

Abstract
Background
Although antidepressants are often a first‐line treatment for adults with moderate to severe depression, many people do not respond adequately to medication, and are said to have treatment‐resistant depression (TRD). Little evidence exists to inform the most appropriate 'next step' treatment for these people.
Objectives
To assess the effectiveness of standard pharmacological treatments for adults with TRD.
Search methods
We searched the Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR) (March 2016), CENTRAL, MEDLINE, Embase, PsycINFO and Web of Science (31 December 2018), the World Health Organization trials portal and ClinicalTrials.gov for unpublished and ongoing studies, and screened bibliographies of included studies and relevant systematic reviews without date or language restrictions.
Selection criteria
Randomised controlled trials (RCTs) with participants aged 18 to 74 years with unipolar depression (based on criteria from DSM‐IV‐TR or earlier versions, International Classification of Diseases (ICD)‐10, Feighner criteria or Research Diagnostic Criteria) who had not responded to a minimum of four weeks of antidepressant treatment at a recommended dose. Interventions were:
(1) increasing the dose of antidepressant monotherapy;
(2) switching to a different antidepressant monotherapy;
(3) augmenting treatment with another antidepressant;
(4) augmenting treatment with a non‐antidepressant.
All were compared with continuing antidepressant monotherapy. We excluded studies of non‐standard pharmacological treatments (e.g. sex hormones, vitamins, herbal medicines and food supplements).
Data collection and analysis
Two reviewers used standard Cochrane methods to extract data, assess risk of bias, and resolve disagreements. We analysed continuous outcomes with mean difference (MD) or standardised mean difference (SMD) and 95% confidence interval (CI). For dichotomous outcomes, we calculated a relative risk (RR) and 95% CI. Where sufficient data existed, we conducted meta‐analyses using random‐effects models.
Main results
We included 10 RCTs (2731 participants). Nine were conducted in outpatient settings and one in both in‐ and outpatients. Mean age of participants ranged from 42 ‐ 50.2 years, and most were female.
One study investigated switching to, or augmenting current antidepressant treatment with, another antidepressant (mianserin). Another augmented current antidepressant treatment with the antidepressant mirtazapine. Eight studies augmented current antidepressant treatment with a non‐antidepressant (either an anxiolytic (buspirone) or an antipsychotic (cariprazine; olanzapine; quetiapine (3 studies); or ziprasidone (2 studies)). We judged most studies to be at a low or unclear risk of bias. Only one of the included studies was not industry‐sponsored.
There was no evidence of a difference in depression severity when current treatment was switched to mianserin (MD on Hamilton Rating Scale for Depression (HAM‐D) = ‐1.8, 95% CI ‐5.22 to 1.62, low‐quality evidence)) compared with continuing on antidepressant monotherapy. Nor was there evidence of a difference in numbers dropping out of treatment (RR 2.08, 95% CI 0.94 to 4.59, low‐quality evidence; dropouts 38% in the mianserin switch group; 18% in the control).
Augmenting current antidepressant treatment with mianserin was associated with an improvement in depression symptoms severity scores from baseline (MD on HAM‐D ‐4.8, 95% CI ‐8.18 to ‐1.42; moderate‐quality evidence). There was no evidence of a difference in numbers dropping out (RR 1.02, 95% CI 0.38 to 2.72; low‐quality evidence; 19% dropouts in the mianserin‐augmented group; 38% in the control). When current antidepressant treatment was augmented with mirtazapine, there was little difference in depressive symptoms (MD on Beck Depression Inventory (BDI‐II) ‐1.7, 95% CI ‐4.03 to 0.63; high‐quality evidence) and no evidence of a difference in dropout numbers (RR 0.50, 95% CI 0.15 to 1.62; dropouts 2% in mirtazapine‐augmented group; 3% in the control).
Augmentation with buspirone provided no evidence of a benefit in terms of a reduction in depressive symptoms (MD on Montgomery and Asberg Depression Rating Scale (MADRS) ‐0.30, 95% CI ‐9.48 to 8.88; low‐quality evidence) or numbers of drop‐outs (RR 0.60, 95% CI 0.23 to 1.53; low‐quality evidence; dropouts 11% in buspirone‐augmented group; 19% in the control).
Severity of depressive symptoms reduced when current treatment was augmented with cariprazine (MD on MADRS ‐1.50, 95% CI ‐2.74 to ‐0.25; high‐quality evidence), olanzapine (MD on HAM‐D ‐7.9, 95% CI ‐16.76 to 0.96; low‐quality evidence; MD on MADRS ‐12.4, 95% CI ‐22.44 to ‐2.36; low‐quality evidence), quetiapine (SMD ‐0.32, 95% CI ‐0.46 to ‐0.18; I2 = 6%, high‐quality evidence), or ziprasidone (MD on HAM‐D ‐2.73, 95% CI ‐4.53 to ‐0.93; I2 = 0, moderate‐quality evidence) compared with continuing on antidepressant monotherapy.
However, a greater number of participants dropped out when antidepressant monotherapy was augmented with an antipsychotic (cariprazine RR 1.68, 95% CI 1.16 to 2.41; quetiapine RR 1.57, 95% CI: 1.14 to 2.17; ziprasidone RR 1.60, 95% CI 1.01 to 2.55) compared with antidepressant monotherapy, although estimates for olanzapine augmentation were imprecise (RR 0.33, 95% CI 0.04 to 2.69). Dropout rates ranged from 10% to 39% in the groups augmented with an antipsychotic, and from 12% to 23% in the comparison groups. The most common reasons for dropping out were side effects or adverse events.
We also summarised data about response and remission rates (based on changes in depressive symptoms) for included studies, along with data on social adjustment and social functioning, quality of life, economic outcomes and adverse events.
Authors' conclusions
A small body of evidence shows that augmenting current antidepressant therapy with mianserin or with an antipsychotic (cariprazine, olanzapine, quetiapine or ziprasidone) improves depressive symptoms over the short‐term (8 to 12 weeks). However, this evidence is mostly of low or moderate quality due to imprecision of the estimates of effects. Improvements with antipsychotics need to be balanced against the increased likelihood of dropping out of treatment or experiencing an adverse event. Augmentation of current antidepressant therapy with a second antidepressant, mirtazapine, does not produce a clinically important benefit in reduction of depressive symptoms (high‐quality evidence). The evidence regarding the effects of augmenting current antidepressant therapy with buspirone or switching current antidepressant treatment to mianserin is currently insufficient.
Further trials are needed to increase the certainty of these findings and to examine long‐term effects of treatment, as well as the effectiveness of other pharmacological treatment strategies.
Plain language summary
Are there effective medications for treating depression that does not improve with the first medication used?
Background
Depression is a common problem often treated with antidepressant medication. However, many people do not get better with antidepressant treatment and have 'treatment‐resistant depression' (TRD). Several different treatment approaches can be tried ‐ such as increasing the dose of the current medication, adding another medication, or switching to a different antidepressant.
Cochrane reviewers looked at the available evidence to see which of these options may be the best treatment for people with TRD.
Search date
In December 2018, we searched eight medical databases for suitable clinical trials.
Study characteristics
We included 10 randomised controlled trials (RCTs) with 2731 participants (RCTs produce the most robust evidence). These trials investigated three different treatment strategies:
1. changing to a different antidepressant,
2. adding a second antidepressant to the current antidepressant treatment, or
3. adding a different type of medication to current antidepressant treatment ‐ an anti‐anxiety medication or an antipsychotic
We found no trials of increasing the dose of current antidepressant medication. Nine of the 10 studies included in this review were sponsored by pharmaceutical companies.
Key results
One small study investigated changing current antidepressant treatment to a different antidepressant (mianserin) or adding mianserin to current treatment. We are uncertain about the effect changing treatment to mianserin has on depressive symptoms or the likelihood of dropping out of treatment. People who added mianserin to their current antidepressant treatment showed fewer depressive symptoms, but the likelihood of dropping out was not clear.
Adding the antidepressant mirtazapine to current antidepressant treatment had little or no effect on depressive symptoms or on the likelihood of dropping out of treatment.
The effect of adding an anti‐anxiety medication (buspirone) to ongoing antidepressant treatment on depressive symptoms or dropping out is currently uncertain. These findings were based on one small study.
Most studies looked at the effects of adding an antipsychotic medication (cariprazine, quetiapine, ziprasidone or olanzapine) to current antidepressant treatment. These suggested that adding cariprazine results in a small reduction in depressive symptoms; adding quetiapine reduces depressive symptoms; and adding ziprasidone probably results in a small reduction in depressive symptoms. However, our results also suggest that adding these medicines to current treatment probably increases the likelihood of dropping out of treatment. The most common reasons for dropping out were side effects or adverse events. Adding olanzapine to ongoing treatment may reduce depressive symptoms, but the effects on dropping out are uncertain (findings based on one small study).
Nearly all (9/10) of the studies assessed the effects of treatment in the short‐term – six or eight weeks after beginning the new treatment – so the longer term effects of most treatments are unknown.
Quality of the evidence
We judged the quality of the evidence as high, moderate, or low for different outcomes. The main limitation we identified was that the evidence for many of the treatment options investigated came from only one study, and some of these studies also had few participants.
We rated the evidence to be low quality for:
1. switching from the current antidepressant treatment to another antidepressant (mianserin);
2. supplementing current antidepressant treatment with a second antidepressant (mianserin), or with an anti‐anxiety medication (buspirone), or with the antipsychotic olanzapine.
This means we are uncertain about the effects of these treatments on depression symptoms or the likelihood of dropping out of treatment.
We rated the quality of the evidence for adding mirtazapine (an antidepressant), cariprazine (an antipsychotic) or quetiapine (an antipsychotic) to ongoing antidepressant treatment on depressive symptoms as high, meaning we are very confident in the effect of these treatment strategies.
We rated the quality of the evidence for adding the antipsychotic ziprasidone to ongoing antidepressant treatment on the effect on depressive symptoms as moderate, which means the true effect may be different from what we found, although findings are likely to be close.
We rated the quality of the evidence for adding mirtazapine (an antidepressant) to ongoing antidepressant treatment on the likelihood of dropping out of treatment as high. We rated the quality of the evidence for adding cariprazine, olanzapine or ziprasidone (all antipsychotics) on the likelihood of dropping out as moderate.
Summary of findings
Background
Description of the condition
Depression is a common mental disorder, characterized by sadness, loss of interest or pleasure, feelings of guilt or low self‐worth, disturbed sleep or appetite, feelings of tiredness, and poor concentration. By 2030, depression is predicted to be the leading cause of disability in high‐income countries (Mathers 2005). Severity of depression can be classified using the Diagnostic and Statistical Manual of Mental Disorders, fourth edition (DSM‐IV) criteria as mild (five or more symptoms with minor functional impairment), moderate (symptoms or functional impairment are between 'mild' and 'severe') and severe (most symptoms present and interfere with functioning) (NICE 2009).
Antidepressants are often prescribed as the first‐line treatment for adults with moderate to severe depression (NICE 2009). In England in 2014, 57.1 million prescriptions for antidepressants were issued at a cost of GBP 265 million (NHS Digital 2015). However, two‐thirds of people do not respond fully to such pharmacotherapy (Trivedi 2006). Such nonresponse may be because of intolerance to the prescribed medication or non‐adherence to the treatment regimen, but may also be the result of treatment 'resistance', where an adequate dose and duration of treatment has been prescribed. The earliest definition of treatment‐'resistant' depression was given by the World Psychiatric Association; it defined 'resistance' as "an absence of clinical response to treatment with a tricyclic antidepressant at a minimum dose of 150 mg per day of imipramine (or equivalent drug) for 4 to 6 weeks" (WPA 1974). Subsequently, others have suggested more complex classification systems based on nonresponse to multiple courses of treatment (Fava 2005; Fekadu 2009; Thase 1997), and used other terms such as 'treatment refractory' depression and 'antidepressant resistant' depression to describe this condition. For the purpose of this review, we will use the term 'treatment‐resistant depression' as this is the descriptor that has, generally, represented the broadest definition of the condition.
Globally, the prevalence of major depressive disorder (MDD) has been estimated to be 4.7 % (Ferrari 2013), with a lifetime prevalence as high as 17% (Kessler 2013). Depression imposes an economic burden on society in terms of both the direct costs of treatment and indirectly through its effects on productivity (Wang 2003). Healthcare utilisation and related costs of TRD are higher than for MDD (Gibson 2010; Mahlich 2018). If up to one‐third of patients have 'treatment‐resistant' depression (TRD), it is thus clear that this condition represents a considerable burden to patients, health systems and society.
Description of the intervention
Antidepressants are commonly used as the first‐line treatment for adults with moderate to severe depression (NICE 2009). There are five main types of antidepressants: tricyclic (TCAs) and related antidepressants; monoamine‐oxidase inhibitors (MAOIs); selective serotonin re‐uptake inhibitors (SSRIs); serotonin and noradrenaline reuptake inhibitors (SNRIs); and noradrenergic and specific serotonin antidepressants (NaSSAs). SSRIs are generally better tolerated than other classes of antidepressants and are safer with regards to overdose than TCAs. It is, therefore, not surprising that SSRIs are the most commonly prescribed antidepressants for treating depression (NHS Information Centre 2011).
There is no standard approach to the treatment of those whose depression does not respond to antidepressant medication. Guidance published by the American Psychiatric Association (APA 2010) and the National Institute for Health and Clinical Excellence (NICE 2009) suggests that the potential 'next step' may include increasing the dose of the antidepressant medication or switching to another antidepressant (within the same or a different pharmacological class) or augmentation with another pharmacological or psychological treatment. In terms of combination treatment with an additional pharmacological agent, this may comprise either: (1) another antidepressant or (2) a non‐antidepressant medication, which may include (i) lithium, (ii) an antipsychotic (e.g. olanzapine), (iii) pindolol, (iv) triiodothyronine (T3) or (v) buspirone.
How the intervention might work
Most antidepressants work by inhibiting the uptake of monoamine neurotransmitters into neurons. The SSRI group inhibit the uptake of serotonin, and many of the older tricyclics have actions on both serotonin and noradrenaline (along with other pharmacological actions not thought to be important in depression).
For those people whose depression has not responded to initial treatment with an antidepressant, increasing the dose or switching medication may improve their outcome through the associated pharmacological actions, for example, increasing levels of neurotransmitters such as serotonin and noradrenaline. Increased serotonin neurotransmission may also be expected when two serotonergic drugs with different modes of action are combined (NICE 2009). For example, mirtazapine is a noradrenergic and specific serotonergic antidepressant (NaSSA) that acts by antagonising the adrenergic alpha2‐autoreceptors and alpha2‐heteroreceptors as well as by blocking 5‐HT2 and 5‐HT3 receptors (Anttila 2001). In general, the rationale for combination therapy with two different antidepressants or augmentation with a non‐antidepressant medication (such as lithium), is that such treatment broadens the pharmacological actions involved (Anderson 2008).
Why it is important to do this review
Antidepressants continue to be the first‐line treatment for many people with depression. However, two‐thirds of people prescribed antidepressants for depression do not respond fully to such medication (Trivedi 2006). It is, therefore, important to summarise the evidence on the effectiveness of pharmacological interventions for people with TRD in order to establish the best 'next step' treatment for this patient group.
