

Abstract
The ventricular–subventricular zone (V‐SVZ) of the mammalian brain is a site of adult neurogenesis. Within the V‐SVZ reside type B neural stem cells (NSCs) and type A neuroblasts. The V‐SVZ is also a primary site for very aggressive glioblastoma (GBM). Standard‐of‐care therapy for GBM consists of safe maximum resection, concurrent temozolomide (TMZ), and X‐irradiation (XRT), followed by adjuvant TMZ therapy. The question of how this therapy impacts neurogenesis is not well understood and is of fundamental importance as normal tissue tolerance is a limiting factor. Here, we studied the effects of concurrent TMZ/XRT followed by adjuvant TMZ on type B stem cells and type A neuroblasts of the V‐SVZ in C57BL/6 mice. We found that chemoradiation induced an apoptotic response in type A neuroblasts, as marked by cleavage of caspase 3, but not in NSCs, and that A cells within the V‐SVZ were repopulated given sufficient recovery time. 53BP1 foci formation and resolution was used to assess the repair of DNA double‐strand breaks. Remarkably, the repair was the same in type B and type A cells. While Bax expression was the same for type A or B cells, antiapoptotic Bcl2 and Mcl1 expression was significantly greater in NSCs. Thus, the resistance of type B NSCs to TMZ/XRT appears to be due, in part, to high basal expression of antiapoptotic proteins compared with type A cells. This preclinical research, demonstrating that murine NSCs residing in the V‐SVZ are tolerant of standard chemoradiation therapy, supports a dose escalation strategy for treatment of GBM. stem cells 2019;37:1629–1639
Keywords: Neural stem cells, Temozolomide, Ionizing radiation, Glioblastoma, Subventricular zone, Apoptosis
The brain's ventricular–subventricular zone is a site of type B neural stem cell‐mediated neurogenesis and unfortunately for very aggressive glioblastoma. Treatment for glioblastoma consists of resection, concurrent temozolomide, and X‐irradiation, followed by adjuvant temozolomide. We show that concurrent temozolomide/X‐irradiation followed by adjuvant temozolomide induces apoptosis in type A neuroblasts. In contrast, quiescent neural stem cells, which express high levels of Bcl2 and Mcl1, survive and repopulate type A neuroblasts.
Significance Statement.
Evidence suggests that glioblastoma (GBM) contacting the ventricular–subventricular zone (V‐SVZ) is resistant to concurrent temozolomide (TMZ) plus X‐irradiation (XRT). An important question is whether therapeutic escalation of dose to counter aggressive GBM can be limited by a failure of type B neural stem cells (NSCs) that reside in the V‐SVZ to survive TMZ plus XRT and contribute to repair of treatment‐induced injury. Studying the molecular responses to DNA damage in the intact in vivo microenvironment revealed that NSCs over express the antiapoptotic proteins Bcl2 and Mcl1 and do not undergo apoptosis in response to concurrent chemoradiation therapy. Stem cell resistance sustains neurogenesis and suggests targeting the V‐SVZ for the treatment of GBM would not result in unacceptable toxicity.
Introduction
The ventricular–subventricular zone (V‐SVZ) located along the walls of the lateral ventricles is the largest source of neural stem cells (NSCs) in the adult brain 1, containing self‐renewing type B NSCs that generate type C transit amplifying progenitor cells, which in turn generate lineage‐committed progeny. These progeny are primarily type A neuroblasts, which migrate from the V‐SVZ into the rostral migratory stream (RMS) and finally differentiate into neurons as well as immature oligodendrocyte precursor cells 2, 3, 4.
The V‐SVZ is also one potential site of origin, as well as a site of invasion for World Health Organization grade IV astrocytoma (glioblastoma [GBM]) tumors 5. These tumors consist of multiple fractions of proliferative and/or quiescent stem‐like cells thought to be lineally related. GBM located adjacent to the V‐SVZ is very resistant to standard of care therapy 6. The clinical literature is conflicted with regard to the outcome of patients treated with radiation to the V‐SVZ, with one group reporting worse outcomes with higher doses 7, whereas the other groups have reported improved outcomes 8, 9, 10. Importantly, tumors contacting the V‐SVZ have been noted to have a worse outcome compared with noncontacting tumors, and on meta‐analysis, V‐SVZ contact is independent of other outcome predictors 10, 11.
Standard of care for treatment of GBM can involve concurrent temozolomide (TMZ)/radiation therapy (XRT, 1.8–2 Gy/fraction) followed by adjuvant TMZ chemotherapy 12. The impact of this therapeutic regimen on NSCs and their ability to generate progeny is not well characterized. Furthermore, the majority of NSCs in the V‐SVZ are quiescent 13, 14, particularly in the human brain, and the mechanisms of quiescent cell resistance are not as well studied as those of proliferating cells.
Preclinical murine models that used dose fractionation schemes consisting of 10 Gy in a single fraction or 15 Gy in five fractions resulted in loss of type B NSCs and a failure of these cells to differentiate into type C and A cells 15, 16. Similarly, administering two TMZ doses (100 mg/kg per dose) given 12 days apart resulted in significant loss of type B cells 35 days after exposure 3. Such results suggest that TMZ and/or radiation injury can compromise the ability of V‐SVZ NSCs to sustain the neurogenic niche 17. Although insightful to the underlying biology, none of these regimens accurately reflect clinical practice treatment paradigms. Additionally, we considered that a fundamental understanding of the response of quiescent NSCs to TMZ/XRT would be informative and aid in understanding clinical outcomes.
The major lethal lesions generated by ionizing radiation are double‐strand breaks 18. In quiescent type B NSCs, repair of double‐strand break defaults to the nonhomologous end joining pathway (NHEJ) 14. Ionizing radiation also generates large numbers of single‐strand breaks and these are repaired by a base excision repair subpathway 19. As stated above, Achanta et al. reported the loss of type B cells following a single fraction of 10 Gy 15. One gray of photons is estimated to generate 35 double‐strand breaks and 1,000 single‐strand breaks per cell 20. Thus, 10 Gy would be expected to generate 350 double‐strand breaks and 10,000 single‐strand breaks. The knowledge that DNA strand breaks induce p53‐mediated apoptosis 21 and that irradiation of NSCs induces p53 expression 22 suggests that type B NSCs may be susceptible to apoptotic death following accumulation of sufficient DNA damage. Although Sarosiek et al. 23 found that the majority of cells in the brain are resistant to radiation‐induced apoptosis, their studies did not focus on stem or progenitor cells. TMZ‐induced cytotoxicity is due in large part to loss of Bcl2 and activation of caspase 9 and caspase 3 directed apoptosis, driven by unrepaired O6‐methylguanines that degenerate into DNA double‐strand breaks 24.
Herein, we addressed the question of whether concurrent TMZ/XRT followed by adjuvant TMZ, a more accurate reflection of human clinical treatment, disrupted the ability of type B cells in the V‐SVZ to maintain neurogenesis. Furthermore, we quantitatively compared the response of type B NSCs to type A neuroblasts with reference to repair of DNA double‐strand breaks and the execution of apoptosis. Whereas concurrent TMZ/XRT induced significant apoptosis in neuroblasts, NSCs were very resistant. Resistance was not a consequence of differential DNA repair; DSB rate of repair was the same in both cell types. Rather, the antiapoptotic proteins Bcl2 and Mcl1 were expressed to a greater extent in NSCs compared with neuroblasts. We found that type B NSC resiliency allowed these cells to repopulate type A cells following concurrent TMZ/XRT and adjuvant TMZ.
Materials and Methods
Mice
Eight‐ to 10‐week‐old male and female C57BL/6J mice were maintained under specific pathogen‐free conditions. They were maintained in a 12 hour light–dark cycle and had access to food and water ad libitum. All procedures performed on animals were approved by the Institutional Animal Care and Use Committee at Vanderbilt University.
