with defined medium (Red Hi+YE basic medium) supplied with 100 μg/ml ampicillin for further screening. The validation screening was performed as described above and included eight wells with E. coli producing the WT ScLPMO10C enzyme and four wells with E. coli without plasmid as controls. After the validation screening, 3 of the 42 initially-selected transformants were discarded as they did not show chitinolytic activity.
Sequence analysis of selected variants
Single colonies of the 39 selected variants were inoculated in LB medium supplemented with 100 μg/ml ampicillin and incubated at 37 °C and 200 rpm overnight. Glycerol stocks were prepared and stored at −80 °C, and plasmids were isolated using a NucleoSpin plasmid kit (Macherey-Nagel, Düren, Germany) and sent for Sanger sequencing (Eurofins GATC, Cologne, Germany). The sequence data were analyzed in BioEdit (www.mbio.ncsu.edu/BioEdit/bioedit.html).4 Two variants contained unintended mutations, leaving 37 variants for further studies. After removing redundant sequences, 27 unique variants remained (see under “Results” for details).
Generation of additional ScLPMO10C mutants
The gene encoding the M18 mutant (Tyr-79/Asp-80/Ala-82/Phe-111/Gln-141) was transferred from the XylS/Pm-controlled plasmid pWW0 to the pRSETB expression vector using the InFusion HD cloning kit (Clontech), as described previously for WT ScLPMO10C (34). Transfer to this smaller plasmid was necessary to facilitate cloning of additional variants based on the M18 construct using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies). Then, two variants of M18 were produced to partially revert M18 to ScLPMO10C-WT, M18_D80N and M18_Q141W. After verification of the sequences of the mutated plasmids by Sanger sequencing (see above), the plasmids were transformed into One Shot® BL21 StarTM (DE3) chemically-competent E. coli cells (Invitrogen) for protein expression (see below).
Enzyme production
Expression with the XylS/Pm system (pWW0 vector)
A subset of the mutants was selected for production and produced essentially as described in Ref. 49. Briefly, E. coli XL10 gold carrying a mutant plasmid was inoculated from a glycerol stock in 333 ml of a defined basic medium (Red Hi + YE basic medium) supplied with 100 μg/ml ampicillin and incubated overnight at 37 °C and 200 rpm in a shake flask. After cooling the cultures on ice for 5 min, 167 ml of a defined induction medium (Red Hi + YE induction medium) with 100 μg/ml ampicillin and 1.5 mm m-toluate (the inducer molecule of the XylS/Pm expression system) were added, and the incubation was continued at 16 °C and 200 rpm for another 24 h. Cell pellets were harvested by centrifugation at 5000 × g for 15 min at 4 °C (Beckman Coulter Brea, CA), after which a periplasmic extract was prepared using an osmotic shock method (50). All LPMOs were expressed with a signal peptide that directs the proteins to the periplasmic space (34). During secretion the signal peptide is cleaved off, which results in mature enzymes that possess an N-terminal histidine that can coordinate the catalytic copper.
Expression with the pRSETB vector
For production of mutants M18_D80N and M18_Q141W generated in the pRSETB vector, cells from a glycerol stock were used to inoculate 500 ml of Terrific Broth (TB) supplemented with 100 μg/ml ampicillin, followed by incubation at 37 °C for ∼20 h in a Harbinger system (Harbinger Biotechnology & Engineering, Markham, Ontario, Canada). Note that expression was driven by leakiness of the T7 promoter and that no inducer molecule was added. Cells were harvested, and a periplasmic extract was prepared as described above.