There have been several narrative reviews of the evidence on the treatment of people whose depression has not responded to antidepressant medication alone (e.g. Carvalho 2008; Nierenberg 2007; Papakostas 2009). Systematic reviews of the effectiveness of combination treatment for people with depression have not examined the evidence for the treatment‐resistant population (Friedman 2004; Pampallona 2004). Others have summarised the evidence for the effectiveness of particular treatment strategies for those who have not responded to antidepressants: (1) augmentation (Carvalho 2007) with lithium (Bauer 1999) or atypical antipsychotics (Shelton 2008); and (2) within‐ or between‐class switches (Papakostas 2008), and one review focused on interventions for older people (≥ 55 years) (Cooper 2011). However, a number of these reviews included uncontrolled studies or non‐randomised studies or both, as well as randomised controlled trials (RCTs) (Carvalho 2007; Cooper 2011; Shelton 2008).
A previous systematic review of RCTs investigating pharmacological and psychological treatments for people with TRD failed to find strong evidence to guide the management of such people (Stimpson 2002). However this review, along with others (e.g. Bauer 1999, which summarised the evidence for lithium up to June 1997) is out‐of‐date and a number of relevant RCTs have been published subsequently. These include a review by Edwards 2013 that has a narrow focus both in terms of the interventions to be examined (lithium or an atypical antipsychotic) and the definition of treatment resistance. The latter is defined as "failure to respond to at least two previous antidepressants in the current episode of depression". While this definition of treatment resistance has been frequently used, we know that many people do not respond to an initial course of antidepressants and there is little evidence to inform the most appropriate 'next step' treatment for this group (NICE 2009). Therefore, given the continued reliance upon antidepressants as a first‐line treatment for depression, we propose using an inclusive definition of treatment resistance (based on nonresponse to at least four weeks of antidepressant medication) in order to help establish the best 'next step' treatment for the large number of people whose depression does not respond to antidepressants. The rise in the number of prescriptions for antidepressants in recent years means that a review of the evidence for the effectiveness of pharmacological interventions for people with TRD is timely (Pincus 1998;McManus 2000;Middleton 2001).
Studies examining psychological interventions for TRD will be excluded as these are the focus of another review (Ijaz 2018). Together, the evidence from these two linked reviews will provide a comprehensive review of the main interventions for the management of TRD, which will inform clinical decision‐making with regards to the best 'next step' for adults whose depression has not responded to first‐line treatment with medication.
Objectives
To assess the effectiveness of pharmacological interventions for TRD in adults.
Methods
Criteria for considering studies for this review
Types of studies
The methods used in our review were previously described in a published protocol (Williams 2013).
Randomised controlled trials (RCTs) were eligible for inclusion in the review. Trials employing a cross‐over design could be included in the review, using data from the first active treatment stage only. Cluster RCTs were also eligible for inclusion.
Any other study design, including quasi‐randomised studies and non‐randomised studies, were excluded from this review.
Types of participants
Age range
Participants were adults aged 18 to 74 years.
If the study included some participants that were aged under 75 years and some over the age of 74 years, we excluded the study if the mean age of participants was over 74 years. Similarly, if the study included some participants that were aged under 18 years and some aged 18 years or older, we excluded the study if the mean age of participants was less than 18 years.
Definition of treatment‐resistant depression
For a study to be eligible for inclusion in this review, all of the participants had to meet our criteria for treatment‐resistant depression at the point at which they were randomly allocated to treatment. We included studies that began with an open‐label lead‐in phase to confirm treatment resistance provided that only participants meeting our definition of TRD continued into the randomised treatment phase.
We defined treatment‐resistant depression as a primary diagnosis of unipolar depression that had not responded (or had only partially responded) to a minimum of four weeks of antidepressant treatment at a recommended dose (at least 150 mg/day imipramine or equivalent antidepressant (e.g. 20 mg/day citalopram)).
We excluded studies that included participants who had not responded because of intolerance to antidepressant medication.
While there have been initiatives to improve access to psychological therapies in England and elsewhere, access to psychological treatment is still limited and antidepressants are often the first‐line treatment for adults with depression. Therefore, this review did not include studies of interventions intended for those who had not responded to psychological treatment.
Diagnosis
Acceptable diagnoses of unipolar depression included those based on criteria from DSM‐IV‐TR or earlier versions (APA 2000), International Classification of Diseases (ICD)‐10 (WHO 1992), Feighner criteria (Feighner 1972) or Research Diagnostic Criteria (Spitzer 1978). Studies that had not used standardised diagnostic criteria were excluded.
Comorbidity
Studies of participants with comorbid schizophrenia or bipolar disorder were excluded.
Studies including both unipolar and bipolar participants were excluded unless data were available for the subgroup of unipolar participants.
This review included studies involving participants with comorbid physical conditions or other psychological disorders (e.g. anxiety) for whom the pharmacological therapy was not being primarily used to manage the physical illness, in other words, the focus of treatment was TRD ‐ not the comorbidity.
Types of interventions
Experimental interventions
The experimental interventions were based on the 'next step' approach to the management of depression that had not responded to treatment with antidepressants:
increasing the dose of antidepressant monotherapy;
switching to a different antidepressant monotherapy;
augmenting treatment with another antidepressant;
augmenting treatment with a non‐antidepressant.
Antidepressants can be grouped as TCAs, MAOIs, SSRIs, SNRIs and NaSSAs.
Non‐antidepressant medications used as augmentors included antipsychotics (e.g. olanzapine), anxiolytics (e.g. buspirone), antimania drugs (e.g. lithium) and beta‐blockers (e.g. pindolol).
Studies examining non‐standard pharmacological approaches for treating TRD (e.g. sex hormones, vitamins, herbal medicines and food supplements) were excluded. Studies examining psychological interventions given in addition to antidepressant medication for individuals with TRD are included in another review (Ijaz 2018).
Comparator interventions
The control comparison was continuing on the initial antidepressant monotherapy.
Full details of comparisons made can be found in the Data extraction and management section.
Types of outcome measures
Primary outcomes
1. Change in depressive symptoms as measured on rating scales for depression, either clinician‐rated (e.g. Hamilton Rating Scale for Depression (HAM‐D; Hamilton 1960) or Montgomery‐Asberg Depression Rating Scale (MADRS; Montgomery 1979)), or self‐report (e.g. Beck Depression Inventory (BDI; Beck 1961; Beck 1996) (or other validated measures)). Data on observer‐rated and self‐report outcomes were analysed separately.
2. Number of dropouts from study or treatment (all‐cause dropout) within the trials. Where available, we extracted data on reasons for dropout and summarised these in narrative form.
Secondary outcomes
3. Response or remission rates, or both, based on changes in depression measures ‐ either clinician‐rated (e.g. HAM‐D; Hamilton 1960) or self‐report (e.g. BDI; Beck 1961; Beck 1996) or other validated measures. Response is frequently quantified as at least a 50% reduction in symptoms on the HAM‐D or BDI but we accepted the study's original definition. Remission is based on the absolute score on the depression measure. Examples of definitions of remission include 7 or less on the HAM‐D and 10 or less on BDI. Again, we accepted the study authors' original definition.
4. Improvement in social adjustment and social functioning including the Global Assessment of Function (Luborsky 1962) scores, where reported, were summarised in narrative form.
5. Improvement in quality of life as measured on the Short Form (SF)‐36 (Ware 1993), Health of the Nation Outcome Scales (HoNOS) (Wing 1994), or World Health Organization Quality of Life (WHOQOL) (WHOQOL 1998) or similar scale, where reported, were summarised in narrative form.
6. Economic outcomes (e.g. days of work absence/ability to return to work, number of appointments with primary care physician, number of referrals to secondary services, use of additional treatments), where reported were summarised in narrative form.
7. Adverse effects (e.g. completed/attempted suicides), where reported, were summarised in narrative form.
Timing of outcome assessment
Outcomes at each reported follow‐up point were summarised. Where appropriate and if the data allowed, outcomes were categorised as short‐term (up to 12 weeks) and longer‐term (longer than 12 weeks).
Search methods for identification of studies
Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR)
The Cochrane Common Mental Disorders (CCMD) Group maintains an archived register of RCTs, the CCMDCTR. This register contains over 40,000 reference records (reports of RCTs) for anxiety and depressive disorders, bipolar disorder, eating disorders, self‐harm and other mental disorders within the scope of this Group. The CCMDCTR is a partially studies‐based register, with 50% of the reference records tagged to over 12,000 individually PICO‐coded study records. Reports of trials for inclusion in the register were routinely collated from generic searches of MEDLINE (1950‐), Embase (1974‐) and PsycINFO (1967‐), quarterly searches of the Cochrane Central Register of Controlled Trials (CENTRAL) and review‐specific searches of additional databases (to June 2016). Reports of trials were also sourced from international trial registries, drug companies, the handsearching of key journals, conference proceedings and other (non‐Cochrane) systematic reviews and meta‐analyses. Details of the CCMD Group's core search strategies (used to identify RCTs) can be found on the Group's website, with an example of the core MEDLINE search displayed in Appendix 1.
The Group’s Specialised Register had fallen out of date with the Editorial Group’s move from Bristol to York in the summer of 2016.
Electronic searches
We searched the CCMDCTR‐Studies Register using the following terms: Condition = ((depressi* or "affective disorder" or "mood disorder*") and ("treatment‐resistant" or recurrent))
Additionally we searched the CCMDCTR‐References Register using a more sensitive set of terms (keywords and subject headings) to identify additional untagged/uncoded references:
1. depressi* [Ti, Ab, KW] 2. (*refractory* or *resistan* or *recurren*) [Ti, Ab] 3. (augment* or potentiat*) [Ti, Ab] 4. (chronicity or "chronic depress*" or "chronically depress*" or "depressed chronic*" or "chronic major depressi*" or "chronic affective disorder*" or "chronic mood disorder*" or (chronic* and (relaps* or recurr*))) [Ti, Ab, KW] 5. ("persistent depress*" or "persistently depress*" or "depression persist*" or "persistent major depress*" or "persistence of depress*" or "persistence of major depress*") [Ti, Ab] 6. (nonrespon* or non‐respon* or "non respon*" or "not respon*" or "no respon*" or "partial respon*" or "partially respon*" or "incomplete respon*" or "incompletely respon*" or unrespon*) [Ti, Ab] 7. ("failed to respond" or "failed to improve" or "failure to respon*" or "failure to improve" or "failed medication*" or "antidepressant fail*" or "treatment fail*") [Ti, Ab] 8. (inadequate* and respon*) [Ti, Ab] 9. "treatment‐resistant depression" [KW] 10. (recurrence or "recurrent depression" or "recurrent disease") [KW] 11. "drug resistance" [KW] 12. "treatment failure" [KW] 13. "drug potentiation" [KW] 14. augmentation [KW] 15. or/2‐14 16. (1 and 15)
We applied no date or language restrictions to our search. Our search of the CCMDCTR was up‐to‐date as of 18 March 2016.
We ran additional searches via the following biomedical databases (1 January 2016 to 31 December 2018) (Appendix 2):
MEDLINE/Premedline (Ovid);
Embase (Ovid);
Cochrane Central Register of Controlled Trials (CENTRAL);
PsycINFO (Ovid);
Web of Science Core Collection (Clarivate Analytics).
We used the term 'treatment‐resistant' or 'treatment refractory' depression to search international trials registries, including the WHO trials portal (ICTRP) and ClinicalTrials.gov (to 31 December 2018) (Appendix 2), to identify any additional ongoing and unpublished studies. We contacted principal Investigators, when necessary, to request further details of ongoing/unpublished studies or trials reported as conference abstracts only. These searches are up‐to‐date as of 31 December 2018.
Searching other resources
Reference lists of all included studies and other relevant systematic reviews were searched for papers that might meet the inclusion criteria. Subject experts were also contacted to ensure that all relevant published and unpublished studies were considered for inclusion.
Data collection and analysis
Selection of studies
One review author (NW, PD or SI) examined titles and abstracts to remove obviously irrelevant reports and then screened study abstracts against inclusion criteria using a standardised abstract screening form. In any case of uncertainty, an over‐inclusive approach was taken and the full paper was obtained along with those for the studies assessed as meeting the inclusion criteria. Two review authors (of CW, MT, NW, GL, DK, PD and SI) screened each paper for inclusion or exclusion from the review. If any disagreements arose, these were discussed with a third review author. If it was not possible to determine eligibility for a study, it was added to the list of those awaiting assessment and the authors were contacted for further information or clarification.
The study selection process was documented using a PRISMA study selection flow diagram.
Data extraction and management
Data regarding participants, interventions and their comparators, methodological details, and treatment effects, including dropouts and possible biases, were independently extracted by two review authors (PD and SI) using a standardised data extraction form. If any disagreements arose, these were discussed with a third review author. The data extraction form was piloted during the first phase of data extraction.
Information relating to study population, definition of TRD, sample size, interventions, comparators, potential biases in the conduct of the trial, outcomes, follow‐up and methods of statistical analysis was abstracted.
Main planned comparisons
Increasing the dose of antidepressant monotherapy compared with continuing on an antidepressant.
Switching to a different antidepressant monotherapy compared with continuing on an antidepressant.
Augmenting treatment with another antidepressant compared with continuing on an antidepressant.
Augmenting treatment with a non‐antidepressant compared with continuing on an antidepressant.
Within each of these strategies, where there was sufficient data, this review summarised the evidence for each drug individually. For example, taking the fourth approach of augmenting with a non‐antidepressant medication, the augmentor could have been lithium, olanzapine, buspirone or pindolol. In this case, the evidence for each of these four drugs would be presented separately, rather than combined, to summarise the evidence for a particular 'treatment approach'. This was done to maximise the clinical relevance of the findings.
Given the large number of possible combinations of medications that could be evaluated, it was not possible to provide an exhaustive list of all the potential comparisons. However, one example for each of the different approaches have been given below.
Citalopram 40 mg/day compared with remaining on citalopram 20 mg/day.
Switching to mianserin compared with remaining on fluoxetine.
Addition of mianserin to fluoxetine compared with remaining on fluoxetine alone.
Nortriptyline plus lithium compared with remaining on nortriptyline alone.
Assessment of risk of bias in included studies
Two review authors independently assessed risk of bias for each included study using Cochrane's 'Risk of bias' tool (Higgins 2011a). If any disagreements arose, these were discussed with a third review author. The following criteria were assessed:
sequence generation: was the allocation sequence adequately generated?
allocation concealment: was allocation adequately concealed?
blinding of participants, study personnel and outcome assessors for each outcome: was knowledge of the allocated treatment adequately prevented during the study?
incomplete outcome data for each main outcome or class of outcomes: were incomplete outcome data adequately addressed?
selective outcome reporting: are reports of the study free of suggestion of selective outcome reporting?
other sources of bias: was the study apparently free of other problems that could put it at a high risk of bias?
A description of what was reported to have happened in each study was generated and a judgement on the risk of bias was made for each domain within and across studies, based on the following three categories: low risk of bias; unclear risk of bias; high risk of bias.
Where studies provided little or no detail about randomisation, the authors were contacted to seek clarification.
All risk of bias data were presented graphically and described in the text.