TMZ/Irradiation
The treatment schedules for all cohorts (n = 6 per cohort) can be found in Supporting Information Figure S1. For 5 consecutive days, isoflurane anesthetized mice in cohort 1 were injected with vehicle alone dimethyl sulfoxide (DMSO). One hour after injection, the mice were subjected to sham irradiation. Mice in cohort 1 were euthanized on day 6. For 5 consecutive days, isoflurane anesthetized mice in cohort 2 were injected with vehicle alone (DMSO) and then subjected to sham irradiation 1 hour after injection. On days 19–22, mice in cohort 2 were again injected with DMSO and then 1 hour later subjected to sham irradiation. Mice in cohort 2 were euthanized on day 82. For 5 consecutive days, isoflurane anesthetized mice in cohort 3 were injected with 50 mg/kg of TMZ. Mice in cohort 3 were euthanized on day 6. For 5 consecutive days, the brains of isoflurane anesthetized mice in cohort 4 were administered 2 Gy. Mice in cohort 4 were euthanized on day 6. For 5 consecutive days, isoflurane anesthetized mice in cohort 5 were injected with 50 mg/kg TMZ. One hour after injection, the brains of these mice were administered 2 Gy. Mice in cohort 5 were euthanized on day 6. For 5 consecutive days, isoflurane anesthetized mice in cohort 6 were injected with 50 mg/kg TMZ. One hour after injection, the brains of these mice were administered 2 Gy. On days 19–22, mice in cohort 6 were injected with 100 mg/kg TMZ. Mice in cohort 6 were euthanized on day 82. Isoflurane anesthetized mice in cohort 7 were injected with 50 mg/kg TMZ whereas mice in cohort 8 were injected with DMSO. One hour after injection, the brains of mice in cohort 7 were administered 1 Gy whereas the mice in cohort 8 underwent sham treatment. Mice were euthanized 0.1, 0.18, 0.266, 0.6, 1.1, 3, 5, and 24 hours later. A dose of 1 Gy was used so that distinct 53BP1 foci could be quantified.
Brains were irradiated using a 300 kVp/10 mA x‐ray machine at a dose rate of 1.88 Gy per min. With the exception of the brain, the entire animal was shielded by a custom lead block 2.5 cm thick. Radiation dosimetry was performed using GAF chromic EBT‐XD film whose calibration was traceable to a National Bureau Standards measurement standard.
Tumor Model
The brains of 8‐week‐old female C57BL/6 mice were injected with 200,000 glioma 261 (GL261, 25) cells in volume of 1 μl at coordinates 1 mm anterior, 1.5 mm lateral to the bregma, and 2.3 mm deep from the pial surface. Two weeks after tumor cell injection, mice were randomly assigned to cohort 5 (n = 5) or to cohort 1 (n = 5).
Immunofluorescence and Antibodies
Anesthetized mice underwent transcardial perfusion with saline and then with 10% buffered formalin. Brains were removed, fixed in 10% buffered formalin, and processed into paraffin. Ten micrometer coronal sections were obtained and processed for immunofluorescence. The following antibodies were used. Chicken anti‐glial fibrillary acidic protein (GFAP) (Abcam, Cambridge, MA, USA, ab4647, 1:3,000), rabbit anti‐Sox2 (Cell Signaling, Danvers, MA, USA, 3579p, 1:200), rat anti‐Sox2 (eBioscience, Thermo Fisher, Waltham, MA, USA, 14‐9811‐80, 1:100), rabbit anti‐Ki67 (Abcam, ab16667, 1:100), rabbit anti‐Dcx (Cell Signaling, 4604S, 1:500), rabbit anticleaved caspase 3 (Cell Signaling, 966, 1:100), rabbit anti‐Mcl1 (Thermo Fisher, PA5‐27597), anti‐53BP1 (Novus Biologicals, Centennial, CO, US, NB100‐304, 1:3,000), rabbit anti‐p53 (Leica Biosystems, Buffalo Grove, IL, USA, NCL‐L‐p53‐CM5p, 1/500), rabbit anti‐Bax (Abcam, ab32503, 1:100), and rabbit anti‐Bcl2 (LSBIO LifeSpan BioSciences, Seattle, WA, USA, LS‐C148229‐50. The following secondary antibodies were used. Goat anti‐chicken (Life Sciences, Invitrogen, Thermo Fisher, Waltham, MA, USA, A11041, Alexa Fluor 568, 1:2,000), goat anti‐rabbit (Life Sciences, Invitrogen, Thermo Fisher, A21244, Alex Fluor 647, 1:2,000), goat anti‐rabbit (Life Sciences, Invitrogen, Thermo Fisher, A11011, Alex Fluor 568, 1:2,000), donkey anti‐rabbit (Thermo Fisher, SA5‐10041, DyLight 650, 1:2,000), donkey anti‐rat (Thermo Fisher, SA510029, DyLight 650, 1:2,000), and donkey anti‐rabbit (Life Sciences, Invitrogen, Thermo Fisher, A21206, Alexa Fluor 488, 1:2,000). Representative images of slides stained solely with the secondary antibodies used for Figures 1, 2, 3, 4, 5, 6 are shown in Supporting Information Figure S2. Counterstaining was with DAPI. Whole slide immunofluorescence imaging was performed in the Digital Histology Shared Resource at Vanderbilt University Medical Center using a Leica Aperio Versa 200 whole slide scanner that provides high resolution wide field images of ranging from ×0.05 to ×40. Confocal images of immunofluorescent staining were acquired using an Olympus FV‐1000 inverted confocal microscope using either a ×40 or a ×60 oil immersion objective lens.
Figure 1.

Type B neural stem cells (NSCs) but not type A neuroblasts are resistant to concurrent temozolomide (TMZ)/XRT. Representative image of GFAP‐expressing type B NSCs (false red) located within 30 μm of a dorsal wall of a ventricle (V) obtained from nontumor‐bearing DMSO/sham‐treated mice (A) or nontumor‐bearing mice treated with concurrent TMZ/XRT (B). (C): Quantification of GFAP positive nuclei within 30 μm of a ventricle in nontumor‐bearing mice. GFAP‐expressing type B NSCs (false red) located within 30 μm of a dorsal wall of a ventricle obtained from glioblastoma (GBM)‐bearing DMSO/sham‐treated mice (D) or GBM‐bearing mice treated with concurrent TMZ/XRT (E). (F): Quantification of GFAP positive nuclei within 30 μm of a ventricle in GBP‐bearing mice. Representative image of Dcx‐expressing type A neuroblasts (false red) along a ventricle and in the rostral migratory stream (RMS) from nontumor‐bearing sham‐treated (G, left panel) and TMZ/XRT‐treated mouse (G, right panel). (H): Quantification of Dcx‐expressing cells in nontumor‐bearing mice. Representative image of Dcx‐expressing type A neuroblasts (false red) along a ventricle and in the RMS from GBM tumor‐bearing sham‐treated (I, left panel) and TMZ/XRT‐treated mouse (I, right panel). (J): Quantification of Dcx‐expressing cells in a GBM tumor‐bearing mice. Nuclei are stained with DAPI (false white). White bars = 100 μm. Images obtained using a Leica Aperio versa 200 whole slide scanner.
Figure 2.

Ki67 positive cells along a ventricular–subventricular zone (V‐SVZ) in nontumor‐bearing, sham‐treated mice. (A): Ki67 immunostaining (false green) of proliferating cells along a V‐SVZ. DAPI staining (false white). Images obtained using a Leica Aperio versa 200 whole slide scanner (B) coimmunostaining of Ki67 (false green) and Sox2 (false red) along a V‐SVZ. Nuclei are stained with DAPI (blue). White bar = 100 μm. Images obtained using an Olympus FV‐1000 inverted confocal microscope. Abbreviation: V, ventricle.
Figure 3.