Protein purification
Periplasmic extracts containing the mature (i.e. signal peptide-free) proteins were filtered using 0.45-μm syringe filters (Sarstedt, Nümbrecht, Germany) and adjusted to 50 mm Tris-HCl (pH 7.5) after which the proteins were purified by anion-exchange chromatography using an ÄKTA pure chromatography system (GE Healthcare) equipped with a 5-ml HiTrap DEAE FF column (GE Healthcare), with 50 mm Tris-HCl (pH 7.5) as running buffer. The proteins were eluted by applying a linear salt gradient (0–500 mm NaCl) over 60 column volumes (CV). Protein fractions were examined by SDS-PAGE (Bio-Rad), and relevant fractions were pooled and concentrated to 1 ml using 10,000 molecular weight cutoff (MWCO) Vivaspin ultrafiltration tubes (Sartorius, Göttingen, Germany). The proteins were further purified by size-exclusion chromatography using a HiLoad 16/60 Superdex 75 column with a running buffer consisting of 50 mm Tris-HCl (pH 8.0) and 200 mm NaCl. Fractions containing the LPMO were concentrated using 10,000 MWCO Vivaspin ultrafiltration tubes (Sartorius) with simultaneous buffer exchange to 20 mm sodium phosphate buffer (pH 6.0). Protein purity was assessed by SDS-PAGE. Before being employed in enzymatic reactions, the purified LPMOs were saturated with Cu(II) by a 30-min incubation with CuSO4 in a 1:3 molar ratio (LPMO/Cu2+), followed by desalting using PD Midi-Trap G-25 columns (GE Healthcare) equilibrated with 20 mm sodium phosphate (pH 6.0) to remove excess Cu2+, as described previously (34). Protein concentrations were determined by measuring A280 (absorbance at 280 nm) in a spectrophotometer (Eppendorf Biophotometer, Hamburg, Germany), and absorbances were converted to protein concentrations using theoretical extinction coefficients calculated with the ExPASy ProtParam tool. The purified and copper saturated LPMOs were stored at 4 °C.
Additional enzymes
The S. marcescens GH18 exo-chitinase SmChi18A, endo-chitinase SmChi18C, its GH20 chitobiase (SmGH20), and its chitin-oxidizing AA10 LPMO (SmLPMO10A, also known as CBP21) were expressed and purified, as described previously (5, 44, 51). Full-length LPMO10C from S. coelicolor A3(2) (ScLPMO10C) and a truncated variant lacking the CBM2 (ScLPMO10C-CD) were produced and purified, as described previously (34). Purified Thermobifida fusca GH5 endoglucanase TfCel5A (52) was provided as a kind gift from the late Prof. David Wilson (Cornell University, New York).
Determination of apparent melting temperature (Tm)
The apparent Tm of the proteins was determined according to a protein thermal shift assay (Thermo Fisher Scientific) based on using the fluorescent dye SYPRO Orange to monitor protein unfolding (39). The quantum yield of the dye is significantly increased upon binding to hydrophobic regions of the protein that become accessible as the protein unfolds. The fluorescence emission was monitored using a StepOnePlus real-time PCR machine (Thermo Fisher Scientific). 0.1 g/liter LPMO in 50 mm sodium phosphate buffer (pH 6.0) was heated in the presence of the dye in a 96-well plate from 25 to 99 °C over 50 min. For each protein, the experiment was carried out in quadruplicate (i.e. n = 4).
Substrates
Activity was assessed on β-chitin and PASC. β-Chitin (extracted from squid pen and deproteinized, batch 20140101, France Chitin, Orange, France) was ball-milled and sieved in order to produce particles with a size <0.85 mm. PASC was prepared from Avicel PH-101 (microcrystalline cellulose) as described by Wood (53).
Substrate-binding assay
Protein binding to β-chitin was evaluated using a slightly-modified variant of the A280 method described previously (44). The binding mixtures contained 10 mg/ml β-chitin and 0.1 mg/ml protein in 50 mm sodium phosphate (pH 6.0) and were incubated at 22 °C and 1000 rpm in an Eppendorf thermomixer (Eppendorf, Hamburg, Germany). Substrate binding was monitored by measuring the A280 of the supernatant at various time points, using aliquots that were taken from the binding reactions and immediately vacuum-filtered over a 0.45-μm filter to separate unbound protein from protein bound to substrate. Theoretical extinction coefficients calculated with the ExPASy ProtParam tool were used to determine the protein concentration.
Chitin degradation experiments
Activity assays
Unless stated otherwise, reactions were performed in 50 mm sodium phosphate buffer (pH 6.0) in the presence of 1 mm ascorbic acid (reductant) at 40 °C and 800 rpm in an Eppendorf thermomixer. All reactions were performed in triplicate.
Qualitative analysis of chitinolytic activity
For the initial verification of chitinolytic activity, the mutants were incubated at 10 μm with 10 g/liter β-chitin. WT ScLPMO10C and chitin-active SmLPMO10A (CBP21) were included in the experiment as controls. After separation of the solid material by vacuum filtering through a 0.45-μm filter, supernatants were analyzed by MALDI-ToF (see below) for detection of oxidized chito-oligomers.