Measures of treatment effect
Continuous outcomes were analysed by calculating the mean difference (MD) between groups, where studies used the same outcome measure for comparison. Where different outcome measures were used to assess the same outcome, the standardised mean difference (SMD) and 95% confidence intervals (CI) were calculated. The SMD was interpreted as follows: 0.2 represents a small effect, 0.5 a moderate effect, and 0.8 a large effect (Cohen 1988).
We calculated risk ratios (RRs) for dichotomous outcomes. When overall risks were significant, the number needed to treat for an additional beneficial outcome (NNTB) or for an additional harmful outcome (NNTH) to produce one outcome was calculated by combining the overall RR with an estimate of prevalence of the event in the control group of the trials.
Unit of analysis issues
Cluster‐randomised trials
We planned to incorporate results from cluster RCTs into the review using generic inverse variance methods (Higgins 2011). With cluster RCTs, it is important to ensure that the data were analysed taking into account the clustered nature of the data. The intracluster correlation coefficient (ICC) would be extracted for each trial. Where no such data were reported, this information was requested from study authors. If this was not available, in line with the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), we planned to use estimates from similar studies in order to 'correct' data for clustering, where this had not been done. We did not, however, identify any cluster RCTs that met the inclusion criteria for our review.
Cross‐over trials
For cross‐over trials, only results from the first randomised treatment period were to be included in the analysis. We did not, however, identify any cross‐over trials that met the inclusion criteria for our review.
Studies with multiple treatment groups
Trials that have more than two arms (e.g. pharmacological intervention (A); pharmacological intervention (B); and control) can cause problems in pairwise meta‐analysis. Where we identified studies with two or more active treatment arms, the following approach was undertaken (dependent on whether the outcome was continuous or dichotomous).
For a continuous outcome: means, standard deviations (SDs) and the number of participants for each active treatment group were pooled across treatment arms as a function of the number of participants in each arm to be compared against the control group (Higgins 2011).
For a dichotomous outcome: active treatment groups were combined into a single arm for comparison against the control group (in terms of the number of people with events and sample sizes), or the control group was split equally (Higgins 2011).
Dealing with missing data
Where there were missing data, study authors were contacted to obtain data. If an outcome was missing for more than 50% of participants, this study was excluded from the analysis. When available, we used intention‐to‐treat (ITT) analyses from study reports and wrote to study authors to request relevant unreported analyses.
Assessment of heterogeneity
Heterogeneity was assessed using the Chi2 test, which provides evidence of variation in effect estimates beyond that of chance. The Chi2 test has low power to assess heterogeneity when there are few included studies or small numbers of participants, so the P value was conservatively set at 0.1. Heterogeneity was also quantified using the I2 statistic, which calculates the percentage of variability due to heterogeneity rather than chance. We expected, a priori, that there would be considerable clinical heterogeneity between studies, therefore I2 values between 50% and 90% were considered to represent substantial statistical heterogeneity and were explored further. However, the importance of the observed I2 value depended on the magnitude and direction of treatment effects and the strength of evidence for heterogeneity (Higgins 2011).
Assessment of reporting biases
Reporting bias was managed by undertaking comprehensive searches for papers that were not restricted to the English language. Outcome reporting bias was determined for all included studies and trial protocols were sought wherever possible. If outcome data were missing, these were requested from authors. We had planned to use funnel plots to help detect reporting biases if at least 10 studies were included for a comparison (Higgins 2011) and undertake formal assessment of asymmetry in the funnel plot using the Egger test (Egger 1997).
Data synthesis
Given the potential for heterogeneity in the included interventions, we used a random‐effects model for all analyses. This approach incorporated the assumption that the different studies were estimating different, yet related, intervention effects and it took into account differences between studies even if there was no statistically significant heterogeneity. Heterogeneity was tested formally using both the Chi2 test and I2 statistic (as outlined above). We sought clinical advice in terms of combining treatment groups in order to ensure that findings were clinically meaningful. Where studies reported both short‐term and long‐term outcomes, separate meta‐analyses were conducted for each of these time points.
Where meta‐analysis was not possible (e.g. due to insufficient data or substantial heterogeneity), a narrative assessment of the evidence was given. This assessment summarised the evidence according to intervention type.
Subgroup analysis and investigation of heterogeneity
A priori, we considered the degree of treatment resistance recorded at the point of entry to the trial as a potential effect modifier. Therefore, the following subgroup analyses were planned:
severity of depression: classifying participants as non‐responders or partial responders at baseline;
Length of acute treatment phase (before trial entry): four weeks or longer; 12 weeks or longer and six months or longer.
Such subgroup analyses were only to be conducted when there were data from at least 10 studies to be included (Higgins 2011).
Sensitivity analysis
Sensitivity analyses were planned to explore how much of the variation between studies comparing pharmacological interventions for TRD was accounted for by between‐study differences in:
study quality: allocation concealment was used as a marker of trial quality. Studies that had not used allocation concealment were to be excluded;
blinding: studies that were unblinded (participants, study personnel or outcome assessors) were excluded;
attrition: studies with more than 20% dropout were excluded;
missing data: studies that had imputed missing data were excluded;
funding source: studies funded by pharmaceutical companies were excluded;
publication type: studies not published in full (conference abstract/proceedings, doctoral dissertation) were excluded.
Sensitivity analysis was planned by excluding the studies above and comparing each of the results with the full analysis which included all trials.
Summary of findings tables
'Summary of findings' tables were prepared for all of the comparisons in this review for the following outcomes: depressive symptoms, dropouts, response to treatment, remission from depression and quality of life. Where studies reported outcome data based on more than one method of measurement (e.g. multiple depression severity scales) we reported the study's own choice of primary outcome in the 'Summary of findings' table. Similarly, where studies reported data for more than one time point, we chose the time point corresponding to the primary outcome to include in the summary of findings.
Results
Description of studies
Results of the search
We identified 7969 references through our searches of bibliographic databases and trial registries and 19 references from additional sources. After removing duplicates, we screened 7342 titles and abstracts, of which we excluded 6728 as not relevant. Full‐text reports of 614 references were obtained and screened against our eligibility criteria. We excluded 573 articles and provided reasons for exclusion in Figure 1. We used a hierarchy of reasons for exclusion, shown in Table 9, which also reports the number and frequency of articles excluded for each reason. The most common reasons for exclusions were: that the intervention evaluated was not a standard pharmacological treatment (n = 131, 22.9%), study participants did not meet the definition of treatment‐resistant depression (n = 112, 19.5%), the comparison made in the study was not one of interest to the review (for example, there was no control group that continued on the original antidepressant therapy) (n = 100, 17.5%) or the study was not randomised (n = 106, 18.5%).
1.
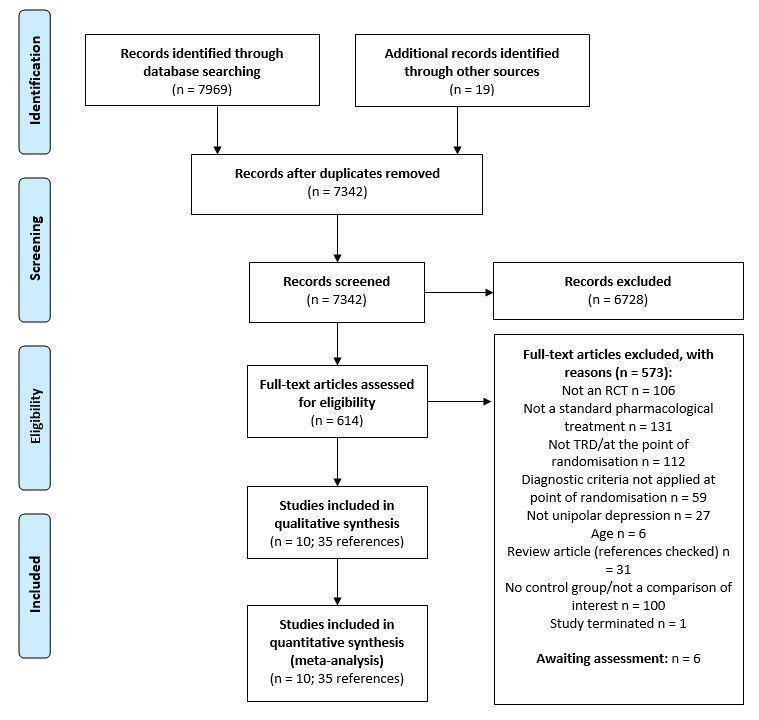
Flow diagram
1. Hierarchy of reasons for exclusion.
| Reason for Exclusion | No of articles excluded (%) |
| Not an RCT | 106 (18.5) |
| Not a standard pharmacological treatment | 131 (22.9) |
| No control group/not a comparison of interest | 100 (17.5) |
| Not TRD/at point of randomisation | 112 (19.5) |
| Not unipolar depression | 27 (4.7) |
| Diagnostic criteria not applied at point of randomisation | 59 (10.3) |
| Age | 6 (1.0) |
| Trial terminated | 1 (0.2) |
| Review article ‐ refs checked | 31 (5.4) |
| Total | 573 (100) |
RCT: randomised controlled trial TRD: Treatment‐resistant depression
Eligibility could not be determined for six references and no response was received from requests to study authors for further clarification (see Characteristics of studies awaiting classification). In total, 35 references reporting ten studies were included in this review. All studies contributed data to meta‐analyses. We contacted the authors of 57 articles to clarify details of their study and received a response from 11. Full details of the study flow are given in a PRISMA flow diagram in Figure 1.
Included studies
Included studies
We identified ten studies (35 references) that were eligible for inclusion in the review (Appelberg 2001; Bauer 2009 (ONYX); Dunner 2007; Durgam 2016; El‐Khalili 2010 (PEARL); Ferreri 2001; Kessler 2018; McIntyre 2007; Papakostas 2015; Shelton 2001). Further information on each study can be found in the Characteristics of included studies table.
Design
All of the included studies were parallel‐group randomised controlled trials and the unit of allocation was the individual participant. Eight studies were multisite (number of sites ranged from two to 84). One study had a single site (McIntyre 2007). The number of study sites was not reported in Dunner 2007.
Five studies were three‐arm trials (Bauer 2009 (ONYX); Dunner 2007; Durgam 2016; El‐Khalili 2010 (PEARL); Ferreri 2001).
Length of follow‐up was short‐term (12 weeks or less) in nine studies. Only one study (Kessler 2018) reported data for longer‐term outcomes (24 and 52 weeks).
Sample sizes
Sample size ranged from 20 (Shelton 2001) to 819 (Durgam 2016) with a median of 122 participants (IQR: 71, 472).
Setting and date
All studies were carried out in high‐income countries. Four studies were conducted in the US (Dunner 2007; El‐Khalili 2010 (PEARL); Papakostas 2015; Shelton 2001); one each in Canada (McIntyre 2007), England (Kessler 2018) Finland (Appelberg 2001) and France (Ferreri 2001); and two in multiple countries across Europe, North America and Australia (Bauer 2009 (ONYX); Durgam 2016).
Nine studies were conducted solely in outpatient medical settings and one (Ferreri 2001) included both in‐ and outpatients. One study (Kessler 2018) was conducted in primary care. Six studies were conducted in the decade 2000 to 2009 (Appelberg 2001; Bauer 2009 (ONYX); Dunner 2007; Shelton 2001; Ferreri 2001; McIntyre 2007) and four since 2010 (Durgam 2016; El‐Khalili 2010 (PEARL); Papakostas 2015; Kessler 2018).
Participants
The mean age of participants was very similar across the included studies, ranging from 42 years (Shelton 2001) to 50.2 years (Kessler 2018). The majority of participants in the included studies were women (51.7% in Dunner 2007 to 75% in Shelton 2001).
Definition of treatment‐resistant depression
Participants had a diagnosis of major depressive disorder (MDD) according to DSM III‐R (Ferreri 2001), DSM‐IV (Appelberg 2001; Bauer 2009 (ONYX); Dunner 2007; El‐Khalili 2010 (PEARL); McIntyre 2007; Papakostas 2015; Shelton 2001) DSM‐IV‐TR (Durgam 2016), or ICD‐10 (Kessler 2018) criteria. Participants were additionally required to have a HAM‐D score of at least 18 (McIntyre 2007), 20 (Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL); Shelton 2001) or 25 (Ferreri 2001); a MADRS score of 22 or more (Durgam 2016); a BDI‐II score of 14 or more (Kessler 2018); or a score of greater than four on the clinical global impressions (CGI) scale (Dunner 2007). MDD was recurrent for all participants in Shelton 2001.
In nine studies, participants had TRD at the time of enrolment (Appelberg 2001; Bauer 2009 (ONYX); Dunner 2007; Durgam 2016; El‐Khalili 2010 (PEARL); Ferreri 2001; McIntyre 2007; Shelton 2001; Kessler 2018). In one study (Papakostas 2015), participants were recruited with Major Depressive Disorder. Following an 8‐week open‐label treatment with fluoxetine (10 mg/d or greater), participants who continued to meet DSM criteria were considered to be treatment‐resistant and randomised to adjunctive treatment with an antipsychotic.
In eight of the nine studies where participants had TRD at the time of enrolment, participants had had an inadequate response to at least one antidepressant at the manufacturers recommended minimum dose (or above) for a minimum of four (Bauer 2009 (ONYX); Dunner 2007) or six (Appelberg 2001; Durgam 2016; El‐Khalili 2010 (PEARL); Ferreri 2001; Kessler 2018; McIntyre 2007) weeks. Two of these studies (Bauer 2009 (ONYX) and El‐Khalili 2010 (PEARL)) had a further requirement of at least one dose increase as permitted by the label. In the ninth trial (Shelton 2001), participants had failed to respond to antidepressants of two different classes (each given for a minimum of four weeks).
Two of the studies in which participants had TRD at enrolment had an open‐label lead‐in phase in which participants had a trial of a further antidepressant at or above the recommended minimum dose for six weeks (Dunner 2007; Shelton 2001). Only participants with an inadequate response to this were then randomised into the main trial.
Two studies had exclusion criteria based on resistance to prior antidepressant treatments. Durgam 2016 and Papakostas 2015 both excluded individuals with an inadequate response to three or more antidepressants at sufficient doses during the current depressive episode.
Further details of previous antidepressant treatments can be found in the Characteristics of included studies table.
Interventions
There were no studies of increasing the dose of antidepressant monotherapy that met the inclusion criteria for the review.
One study looked at the effectiveness of switching from current antidepressant therapy (fluoxetine) to another antidepressant (mianserin, Ferreri 2001) compared with continuing on the original antidepressant therapy.
Two studies examined the effects of augmenting current antidepressant treatment with another antidepressant compared with continuing on antidepressant monotherapy. The adjunctive treatment was mianserin in one study (Ferreri 2001, a three‐arm trial) and mirtazapine in the other (Kessler 2018).
Eight studies examined the effectiveness of augmenting treatment with a non‐antidepressant medication compared with continuing on an antidepressant. The adjunctive treatment was an anxiolytic (buspirone) in one study (Appelberg 2001) and an antipsychotic in seven studies (Bauer 2009 (ONYX); Dunner 2007; Durgam 2016; El‐Khalili 2010 (PEARL); McIntyre 2007; Papakostas 2015; Shelton 2001). Three studies examined the effectiveness of antipsychotic quetiapine (Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL); McIntyre 2007), two studied ziprasidone (Papakostas 2015; Dunner 2007), and there was a single study for cariprazine (Durgam 2016) and olanzapine (Shelton 2001). Four studies were three‐arm trials which investigated higher and lower doses of the augmented antipsychotic (quetiapine ‐ Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL); cariprazine ‐ Durgam 2016; ziprasidone ‐ Dunner 2007). Length of treatment was six weeks in four studies (Appelberg 2001; Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL);Ferreri 2001), eight weeks in four studies (Dunner 2007; Durgam 2016; McIntyre 2007; Papakostas 2015; Shelton 2001) and up to 52 weeks in one study (Kessler 2018).