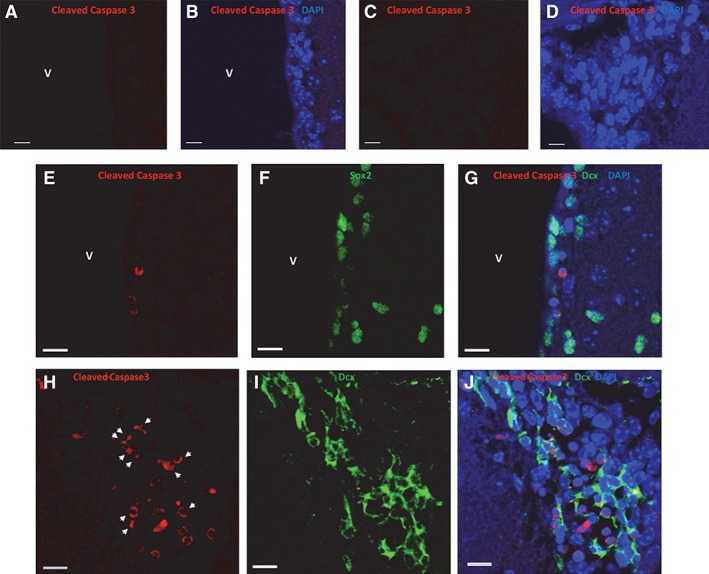
Concurrent temozolomide (TMZ)/XRT induces apoptosis in Dcx‐expressing type A neuroblasts but not in Sox2 neural stem cells (NSCs). Cleaved caspase 3 immunostaining (false red) in the dorsolateral ventricle (A, B) and in the rostral migratory stream (RMS; C, D) of a 10‐week‐old nontumor‐bearing sham‐treated female mouse. DAPI staining blue. (E–J): Ten‐week‐old female mice were injected i.p. with 50 mg/kg TMZ. One hour later, 1 Gy was administered to whole brain. Mice were euthanized 5 hours after irradiation. Panels (E)–(G) illustrate a representative dorsolateral ventricle immunostained for cleaved caspase 3 (false red) and Sox2 (false green). Nuclei are stained with DAPI (blue). Panels (H)–(J) illustrate a representative RMS immunostained for cleaved caspase 3 (false red) and Dcx (false green). Cells costaining for cleaved caspase 3 and Dcx are denoted by white arrow heads. Nuclei are stained with DAPI. White bars = 10 μm. Images obtained using an Olympus FV‐1000 inverted confocal microscope. Abbreviation: V, ventricle.
Figure 4.
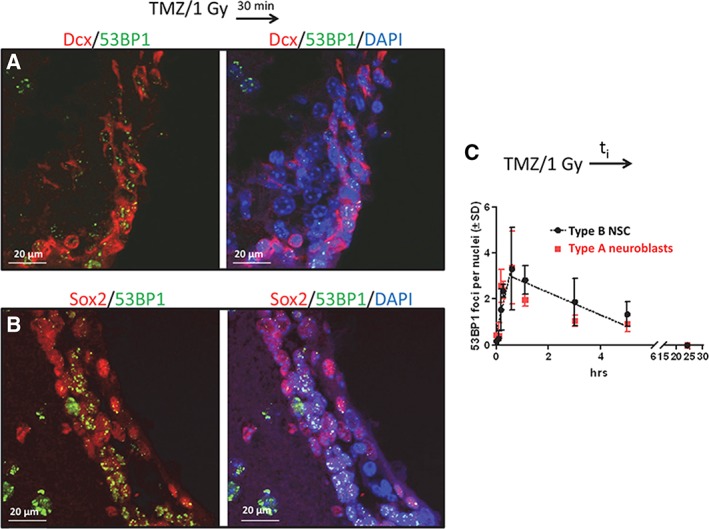
Formation and resolution of 53BP1 foci in type B neural stem cells (NSCs) and in type A neuroblasts. Nontumor‐bearing female mice were injected i.p. with 50 mg/kg temozolomide. One hour later, 1 Gy was administered to whole brain. Transcardial perfusion was initiated 1, 5, 10, 30, 60, 180, 300, or 1,440 minutes after irradiation (cohort 7, Supporting Information Fig. S1). Fixation of brain tissue was completed within 6 minutes of the start of perfusion. (A): 53BP1 foci (false green) in Dcx‐expressing cells (false red). (B): 53BP1 foci (false green) in Sox2 expressing cells (false red) located within 30 μm of a ventricle. (C): The number of 53BP1 foci per nuclei (±SD) in type B NSCs and type A neuroblasts as a function of time after irradiation. Nuclei are stained with DAPI (blue). White bar = 20 μm. Images acquired by confocal microscopy.
Figure 5.
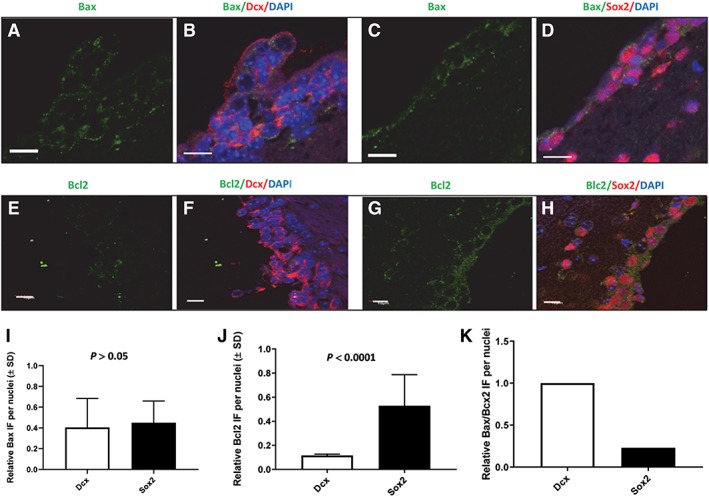
Bax and Bcl2 expression in Dcx neuroblasts and in Sox2 neural stem cells (NSCs) located within 30 μm of a ventricle of the ventricular–subventricular zone obtained from nontumor‐bearing sham‐treated female mice. Panels (A)–(D) illustrate coimmunostaining for Bax (false green, Cy2), Dcx (false red, Cy5), and Sox2 (false red, Cy7). Panels (A) and (B) illustrate Bax and Dcx immunostaining. Panels (C) and (D) illustrate Bax and Sox2 immunostaining. Panels (E)–(H) illustrate coimmunostaining for Bcl2 (false green, Cy2), Dcx (false red, Cy5), and Sox2 (false red, Cy7). Panels (E) and (F) illustrate Bcl2 and Dcx immunostaining. Panels (G) and (H) illustrate Bcl2 and Sox2 immunostaining. Panel (I) illustrates quantification of Bax expression in Dcx‐expressing and Sox2‐expressing cells. Panel (J) illustrates quantification of Bcl2 expression in Dcx‐expressing and Sox2‐expressing cells. Panel (K) illustrates relative Bax/Bcl2 ratios for Dcx and Sox2‐expressing cells. DAPI staining is blue. White bar = 10 μm. The confocal images illustrate immunofluorescence throughout 4.92 μm of the Z axis.
Figure 6.
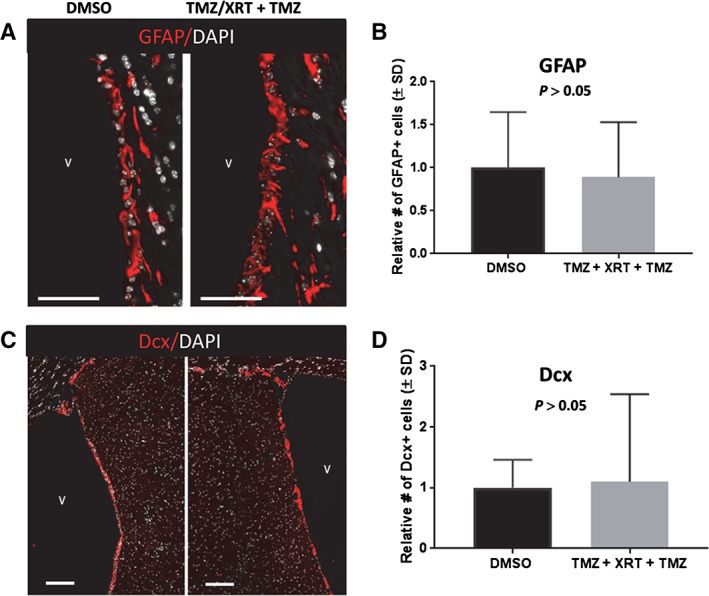
Type B neural stem cells (NSCs) are resistant to concurrent temozolomide (TMZ)/XRT followed by adjuvant TMZ. (A): GFAP‐expressing type B NSCs located within 30 μm of a dorsal wall of a ventricle obtained from sham‐treated (cohort 2) or concurrent/adjuvant treatment, cohort 6, Supporting Information Figure S1. (A): Representative images of GFAP expressing cells (false red) imaged using a Leica Aperio versa 200 slide scanning microscope. Nuclei were stained with DAPI (false white). (B): Quantification of GFAP positive nuclei within 30 μm of a ventricle. (C): Representative images of Dcx‐expressing neuroblasts (false red) in the ventricular–subventricular zone. The white bar = 100 μm. Nuclei are stained with DAPI (false white). (D): Quantification of Dcx‐expressing cells. Images obtained using a Leica Aperio versa 200 microscope. The magnification in (C) is at ×4 so that repopulation of Dcx cells is apparent.