Quantitative analysis of β-chitin activity
For quantitative analysis of the activity toward β-chitin, the LPMOs were incubated at 1 μm with 10 g/liter β-chitin for up to 24 h. A purified endo-chitinase (ChiC from S. marcescens; SmChiC) was added to all reactions at 0.25 μm to facilitate solubilization of oxidized products, as low LPMO activity alone may not be sufficient to generate oxidized products that are sufficiently short to become soluble (13). Aliquots were withdrawn at selected time points and filtered before the soluble fraction was merged with an identical volume of a 1 μm solution of chitobiase from S. marcescens (SmCHB) in 5 mm Tris-HCl (pH 8.0), followed by overnight incubation at 37 °C. The chitobiase, a GH20 β-hexosaminidase, cleaves off single GlcNAc units from chito-oligosaccharides until GlcNAc is the final product. If the chito-oligosaccharide is oxidized at the C1 position, the final product is an oxidized dimer (chitobionic acid) (5). As a result of this procedure, all soluble oxidized products are converted to chitobionic acid, which can easily be quantified. The products were analyzed using an UHPLC system (see below). In some cases, the total amount of oxidized sites was determined, as described under “LPMO reactions with H2O2 feeding.”
Probing the cause of enzyme inactivation
Reactions were set up as above, and samples were collected and filtered after 4 and 6 h. At 6 h, the remaining reaction mixture was divided into three identical fractions, where fresh reductant (1 mm) was added to one fraction, fresh reductant and fresh substrate (10 g/liter) to another, and fresh reductant and fresh LPMO (1 μm) to the third fraction. The incubation was continued overnight before stopping the reactions by filtration. All supernatants were subjected to a chitobiase treatment, as above, before product quantification using an UHPLC system (see below).
LPMO reactions with H2O2 feeding feeding
The LPMOs were preincubated at 1 μm in 50 mm sodium phosphate (pH 6.0) containing 10 g/liter β-chitin and at 1000 rpm for 10 min to reach 40 °C. This preincubation was followed by addition of ascorbic acid to a final concentration of 100 μm to reduce the catalytic copper from the Cu(II) to the Cu(I) state. Addition of ascorbic acid was immediately followed by addition of H2O2 to a final concentration of 15 μm. Control reactions without added H2O2 were also included. Aliquots were withdrawn and immediately vacuum-filtered every 7.5 min, and every 15 min a fresh batch of ascorbic acid and H2O2 was added subsequently to withdrawing an aliquot from the reaction. The H2O2-driven reactions were also carried out using different ascorbic acid concentrations (0–100 μm) and a higher H2O2 concentration (100 μm added every 20 min for 60 min). Reaction supernatants were subjected to chitobiase treatment, as above. At the final time point (60 min), next to preparing a product-containing supernatant by the usual filtration procedure, part of the (nonfiltered) reaction mixture (containing both soluble and insoluble products) was boiled for 10 min at 98 °C to stop the LPMO reaction and merged with an identical volume of a chitinase mixture (10 μm SmChiA, 6 μm SmChiC, 1 μm SmCBH), followed by overnight incubation at 37 °C and 600 rpm. This treatment is sufficient to completely degrade all chitin and converts all oxidized products to chitobionic acid, as described above. The resulting hydrolysates were used to assess the total amount of oxidized products using UHPLC (see below).
Cellulose degradation experiments
The LPMOs were incubated at 1 μm with 5 g/liter PASC, prepared from Avicel essentially as described in Ref. 53, and aliquots were taken and filtered after 1 and 4 h. To facilitate quantification of the solubilized oxidized products, these products were treated with a cellulase that converts oxidized products of varying length to a mixture of shorter fragments, including oxidized dimers (GlcGlc1A) and trimers (Glc2Glc1A). This conversion was achieved by an overnight incubation of solubilized cello-oligosaccharides with 0.5 μm of an endoglucanase (TfCel5A from T. fusca) at 37 °C. Subsequently, oxidized dimers (GlcGlc1A) and trimers (Glc2Glc1A) were quantified by HPAEC-PAD (see below). An aliquot not degraded with endoglucanase was also analyzed.
Product analysis
MALDI-ToF analysis
Reaction supernatants were assayed qualitatively using a MALDI-ToF UltrafleXtreme mass spectrometer (Bruker Daltonics GmbH, Bremen, Germany) equipped with a nitrogen 337-nm laser. Reaction products (1 μl) were applied to an MTP 384 ground steel target plate TF (Bruker Daltonics) and merged with 2 μl of 9 mg/ml 2,5-dihydroxybenzoic acid dissolved in 30% acetonitrile, followed by air-drying. Data collection and analysis were carried out using the Bruker FlexAnalysis software.