Comparisons
Participants in the comparator arm continued to receive the same dose of antidepressant monotherapy in all of the trials. In all but one trial (Dunner 2007), a placebo was given in addition to the continued antidepressant treatment.
Primary outcomes
1). Change in depressive symptoms
All studies reported one or more measures of depressive symptoms measured on a severity rating scale. Six studies used the Montgomery‐Asberg Depression Scale (Appelberg 2001; Bauer 2009 (ONYX); Dunner 2007; Durgam 2016; El‐Khalili 2010 (PEARL); Shelton 2001), six the Hamilton Depression Rating Scale (Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL); Ferreri 2001; McIntyre 2007; Papakostas 2015; Shelton 2001), one the Beck Depression Inventory‐II (BDI‐II) and Patient Health Questionnaire‐9 (PHQ‐9) (Kessler 2018), and one the Quick Inventory of Depressive Symptomatology ‐ self‐report version (QIDS‐SR, Papakostas 2015). Five studies reported mean change from baseline in each study arm (Ferreri 2001; Kessler 2018; McIntyre 2007; Papakostas 2015; Shelton 2001), one reported mean percentage change from baseline (Appelberg 2001) and four studies reported a least squares mean change estimated from an analysis of covariance (ANCOVA) incorporating baseline depression severity as a covariate in the model (Bauer 2009 (ONYX); Dunner 2007; Durgam 2016; El‐Khalili 2010 (PEARL).
2). Number of dropouts
All studies reported the number of participants in each arm who dropped out during the treatment period for any reason.
Secondary outcomes
3). Response or remission rates
Nine studies reported a dichotomous outcome of response to treatment, defined as a reduction of 50% or more from baseline to end of treatment in score on either the MADRS (Bauer 2009 (ONYX); Dunner 2007; Durgam 2016; El‐Khalili 2010 (PEARL); Shelton 2001), HAM‐D (Papakostas 2015), BDI‐II (Kessler 2018) or QIDS‐SR (Papakostas 2015).
Eight studies reported remission from depression using a cut‐off score of less than or equal to eight (Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL)); or ten (Dunner 2007; Durgam 2016) on the MADRS, seven (McIntyre 2007; Papakostas 2015) or eight (Ferreri 2001) on the HAM‐D, nine on the BDI‐II (Kessler 2018) or five on the QIDS‐SR (Papakostas 2015).
4). Social Adjustment and Social functioning scales
No studies reported data on either social adjustment or social functioning.
5). Improvement in Quality of Life Measure
Three studies reported data on quality of life. Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL) reported mean change from baseline in quality of life using the Quality of Life Enjoyment and Satisfaction Questionnaire short‐form (Q‐LES‐Q‐SF). Kessler 2018 reported mean scores on the five‐level EQ‐5D (EQ‐5D‐5L) scale, and the aggregate mental functioning and aggregate physical functioning scales of the SF‐12 at follow‐up.
6). Economic outcomes
One study (Kessler 2018) reported direct costs to health and social services, costs to patients and carers, and time off work for patients and carers.
7). Adverse Events
Eight studies reported arm‐level adverse event data (Bauer 2009 (ONYX); Dunner 2007; Durgam 2016; El‐Khalili 2010 (PEARL); Ferreri 2001; Kessler 2018; McIntyre 2007; Papakostas 2015). Information on adverse events was incompletely reported in two studies (Appelberg 2001; Shelton 2001).
Excluded studies
In total, we excluded 573 references at full‐text screening. In accordance with the guidance in the Cochrane Handbook, we have provided a list of those studies that almost met our eligibility criteria and further details of their reason for exclusion (see table of Excluded studies). Of these 105 studies, the primary reasons for exclusion were as follows: 45 did not have a comparator arm where participants continued with current antidepressant medication only (with or without placebo); 33 did not meet our criteria for treatment‐resistant depression at the point of randomisation and in 26 studies no formal diagnostic criteria for diagnosis of depression were applied at the point of randomisation.
The STAR*D trial (STAR*D 2004) was one of our excluded studies. This study evaluated a sequence of different treatment options for depression. Participants who failed to respond to treatment were randomised into the next level of the study. Participants enrolled in level one had MDD, not TRD (therefore, this level was not eligible for our review). Each successive level involved a switching to a different treatment strategy or augmenting current treatment with additional medication. As none of these later levels included a group that continued on current antidepressant medication alone they were also not eligible for inclusion.
Ongoing studies
We did not identify any ongoing studies.
Studies awaiting classification
There was insufficient information in the study reports for six studies (Cao 2005; Clunie 2001; Euctr‐002130‐11‐Es 2007; Gulrez 2012; Moica 2018; Zhu 2003) to determine eligibility for inclusion in our review. Two of these reports were trial protocols (Clunie 2001; Euctr‐002130‐11‐Es 2007) and no publications relating to the studies could be identified.
We were unable to contact the authors for further information for two of these (Euctr‐002130‐11‐Es 2007; Zhu 2003) and did not receive a response for the remainder (Cao 2005; Clunie 2001; Gulrez 2012; Moica 2018). Further information on these studies can be found in the Characteristics of studies awaiting classification table.
Risk of bias in included studies
Full details of the risk of bias for the included studies are given in the Characteristics of included studies table. Graphical representations of the overall risk of bias in included studies are presented for each risk of bias item (Figure 2) and for each study (Figure 3). Given the small number of studies included in the various comparisons, no formal assessment of reporting bias using a funnel plot was undertaken.
2.
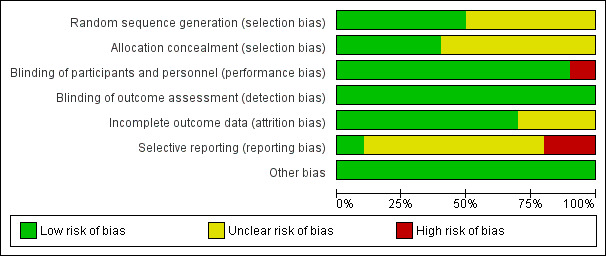
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.
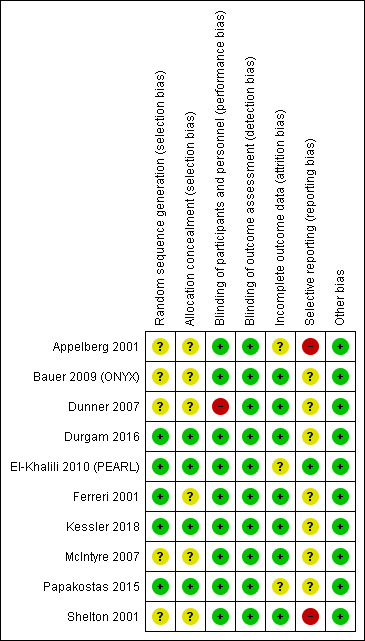
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Random Sequence Generation (Selection Bias)
All included studies described themselves as randomised. Five studies did not provide any information on how the randomisation sequence was generated (Appelberg 2001; Bauer 2009 (ONYX); Dunner 2007; McIntyre 2007; Shelton 2001) and were rated as being at an unclear risk of bias. Four studies described the use of a computer or web‐based software (Durgam 2016; El‐Khalili 2010 (PEARL); Kessler 2018; Papakostas 2015) or a randomisation schedule (Ferreri 2001) and were judged to be at low risk.
Allocation Concealment (Selection Bias)
Allocation concealment was not described and was therefore rated as unclear in six studies (Appelberg 2001; Bauer 2009 (ONYX); Dunner 2007; Ferreri 2001; McIntyre 2007; Shelton 2001). Three studies describing a centralised allocation system (Durgam 2016; Kessler 2018; Papakostas 2015) were assessed as low risk. The final study was judged to be at low risk of bias for allocation concealment due to the use of a computer‐generated randomisation sequence in combination with study treatments reported to be administered in identical packaging (El‐Khalili 2010 (PEARL).
Blinding
Performance Bias (blinding of participants and those delivering the intervention)
Nine studies reported the use of a placebo intervention in the comparator arm identical in form and appearance to the active intervention and were therefore judged to be at a low risk of bias (Appelberg 2001; Bauer 2009 (ONYX); Durgam 2016; El‐Khalili 2010 (PEARL); Ferreri 2001; Kessler 2018; McIntyre 2007; Papakostas 2015; Shelton 2001).One study was described as an open‐label trial and therefore assigned a high risk of bias rating for this domain (Dunner 2007).
Detection Bias (blinding of outcome assessors).
All of the included studies were judged to be at a low risk of bias for this domain. Eight studies were described as double‐blind and reported the use of a placebo identical in appearance to the active intervention (Appelberg 2001; Bauer 2009 (ONYX); Durgam 2016; El‐Khalili 2010 (PEARL); Papakostas 2015; Shelton 2001) and we regarded this as sufficient information to conclude that outcome assessors were blinded to treatment allocation. Two studies (Dunner 2007; Kessler 2018) explicitly stated that outcome assessors were blind to treatment allocation. None of the included studies evaluated the success of blinding.
Incomplete outcome data
Overall, attrition was less than 20% in four studies (Appelberg 2001, 14.8%; Bauer 2009 (ONYX), 14%; Durgam 2016, 17.3%; Kessler 2018, 4%), between 20 and 30% in four studies (El‐Khalili 2010 (PEARL), 22.9%; Ferreri 2001, 24.3%; Papakostas 2015, 26.6%; Shelton 2001, 20%) and over 40% in two studies (Dunner 2007, 40.6%; McIntyre 2007, 41.4%).
Eight studies (Appelberg 2001; Bauer 2009 (ONYX); Dunner 2007; Durgam 2016; El‐Khalili 2010 (PEARL); Ferreri 2001; McIntyre 2007; Shelton 2001) described modified intention‐to‐treat analyses including all participants with at least one post‐baseline efficacy assessment. Last observation carried forward (LOCF) was used to account for missing data and no significant imbalances in missing data between trial arms were noted. Each of these studies, therefore, was judged to be at low risk of bias for this domain. One study (Papakostas 2015) reported an intention‐to‐treat analysis based on all originally randomised patients but provided no information about how missing data were handled (37 of 139 randomised participants dropped out of the study), so was judged to be at an unclear risk of bias. The primary analyses of Kessler 2018 compared the treatment groups as randomised without imputing missing values. Sensitivity analyses were conducted to investigate the influence of missing data.
Selective reporting
One study (El‐Khalili 2010 (PEARL)) was judged to be at a low risk of selective outcome reporting bias as all outcomes of interest to the review that were described in the study protocol were reported. Risk of bias for this domain was rated as unclear for seven studies (Bauer 2009 (ONYX); Dunner 2007; Durgam 2016; Ferreri 2001; Kessler 2018; McIntyre 2007; Papakostas 2015). No protocol was available for two studies (Dunner 2007; Ferreri 2001) and the study protocol did not provide any details of the outcomes to be assessed in another study (McIntyre 2007). Three studies reported additional outcome data not detailed in the study protocol (Bauer 2009 (ONYX), Durgam 2016, Papakostas 2015). Kessler 2018 administered the PHQ‐9 questionnaire at 12, 24 and 52 weeks but only reported data for the 12‐week time point. Two studies were judged to be at a high risk of bias for selective outcome reporting. Shelton 2001 listed depression severity as measured by the HAM‐D as the primary outcome measure. However, dichotomous outcomes of response and remission were determined using MADRS scores. Appelberg 2001 did not report results in full for the primary outcome of depression severity for all time points measured.
Other potential sources of bias
No other potential sources of bias were identified.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8
Summary of findings for the main comparison. Switching to mianserin (60 mg/d) compared to continuing on current antidepressant (fluoxetine 20 mg/d) for treatment‐resistant depression in adults.
| Switching to mianserin (60 mg/d) compared to continuing on current antidepressant (fluoxetine 20 mg/d) for treatment‐resistant depression in adults | |||||
| Patient or population: treatment‐resistant depression in adults Setting: inpatient and outpatient Intervention: switching to mianserin (60 mg/d) Comparison: continuing on current antidepressant (fluoxetine 20 mg/d) | |||||
| Outcomes | № of participants (studies) Follow‐up | Certainty of the evidence (GRADE) | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | |
| Risk with continuing on current antidepressant (fluoxetine 20 mg/d) | Risk difference with switching to mianserin (60 mg/d) | ||||
| Depressive symptoms (HAM‐D) Scale from: 0 to 54 (worse) follow‐up: 6 weeks | 71 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | MD ‐ 1.8 (‐5.23 to 1.63) | The mean depressive symptom score (HAM‐D) was 15.6 | MD 1.8 lower (5.23 lower to 1.63 higher) |
| Dropouts follow‐up: 6 weeks | 72 (1 RCT) | ⊕⊕⊝⊝ LOW 2 | RR 2.08 (0.94 to 4.59) | Study population | |
| 184 per 1,000 | 199 more per 1,000 (11 fewer to 661 more) | ||||
| Response (≥ 50% improvement in HAM‐D score) follow‐up: 6 weeks | 71 (1 RCT) | ⊕⊕⊝⊝ LOW 2 | RR 1.32 (0.76 to 2.27) | Study population | |
| 368 per 1,000 | 118 more per 1,000 (88 fewer to 468 more) | ||||
| Remission (HAM‐D total score ≤ 8) follow‐up: 6 weeks | 71 (1 RCT) | ⊕⊕⊝⊝ LOW 3 | RR 1.97 (0.88 to 4.42) | Study population | |
| 184 per 1,000 | 179 more per 1,000 (22 fewer to 630 more) | ||||
| Quality of life ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | |||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||
1 Downgraded two levels for imprecision (broad confidence interval that crossed the null)
2 Downgraded two levels for imprecision (very broad confidence interval that crossed the null)
3 Downgraded two levels for imprecision (very broad confidence interval)
Summary of findings 2. Augmentation of current antidepressant (fluoxetine 20 mg/d) with mianserin (60 mg/d) compared to augmentation of current antidepressant (fluoxetine 20 mg/d) with placebo for treatment‐resistant depression in adults.