Image Quantification
Two microscopy techniques were used: (1) fluorescent confocal microscopy: primary antibody‐specific immunofluorescence intensity per cell was quantified using ImageJ (NIH) and (2) fluorescent whole slide microscopy: the software package CellProfiler (http://cellprofiler.org) 26, 27 was used to quantify cells expressing GFAP or Dcx within 30 μm of a dorsal or dorsolateral ventricle. Briefly, a threshold was applied to a whole tissue signal in order to produce a tissue mask. The borders of this mask were then eroded to remove ∼30 μm of the surface area, and the inverse of the two was used as a mask of cells localized near the dorsal and dorsolateral ventricles. DAPI signal was used to identify individual nuclei within a relevant size range. Cells positive for GFAP and Dcx were identified similarly. All GFAP and Dcx identified objects were filtered for the presence of DAPI so that only whole cells were counted (CellProfiler pipeline available).
Statistical Analysis
GraphPad Prism Version 7.02 was used to perform all statistical analyses. Nonparametric Mann–Whitney U tests with Welch's correction were used to analyze differences between groups. p < .05 was considered statistically significant.
Results
Type B NSCs but not type A neuroblasts are resistant to concurrent TMZ/XRT. For 5 consecutive days, male and female C57BL/6 mice 8–10 weeks of age were injected with 0 (DMSO‐solvent control, cohort 1) or 50 mg/kg of TMZ. One hour later, the mice were administered a whole brain dose of 0 or 2 Gy (denoted as TMZ/XRT, cohort 5, Supporting Information Fig. S1). The remainder of the body was shielded in lead. Mice were euthanized on day 6. Control mice received five daily injections of DMSO (cohort 1, Supporting Information Fig. S1).
Barazzuol et al. 14 found that more than 90% of type B NSCs are located within 30 μm of a lateral ventricle surface (as defined in Excel file S1, Data, Table S2D, and reference 14) costained for GFAP, Sox2, and Nestin, an observation that is consistent with other reports 3, 14, 15, 28. Therefore, to increase clarity of NSC identification, Barazzuol et al. 14 identified type B NSCs as GFAP immunostained cells located within 30 μm of lateral V‐SVZ ventricles. As we too observed a high concordance of GFAP and Sox2 expression in cells located within 30 μm of lateral V‐SVZ (Supporting Information Fig. S3), we used these criteria of Barazzuol et al. 14 to identify type B NSCs. Figure 1A, 1B illustrates representative dorsal images from the V‐SVZ following sham‐treatment or TMZ/XRT. Quantification of the total number of cells and the number of GFAP‐expressing type B cells 14, 15, 29 located within 30 μm of the dorsal or dorsolateral ventricle was undertaken as described in Materials and Methods. TMZ/XRT treatment did not affect the total number of nuclei located in either the dorsolateral or dorsal V‐SVZ subdomains compared with sham treatment (p > .05) nor did the treatment affect the number of GFAP positive cells (p = .42, Fig. 1C), as assessed by quantifying type B cells (n = 2,410 for sham treatment and n = 2,704 for TMZ/XRT) located in the V‐SVZ of both hemispheres per section, two sections per mouse, and three mice per sex. Furthermore, the results were independent of sex (p > .05, data not shown).
This experiment was then repeated in female mice harboring a syngeneic/orthotopic GBM injected adjacent to a V‐SVZ. Fourteen days after injection of GBM tumor cells into brain mice were randomly assigned to either cohort 1 or 5. A representative H&E stained tumor section is shown in Supporting Information Figure S3B. In all mice, GBM tumor growth destroyed or distorted the V‐SVZ that was adjacent to it. Figure 1D, 1E illustrate representative dorsal images of a V‐SVZ following sham‐treatment or TMZ/XRT of tumor‐bearing mice. Concurrent TMZ/XRT treatment (cohort 5) did not produce a statistically significant change in type B NSC cells in the proximal contralateral V‐SVZ compared with sham/DMSO control (Fig. 1F, p = .29, n = 457 for sham treatment and n = 369 for TMZ/XRT, one V‐SVZ per section, two coronal sections per mouse, and five mice per point).
Type A neuroblasts are located both in the V‐SVZ and in the RMS and are distinguished by expression of Dcx 13 (Fig. 1G, left panel). Strikingly, 5 consecutive days of concurrent TMZ/XRT resulted in a severe loss of Dcx‐expressing cells in the V‐SVZ and in the RMS (Fig. 1G, right panel), suggesting significant neuroblast cell death (Fig. 1H, p = .0001 compared with DMSO/sham treatment). These results were independent of gender (p > .05, as assessed by quantifying 2,242 nuclei in the V‐SVZ in both hemispheres of a section, two sections per mouse, three mice per gender for TMZ/XRT treatment, and 1,478 nuclei for sham treatment). Representative images from a male mouse and a female mouse are shown in Supporting Information Figure S4.
Quantification of Dcx‐expressing cells in the brains of mice bearing GBM was also undertaken (Fig. 1). Representative images of Dcx‐expressing cells in sham‐treated and TMZ/XRT‐treated mice are shown in panel (I). The presence of a GBM did not protect Dcx cells from the TMZ/XRT treatment (p < .001, Fig. 1J).
Dcx‐expressing neuroblasts are a major source of GABA that activate GABAA receptors in type B NCSs, instructing the cells to remain quiescent 30, 31. Loss of neuroblasts following concurrent TMZ/XRT might be expected to remove a proliferation inhibitory signal, allowing NSCs to enter the cell cycle for the purpose of repopulating type C and A cells 31. To determine if this was the case, we inspected the V‐SVZ for Ki67 positive cells in the brains of nontumor‐bearing DMSO/sham‐treated mice and in nontumor‐bearing mice following completion of the 5 days of concurrent TMZ/XRT. The representative image shown in Figure 2A illustrates Ki67 immunostaining along a V‐SVZ and an RMS, while the images shown in Figure 2B illustrate Ki67 immunostaining in Sox2‐expressing cells located along the V‐SVZ of DMSO/sham‐treated brain. Under the experimental conditions used in this investigation, 2% of 155 type B NSCs located along seven V‐SVZ obtained from the brains of DMSO/sham‐treated mice were Ki67 positive. This is consistent with the report from Ponti et al. 13, who found 4% of NSCs were proliferative. We inspected the V‐SVZ from seven 10 μm coronal sections, both hemispheres per section, obtained from three mice following completion of 5 days of concurrent TMZ/XRT and did not observe a single Ki67 positive Sox2‐expressing cell (a representative V‐SVZ is shown in Supporting Information Fig. S5) nor did we observe any Ki67 positive cells immediately following 5 days of concurrent treatment (data not shown).
Next, we assessed the effects of independently administering 50 mg/kg of TMZ for 5 consecutive days (cohort 3) and the effects of independently administering 2 Gy/day for 5 consecutive days on type A neuroblasts and type B NSCs (cohort 4, Supporting Information Fig. S1). Either treatment alone resulted in loss of the Dcx‐expressing cells in nontumor‐bearing mice when compared with sham treatment (Supporting Information Fig. S6). Thus, both TMZ and irradiation are independently cytotoxic to type A cells. However, neither treatment affected the NSCs.
It was of interest to determine if the loss of Dcx‐expressing neuroblasts following concurrent TMZ/XRT was due in part to induction of apoptosis. Because sex did not affect the response to concurrent TMZ/XRT, we chose to perform the experiments using nontumor‐bearing female mice. In 10‐week‐old sham‐treated female mice (cohort 8, Supporting Information Fig. S1), we could not detect cleaved caspase 3 immunostaining in dorsal (not shown) or dorsolateral ventricles (Fig. 3A, 3B) or in the RMS (Fig. 3C, 3D; n = 228 cells). In contrast, cleaved caspase 3 immunostaining (Fig. 3E–3J) was observed 5 hours after 10‐week‐old female mice were injected i.p. with 50 mg/kg TMZ and the brain irradiated 1 hour later with 1 Gy (cohort 7, Supporting Information Fig. S1). This time frame was chosen because Barazzuol et al. 14 have shown that this is an optimal time to quantify apoptosis in irradiated V‐SVZ. Although cleaved caspase immunostaining was not observed in Sox2 NSCs (n = 143, panels E–G), immunostaining was observed in Dcx‐expressing cells (n = 147; Fig. 3H–3J). Thus, there exists a significant difference in how type B NSCs and type A neuroblasts respond to concurrent TMZ/XRT.