UHPLC analysis
Quantification of oxidized chitobiose was achieved using a Dionex Ultimate 3000 UHPLC system (DionexCorp., Sunnyvale, CA) equipped with a Rezex RFQ-Fast Acid H+ (8%) 7.8 × 100-mm column (Phenomenex, Torrance, CA) operated at 85 °C. Sample components were eluted isocratically using 5 mm sulfuric acid as mobile phase and were detected using UV absorption at 194 nm. Data collection and analysis were carried out with the Chromeleon 7.0 software. Standards were generated in-house by complete oxidation of N-acetyl-chitobiose (Megazyme; 95% purity) with a chitooligosaccharide oxidase from Fusarium graminearum (ChitO (54)) as described previously (5).
HPAEC-PAD analysis
Oxidized products generated from cellulosic substrates were analyzed by HPAEC using a DionexTM ICS5000 system (Thermo Fisher Scientific, Sunnyvale, CA) equipped with a disposable electrochemical gold electrode and a CarboPac PA1 column (Dionex) operated with 0.1 m NaOH (eluent A) at a column temperature of 30 °C. A multistep linear gradient with increasing amounts of eluent B (0.1 m NaOH + 1 m NaOAc) was used to elute the products, going from 0 to 10% B over 10 min; 10–30% B over 25 min; 30–100% B over 5 min; 100–0% B over 1 min; and 0% B (reconditioning) for 9 min. Data collection and analysis were carried out with the Chromeleon 7.0 software. For analysis of endoglucanase-treated samples containing only two oxidized products, oxidized cellobiose and cellotriose, a steeper gradient of eluent B was used as follows: 0–10% B over 10 min; 10–14% B over 5 min; 14–30% B over 1 min; 30–100% B over 2 min; 100–0% B over 0.1 min; and 0% B (reconditioning) for 10.9 min. Cellobiose (Sigma) and cellotriose (Megazyme) were used as substrates for production of the C1-oxidized standards cellobionic acid (GlcGlc1A) and cellotrionic acid (Glc2Glc1A), respectively, by incubation with cellobiose dehydrogenase from Myriococcum thermophilum (MtCDH) (55).
Author contributions
M. S. J., G. K., G. K. N., and Z. F. investigation; M. S. J. and Z. F. visualization; M. S. J., G. K., P. C., H. F. K., G. K. N., H. S., and Z. F. methodology; M. S. J., G. K., Z. F., and V. G. H. E. writing-original draft; G. K., B. B., Z. F., and V. G. H. E. conceptualization; G. K., H. F. K., and G. K. N. data curation; B. B. and G. V.-K. writing-review and editing; G. V.-K., Z. F., and V. G. H. E. supervision; V. G. H. E. funding acquisition; V. G. H. E. validation; V. G. H. E. project administration.
Supplementary Material
Acknowledgments
Infrastructure was supported in part by NorBioLab Grant 226247 provided by the Research Council of Norway. We thank Lasse Fredriksen for helpful discussions.
This work was supported by the Research Council of Norway through the NorZymeD Project No. 221568. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S6, Table S1, supporting Methods, and supporting Refs. 1–4
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- LPMO
- lytic polysaccharide monooxygenase
- GlcGlc1A
- aldonic acid of cellobiose
- GlcNAcGlcNAc1A
- aldonic acid of chitobiose
- AscA
- ascorbic acid
- AA
- auxiliary activity
- CBM
- carbohydrate-binding module
- DP
- degree of polymerization
- GH
- glycoside hydrolase
- PASC
- phosphoric acid swollen cellulose
- MRM
- multiple-reaction monitoring
- HPAEC
- high-performance anion-exchange chromatography
- HPAEC-PAD
- high performance anion-exchange chromatography with pulsed amperometric detection
- PDB
- Protein Data Bank
- ESI
- electrospray ionization
- MWCO
- molecular weight cutoff
- MD
- molecular dynamics.