| Augmentation of current antidepressant (fluoxetine 20 mg/d) with mianserin (60 mg/d) compared to augmentation of current antidepressant (fluoxetine 20 mg/d) with placebo for treatment‐resistant depression in adults | |||||
| Patient or population: treatment‐resistant depression in adults Setting: inpatient and outpatient Intervention: augmentation of current antidepressant (fluoxetine 20 mg/d) with mianserin (60 mg/d) Comparison: augmentation of current antidepressant (fluoxetine 20 mg/d) with placebo | |||||
| Outcomes | № of participants (studies) Follow‐up | Certainty of the evidence (GRADE) | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | |
| Risk with augmentation of current antidepressant (fluoxetine 20 mg/d) with placebo | Risk difference with augmentation of current antidepressant (fluoxetine 20 mg/d) with mianserin (60 mg/d) | ||||
| Depressive symptoms (HAM‐D) Scale from: 0 to 54 (worse) follow‐up: 6 weeks | 70 (1 RCT) | ⊕⊕⊕⊝ MODERATE 1 | ‐ | The mean depressive symptom score (HAM‐D) was 15.6 | MD 4.8 lower (8.18 lower to 1.42 lower) |
| Dropouts follow‐up: 6 weeks | 70 (1 RCT) | ⊕⊕⊝⊝ LOW 2 | RR 1.02 (0.38 to 2.72) | Study population | |
| 184 per 1,000 | 4 more per 1,000 (114 fewer to 317 more) | ||||
| Response (≥ 50% reduction in HAM‐D score) follow‐up: 6 weeks | 70 (1 RCT) | ⊕⊕⊝⊝ LOW 3 | RR 1.70 (1.03 to 2.78) | Study population | |
| 368 per 1,000 | 258 more per 1,000 (11 more to 656 more) | ||||
| Remission (HAM‐D total score ≤ 7) follow‐up: 6 weeks | 70 (1 RCT) | ⊕⊕⊝⊝ LOW 3 | RR 2.38 (1.09 to 5.16) | Study population | |
| 184 per 1,000 | 254 more per 1,000 (17 more to 766 more) | ||||
| Quality of life ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | |||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||
1 Downgraded one level for imprecision (broad confidence interval)
2 Downgraded two levels for imprecision (broad confidence interval that crossed the null)
3 Downgraded two levels for imprecision (very broad confidence interval)
Summary of findings 3. Augmentation of current antidepressant (SSRI/SNRI) with mirtazapine (30 mg/d) compared to augmentation of current antidepressant (SSRI/SNRI) therapy with placebo for treatment‐resistant depression in adults.
| Augmentation of current antidepressant (SSRI/SNRI) with mirtazapine (30 mg/d) compared to augmentation of current antidepressant (SSRI/SNRI) therapy with placebo for treatment‐resistant depression in adults | |||||
| Patient or population: treatment‐resistant depression in adults Setting: outpatients Intervention: augmentation of current antidepressant (SSRI/SNRI) with mirtazapine (30 mg/d) Comparison: augmentation of current antidepressant (SSRI/SNRI) therapy with placebo | |||||
| Outcomes | № of participants (studies) Follow‐up | Certainty of the evidence (GRADE) | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | |
| Risk with augmentation of current antidepressant (SSRI/SNRI) therapy with placebo | Risk difference with augmentation of current antidepressant (SSRI/SNRI) with mirtazapine (30 mg/d) | ||||
| Depressive symptoms (BDI‐II) Scale from: 0 to 64 (worse) follow‐up: 12 weeks | 431 (1 RCT) | ⊕⊕⊕⊕ HIGH | ‐ | The mean depressive symptom score (BDI‐II) was 19.7 | MD 1.7 lower (4.03 lower to 0.63 higher) |
| Dropouts follow‐up: 12 weeks | 480 (1 RCT) | ⊕⊕⊕⊕ HIGH | RR 0.50 (0.15 to 1.62) | Study population | |
| 33 per 1,000 | 17 fewer per 1,000 (28 fewer to 21 more) | ||||
| Response (≥ 50% improvement in BDI‐II score) follow‐up: 12 weeks | 431 (1 RCT) | ⊕⊕⊕⊕ HIGH | RR 1.22 (0.97 to 1.54) | Study population | |
| 359 per 1,000 | 79 more per 1,000 (11 fewer to 194 more) | ||||
| Remission (BDI‐II total score ≤ 9) follow‐up: 12 weeks | 431 (1 RCT) | ⊕⊕⊕⊕ HIGH | RR 1.21 (0.88 to 1.65) | Study population | |
| 244 per 1,000 | 51 more per 1,000 (29 fewer to 159 more) | ||||
| Quality of life (EQ‐5D‐5L) ‐ 12 weeks | 447 (1 RCT) | ⊕⊕⊕⊕ HIGH | ‐ | The mean quality of life (EQ‐5D‐5L) at 12 weeks was 0.73 | MD 0.01 lower (0.06 lower to 0.04 higher) |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | |||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||
Summary of findings 4. Augmentation of current antidepressant (SSRI) with busiprone (20 mg/d to 60 mg/d) compared to augmentation of current antidepressant (SSRI) with placebo for treatment‐resistant depression in adults.
| Augmentation of current antidepressant (SSRI) with busiprone (20 mg/d to 60 mg/d) compared to augmentation of current antidepressant (SSRI) with placebo for treatment‐resistant depression in adults | |||||
| Patient or population: treatment‐resistant depression in adults Setting: outpatient Intervention: augmentation of current antidepressant (SSRI) with busiprone (20 mg/d to 60 mg/d) Comparison: augmentation of current antidepressant (SSRI) with placebo | |||||
| Outcomes | № of participants (studies) Follow‐up | Certainty of the evidence (GRADE) | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | |
| Risk with augmentation of current antidepressant (SSRI) with placebo | Risk difference with augmentation of current antidepressant (SSRI) with busiprone (20 mg/d to 60 mg/d) | ||||
| Depressive symptoms (MADRS) Scale from: 0 to 60 (worse) follow‐up: 6 weeks | 102 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | ‐ | The mean depressive symptom score (MADRS) was 30.5 % reduction from baseline | MD 0.3 % reduction from baseline lower (9.48 lower to 8.88 higher) |
| Dropouts follow‐up: 6 weeks | 108 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | RR 0.60 (0.23 to 1.53) | Study population | |
| 185 per 1,000 | 74 fewer per 1,000 (143 fewer to 98 more) | ||||
| Response to treatment ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ |
| Remission ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ |
| Quality of life ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | |||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||
1 Downgraded two levels for imprecision (very broad confidence interval that crossed the null)
Summary of findings 5. Augmentation of current antidepressant (various) with cariprazine (1 mg/d to 4.5 mg/d) compared to augmentation of current antidepressant (various) with placebo for treatment‐resistant depression in adults.
| Augmentation of current antidepressant (various) with cariprazine (1 mg/d to 4.5 mg/d) compared to augmentation of current antidepressant (various) with placebo for treatment‐resistant depression in adults | |||||
| Patient or population: treatment‐resistant depression in adults Setting: outpatient Intervention: augmentation of current antidepressant (various) with cariprazine (1 mg/d to 4.5 mg/d) Comparison: augmentation of current antidepressant (various) with placebo | |||||
| Outcomes | № of participants (studies) Follow‐up | Certainty of the evidence (GRADE) | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | |
| Risk with augmentation of current antidepressant (various) with placebo | Risk difference with augmentation of current antidepressant (various) with cariprazine (1 mg/d to 4.5 mg/d) | ||||
| Depressive symptoms (MADRS) ‐ Any dose Scale from: 0 to 60 (worse) follow‐up: 8 weeks | 808 (1 RCT) | ⊕⊕⊕⊕ HIGH | ‐ | The mean depressive symptom score (MADRS) ‐ any dose ‐ was 16.4 points | MD 1.5 points lower (2.74 lower to 0.25 lower) |
| Dropouts ‐ any dose follow‐up: 8 weeks | 821 (1 RCT) | ⊕⊕⊕⊝ MODERATE 1 | RR 1.68 (1.16 to 2.41) | Study population | |
| 119 per 1,000 | 81 more per 1,000 (19 more to 168 more) | ||||
| Response (≥ 50% improvement in MADRS score) ‐ any dose follow‐up: 8 weeks | 808 (1 RCT) | ⊕⊕⊕⊝ MODERATE 1 | RR 1.27 (1.07 to 1.52) | Study population | |
| 383 per 1,000 | 103 more per 1,000 (27 more to 199 more) | ||||
| Remission (MADRS total score ≤ 10) ‐ any dose follow‐up: 8 weeks | 808 (1 RCT) | ⊕⊕⊕⊝ MODERATE 2 | RR 1.07 (0.86 to 1.33) | Study population | |
| 299 per 1,000 | 21 more per 1,000 (42 fewer to 99 more) | ||||
| Quality of life ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | |||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||
1 Downgraded one level for imprecision (broad confidence interval)
2 Downgraded one level for imprecision (broad confidence interval that crossed the null)
Summary of findings 6. Augmentation of current antidepressant (fluoxetine 20 mg/d to 60 mg/d) with olanzapine (5 mg/d to 20 mg/d) compared to augmentation of current antidepressant (fluoxetine 20 mg/d to 60 mg/d) with placebo for treatment‐resistant depression in adults.
| Augmentation of current antidepressant (fluoxetine 20 mg/d to 60 mg/d) with olanzapine (5 mg/d to 20 mg/d) compared to augmentation of current antidepressant (fluoxetine 20 mg/d to 60 mg/d) with placebo for treatment‐resistant depression in adults | |||||
| Patient or population: treatment‐resistant depression in adults Setting: outpatient Intervention: augmentation of current antidepressant (fluoxetine 20 mg/d to 60 mg/d) with olanzapine (5 mg/d to 20 mg/d) Comparison: augmentation of current antidepressant (fluoxetine 20 mg/d to 60 mg/d) with placebo | |||||
| Outcomes | № of participants (studies) Follow‐up | Certainty of the evidence (GRADE) | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | |
| Risk with augmentation of current antidepressant (fluoxetine 20 mg/d to 60 mg/d) with placebo | Risk difference with augmentation of current antidepressant (fluoxetine 20 mg/d to 60 mg/d) with olanzapine (5 mg/d to 20 mg/d) | ||||
| Depressive symptoms (HAM‐D) Scale from: 0 to 52 (worse) follow‐up: 8 weeks | 20 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | ‐ | The mean depressive symptom score (HAM‐D) was ‐3.8 | MD 7.9 lower (16.76 lower to 0.96 higher) |
| Depressive symptoms (MADRS) Scale from: 0 to 60 (worse) follow‐up: 8 weeks | 20 (1 RCT) | ⊕⊕⊝⊝ LOW 2 | ‐ | The mean depressive symptom score (MADRS) was ‐1.2 | MD 12.4 lower (22.44 lower to 2.36 lower) |
| Dropouts follow‐up: 8 weeks | 20 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | RR 0.33 (0.04 to 2.69) | Study population | |
| 300 per 1,000 | 201 fewer per 1,000 (288 fewer to 507 more) | ||||
| Response (≥50% reduction in MADRS score) follow‐up: 8 weeks | 20 (1 RCT) | ⊕⊕⊝⊝ LOW 2 | RR 6.00 (0.87 to 41.21) | Study population | |
| 100 per 1,000 | 500 more per 1,000 (13 fewer to 4,021 more) | ||||
| Remission ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ |
| Quality of life ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | |||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||
1 Downgraded two levels for imprecision (very broad confidence interval that crossed the null)
2 Downgraded two levels for imprecision (very broad confidence interval)
Summary of findings 7. Augmentation of current antidepressant (various) with quetiapine (150 mg/d to 300 mg/d) compared to augmentation of current antidepressant (various) with placebo for treatment‐resistant depression in adults.
| Augmentation of current antidepressant (various) with quetiapine (150 mg/d to 300 mg/d) compared to augmentation of current antidepressant (various) with placebo for treatment‐resistant depression in adults | |||||
| Patient or population: treatment‐resistant depression in adults Setting: outpatient Intervention: augmentation of current antidepressant (various) with quetiapine (150 mg/d to 300 mg/d) Comparison: augmentation of current antidepressant (various) with placebo | |||||
| Outcomes | № of participants (studies) Follow‐up | Certainty of the evidence (GRADE) | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | |
| Risk with augmentation of current antidepressant (various) with placebo | Risk difference with augmentation of current antidepressant (various) with quetiapine (150 mg/d to 300 mg/d) | ||||
| Depressive symptoms (MADRS or HAM‐D) ‐ Any dose | 977 (3 RCTs) | ⊕⊕⊕⊕ HIGH | ‐ | The mean depressive symptom score (MADRS or HAM‐D) ‐ any dose was not calculable | SMD 0.32 lower (0.46 lower to 0.18 lower) |
| Dropouts ‐ Any dose | 997 (3 RCTs) | ⊕⊕⊕⊝ MODERATE 1 | RR 1.33 (0.90 to 1.95) | Study population | |
| 159 per 1,000 | 52 more per 1,000 (16 fewer to 151 more) | ||||
| Response (≥ 50% reduction in MADRS or HAM‐D score) ‐ Any dose | 977 (3 RCTs) | ⊕⊕⊕⊝ MODERATE 2 | RR 1.25 (1.09 to 1.44) | Study population | |
| 442 per 1,000 | 110 more per 1,000 (40 more to 194 more) | ||||
| Remission (MADRS score ≤ 8/HAM‐D score ≤ 7) ‐ Any dose | 977 (3 RCTs) | ⊕⊕⊕⊝ MODERATE 2 | RR 1.53 (1.23 to 1.90) | Study population | |
| 233 per 1,000 | 123 more per 1,000 (54 more to 210 more) | ||||
| Quality of life (% max score of Q‐LES‐Q‐SF) ‐ Any dose Scale from: 0 to 100 (better) follow‐up: 6 weeks | 884 (2 RCTs) | ⊕⊕⊕⊝ MODERATE 1 | ‐ | The mean quality of life (% max score of Q‐LES‐Q‐SF) ‐ any dose was 55% | MD 0.57 higher (1.52 lower to 2.65 higher) |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | |||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||
1 Downgraded one level for imprecision (broad confidence interval which crossed the null)
2 Downgraded one level for imprecision (broad confidence interval)
Summary of findings 8. Augmentation of current antidepressant (SSRI) with ziprasidone (40 mg/d to 160 mg/d) compared to augmentation of current antidepressant (SSRI) with placebo or continue on SSRI monotherapy for treatment‐resistant depression in adults.
| Augmentation of current antidepressant (SSRI) with ziprasidone (40 mg/d to 160 mg/d) compared to augmentation of current antidepressant (SSRI) with placebo or continue on SSRI monotherapy for treatment‐resistant depression in adults | |||||
| Patient or population: treatment‐resistant depression in adults Setting: outpatient Intervention: augmentation of current antidepressant (SSRI) with ziprasidone (40 mg/d to 160 mg/d) Comparison: augmentation of current antidepressant (SSRI) with placebo or continue on SSRI monotherapy | |||||
| Outcomes | № of participants (studies) Follow‐up | Certainty of the evidence (GRADE) | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | |
| Risk with augmentation of current antidepressant (SSRI) with placebo or continue on SSRI monotherapy | Risk difference with Augmentation of current antidepressant (SSRI) with ziprasidone (40 mg/d to 160 mg/d) | ||||
| Depressive symptoms (HAM‐D) ‐ Any dose Scale from: 0 to 54 (worse) follow‐up: 6 to 8 weeks | 199 (2 RCTs) | ⊕⊕⊕⊝ MODERATE 1 | ‐ | The mean depressive symptom score (HAM‐D) ‐ any dose was 14.87 points | MD 2.73 points lower (4.53 lower to 0.93 lower) |
| Dropouts ‐ Any dose follow‐up: 6‐8 | 199 (2 RCTs) | ⊕⊕⊕⊝ MODERATE 1 | RR 1.60 (1.01 to 2.55) | Study population | |
| 227 per 1,000 | 136 more per 1,000 (2 more to 352 more) | ||||
| Response (≥ 50% reduction in MADRS/HAM‐D score) ‐ Any dose follow‐up: 6‐8 weeks | 199 (2 RCTs) | ⊕⊕⊕⊝ MODERATE 1 | RR 1.80 (1.07 to 3.04) | Study population | |
| 182 per 1,000 | 145 more per 1,000 (13 more to 371 more) | ||||
| Remission (clinician‐rated) ‐ Any dose follow‐up: 6‐8 weeks | 199 (2 RCTs) | ⊕⊕⊕⊝ MODERATE 2 | OR 1.46 (0.75 to 2.86) | Study population | |
| 250 per 1,000 | 77 more per 1,000 (50 fewer to 238 more) | ||||
| Quality of life ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | |||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||
1 Downgraded one level for imprecision (broad confidence interval)
2 Downgraded one level for imprecision (broad confidence interval that crossed the null)
Comparison 1 ‐ Increasing the dose of antidepressant monotherapy compared with continuing on an antidepressant
None of the included studies compared increasing the dose of antidepressant monotherapy with continuing on an antidepressant.