One possibility for the differential sensitivity observed following concurrent TMZ/XRT is a differential repair of radiation damage in type A neuroblasts versus type B NSCs. Barazzuol et al. 14 found that 53BP1 foci formation, which is considered linearly related to the number of DNA double‐strand breaks 32, was a well‐validated marker for quantifying DNA double‐strand breaks in type B NSCs and in Dcx‐expressing neuroblasts. Therefore, 53BP1 foci formation and resolution was quantified in Dcx‐expressing neuroblasts cells located within 30 μm of a lateral ventricle (Fig. 4A) and in Sox2‐expressing NSCs (Fig. 4B). This was accomplished by coimmunostaining each slide for 53BP1, Sox2, and Dcx following exposure to TMZ plus 1 Gy. A dose of 1 Gy was chosen so that distinct foci could be quantified. Radiation‐induced DNA damage such as DNA double‐strand breaks generated in yeast or eukaryotic cells is linearly related to dose and the repair pathways used are not known to be dependent on dose, at least between 0.5 and 20 Gy 33, 34, 35, 36. Although both Dcx‐expressing cells and Sox2‐expressing cells from nontumor‐bearing sham‐treated mice expressed 53BP1 protein, there were no foci present (Supporting Information Fig. S7). In mice treated with concurrent TMZ/XRT, there was no statistically significant difference between Sox2‐expressing and Dcx‐expressing cells with regard to formation or resolution of foci as a function of time after irradiation (p > .05, Fig. 4C). The number of nuclei counted per time point is shown in Supporting Information Figure S7C. Therefore, we concluded that repair of TMZ/XRT‐induced DNA damage could not account for the differences in sensitivity.
DNA damage‐induced apoptosis in brain can be initiated by p53 37. Consistent with this knowledge, p53 expression was found to be elevated 3 hours after a TMZ + 1 Gy exposure in both Sox2‐expressing cells and in Dcx‐expressing cells (Supporting Information Fig. S8). The question of whether apoptosis is executed depends, in part, on the differential expression of proapoptotic and antiapoptotic proteins 23. We examined the expression of Bax or Bcl2 in Sox2‐expressing and in Dcx‐expressing cells. Slides containing coronal sections obtained from nontumor‐bearing sham‐treated mice were coimmunostained for Bax, Dcx, and Sox2 or Bcl2, Dcx, and Sox2. Figure 5A–5D illustrates representative images of Bax expression in type B NSCs located within 30 μm of lateral V‐SVZs (n = 83 nuclei) and in Dcx‐expressing type A cells (n = 122 nuclei). Quantification of Bax expression in both hemispheres per 10 μm coronal sections, two sections per mouse, three female mice per point indicated that there was not a statistically significant difference in Bax expression in the two cell types (Fig. 5I, p > .05). In contrast, quantification of Bcl2 positive Dcx‐expressing type A cells (Fig. 5E, 5F) and Bcl2‐positive Sox2‐expressing cells (Fig. 5G, 5H) revealed that Sox2‐expressing cells (n = 90) located within 30 μm of lateral V‐SVZs harbored 4.5‐fold more Bcl2 compared with Dcx‐expressing cells (n = 85, Fig. 5J, p < .001). Figure 5K illustrates the relative Bax/Bcl2 ratio in Dcx and Sox2 expressing cells. Based on the knowledge that 98% of Sox2‐expressing NSCs were quiescent (i.e., Ki67 negative), elevated Blc2 expression measured in 90 NSCs occurred predominately in nonproliferating cells.
The antiapoptotic protein Mcl1 has been implicated in neuroblast survival 38. Thus, it was of interest to determine Mcl1 expression in type B NSCs and neuroblasts. Slides containing coronal sections obtained from nontumor‐bearing sham‐treated mice were coimmunostained for Mcl1, Dcx, and Sox2. Quantification of Mcl1‐positive Dcx‐expressing cells (Supporting Information Fig. S9A, S9B) and Mcl1‐positive Sox2‐expressing cells (Supporting Information Fig. S9C, S9D) in three female mice revealed that Sox2‐expressing cells (n = 147) located within 30 μm of dorsal and dorsolateral V‐SVZs expressed greater than 10‐fold more Mcl1 than Dcx‐expressing cells (n = 188, Supporting Information Fig. S9E, p < .0002). The increased expression of Bcl2 and Mcl1 in Sox2 cells compared with Dcx cells is consistent with the observation that Sox2 NSCs do not undergo apoptosis following concurrent TMZ/XRT.
As adjuvant TMZ following concurrent TMZ/XRT is commonly administered to patients as clinical standard of care, we explored the consequence of administering adjuvant TMZ to mice previously exposed to concurrent TMZ/XRT (cohort 6, Supporting Information Fig. S1). Male (n = 3) and female (n = 3) nontumor‐bearing C57BL/6 mice were administered sham treatment (cohort 2) or concurrent TMZ/XRT. Then on days 19–22 the mice were injected with TMZ (100 mg/kg, i.p., cohort 6, Supporting Information Fig. S1). The adjuvant TMZ dose administered was double that of the concurrent dose, mimicking clinical practice. Use of 100 mg/kg as an adjuvant dose was based on the work of Mich et al. 3. Mice were euthanized 60 days after the last TMZ injection in order to provide sufficient time for cell renewal. The number of type B NSCs and type A neuroblasts present in a dorsal or dorsolateral V‐SVZ was imaged by slide scanning microscopy and quantified as detailed in Materials and Methods. The analysis demonstrated that type B NSCs cells exhibit significant resiliency to radiation‐induced DNA damage and TMZ‐mediated DNA methylation. Sixty days after the last TMZ exposure, the number of type B GFAP‐expressing NSCs (n = 3,524) present within 30 μm of a ventricle, two hemispheres per coronal section, two sections per mouse, and six mice, was not significantly different from sham treatment (p > .05, n = 2,953, two hemispheres per coronal section, two sections per mouse, six mice, Fig. 6A, 6B). Importantly, these cells were able to repopulate and sustain a population of type A Dcx‐expressing neuroblasts (n = 3,079), whose numbers were not significantly different from those found in sham‐treated brain (n = 2,519, p > .05, Fig. 6C, 6D).
Discussion
Patients with GBM tumors located within the V‐SVZ exhibit shortened progression‐free survival that significantly diminishes overall survival 39, 40. Treating these tumors results in the V‐SVZ being subjected to concurrent TMZ/XRT followed by adjuvant TMZ therapy.
The V‐SVZ is a nourishing stem cell niche and several studies have led investigators to conclude that NSCs in the adult human V‐SVZ maintain neurogenesis 41, 42, albeit, at a greatly diminished rate 43. Thus, an important, unanswered question is whether type B NSCs can survive and function following cytotoxic standard of care therapy. Previous studies focused on the independent effects of radiation alone (reviewed in reference 17 or TMZ alone 3, but not their combined effect. Although these treatment regimens are relevant to certain clinical scenarios of patients with poor performance status, they do not reflect the broader group of patients treated with daily fractionated radiation (1.8–2 Gy per fraction) and TMZ. In order to more closely mimic current standard of care therapy used to treat GBM 44, we used a preclinical murine model in which type B NCSs were subjected to concurrent TMZ/XRT followed by adjuvant TMZ in their in vivo self‐sustaining microenvironment. To the best of our knowledge, this is the first report on NSC outcome following TMZ plus XRT treatment.