References
- 1. Vaaje-Kolstad G., Westereng B., Horn S. J., Liu Z., Zhai H., Sørlie M., and Eijsink V. G. H. (2010) An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330, 219–222 10.1126/science.1192231 [DOI] [PubMed] [Google Scholar]
- 2. Levasseur A., Drula E., Lombard V., Coutinho P. M., and Henrissat B. (2013) Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 6, 41 10.1186/1754-6834-6-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Agostoni M., Hangasky J. A., and Marletta M. A. (2017) Physiological and molecular understanding of bacterial polysaccharide monooxygenases. Microbiol. Mol. Biol. Rev. 81, e–00015 10.1128/MMBR.00015-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Paspaliari D. K., Loose J. S., Larsen M. H., and Vaaje-Kolstad G. (2015) Listeria monocytogenes has a functional chitinolytic system and an active lytic polysaccharide monooxygenase. FEBS J. 282, 921–936 10.1111/febs.13191 [DOI] [PubMed] [Google Scholar]
- 5. Loose J. S., Forsberg Z., Fraaije M. W., Eijsink V. G. H., and Vaaje-Kolstad G. (2014) A rapid quantitative activity assay shows that the Vibrio cholerae colonization factor GbpA is an active lytic polysaccharide monooxygenase. FEBS Lett. 588, 3435–3440 10.1016/j.febslet.2014.07.036 [DOI] [PubMed] [Google Scholar]
- 6. Vaaje-Kolstad G., Horn S. J., van Aalten D. M., Synstad B., and Eijsink V. G. H. (2005) The non-catalytic chitin-binding protein CBP21 from S. marcescens is essential for chitin degradation. J. Biol. Chem. 280, 28492–28497 10.1074/jbc.M504468200 [DOI] [PubMed] [Google Scholar]
- 7. Harris P. V., Welner D., McFarland K. C., Re E., Navarro Poulsen J. C., Brown K., Salbo R., Ding H., Vlasenko E., Merino S., Xu F., Cherry J., Larsen S., and Lo Leggio L. (2010) Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: structure and function of a large, enigmatic family. Biochemistry 49, 3305–3316 10.1021/bi100009p [DOI] [PubMed] [Google Scholar]
- 8. Vermaas J. V., Crowley M. F., Beckham G. T., and Payne C. M. (2015) Effects of lytic polysaccharide monooxygenase oxidation on cellulose structure and binding of oxidized cellulose oligomers to cellulases. J. Phys. Chem. B 119, 6129–6143 10.1021/acs.jpcb.5b00778 [DOI] [PubMed] [Google Scholar]
- 9. Eibinger M., Ganner T., Bubner P., Rošker S., Kracher D., Haltrich D., Ludwig R., Plank H., and Nidetzky B. (2014) Cellulose surface degradation by a lytic polysaccharide monooxygenase and its effect on cellulase hydrolytic efficiency. J. Biol. Chem. 289, 35929–35938 10.1074/jbc.M114.602227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eibinger M., Sattelkow J., Ganner T., Plank H., and Nidetzky B. (2017) Single-molecule study of oxidative enzymatic deconstruction of cellulose. Nat. Commun. 8, 894 10.1038/s41467-017-01028-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Johansen K. S. (2016) Discovery and industrial applications of lytic polysaccharide mono-oxygenases. Biochem. Soc. Trans. 44, 143–149 10.1042/BST20150204 [DOI] [PubMed] [Google Scholar]
- 12. Müller G., Chylenski P., Bissaro B., Eijsink V. G. H., and Horn S. J. (2018) The impact of hydrogen peroxide supply on LPMO activity and overall saccharification efficiency of a commercial cellulase cocktail. Biotechnol. Biofuels 11, 209 10.1186/s13068-018-1199-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Courtade G., Forsberg Z., Heggset E. B., Eijsink V. G. H., and Aachmann F. L. (2018) The carbohydrate-binding module and linker of a modular lytic polysaccharide monooxygenase promote localized cellulose oxidation. J. Biol. Chem. 293, 13006–13015 10.1074/jbc.RA118.004269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boraston A. B., Bolam D. N., Gilbert H. J., and Davies G. J. (2004) Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem. J. 382, 769–781 10.1042/BJ20040892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hervé C., Rogowski A., Blake A. W., Marcus S. E., Gilbert H. J., and Knox J. P. (2010) Carbohydrate-binding modules promote the enzymatic deconstruction of intact plant cell walls by targeting and proximity