Comparison 2 ‐ Switching to a different antidepressant monotherapy compared with continuing on an antidepressant
One study examined the effects of switching to a different antidepressant monotherapy compared with continuing on an antidepressant (Ferreri 2001). Participants who had failed to respond to at least six weeks of treatment with fluoxetine were randomised to continue this treatment or switch to mianserin at a dose of 60 mg per day. The study was a three arm trial ‐ in the third arm participants had their fluoxetine treatment augmented with mianserin. Participants in the two monotherapy arms (fluoxetine only or mianserin only) also received a placebo in addition to their antidepressant.
Primary Outcomes
Change in depressive symptoms
Clinician‐rated measures
One study (Ferreri 2001, n = 71) found no evidence of a difference in mean change from baseline in HAM‐D 17 scores when current antidepressant medication (fluoxetine) was switched to mianserin compared with continuing on current treatment (MD ‐1.8, 95% CI ‐5.23 to 1.63; low‐quality evidence; Analysis 1.1).
1.1. Analysis.
Comparison 1 Switch to mianserin, Outcome 1 Depressive symptoms (HAM‐D).
Self‐report measures
No self‐report measures of depressive symptoms were made in the study.
Dropouts
There was no evidence of a difference in the numbers of participants dropping out when antidepressant medication was switched to mianserin than when current medication was continued (one study, Ferreri 2001, n = 72; RR 2.08, 95% CI 0.94 to 4.59; low‐quality evidence; Analysis 1.2)
1.2. Analysis.
Comparison 1 Switch to mianserin, Outcome 2 Dropouts.
Reasons for dropping out
Participants' reasons for discontinuation are reported in Table 10. The main reasons for dropping out were intolerance to medication or adverse events in the mianserin arm and ineffectiveness of treatment in the group that continued current antidepressant medication.
2. Reasons for dropouts.
| Study | N | time point | intervention | Reasons for drop out | |
| Intervention | Control (continue on AD monotherapy) | ||||
| Appelberg 2001 | 108 | 6 weeks | Busiprone (augmentation of AD) | Protocol violation n = 3 Side effects n = 1 Moved house n = 1 Did not attend final assessment n = 1 |
Protocol violation n = 3 Did not attend final assessment n = 2 Stopped taking study medication n = 2 Spouse threw away study medication n = 1 Suspected angina n = 1 Heavy alcohol misuse n = 1 |
| Dunner 2007 | 64 | 8 weeks | Ziprasidone (augmentation of AD) | 80 mg/d: Treatment‐related adverse event n = 9 Withdrew consent n = 1 Miscellaneous n = 1 160 mg/d: Treatment‐related adverse event n = 7 Withdrew consent n = 1 Miscellaneous n = 2 |
Withdrew consent n = 4 Miscellaneous n = 1 |
| Durgam 2016 | 819 | 8 weeks | Cariprazine (augmentation of AD) | 1 mg/d to 2 mg/d: Adverse event n = 18 Insufficient response n = 4 Protocol violation n = 10 Withdrew consent n = 13 Lost to follow‐up n = 2 2 mg/d to 4.5 mg/d: Adverse event n = 36 Protocol violation n = 9 Withdrew consent n = 14 Lost to follow‐up n = 4 |
Adverse event n = 8 Insufficient response n = 2 Protocol violation n = 6 Withdrew consent n = 11 Lost to follow‐up n = 2 Other n = 2 |
| El‐Khalili 2010 (PEARL) | 446 | 6 weeks | Quetiapine (augmentation of AD) | 150 mg/d: Adverse event n = 16 Lack of therapeutic response n = 2 Severe non‐compliance with study protocol n = 2 Did not complete ≥ 36 days of study treatment n = 1 Lost to follow‐up n = 8 Not willing to continue n = 4 Eligibility criteria not fulfilled n = 1 300 mg/d: Adverse event n = 27 Did not complete ≥ 36 days of study treatment n = 1 Lost to follow‐up n = 7 Not willing to continue n = 6 Eligibility criteria not fulfilled n = 1 Other n = 3 |
Adverse event n = 1 Lack of therapeutic response n = 4 Lost to follow‐up n = 10 Not willing to continue n = 8 |
| Ferreri 2001 | 104 | 6 weeks | Mianserine (switched from AD) Mianersine (augmentation of AD) |
Switched to mianserin alone: Ineffectiveness n = 1 Lost to follow‐up n = 2 Intolerance/ adverse events n = 8 Substantial improvement n = 0 Personal reason n = 1 Augmented with mianserin: Ineffectiveness n = 1 Lost to followup n = 2 Intolerance/ adverse events n = 2 Substantial improvement n = 1 Personal reason n = 1 |
Ineffectiveness n = 6
Lost to follow‐up n = 1 Intolerance/ adverse events n = 0 Substantial improvement n = 0 Personal reason n = 1 |
| Kessler 2018 | 480 | 52 weeks | Mirtazapine (augmentation of AD) | Withdrew from study n = 6 Not willing to take part n = 2 Health n = 1 Other n = 3 Lost to follow‐up n = 44 | Withdrew from study n = 12 Not willing to take part n = 8 Health n = 2 Other n = 2 Lost to follow‐up n = 28 |
| McIntyre 2007 | 58 | 6 weeks | Quetiapine (augmentation of AD) | Adverse events n = 8 Consent withdrawal n = 2 Protocol violation n = 1 |
Lack of efficacy n = 9
Adverse events n = 2 Lost to follow‐up n = 2 |
| Papakostas 2015 | 139 | 8 weeks | Ziprasidone (augmentation of AD) | Intolerance n = 10 Inefficacy n = 3 Lost to follow‐up n = 3 Other n = 6 |
Inefficacy n = 1 Lost to follow‐up n = 7 Other n = 7 |
| Shelton 2001 | 20 | 8 weeks | Olanzapine (augmentation of AD) | Protocol violation n = 1 | Lack of efficacy n = 1 Personal conflict n = 1 Protocol violation n = 1 |
AD: antidepressant
Secondary Outcomes
Response to treatment
Clinician‐rated measures
One study (Ferreri 2001, n = 71) found no evidence of a difference in response to treatment between participants whose antidepressant was switched to mianserin and those who continued on their current medication (RR 1.32, 95% CI 0.76 to 2.27; low‐quality evidence; Analysis 1.3).
1.3. Analysis.
Comparison 1 Switch to mianserin, Outcome 3 Response (≥ 50% improvement in HAM‐D score).
Self‐report measures
No self‐report measures of response to treatment were made in the study.
Remission
Clinician‐rated measures
There was no evidence of a difference in the numbers of participants experiencing remission from depression when antidepressant medication was switched to mianserin than when current medication was continued (one study, Ferreri 2001, n = 71; RR 1.97, 95% CI 0.88 to 4.42; low‐quality evidence; Analysis 1.4).
1.4. Analysis.
Comparison 1 Switch to mianserin, Outcome 4 Remission (HAM‐D total score ≤ 8).
Self‐report measures
No self‐report measures of remission from depression were made in the study.
Social adjustment and social functioning
No data on social adjustment or functioning were reported.
Quality of life
No data on quality of life were reported.
Economic outcomes
No data on any economic outcomes were reported.
Adverse effects
One study (Ferreri 2001, n = 72) reported arm‐level adverse event data, including the number of participants in each arm that experienced at least one adverse event and the numbers of each type of adverse event occurring in each arm. More participants reported adverse events after switching to mianserin than continuing fluoxetine (70.5% versus 28.1%). The most commonly reported adverse events were dizziness and drowsiness in the mianserin arm and headache and asthenia in the fluoxetine arm.
Further details can be found in Table 11. The number of participants who discontinued medication due to adverse events is reported in Table 10 and described under the primary outcome of dropouts.
3. Adverse events.
| Study (intervention) | All AEs | Serious AEs | Serious AE details/notes | ||
| Intervention | Control | Intervention | Control | ||
|
Appelberg 2001 (augment with busiprone) |
n.r. | n.r. | 0/54 (0%) | 0/54 (0%) | "No serious adverse events were observed. No statistically significant differences were observed in UKU scores between treatment groups throughout the study" (p.450, Safety). 1 participant in the busiprone arm was reported to have dropped out of the study due to side effects. |
|
Bauer 2009 (ONYX) (augment with quetiapine) |
150 mg/d: 108/166 (65%) 300 mg/d: 121/161 (75%) |
86/160 (54%) | 150 mg/d: 2/166 (1.2%) 300 mg/d: 3/161 (1.8%) |
3/160 (1.9%) | Serious AEs were investigator‐defined. No further detail provided |
|
Durgam 2016 (augment with cariprazine) |
1 mg/d to 2 mg/d: 189/273 (69.2%) 2 mg/d to 4.5 mg/d: 214/273 (78.4%) |
157/266 (59%) | 1 mg/d to 2 mg/d: 0/273 (0%) 2 mg/d to 4.5 mg/d: 2/273 (0.7%) |
1/266 (0.4%) | Serious AEs were depression (placebo); agitation (cariprazine 2 mg/d to 4.5 mg/d); and panic attack, dyspnoea and noncardiac chest pain (one patient experienced three events, cariprazine 2 mg/d to 4.5 mg/d). |
|
Dunner 2007 (augment with ziprasidone) |
80 mg/d: 21/21 (100%) 160 mg/d: 16/19 (84.2%) |
8/20 (40%) | 80 mg/d: 0/21 160 mg/d: 0/19 |
0/20 | "No serious adverse events were reported" (p.1071). |
|
El‐Khalili 2010 (PEARL) (augment with quetiapine) |
150 mg/d: 122/148 (82.4%) 300 mg/d: 130/149 (87.2%0 |
99/148 (66.9%) | 150 mg/d: 0/148 300 mg/d: 0/149 |
0/148 | "Two serious AEs were reported during the study period. One patient experienced a transient ischaemic attack (placebo) and one patient experienced worsening of cervical spondylitis (quetiapine XR 150 mg/d); neither was considered treatment related by the investigator." (p.925) |
|
Ferreri 2001 (switch to/augment with mianserin) |
switch: 24/34 (70.5%); augment:17/38 (44.7%) | 9/32 (28.1%) | n.r. | n.r. | No information on serious adverse events reported |
|
Kessler 2018 augment with mirtazapine) |
12 weeks: 167/241 (69.3%) | 12 weeks: 91/239 (38.1%) | At 12 weeks: 8/241 (3.32%) | At 12 weeks: 3/239 (1.25%) | Serious SAEs at 12 weeks: Mirtazapine: central nervous system/transient ischaemic attack (TIA) (n = 1), cardiovascular (n = 1), dental (n = 1), gynaecological (n = 1), pancreatitis (n = 1), psychiatric (n = 2) and respiratory (n = 1). Four serious SAEs in this group were considered to be not related to the intervention, two unlikely to be related and two possibly related. Placebo: infection (n = 1) and musculoskeletal trauma (n = 2). (all three serious SAEs in this group were considered to be not related to the intervention). Adverse events beyond 12 weeks were reported by frequency of event. Numbers of patients experiencing ≥ 1 adverse event not provided |
|
McIntyre 2007 (augment with quetiapine) |
n.r. | n.r. | 0/29 (0%) | 0/29 (0%) | "No serious AEs were reported." (p.491, Safety and tolerability). Most commonly occurring adverse events reported in table 2 (type and frequency broken down by treatment) but numbers of participants experiencing ≥ 1 adverse event not reported |
|
Papakostas 2015 (augment with ziprasidone) |
n.r. | n.r. | 2/71 (2.8%) | 2/68 (2.9%) | Serious AEs: Ziprasidone (one hospitalisation because of treatment‐emergent suicidal thoughts and one because of a fall). Placebo (hospitalisations for treatment‐emergent viral meningitis and pneumonia respectively). Data for all adverse events reported by frequency of event. Numbers of patients experiencing ≥ 1 adverse event not reported |
|
Shelton 2001 (augment with olanzapine) |
n.r. | n.r. | n.r. | n.r. | Most commonly occurring adverse events described in the text but numbers (and treatment) of participants experiencing AEs not reported |
AE: adverse event n.r.: Not reported TIA: transient ischaemic attack
Comparison 3 ‐ Augmenting treatment with another antidepressant compared with continuing on an antidepressant
Two studies examined the effects of augmenting antidepressant treatment with a second antidepressant. Augmentation was with mianserin in one study (Ferreri 2001) and mirtazapine in the other study (Kessler 2018).
Primary Outcomes
Change in depressive symptoms
Clinician‐rated measures
Augmentation with mianserin
One study (Ferreri 2001, n = 70) found evidence of a difference in HAM‐D scores when current antidepressant mediation was augmented with mianserin compared with placebo (MD ‐4.8, 95% CI ‐8.18 to ‐1.42; moderate‐quality evidence; Analysis 2.1)
2.1. Analysis.
Comparison 2 Augment current antidepressant with mianserin, Outcome 1 Depressive symptoms (HAM‐D).
Augmentation with mirtazapine
No studies reported change in depressive symptoms based on clinician‐rated scale.
Self‐report measures
Augmentation with mianserin
No studies reported change in depressive symptoms based on a self‐report scale.
Augmentation with mirtazapine
One study (Kessler 2018, n = 480) found insufficient evidence of difference in BDI‐II scores between participants whose current antidepressant medication was augmented with mirtazapine and those who medication was augmented with placebo at 12 weeks (MD ‐1.7, 95% CI ‐4.03 to 0.63; high‐quality evidence; Analysis 3.1), 24 weeks (MD ‐0.9, 95% CI ‐3.39 to 1.59; high‐quality evidence; Analysis 3.1) or 52 weeks (MD 0.1, 95% CI ‐2.38 to 2.58; high‐quality evidence; Analysis 3.1). There was also insufficient evidence of a difference between groups on PHQ‐9 scores obtained at 12 weeks (MD ‐0.89, 95% CI ‐2.08 to 0.30; high‐quality evidence; Analysis 3.2).
3.1. Analysis.
Comparison 3 Augment current antidepressant with mirtazapine, Outcome 1 Depressive symptoms (BDI‐II).
3.2. Analysis.
Comparison 3 Augment current antidepressant with mirtazapine, Outcome 2 Depressive symptoms (PHQ‐9, 12 weeks).