Inspection and quantification of Dcx‐expressing type A neuroblasts following concurrent TMZ/XRT (cohort 5) revealed a few neuroblasts present in either the V‐SVZ or the RMS in either nontumor bearing mice or tumor‐bearing mice (Fig. 1). In contrast, 5 consecutive days of concurrent TMZ/XRT did not diminish the number of type B NCSs located in the V‐SVZ. Type B cells are mostly quiescent. Estimates for the percent type B cells that are quiescent at any one time range from 90% to 96% 13, 14. Under the experimental conditions used in this investigation, approximately 98% of type B NSCs were quiescent (Fig. 2). Interestingly, under basal conditions an NSC proliferation rate of only 4% to 10% is sufficient to sustain the entire population of transient‐amplifying cells and neuroblasts 2. While quiescent NSCs are well known to be resistant to antimitotics such as cytarabine, there is not currently a well characterized understanding of why quiescent NSCs would be resistant to cytotoxic TMZ/XRT treatment.
TMZ is a methylating agent that produces O6—methylguanine lesions 45 that can result in cytotoxicity due to futile mismatch repair 46. Futile mismatch repair can generate DNA double‐strand breaks and in proliferating cells trigger apoptosis 47. Thus, type B quiescent cells would not be expected to be susceptible to TMZ‐induced cell killing. Ionizing radiation also generates DNA double‐strand breaks that can trigger apoptosis 48, 49 as well as mitotic‐linked cell death in proliferating cells, a consequence of formation of lethal chromosomal aberrations 50, 51. Dai et al. 52 found that quiescence, in and of itself, does not impair the stress response induced by ionizing radiation. Wilson and Keng 53 examined the rate of repair of single and double‐strand breaks in quiescent and proliferating cells following γ‐irradiation. They found that repair kinetics were the same for both cell types. These results are consistent with our work that demonstrated similar kinetics for recruitment and resolution of radiation‐induced 53BP1 foci in type B and A cells. It should be mentioned that the number of foci observed in vivo in the V‐SVZ is less than what is observed when measuring foci in cell culture 54. However, it is an accurate and accepted surrogate for estimating DNA damage‐induced NHEJ in vivo 14, 54. 53BP1 foci serve several functions: they act as molecular scaffolds for recruitment of proteins such as the chromatin modulator Expand1 and they amplify ATM activity 55. The observation that 53BP1 foci formation was similar in type B and A cells suggests that NHEJ repair capacity was similar in the two cell types.
Radiation and TMZ‐mediated DNA damage can activate the intrinsic apoptotic pathway 49, 56, triggered by oligomerization of proapoptotic proteins such as Bax or Bak, with subsequent mitochondrial outer membrane permeabilization 23 that ultimately results in activation of caspases 3 and 7. The TMZ/XRT exposure was found to induce caspase 3 activation within 5 hours in proliferating type A neuroblasts but not in type B NSCs. Thus, our data and that of others support the hypothesis that the loss of neuroblasts was, in large part, a consequence of DNA damage‐induced apoptosis in type A cells and in progenitor type C cells 3, 14, 15, 16, 57. Additionally, some loss may be a consequence of neuroblast migration and terminal differentiation 14.
We determined whether there was differential expression of proapoptotic or antiapoptotic proteins in type B and A cells. Bax expression was quantified in type B NSCs and in type A neuroblasts. We found no significant differences on a per cell basis. However, we found a significant expression differential for the antiapoptotic proteins Bcl2 and Mcl1: quiescent type B NSCs expressed significantly more of these proteins than proliferating type A neuroblasts. Although we recognize that this study does not represent a comprehensive analysis of all antiapoptotic and proapoptotic proteins, it highlights a key question that remains to be addressed: what are the mechanisms that underlie the resistance of NSCs to DNA damaging agents?
We determined whether NSCs exposed to current TMZ/XRT followed by adjuvant TMZ (cohort 6) can continue to sustain neurogenesis. Our data indicate that type B NSCs are able to survive this treatment, and with sufficient recovery time, regenerate type A neuroblasts. One limitation of this study is that we only investigated neurogenesis in the V‐SVZ. We did not determine if the DCX‐expressing cells could form functional neurons in the olfactory bulb after migrating through the RMS.
Conclusion
Our results suggest that type B NSCs are able to sustain neurogenesis following a course of TMZ/XRT due to the survivability of the NSCs. Such a result holds implications for both the normal brain physiology after undergoing radiation and chemotherapy for malignant brain tumors and support dose escalation for treatment of GBM within the V‐SVZ.
Author Contributions
B.D.C., M.L.F.: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; G.T., J.T.R.: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; A.A.B.: tumor model, final approval of manuscript; D.D., L.J.: collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; K.B.: conception and design, collection and/or assembly of data, data analysis and interpretation, final approval of manuscript; R.A.I.: conception and design, data analysis and interpretation, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supporting information
Figure S1 Protocols for treating male and female C57BL/6 mice. Cohort 1: For 5 consecutive days mice were injected with DMSO. Mice were euthanized on Day 6. Cohort 2: For 5 consecutive days mice were injected with DMSO. Mice were again injected with DMSO on days 19–22. Mice were euthanized on Day 82. Cohort 3: For 5 consecutive days mice were injected with 50 mg/kg TMZ. Mice were euthanized on Day 6. Cohort 4: For 5 consecutive days the brains of mice were administered 2 Gy. Mice were euthanized on Day 6. Cohort 5: For 5 consecutive days mice were injected with 50 mg/kg TMZ. One hour after injection the brain was administered 2 Gy. Mice were euthanized on Day 6. Cohort 6: For 5 consecutive days mice were injected with 50 mg/kg TMZ. One hour after injection the brain was administered 2 Gy. On days 19–22 mice were injected with 100 mg/kg TMZ. Mice were euthanized on Day 82. Cohort 7: Mice were injected with 50 mg/kg TMZ. One hour after injection the brain was administered 1 Gy. Mice in cohort 8 were injected with DMSO and represent the controls for mice in cohort 7. Mice were euthanized 0.1 hour to 24 hours after irradiation. Immediately prior to euthanasia all mice in all cohorts underwent transcardial perfusion.
Figure S2 Representative images from slides stained solely with secondary antibodies used in Figures 1, 2, 3, 4, 5, 6. Images shown for the Leica microscope are at 20x. Confocal images are at 40x.
Figure S3 A) The presence of type B neural stem cells expressing GFAP (false red), Sox2 (false green), within 30 μm of the ventricle of the dorsolateral subdomain. Nuclei are stained with DAPI (blue). V = ventricle. Red bar = 50 μM. B) H&E of GBM tumor located in brain following 5 consecutive days of TMZ + XRT. Black arrows denote present of tumor and contralateral V‐SVZ. 1.80X
Figure S4 Representative images of Dcx expressing type A neuroblasts (false red) along a ventricle and in the RMS from a male mouse and a female mouse following exposure to concurrent TMZ/XRT. Nuclei are stained with DAPI (false white). White bars = 100 μm.
Figure S5 Representative images of Ki67 immunostaining (false green) in Sox2‐expressing cells (false red) from a non‐tumor bearing, TMZ/XRT‐treated mouse. DAPI blue. White bar = 100 μm. V = ventricle.
Figure S6 Dcx‐expressing (false green, A ‐ C) and Sox2‐expressing (false red, D ‐ F) cells in sham‐treated mice (A & D) and in mice whose brains were administered 2 Gy every day for 5 days (B & E), and in mice administered TMZ for 5 consecutive days (C & F). Nuclei stained with DAPI (false white). V = ventricle. White bar = 100 μm.
Figure S7 Expression of 53BP1 in Dcx and Sox2‐expressing cells from sham‐treated mice 1 hour after i.p. injection of DMSO (Cohort 8, Supplemental Fig. 1). A) Dcx‐expressing cells (false red), 53BP1 (false green), and DAPI (blue). B) Sox2‐expressing cells (false red), 53BP1 (false green) and DAPI (blue). C) Number of nuclei counted for each time point shown in Figure 4(C).
Figure S8 Expression of p53 (false green) in Sox2‐expresssing (false red, Panels A & B) and Dcx‐expressing cells (false red, Panels C & D) 3 hours after mice were exposed to either sham‐treatment, Cohort 8, Supplemental Fig 1or TMZ (50 mg/kg) followed by administration of 1 Gy to the brain, Cohort 7, Supplemental Figure 7. Nuclei are stained with DAPI. White line = 10 μm.