effects. Proc. Natl. Acad. Sci. U.S.A. 107, 15293–15298 10.1073/pnas.1005732107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Quinlan R. J., Sweeney M. D., Lo Leggio L., Otten H., Poulsen J. C., Johansen K. S., Krogh K. B., Jørgensen C. I., Tovborg M., Anthonsen A., Tryfona T., Walter C. P., Dupree P., Xu F., Davies G. J., and Walton P. H. (2011) Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. U.S.A. 108, 15079–15084 10.1073/pnas.1105776108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kjaergaard C. H., Qayyum M. F., Wong S. D., Xu F., Hemsworth G. R., Walton D. J., Young N. A., Davies G. J., Walton P. H., Johansen K. S., Hodgson K. O., Hedman B., and Solomon E. I. (2014) Spectroscopic and computational insight into the activation of O2 by the mononuclear Cu center in polysaccharide monooxygenases. Proc. Natl. Acad. Sci. U.S.A. 111, 8797–8802 10.1073/pnas.1408115111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Beeson W. T., Phillips C. M., Cate J. H., and Marletta M. A. (2012) Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J. Am. Chem. Soc. 134, 890–892 10.1021/ja210657t [DOI] [PubMed] [Google Scholar]
- 19. Bissaro B., Røhr Å. K., Müller G., Chylenski P., Skaugen M., Forsberg Z., Horn S. J., Vaaje-Kolstad G., and Eijsink V. G. H. (2017) Oxidative cleavage of polysaccharides by monocopper enzymes depends on H2O2. Nat. Chem. Biol. 13, 1123–1128 10.1038/nchembio.2470 [DOI] [PubMed] [Google Scholar]
- 20. Loose J. S., Arntzen M. Ø., Bissaro B., Ludwig R., Eijsink V. G. H., and Vaaje-Kolstad G. (2018) Multipoint precision binding of substrate protects lytic polysaccharide monooxygenases from self-destructive off-pathway processes. Biochemistry 57, 4114–4124 10.1021/acs.biochem.8b00484 [DOI] [PubMed] [Google Scholar]
- 21. Eijsink V. G. H., Petrovic D., Forsberg Z., Mekasha S., Røhr Å. K., Várnai A., Bissaro B., and Vaaje-Kolstad G. (2019) On the functional characterization of lytic polysaccharide monooxygenases (LPMOs). Biotechnol. Biofuels 12, 58 10.1186/s13068-019-1392-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuusk S., Bissaro B., Kuusk P., Forsberg Z., Eijsink V. G. H., Sørlie M., and Väljamäe P. (2018) Kinetics of H2O2-driven degradation of chitin by a bacterial lytic polysaccharide monooxygenase. J. Biol. Chem. 293, 12284 10.1074/jbc.AAC118.004796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hegnar O. A., Petrovic D. M., Bissaro B., Alfredsen G., Várnai A., and Eijsink V. G. H. (2019) pH-dependent relationship between catalytic activity and hydrogen peroxide production shown via characterization of a lytic polysaccharide monooxygenase from Gloeophyllum trabeum. Appl. Environ Microbiol. 85, e02612–18 10.1128/AEM.02612-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kittl R., Kracher D., Burgstaller D., Haltrich D., and Ludwig R. (2012) Production of four Neurospora crassa lytic polysaccharide monooxygenases in Pichia pastoris monitored by a fluorimetric assay. Biotechnol. Biofuels 5, 79 10.1186/1754-6834-5-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Isaksen T., Westereng B., Aachmann F. L., Agger J. W., Kracher D., Kittl R., Ludwig R., Haltrich D., Eijsink V. G. H., and Horn S. J. (2014) A C4-oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello-oligosaccharides. J. Biol. Chem. 289, 2632–2642 10.1074/jbc.M113.530196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tandrup T., Frandsen K. E. H., Johansen K. S., Berrin J. G., and Lo Leggio L. (2018) Recent insights into lytic polysaccharide monooxygenases (LPMOs). Biochem. Soc. Trans. 46, 1431–1447 10.1042/BST20170549 [DOI] [PubMed] [Google Scholar]
- 27. Phillips C. M., Beeson W. T., Cate J. H., and Marletta M. A. (2011) Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem. Biol. 6, 1399–1406 10.1021/cb200351y [DOI] [PubMed] [Google Scholar]