Dropouts
Augmentation with mianserin
The was no evidence of a difference in numbers dropping out of treatment when current antidepressant treatment was augmented with mianserin compared with placebo (one study, Ferreri 2001, n = 70; RR 1.02, 95% CI 0.38 to 2.72; low‐quality evidence; Analysis 2.2).
2.2. Analysis.
Comparison 2 Augment current antidepressant with mianserin, Outcome 2 Dropouts.
Augmentation with mirtazapine
One study (Kessler 2018, n = 480) found no evidence of a difference in the number of participants dropping out of treatment augmented with mirtazapine compared with those who received placebo at 12, 24 or 52 weeks (12 weeks: RR 0.50, 95% CI 0.15 to 1.62; 24 weeks: RR 0.50, 95% CI 0.19 to 1.30; 52 weeks: (RR 0.46, 95% CI 0.18 to 1.18; all high‐quality evidence; Analysis 3.3)
3.3. Analysis.
Comparison 3 Augment current antidepressant with mirtazapine, Outcome 3 Dropouts.
Reasons for dropping out
Participants' reasons for discontinuation are reported in Table 10.
Secondary Outcomes
Response to treatment
Clinician‐rated measures
Augmentation with mianserin
One study (Ferreri 2001, n = 70) found evidence of an increased response to treatment when current antidepressant medication was augmented with mianserin compared with placebo (RR = 1.70, 95% CI 1.03 to 2.78; moderate‐quality evidence; Analysis 2.3).
2.3. Analysis.
Comparison 2 Augment current antidepressant with mianserin, Outcome 3 Response (≥ 50% reduction in HAM‐D score).
Augmentation with mirtazapine
No clinician‐rated measures of response to treatment were made in the study.
Self‐report measures
Augmentation with mianserin
No self‐report measures of response to treatment were made in the study.
Augmentation with mirtazapine
The was weak evidence that participants whose current antidepressant medication was augmented with mirtazapine had an increased response to treatment compared with placebo at 12 weeks (Kessler 2018, n = 480; RR 1.22, 95% CI 0.97 to 1.54; high‐quality evidence; Analysis 3.4). There was no evidence for a difference at 24 or 52 weeks (24 weeks: RR 1.01, 95% CI 0.83 to 1.23; 52 weeks: RR 1.00, 95% CI 0.82 to 1.22; high‐quality evidence; Analysis 3.4).
3.4. Analysis.
Comparison 3 Augment current antidepressant with mirtazapine, Outcome 4 Response (≥ 50% improvement in BDI‐II score).
Remission
Clinician‐rated measures
Augmentation with mianserin
One study (Ferreri 2001, n = 70) found evidence of an increased likelihood of meeting criteria for remission when current antidepressant medication was augmented with mianserin compared with placebo (RR 2.38, 95% CI 1.09 to 5.16; low‐quality evidence; Analysis 2.4).
2.4. Analysis.
Comparison 2 Augment current antidepressant with mianserin, Outcome 4 Remission (HAM‐D total score ≤ 7).
Augmentation with mirtazapine
No studies reported the outcome of remission determined using a clinician‐reported measure.
Self‐report measures
Augmentation with mianserin
No studies reported the outcome of remission determined using a self‐report measure.
Augmentation with mirtazapine
There was no evidence (Kessler 2018, n = 480) of a difference in the numbers of participants meeting the criteria for remission when current antidepressant medication was augmented with mirtazapine compared with placebo at any time point measured (12 weeks; RR 1.21, 95% CI 0.88 to 1.65; 24 weeks; RR 1.16, 95% CI 0.86 to 1.55; 52 weeks; RR 0.98, 95% CI 0.74 to 1.30; high‐quality evidence; Analysis 3.4).
Social adjustment and social functioning
No data on social adjustment or functioning were reported.
Quality of life
Augmentation with mianserin
No quality of life data were reported.
Augmentation with mirtazapine
One study (Kessler 2018, n = 480) measured quality of life using the EQ‐5D‐5L and SF‐12. There was no evidence of a difference in EQ‐5D‐5L scores between participants whose medication was augmented with mirtazapine compared with placebo at any of the time points measured (12 weeks; MD ‐0.01, 95% CI ‐0.06 to 0.04; 24 weeks: MD ‐0.02, 95% CI ‐0.07 to 0.03; 52 weeks: MD ‐0.03, 95% CI ‐0.08 to 0.02; all high‐quality evidence; Analysis 3.6).
3.6. Analysis.
Comparison 3 Augment current antidepressant with mirtazapine, Outcome 6 Quality of life (EQ‐5D‐5L).
On the SF‐12 checklist, there was evidence of an improvement on the aggregate mental functioning subscale in the mirtazapine augmentation group at 12 weeks (MD 3.61, 95% CI 1.23 to 5.99; high‐quality evidence; Analysis 3.7) compared with placebo but not at 24 or 52 weeks (24 weeks: MD 1.98, 95% CI ‐0.64 to 4.60; 52 weeks: MD 1.29, 95% CI ‐1.44 to 4.02; high‐quality evidence; Analysis 3.7). There was no evidence of a difference in scores on the aggregate physical functioning subscale at any of the time points measured (12 weeks: MD ‐1.76, 95% CI ‐4.20 to 0.68; 24 weeks: MD ‐2.49, 95% CI ‐5.04 to 0.06; 52 weeks: MD ‐0.98, 95% CI ‐3.61 to 1.65; all high‐quality evidence; Analysis 3.8).
3.7. Analysis.
Comparison 3 Augment current antidepressant with mirtazapine, Outcome 7 Quality of life (SF‐12 ‐ aggregate mental functioning).
3.8. Analysis.
Comparison 3 Augment current antidepressant with mirtazapine, Outcome 8 Quality of life (SF‐12 ‐ aggregate physical functioning).
Economic outcomes
Augmentation with mianserin
No economic outcomes were reported.
Augmentation with mirtazapine
One study (Kessler 2018, n = 480) reported direct costs to health and social services, costs to patients and carers, and time off work for patients and carers. There was no evidence of a difference in resource use cost when current antidepressant treatment was augmented with mirtazapine compared with placebo for any of the resources considered. Cost‐effectiveness analyses were also reported. No clinically important differences in quality adjusted life years (QALYs) or costs were observed between the two groups at 12 weeks follow‐up (difference of 0.002, (95% CI –0.002 to 0.005) QALYs); (difference in costs £2 (95% CI –£27 to £31)) or at 52 weeks follow‐up (difference of 0.009 (95% CI –0.016 to 0.035) QALYs); (difference in costs £69 (95% CI –£74 to £206)). There was, therefore, no evidence that mirtazpine was a cost‐effective use of NHS resources.
Adverse effects
All of the included studies reported some information about adverse events. Further details can be found in Table 11.
Two studies of augmentation of current antidepressant therapy with an additional antidepressant reported the number of participants in each arm that experienced an adverse event. The incidence of adverse events was higher when augmenting with mianserin (44.7%, Ferreri 2001) or mirtazapine (69.3%, Kessler 2018) than placebo (28.1% and 38.1% in the respective control groups). Both studies also reported the frequency of each type of adverse event experienced. The most commonly reported adverse events were dizziness, somnolence, dry mouth, asthenia, and weight increase.
One study (Kessler 2018) reported the number of participants experiencing serious adverse events (SAEs). The overall incidence of SAEs was low and the majority were judged by the study investigators to be unrelated (or unlikely to be related) to study medication. The incidence of adverse events was 3.32% in participants whose current antidepressant therapy was augmented with mirtazapine compared with 1.25% when augmented with placebo.
The number of participants who discontinued in studies due to adverse events are reported in Table 10 and described under the primary outcome of dropouts.
Comparison 4 ‐ Augmenting treatment with a non‐antidepressant compared with continuing on an antidepressant
Eight studies examined the effects of augmenting antidepressant treatment with a non‐antidepressant. Augmentation was with an anxiolytic (buspirone) in one study (Appelberg 2001) and an antipsychotic (cariprazine, olanzapine, quetiapine or ziprasidone) in seven studies (Bauer 2009 (ONYX); Dunner 2007; Durgam 2016; El‐Khalili 2010 (PEARL); McIntyre 2007; Papakostas 2015; Shelton 2001).
Primary Outcomes
Change in depressive symptoms
Clinician‐rated measures
Augmentation with buspirone
One study (Appelberg 2001, n = 102) found no evidence of a difference in mean change from baseline in MADRS scores when augmenting current antidepressant medication with the anxiolytic buspirone compared with placebo (MD ‐0.30, 95% CI ‐9.48 to 8.88; low‐quality evidence; Analysis 4.1).
4.1. Analysis.
Comparison 4 Augment current antidepressant with buspirone (anxiolytic), Outcome 1 Depressive symptoms (MADRS).
Augmentation with cariprazine
One three‐arm, study (Durgam 2016, n = 808) found a reduction in MADRS scores from baseline (the primary outcome for the study) when antidepressant treatment was augmented with the antipsychotic cariprazine at a dose of 2 mg/d to 4.5 mg/d (MD ‐2.1, 95% CI ‐3.63 to ‐0.57; Analysis 5.1) compared with placebo, but there was only weak evidence of an effect at the lower dose of 1 mg/d to 2 mg/d (MD ‐0.90 , 95% CI ‐2.29 to 0.49; Analysis 5.1). Pooling the two active treatment arms showed an overall effect for cariprazine (MD ‐1.50, 95% CI ‐2.74 to ‐0.25; high‐quality evidence; Analysis 5.1).
5.1. Analysis.
Comparison 5 Augment current antidepressant with cariprazine (antipsychotic), Outcome 1 Depressive symptoms (MADRS).
Augmentation with olanzapine
One study (Shelton 2001, n = 20) found evidence of a reduction in MADRS scores when augmenting current antidepressant treatment with the antipsychotic olanzapine compared with placebo (MD ‐12.4, 95% CI ‐22.44 to ‐2.36; low‐quality evidence; Analysis 6.1). However, evidence of a difference between groups was weaker based on the HAM‐D rating scale, which was the primary outcome for the study (MD ‐7.9, 95% CI ‐16.76 to 0.96; low‐quality evidence; Analysis 6.2).
6.1. Analysis.
Comparison 6 Augment current antidepressant with olanzapine (antipsychotic), Outcome 1 Depressive symptoms (MADRS).
6.2. Analysis.
Comparison 6 Augment current antidepressant with olanzapine (antipsychotic), Outcome 2 Depressive symptoms (HAM‐D).
Augmentation with quetiapine
Three studies (Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL); McIntyre 2007 n = 977) found evidence of a reduction in depressive symptomology (measured using MADRS ‐ Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL) ‐ or HAM‐D ‐ McIntyre 2007) when augmenting current antidepressant treatment with the antipsychotic quetiapine at a dose of 150 or 200 mg/d (SMD ‐0.34, 95% CI ‐0.53 to ‐0.14; I2 = 28%; Analysis 7.1). Two of these studies (Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL) also examined the effects of adjunctive quetiapine at a higher dose of 300 mg/d and found evidence of a reduction in MADRS scores compared with placebo (MD ‐2.85, 95% CI ‐4.23 to ‐1.47, I2 = 0%; Analysis 7.1). Pooling across doses, there was evidence overall (three studies (Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL); McIntyre 2007 n = 977) of a beneficial effect for quetiapine on symptoms of depression (SMD ‐0.32, 95% CI ‐0.46 to ‐0.18; I2 = 6%, high‐quality evidence; Analysis 7.1).
7.1. Analysis.
Comparison 7 Augment current antidepressant with quetiapine (antipsychotic), Outcome 1 Depressive symptoms (MADRS or HAM‐D).
Augmentation with ziprasidone
Two studies examined the effectiveness of augmenting current antidepressant therapy with the antipsychotic ziprasidone (Dunner 2007; Papakostas 2015, n = 199). Dunner 2007 was a three‐arm study comparing two doses of ziprasidone (80 mg/d or 160 mg/d) with continuing on antidepressant monotherapy alone. Papakostas 2015 compared augmenting current antidepressant therapy with 40 mg/d to 160 mg/d of ziprasidone (mean dose 98 mg) with placebo.
Dunner 2007 found no evidence of a difference in mean change in HAM‐D scores (the primary outcome of the study) for a dose of 80 mg (n = 41, mean difference ‐0.91, 95% CI ‐4.84 to 3.02; Analysis 8.1) or 160 mg per day (n = 39; MD ‐2.56, 95% CI ‐6.72 to 1.60; Analysis 8.1) but confidence intervals were wide. Papakostas 2015 found evidence of a reduction in depressive symptoms on the HAM‐D (MD ‐3.10, 95% CI ‐5.19 to ‐1.01; Analysis 8.1) for a mean dose of 98 mg/d (range 40 mg to 160 mg) of ziprasidone daily. Pooling across doses, there was evidence overall for a greater reduction from baseline in HAM‐D scores for ziprasidone compared with antidepressant monotherapy (MD ‐2.73, 95% CI ‐4.53 to ‐0.93; I2 = 0, moderate‐quality evidence; Analysis 8.1).
8.1. Analysis.
Comparison 8 Augment current antidepressant with ziprasidone (antipsychotic), Outcome 1 Depressive symptoms (HAM‐D).
Dunner 2007 also assessed depression severity using the MADRS scale. There was no evidence of a difference in mean change in MADRS scores at any dose (80 mg/d: MD ‐1.53, 95% CI ‐6.94 to 3.88; 160 mg/d: 3.82, 95% CI ‐9.64 to 2.00; pooled dose: MD ‐2.62, 95% CI ‐7.47 to 2.23; low‐quality evidence; Analysis 8.2).
8.2. Analysis.
Comparison 8 Augment current antidepressant with ziprasidone (antipsychotic), Outcome 2 Depressive symptoms (MADRS).
Self‐report measures
Augmentation with ziprasidone
Papakostas 2015, (n = 139) found evidence of a reduction in scores on the self‐report QIDS‐SR (MD ‐2.50, 95% CI ‐3.83 to ‐1.17; moderate‐quality evidence; Analysis 8.3) for a dose of 40 mg to 160 mg of ziprasidone daily.
8.3. Analysis.
Comparison 8 Augment current antidepressant with ziprasidone (antipsychotic), Outcome 3 Depressive symptoms (QIDS‐SR).
Dropouts
All of the included studies reported the numbers of participants who discontinued in each trial arm. Participants' reasons for discontinuation are reported in Table 10.
Augmentation with buspirone
There was no evidence of a difference in the number of participants dropping out of treatment augmented with the anxiolytic buspirone compared with those who received placebo (one study, Appelberg 2001, n = 108; RR 0.60, 95% CI 0.23 to 1.53; low‐quality evidence; Analysis 4.2).
4.2. Analysis.
Comparison 4 Augment current antidepressant with buspirone (anxiolytic), Outcome 2 Dropouts.
Augmentation with cariprazine
In a three‐arm trial of augmentation with cariprazine (Durgam 2016, n = 821), more participants in the intervention arms dropped out than in the placebo arm (RR 1.68, 95% CI 1.16 to 2.41; moderate‐quality evidence; Analysis 5.2). The evidence was weaker for the lower dose of 1 mg to 2 mg of cariprazine per day (RR 1.43, 95% CI 0.94 to 2.17; Analysis 5.2) than for the higher dose of 2 mg to 4.5 mg per day (RR 1.92, 95% CI 1.30 to 2.84; Analysis 5.2).