Figure S9 Mcl1 expression in Dcx neuroblasts and in Sox2 NSCs located within 30 μm cells of a ventricle of the V‐SVZ obtained from nontumor‐bearing sham‐treated female mice. Slides were coimmunostained for Mcl1 (false green, Cy2), Dcx (false red, Cy5), and Sox2 (false red, Cy7). Panels A & B illustrate Mcl1 and Dcx immunostaining. Panels C & D illustrate Mcl1 and Sox2 immunostaining. Panel E illustrates quantification of Mcl1 expression in Dcx‐expressing and Sox2‐expressing cells. DAPI staining is blue. V = ventricle. White bar = 10 μm. The confocal images illustrate immunofluorescence throughout 4.92 μm of the Z axis.
Acknowledgments
This study was supported in part by NIH/NCI R01CA166492 (M.L.F.), NIH/NINDS R01NS096238 (R.A.I.), the Michael David Greene Brain Cancer Fund/Vanderbilt‐Ingram Cancer Center (R.A.I.), and Vanderbilt‐Ingram Cancer CTR Grant P30 CA68485. Additional support for experiments performed using the VUMC Cell Imaging Shared Resource was provided in part by DK20593, DK58404, HD15052, DK59637 and EY08126.
References
- 1. Lim DA, Alvarez‐Buylla A. The adult ventricular‐subventricular zone (V‐SVZ) and olfactory bulb (OB) neurogenesis. Cold Spring Harb Perspect Biol 2016;8:1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Obernier K, Cebrian‐Silla A, Thomson M et al. Adult neurogenesis is sustained by symmetric self‐renewal and differentiation. Cell Stem Cell 2018;22:221.e228–234.e228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mich JK, Signer RA, Nakada D et al. Prospective identification of functionally distinct stem cells and neurosphere‐initiating cells in adult mouse forebrain. eLife 2014;3:e02669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fuentealba LC, Obernier K, Alvarez‐Buylla A. Adult neural stem cells bridge their niche. Cell Stem Cell 2012;10:698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sinnaeve J, Mobley BC, Ihrie RA. Space invaders: Brain tumor exploitation of the stem cell niche. Am J Pathol 2018;188:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smith AW, Mehta MP, Wernicke AG. Neural stem cells, the subventricular zone and radiotherapy: Implications for treating glioblastoma. J Neurooncol 2016;128:207–216. [DOI] [PubMed] [Google Scholar]
- 7. Muracciole X, El‐Amine W, Tabouret E et al. Negative survival impact of high radiation doses to neural stem cells niches in an IDH‐wild‐type glioblastoma population front oncol. 2018;8:426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen L, Guerrero‐Cazares H, Ye X et al. Increased subventricular zone radiation dose correlates with survival in glioblastoma patients after gross total resection. Int J Radiat Oncol Biol Phys 2013;86:616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee P, Eppinga W, Lagerwaard F et al. Evaluation of high ipsilateral subventricular zone radiation therapy dose in glioblastoma: A pooled analysis. Int J Radiat Oncol Biol Phys 2013;86:609–615. [DOI] [PubMed] [Google Scholar]
- 10. Mistry AM, Hale AT, Chambless LB et al. Influence of glioblastoma contact with the lateral ventricle on survival: A meta‐analysis. J Neurooncol 2017;131:125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mistry AM, Dewan MC, White‐Dzuro GA et al. Decreased survival in glioblastomas is specific to contact with the ventricular‐subventricular zone, not subgranular zone or corpus callosum. J Neurooncol 2017;132:341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oncology NCPGi . Central Nervous System Cancers; 2018.
- 13. Ponti G, Obernier K, Guinto C et al. Cell cycle and lineage progression of neural progenitors in the ventricular‐subventricular zones of adult mice. Proc Natl Acad Sci USA 2013;110:E1045–E1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barazzuol L, Ju L, Jeggo PA. A coordinated DNA damage response promotes adult quiescent neural stem cell activation. PLoS Biol 2017;15:e2001264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Achanta P, Capilla‐Gonzalez V, Purger D et al. Subventricular zone localized irradiation affects the generation of proliferating neural precursor cells and the migration of neuroblasts. Stem Cells 2012;30:2548–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lazarini F, Mouthon MA, Gheusi G et al. Cellular and behavioral effects of cranial irradiation of the subventricular zone in adult mice. PLoS One 2009;4:e7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Capilla‐Gonzalez V, Bonsu JM, Redmond KJ et al. Implications of irradiating the subventricular zone stem cell niche. Stem Cell Res 2016;16:387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stewart RD. Two‐lesion kinetic model of double‐strand break rejoining and cell killing. Radiat Res 2001;156:365–378. [DOI] [PubMed] [Google Scholar]
- 19. Abbotts R, Wilson DM 3rd. Coordination of DNA single strand break repair. Free Radic Biol Med 2017;107:228–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thompson LaL CL. Origin, recognition, signaling and repair of DNA double‐strand breaks in mammalian cells. Landes Biosci 2003;7:1–39. [Google Scholar]
- 21. Nelson WG, Kastan MB. DNA strand breaks: The DNA template alterations that trigger p53‐dependent DNA damage response pathways. Mol Cell Biol 1994;14:1815–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gil‐Perotin S, Marin‐Husstege M, Li J et al. Loss of p53 induces changes in the behavior of subventricular zone cells: Implication for the genesis of glial tumors. J Neurosci 2006;26:1107–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sarosiek KA, Fraser C, Muthalagu N et al. Developmental regulation of mitochondrial apoptosis by c‐Myc governs age‐ and tissue‐specific sensitivity to cancer therapeutics. Cancer Cell 2017;31:142–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ochs K, Kaina B. Apoptosis induced by DNA damage O6‐methylguanine is Bcl‐2 and caspase‐9/3 regulated and Fas/caspase‐8 independent. Cancer Res 2000;60:5815–5824. [PubMed] [Google Scholar]
- 25. Hardee ME, Marciscano AE, Medina‐Ramirez CM et al. Resistance of glioblastoma‐initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor‐beta. Cancer Res 2012;72:4119–4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carpenter AE, Jones TR, Lamprecht MR et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol 2006;7:R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lamprecht MR, Sabatini DM, Carpenter AE. CellProfiler: Free, versatile software for automated biological image analysis. Biotechniques 2007;42:71–75. [DOI] [PubMed] [Google Scholar]
- 28. Lopez‐Juarez A, Remaud S, Hassani Z et al. Thyroid hormone signaling acts as a neurogenic switch by repressing Sox2 in the adult neural stem cell niche. Cell Stem Cell 2012;10:531–543. [DOI] [PubMed] [Google Scholar]
- 29. Doetsch F, Caille I, Lim DA et al. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 1999;97:703–716. [DOI] [PubMed] [Google Scholar]
- 30. Fernando RN, Eleuteri B, Abdelhady S et al. Cell cycle restriction by histone H2AX limits proliferation of adult neural stem cells. Proc Natl Acad Sci USA 2011;108:5837–5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Daynac M, Chicheportiche A, Pineda JR et al. Quiescent neural stem cells exit dormancy upon alteration of GABAAR signaling following radiation damage. Stem Cell Res 2013;11:516–528. [DOI] [PubMed] [Google Scholar]
- 32. Rogakou EP, Boon C, Redon C et al. Megabase chromatin domains involved in DNA double‐strand breaks in vivo. J Cell Biol 1999;146:905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Frankenberg‐Schwager M, Frankenberg D, Blocher D et al. The linear relationship between DNA double‐strand breaks and radiation dose (30 MeV electrons) is converted into a quadratic function by cellular repair. Int J Radiat Biol Relat Stud Phys Chem Med 1980;37:207–212. [DOI] [PubMed] [Google Scholar]
- 34. Paull TT, Rogakou EP, Yamazaki V et al. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 2000;10:886–895. [DOI] [PubMed] [Google Scholar]
- 35. Ward IM, Minn K, Jorda KG et al. Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J Biol Chem 2003;278:19579–19582. [DOI] [PubMed] [Google Scholar]
- 36. Schultz LB, Chehab NH, Malikzay A et al. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double‐strand breaks. J Cell Biol 2000;151:1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chong MJ, Murray MR, Gosink EC et al. Atm and Bax cooperate in ionizing radiation‐induced apoptosis in the central nervous system. Proc Natl Acad Sci USA 2000;97:889–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Malone CD, Hasan SM, Roome RB et al. Mcl‐1 regulates the survival of adult neural precursor cells. Mol Cell Neurosci 2012;49:439–447. [DOI] [PubMed] [Google Scholar]
- 39. Smith AW, Parashar B, Wernicke AG. Subventricular zone‐associated glioblastoma: A call for translational research to guide clinical decision making. Neurogenesis 2016;3:e1225548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Adeberg S, Konig L, Bostel T et al. Glioblastoma recurrence patterns after radiation therapy with regard to the subventricular zone. Int J Radiat Oncol Biol Phys 2014;90:886–893. [DOI] [PubMed] [Google Scholar]
- 41. Wang C, Liu F, Liu YY et al. Identification and characterization of neuroblasts in the subventricular zone and rostral migratory stream of the adult human brain. Cell Res 2011;21:1534–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ernst A, Alkass K, Bernard S et al. Neurogenesis in the striatum of the adult human brain. Cell 2014;156:1072–1083. [DOI] [PubMed] [Google Scholar]
- 43. Sanai N, Nguyen T, Ihrie RA et al. Corridors of migrating neurons in the human brain and their decline during infancy. Nature 2011;478:382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. 1.2018 NCPGiOv . Central Nervous System Cancers 2018.