- 28. Courtade G., Wimmer R., Røhr Å. K., Preims M., Felice A. K., Dimarogona M., Vaaje-Kolstad G., Sørlie M., Sandgren M., Ludwig R., Eijsink V. G. H., and Aachmann F. L. (2016) Interactions of a fungal lytic polysaccharide monooxygenase with beta-glucan substrates and cellobiose dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 113, 5922–5927 10.1073/pnas.1602566113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aachmann F. L., Sørlie M., Skjåk-Braek G., Eijsink V. G. H., and Vaaje-Kolstad G. (2012) NMR structure of a lytic polysaccharide monooxygenase provides insight into copper binding, protein dynamics, and substrate interactions. Proc. Natl. Acad. Sci. U.S.A. 109, 18779–18784 10.1073/pnas.1208822109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bissaro B., Isaksen I., Vaaje-Kolstad G., Eijsink V. G. H., and Røhr Å. K. (2018) How a lytic polysaccharide monooxygenase binds crystalline chitin. Biochemistry 57, 1893–1906 10.1021/acs.biochem.8b00138 [DOI] [PubMed] [Google Scholar]
- 31. Forsberg Z., Bissaro B., Gullesen J., Dalhus B., Vaaje-Kolstad G., and Eijsink V. G. H. (2018) Structural determinants of bacterial lytic polysaccharide monooxygenase functionality. J. Biol. Chem. 293, 1397–1412 10.1074/jbc.M117.817130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Danneels B., Tanghe M., and Desmet T. (2019) Structural features on the substrate-binding surface of fungal lytic polysaccharide monooxygenases determine their oxidative regioselectivity. Biotechnol. J. 14, e1800211 10.1002/biot.201800211 [DOI] [PubMed] [Google Scholar]
- 33. Span E. A., Suess D. L. M., Deller M. C., Britt R. D., and Marletta M. A. (2017) The role of the secondary coordination sphere in a fungal polysaccharide monooxygenase. ACS Chem. Biol. 12, 1095–1103 10.1021/acschembio.7b00016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Forsberg Z., Mackenzie A. K., Sørlie M., Røhr Å. K., Helland R., Arvai A. S., Vaaje-Kolstad G., and Eijsink V. G. H. (2014) Structural and functional characterization of a conserved pair of bacterial cellulose-oxidizing lytic polysaccharide monooxygenases. Proc. Natl. Acad. Sci. U.S.A. 111, 8446–8451 10.1073/pnas.1402771111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Frandsen K. E., Simmons T. J., Dupree P., Poulsen J. C., Hemsworth G. R., Ciano L., Johnston E. M., Tovborg M., Johansen K. S., von Freiesleben P., Marmuse L., Fort S., Cottaz S., Driguez H., Henrissat B., et al. (2016) The molecular basis of polysaccharide cleavage by lytic polysaccharide monooxygenases. Nat. Chem. Biol. 12, 298–303 10.1038/nchembio.2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Crooks G. E., Hon G., Chandonia J. M., and Brenner S. E. (2004) WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Frommhagen M., Westphal A. H., Hilgers R., Koetsier M. J., Hinz S. W. A., Visser J., Gruppen H., van Berkel W. J. H., and Kabel M. A. (2018) Quantification of the catalytic performance of C1-cellulose-specific lytic polysaccharide monooxygenases. Appl. Microbiol. Biotechnol. 102, 1281–1295 10.1007/s00253-017-8541-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Forsberg Z., Røhr A. K., Mekasha S., Andersson K. K., Eijsink V. G. H., Vaaje-Kolstad G., and Sørlie M. (2014) Comparative study of two chitin-active and two cellulose-active AA10-type lytic polysaccharide monooxygenases. Biochemistry 53, 1647–1656 10.1021/bi5000433 [DOI] [PubMed] [Google Scholar]
- 39. Huynh K., and Partch C. L. (2015) Analysis of protein stability and ligand interactions by thermal shift assay. Curr. Protoc. Protein Sci. 79, 28.9.1–14 10.1002/0471140864.ps2809s79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Westereng B., Arntzen M. Ø., Aachmann F. L., Várnai A., Eijsink V. G. H., and Agger J. W. (2016) Simultaneous analysis of C1 and C4 oxidized oligosaccharides, the products of lytic polysaccharide monooxygenases acting on cellulose. J. Chromatogr. A 1445, 46–54 10.1016/j.chroma.2016.03.064 [DOI] [PubMed] [Google Scholar]
- 41. Sabbadin F., Hemsworth G. R., Ciano L., Henrissat B., Dupree P., Tryfona T., Marques R. D. S., Sweeney S. T., Besser K., Elias L., Pesante G., Li Y., Dowle A. A., Bates R., Gomez L. D., et al. (2018) An ancient family of lytic polysaccharide monooxygenases with roles in arthropod development and biomass digestion. Nat. Commun. 9, 756 10.1038/s41467-018-03142-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang B., Johnston E. M., Li P., Shaik S., Davies G. J., Walton P. H., and Rovira C. (2018) QM/MM studies into the H2O2-dependent activity of lytic polysaccharide monooxygenases: evidence for the formation of a caged hydroxyl radical intermediate. ACS Catal. 8, 1346–1351 10.1021/acscatal.7b03888 [DOI] [Google Scholar]
- 43. Simmons T. J., Frandsen K. E. H., Ciano L., Tryfona T., Lenfant N., Poulsen J. C., Wilson L. F., Tandrup T., Tovborg M., Schnorr K., Johansen K. S., Henrissat B., Walton P. H., Lo Leggio L., and Dupree P. (2017) Structural and electronic determinants of lytic polysaccharide monooxygenase reactivity on polysaccharide substrates. Nat. Commun. 8, 1064 10.1038/s41467-017-01247-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vaaje-Kolstad G., Houston D. R., Riemen A. H., Eijsink V. G. H., and van Aalten D. M. (2005) Crystal structure and binding properties of the S. marcescens chitin-binding protein CBP21. J. Biol. Chem. 280, 11313–11319 10.1074/jbc.M407175200 [DOI] [PubMed] [Google Scholar]
- 45. Bissaro B., Várnai A., Røhr. Å. K., and Eijsink V. G. H. (2018) Oxidoreductases and reactive oxygen species in conversion of lignocellulosic biomass. Microbiol. Mol. Biol. Rev. 82, e00029–18 10.1128/MMBR.00029-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Forsberg Z., Sŗlie M., Petroviã D., Courtade G., Aachmann F. L., Vaaje-Kolstad G., Bissaro B., Rḩr Å. K., and Eijsink V. G. H. (2019) Polysaccharide degradation by lytic polysaccharide monooxygenases. Curr. Opin. Struct. Biol. 59, 54–64 10.1016/j.sbi.2019.02.015 [DOI] [PubMed] [Google Scholar]
- 47. Hangasky J. A., Iavarone A. T., and Marletta M. A. (2018) Reactivity of O2 versus H2O2 with polysaccharide monooxygenases. Proc. Natl. Acad. Sci. U.S.A. 115, 4915–4920 10.1073/pnas.1801153115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gawin A., Valla S., and Brautaset T. (2017) The XylS/Pm regulator/promoter system and its use in fundamental studies of bacterial gene expression, recombinant protein production and metabolic engineering. Microb. Biotechnol. 10, 702–718 10.1111/1751-7915.12701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Courtade G., Le S. B., Saetrom G. I., Brautaset T., and Aachmann F. L. (2017) A novel expression system for lytic polysaccharide monooxygenases. Carbohydr. Res. 448, 212–219 10.1016/j.carres.2017.02.003 [DOI] [PubMed] [Google Scholar]
- 50. Manoil C., and Beckwith J. (1986) A genetic approach to analyzing membrane protein topology. Science 233, 1403–1408 10.1126/science.3529391 [DOI] [PubMed] [Google Scholar]
- 51. Mekasha S., Byman I. R., Lynch C., Toupalová H., Anděra L., Næs T., Vaaje-Kolstad G., Eijsink V. G. H. (2017) Development of enzyme cocktails for complete saccharification of chitin using mono-component enzymes from S. marcescens. Proc. Biochem. 56, 132–138 10.1016/j.procbio.2017.02.021 [DOI] [Google Scholar]
- 52. Irwin D. C., Spezio M., Walker L. P., and Wilson D. B. (1993) Activity studies of eight purified cellulases: specificity, synergism, and binding domain effects. Biotechnol. Bioeng. 42, 1002–1013 10.1002/bit.260420811 [DOI] [PubMed] [Google Scholar]
- 53. Wood T. M. (1988) Preparation of crystalline, amorphous, and dyed cellulase substrates. Methods Enzymol. 160, 19–25 10.1016/0076-6879(88)60103-0 [DOI] [Google Scholar]
- 54. Heuts D. P., Winter R. T., Damsma G. E., Janssen D. B., and Fraaije M. W. (2008) The role of double covalent flavin binding in chito-oligosaccharide oxidase from Fusarium graminearum. Biochem. J. 413, 175–183 10.1042/BJ20071591 [DOI] [PubMed] [Google Scholar]
- 55. Zámocký M., Schümann C., Sygmund C., O'Callaghan J., Dobson A. D., Ludwig R., Haltrich D., and Peterbauer C. K. (2008) Cloning, sequence analysis and heterologous expression in Pichia pastoris of a gene encoding a thermostable cellobiose dehydrogenase from Myriococcum thermophilum. Protein Expr. Purif. 59, 258–265 10.1016/j.pep.2008.02.007 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.