- 45. Kaina B, Ziouta A, Ochs K et al. Chromosomal instability, reproductive cell death and apoptosis induced by O6‐methylguanine in Mex‐, Mex+ and methylation‐tolerant mismatch repair compromised cells: Facts and models. Mutat Res 1997;381:227–241. [DOI] [PubMed] [Google Scholar]
- 46. York SJ, Modrich P. Mismatch repair‐dependent iterative excision at irreparable O6‐methylguanine lesions in human nuclear extracts. J Biol Chem 2006;281:22674–22683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roos WP, Batista LF, Naumann SC et al. Apoptosis in malignant glioma cells triggered by the temozolomide‐induced DNA lesion O6‐methylguanine. Oncogene 2007;26:186–197. [DOI] [PubMed] [Google Scholar]
- 48. Vidair CA, Chen CH, Ling CC et al. Apoptosis induced by X‐irradiation of rec‐myc cells is postmitotic and not predicted by the time after irradiation or behavior of sister cells. Cancer Res 1996;56:4116–4118. [PubMed] [Google Scholar]
- 49. Oda E, Ohki R, Murasawa H et al. Noxa, a BH3‐only member of the Bcl‐2 family and candidate mediator of p53‐induced apoptosis. Science 2000;288:1053–1058. [DOI] [PubMed] [Google Scholar]
- 50. Bedford JS, Mitchell JB, Griggs HG et al. Radiation‐induced cellular reproductive death and chromosome aberrations. Radiat Res 1978;76:573–586. [PubMed] [Google Scholar]
- 51. Carrano AV. Chromosome aberrations and radiation‐induced cell death. II. Predicted and observed cell survival. Mutat Res 1973;17:355–366. [DOI] [PubMed] [Google Scholar]
- 52. Dai J, Itahana K, Baskar R. Quiescence does not affect p53 and stress response by irradiation in human lung fibroblasts. Biochem Biophys Res Commun 2015;458:104–109. [DOI] [PubMed] [Google Scholar]
- 53. Wilson KM, Keng PC. Radiation‐induced DNA damage and repair in quiescent and proliferating human tumor cells in vitro. Int J Radiat Biol 1989;55:385–395. [DOI] [PubMed] [Google Scholar]
- 54. Gatz SA, Ju L, Gruber R et al. Requirement for DNA ligase IV during embryonic neuronal development. J Neurosci 2011;31:10088–10100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Panier S, Boulton SJ. Double‐strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 2014;15:7–18. [DOI] [PubMed] [Google Scholar]
- 56. Wang H, Cai S, Ernstberger A et al. Temozolomide‐mediated DNA methylation in human myeloid precursor cells: Differential involvement of intrinsic and extrinsic apoptotic pathways. Clin Cancer Res 2013;19:2699–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Balentova S, Hajtmanova E, Trylcova R et al. Ionizing radiation induced long‐term alterations in the adult rat rostral migratory stream. Acta Histochem 2014;116:265–271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Protocols for treating male and female C57BL/6 mice. Cohort 1: For 5 consecutive days mice were injected with DMSO. Mice were euthanized on Day 6. Cohort 2: For 5 consecutive days mice were injected with DMSO. Mice were again injected with DMSO on days 19–22. Mice were euthanized on Day 82. Cohort 3: For 5 consecutive days mice were injected with 50 mg/kg TMZ. Mice were euthanized on Day 6. Cohort 4: For 5 consecutive days the brains of mice were administered 2 Gy. Mice were euthanized on Day 6. Cohort 5: For 5 consecutive days mice were injected with 50 mg/kg TMZ. One hour after injection the brain was administered 2 Gy. Mice were euthanized on Day 6. Cohort 6: For 5 consecutive days mice were injected with 50 mg/kg TMZ. One hour after injection the brain was administered 2 Gy. On days 19–22 mice were injected with 100 mg/kg TMZ. Mice were euthanized on Day 82. Cohort 7: Mice were injected with 50 mg/kg TMZ. One hour after injection the brain was administered 1 Gy. Mice in cohort 8 were injected with DMSO and represent the controls for mice in cohort 7. Mice were euthanized 0.1 hour to 24 hours after irradiation. Immediately prior to euthanasia all mice in all cohorts underwent transcardial perfusion.
Figure S2 Representative images from slides stained solely with secondary antibodies used in Figures 1, 2, 3, 4, 5, 6. Images shown for the Leica microscope are at 20x. Confocal images are at 40x.
Figure S3 A) The presence of type B neural stem cells expressing GFAP (false red), Sox2 (false green), within 30 μm of the ventricle of the dorsolateral subdomain. Nuclei are stained with DAPI (blue). V = ventricle. Red bar = 50 μM. B) H&E of GBM tumor located in brain following 5 consecutive days of TMZ + XRT. Black arrows denote present of tumor and contralateral V‐SVZ. 1.80X
Figure S4 Representative images of Dcx expressing type A neuroblasts (false red) along a ventricle and in the RMS from a male mouse and a female mouse following exposure to concurrent TMZ/XRT. Nuclei are stained with DAPI (false white). White bars = 100 μm.
Figure S5 Representative images of Ki67 immunostaining (false green) in Sox2‐expressing cells (false red) from a non‐tumor bearing, TMZ/XRT‐treated mouse. DAPI blue. White bar = 100 μm. V = ventricle.
Figure S6 Dcx‐expressing (false green, A ‐ C) and Sox2‐expressing (false red, D ‐ F) cells in sham‐treated mice (A & D) and in mice whose brains were administered 2 Gy every day for 5 days (B & E), and in mice administered TMZ for 5 consecutive days (C & F). Nuclei stained with DAPI (false white). V = ventricle. White bar = 100 μm.
Figure S7 Expression of 53BP1 in Dcx and Sox2‐expressing cells from sham‐treated mice 1 hour after i.p. injection of DMSO (Cohort 8, Supplemental Fig. 1). A) Dcx‐expressing cells (false red), 53BP1 (false green), and DAPI (blue). B) Sox2‐expressing cells (false red), 53BP1 (false green) and DAPI (blue). C) Number of nuclei counted for each time point shown in Figure 4(C).
Figure S8 Expression of p53 (false green) in Sox2‐expresssing (false red, Panels A & B) and Dcx‐expressing cells (false red, Panels C & D) 3 hours after mice were exposed to either sham‐treatment, Cohort 8, Supplemental Fig 1or TMZ (50 mg/kg) followed by administration of 1 Gy to the brain, Cohort 7, Supplemental Figure 7. Nuclei are stained with DAPI. White line = 10 μm.
Figure S9 Mcl1 expression in Dcx neuroblasts and in Sox2 NSCs located within 30 μm cells of a ventricle of the V‐SVZ obtained from nontumor‐bearing sham‐treated female mice. Slides were coimmunostained for Mcl1 (false green, Cy2), Dcx (false red, Cy5), and Sox2 (false red, Cy7). Panels A & B illustrate Mcl1 and Dcx immunostaining. Panels C & D illustrate Mcl1 and Sox2 immunostaining. Panel E illustrates quantification of Mcl1 expression in Dcx‐expressing and Sox2‐expressing cells. DAPI staining is blue. V = ventricle. White bar = 10 μm. The confocal images illustrate immunofluorescence throughout 4.92 μm of the Z axis.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.