5.2. Analysis.
Comparison 5 Augment current antidepressant with cariprazine (antipsychotic), Outcome 2 Dropouts.
Augmentation with olanzapine
One study of augmentation with olanzapine (Shelton 2001, n = 20) found no evidence of a difference in numbers dropping out although the confidence interval was wide (RR 0.33, 95% CI 0.04 to 2.69; low‐quality evidence; Analysis 6.3).
6.3. Analysis.
Comparison 6 Augment current antidepressant with olanzapine (antipsychotic), Outcome 3 Dropouts.
Augmentation with quetiapine
Three trials of augmentation with quetiapine (Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL); McIntyre 2007; n = 977) provided weak evidence of greater dropout in the intervention arms compared with the placebo group (RR 1.33, 95% CI 0.9 to 1.95; I2= 1%, moderate‐quality evidence; Analysis 7.2). When doses of quetiapine were considered separately, those who received the higher dose of 300 mg/d were more likely to drop out compared with the placebo group (two studies, Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL); RR 1.82, 95% CI 1.29 to 2.57; I2 = 0%; Analysis 7.2) but there was no evidence of a difference in dropouts for the lower dose of 150 mg/d or 200 mg/d of quetiapine compared with placebo (three studies, Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL); McIntyre 2007; RR 1.18, 95% CI 0.86 to 1.63; I2 = 42%; moderate‐quality evidence; Analysis 7.2).
7.2. Analysis.
Comparison 7 Augment current antidepressant with quetiapine (antipsychotic), Outcome 2 Dropouts.
Augmentation with ziprasidone
There was evidence from two studies (Dunner 2007, Papakostas 2015, n = 199) that dropout was greater for those who received treatment augmented with ziprasidone compared with antidepressant monotherapy (RR 1.60, 95% CI 1.01 to 2.55, I2 = 0%; moderate‐quality evidence; Analysis 8.4).
8.4. Analysis.
Comparison 8 Augment current antidepressant with ziprasidone (antipsychotic), Outcome 4 Dropouts.
Reasons for dropping out
All of the eight studies that examined the effects of augmenting antidepressant treatment with a non‐antidepressant reported participants' reasons for discontinuation and these are detailed in full in Table 10. Across all of the studies, the most common reason for dropping out was due to an adverse event or an inability to tolerate the side effects of treatment (8.05% of all randomised participants). Greater numbers of participants dropped out for this reason from active intervention arms than from placebo arms and at higher compared with lower doses (in the case of trials with more than one active intervention arm). The proportion of participants dropping out due to adverse events or intolerability was considerably higher for Dunner 2007 than any of the other included studies (n = 16, 26.67%).
Withdrawal of consent or unwillingness to continue was the second most frequently reported reason for dropping out (3.69% of all randomised participants). The proportion of participants dropping out for this reason was higher for Dunner 2007 than any of the other studies (n = 6, 10%).
In four trials, participants dropped out due to protocol violations (Appelberg 2001; Bauer 2009 (ONYX); Durgam 2016; Shelton 2001). Two trials (Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL)) reported dropouts post‐randomisation due to failure to fulfil eligibility criteria.
‘Lost to follow‐up’ was cited as the reason for the dropout of 2.35% of all randomised participants. 1.15% dropped out due to the inefficacy of their treatment and 0.43% because they stopped taking their study medication or due to extreme non‐compliance.
Secondary Outcomes
Response to treatment
Seven studies reported data on the dichotomous outcome of response to treatment (Bauer 2009 (ONYX); Dunner 2007; Durgam 2016; El‐Khalili 2010 (PEARL); McIntyre 2007; Papakostas 2015; Shelton 2001). All measured response using an observer‐rated tool (MADRS or HAM‐D). One study additionally assessed response using a self‐report rating scale (QIDS‐SR; Papakostas 2015).
Clinician‐rated measures
Augmentation with buspirone
No data were reported for the outcome of response to treatment for augmentation of current antidepressant treatment with buspirone.
Augmentation with cariprazine
One three‐arm, study (Durgam 2016, n = 808) found that those whose antidepressant treatment was augmented with the antipsychotic cariprazine at a dose of 2 mg/d to 4.5 mg/d or 1 mg/d to 2 mg/d were more likely to meet criteria for response compared with the placebo group (2 mg/d to 4.5 mg/d: RR 1.29, 95% CI 1.06 to 1.57; 1 mg/d to 2 mg/d: RR 1.25, 95% CI 1.03 to 1.53; Analysis 5.3). When pooling the two active treatment arms, there was a 27% increased likelihood of response to treatment for those who received augmented treatment with cariprazine compared with placebo (RR 1.27, 95% CI 1.07 to 1.52; moderate‐quality evidence; Analysis 5.3).
5.3. Analysis.
Comparison 5 Augment current antidepressant with cariprazine (antipsychotic), Outcome 3 Response (≥ 50% improvement in MADRS score).
Augmentation with olanzapine
The single small study of augmentation with olanzapine (Shelton 2001, n = 20) found that those who received augmented treatment were more likely to meet criteria for response compared with the placebo group but the confidence interval was very wide (RR 6.00, 95% CI 0.87 to 41.21; low‐quality evidence; Analysis 6.4).
6.4. Analysis.
Comparison 6 Augment current antidepressant with olanzapine (antipsychotic), Outcome 4 Response (≥ 50% reduction in MADRS score).
Augmentation with quetiapine
There was evidence from three studies (Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL); McIntyre 2007; n = 977) that those whose antidepressant treatment was augmented with quetiapine (any dose) were more likely to meet criteria for response compared with those receiving placebo (RR = 1.20, 95% CI 1.02 to 1.40; I2 = 0%; moderate‐quality evidence; Analysis 7.3). A benefit in terms of response to treatment was found at both higher (300 mg/d; two studies; Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL); RR 1.29, 95% CI 1.10 to 1.50, I2 = 0%; Analysis 7.3) and lower doses (150 mg/d or 200 mg/d; three studies Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL); McIntyre 2007; RR 1.25, 95% CI 1.09 to 1.43; I2= 0%; Analysis 7.3) compared with placebo.
7.3. Analysis.
Comparison 7 Augment current antidepressant with quetiapine (antipsychotic), Outcome 3 Response (≥ 50% reduction in MADRS or HAM‐D score).
Augmentation with ziprasidone
In a three‐arm trial, Dunner 2007 found no evidence of a difference in response (based on scores on the MADRS) for augmenting with ziprasidone at a daily dose of either 80 mg (RR 1.90, 95% CI 0.39 to 9.28; Analysis 8.5) or 160 mg (RR 3.16, 95% CI 0.72 to 13.76; Analysis 8.5) compared with continuing with antidepressant therapy alone. Papakostas 2015 found weak evidence (RR 1.71, 95% CI 0.97 to 3.00; Analysis 8.5) that those whose antidepressant treatment was augmented with ziprasidone (mean dose: 98 mg/d, range 40 mg to 160 mg) were more likely to meet criteria for response (based on scores of the HAM‐D; with response being the primary outcome for this study) than those who received placebo. Pooling across studies and doses, there was evidence that those whose current antidepressant therapy was augmented with ziprasidone were more likely to meet criteria for response (RR 1.80, 95% CI 1.07 to 3.04; moderate‐quality evidence; Analysis 8.5).
8.5. Analysis.
Comparison 8 Augment current antidepressant with ziprasidone (antipsychotic), Outcome 5 Response (≥ 50% reduction in MADRS/HAM‐D score).
Self‐report measures
Augmentation with ziprasidone
Papakostas 2015 also measured response to treatment on the self‐report SIDS‐SR scale and found evidence that those who received augmentation with ziprasidone were more likely to meet criteria for response to treatment in the ziprasidone augmentation arm compared with the placebo group (n = 139, RR = 2.34, 95% CI 1.16 to 4.72; moderate‐quality evidence; Analysis 8.6).
8.6. Analysis.
Comparison 8 Augment current antidepressant with ziprasidone (antipsychotic), Outcome 6 Response (≥ 50% reduction in QIDS‐SR).
Remission
Six studies reported the outcome of remission from depression at the end of the treatment period (Bauer 2009 (ONYX); Dunner 2007; Durgam 2016; El‐Khalili 2010 (PEARL); McIntyre 2007; Papakostas 2015). All six studies defined remission based on a cut‐off on an observer‐rated depression scale (MADRS or HAM‐D). One study additionally assessed remission using a self‐report rating scale (QIDS‐SR; Papakostas 2015).
Clinician‐rated measures
No data were reported for the outcome of remission for augmentation of current antidepressant treatment with either the anxiolytic buspirone or with the antipsychotic olanzapine.
Augmentation with cariprazine
One three‐arm, study (Durgam 2016, n = 808) found no evidence of a difference in the number meeting criteria for remission amongst participants whose current antidepressant was augmented with either 1 mg/d to 2 mg/d (RR 1.06, 95% CI 0.83 to 1.37; Analysis 5.4) or 4 mg/d to 5 mg/d (RR 1.07, 95% CI 0.83 to 1.38; Analysis 5.4) of cariprazine compared with placebo. Pooling across the cariprazine arms showed no evidence of a difference between groups (RR 1.07, 95% CI 0.86 to 1.33; moderate‐quality evidence; Analysis 5.4).
5.4. Analysis.
Comparison 5 Augment current antidepressant with cariprazine (antipsychotic), Outcome 4 Remission (MADRS total score ≤ 10).
Augmentation with quetiapine
There was evidence of an increased likelihood of meeting criteria for remission in those who received augmentation of current antidepressant therapy with quetiapine at a higher dose (300 mg/d, two studies, Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL); RR 1.53, 95% CI 1.13 to 2.07; I2 = 32%; Analysis 7.5) or lower dose (150 mg/d or 200 mg/d, three studies, Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL); McIntyre 2007; RR 1.51, 95% CI 1.19 to 1.93; I2 = 0%; Analysis 7.5) than those receiving a placebo.
7.5. Analysis.
Comparison 7 Augment current antidepressant with quetiapine (antipsychotic), Outcome 5 Remission (MADRS score ≤ 8/HAM‐D score ≤ 7).
Pooling across the quetiapine doses showed that those who received quetiapine (any dose) in addition to their antidepressant medication had a 50% increased likelihood of meeting criteria for remission compared with those who received placebo (three studies, Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL); McIntyre 2007; RR 1.53, 95% CI 1.22 to 1.90; I2 = 0%; moderate‐quality evidence; Analysis 7.5).
Augmentation with ziprasidone
There was little evidence of a difference in remission outcomes based on clinician‐rated scales for those who received ziprasidone in addition to antidepressant medication compared with those who continued on antidepressant monotherapy (Dunner 2007, Papakostas 2015, n = 199; RR 1.27, 95% CI 0.81 to 2.00, I2 = 0%; moderate‐quality evidence; Analysis 8.7).
8.7. Analysis.
Comparison 8 Augment current antidepressant with ziprasidone (antipsychotic), Outcome 7 Remission (MADRS/HAM‐D).
Self‐report measures
Augmentation with ziprasidone
Papakostas 2015 (n = 139) found evidence that those whose antidepressant treatment was augmented with ziprasidone were more likely to meet criteria for remission when this was determined using a self‐report instrument (RR 2.33, 95% CI 1.03 to 5.25; moderate‐quality evidence; Analysis 8.8).
8.8. Analysis.
Comparison 8 Augment current antidepressant with ziprasidone (antipsychotic), Outcome 8 Remission (QIDS‐SR).
Social adjustment and social functioning
No studies reported data on social adjustment or functioning.
Quality of life
No quality of life data were reported in studies that examined augmentation with buspirone, cariprazine, olanzapine or ziprasidone.
Augmentation with quetiapine
Two three‐arm trials (Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL), n = 884) found no evidence of a difference in terms of quality of life for those whose antidepressant treatment was augmented with the antipsychotic quetiapine compared with placebo (dose 150 mg/d: MD 0.70, 95% CI ‐2.31 to 3.71, I2= 36%; 300 mg/d: MD 0.35, 95% CI ‐2.06 to 2.77, I2= 0; pooled across arms: mean difference 0.57, 95% CI ‐1.52 to 2.65, I2= 0%; Analysis 7.4).
7.4. Analysis.
Comparison 7 Augment current antidepressant with quetiapine (antipsychotic), Outcome 4 Quality of life (% max score of Q‐LES‐Q‐SF).
Economic outcomes
No studies reported data on any economic outcomes.
Adverse effects
All of the included studies reported some information about adverse events. Further details can be found in Table 11.
In the study of augmentation of current antidepressant therapy with the anxiolytic busiprone, Appelberg 2001 reported no serious adverse events and that there were "no statistically significant differences" (p.450) in scores on the Udvalg for Kliniske Undersøgelser (UKU) side effect rating scale (Lingjaerde 1987) between treatment groups during the study.
Six studies of augmentation of current antidepressant therapy with an antipsychotic reported arm‐level adverse event data (Bauer 2009 (ONYX); Dunner 2007; Durgam 2016; El‐Khalili 2010 (PEARL); McIntyre 2007; Papakostas 2015). Four of these studies reported the number of participants in each arm that experienced at least one adverse event (Bauer 2009 (ONYX); Dunner 2007; Durgam 2016; El‐Khalili 2010 (PEARL)). The incidence of adverse events ranged from 40% to 66.9% in the control arms and from 65% to 100% in the intervention arms. All six of the studies reported the frequency of each type of adverse event. A seventh study (Shelton 2001) described the most commonly occurring events across all treatment groups but did not report the numbers experiencing each type of event. Across all six studies, the most commonly reported adverse events were somnolence, dry mouth, akathisia, asthenia, dizziness, headache, nausea, constipation, diarrhoea and changes in appetite or weight.
Six studies of augmentation with an antipsychotic reported the numbers of participants experiencing serious adverse events (SAEs) (Bauer 2009 (ONYX); Dunner 2007; Durgam 2016; El‐Khalili 2010 (PEARL); McIntyre 2007; Papakostas 2015). Overall, the number of serious adverse events reported was low. Further details of the events reported are provided in Table 11.
The number of participants who discontinued in studies due to adverse events are reported in Table 10 and described under the primary outcome of dropouts.
Subgroup analyses
No subgroup analyses were undertaken to explore potential sources of heterogeneity because there were fewer than ten studies available in any comparison.
Sensitivity analysis
There were too few studies to enable pre‐planned sensitivity analyses to be conducted.
Discussion
This review focused on pharmacological approaches for adults with treatment‐resistant depression (TRD). We identified one randomised controlled trial that examined switching current antidepressant treatment to another antidepressant and two trials that augmented current antidepressant therapy with a second antidepressant. We identified eight randomised controlled trials that examined augmentation of current antidepressant therapy with a non‐antidepressant. Augmentation was with an anxiolytic (busiprone) in one study and with an antipsychotic (cariprazine, olanzapine, quetiapine or ziprasidone) in seven studies. We did not identify any studies meeting our eligibility criteria that examined increasing the dose of current antidepressant monotherapy.
Summary of main results
The main findings of the review are summarised in eight key tables (Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8).
The single study that examined switching from current antidepressant treatment to another antidepressant found no evidence of a difference in depressive symptoms, response to treatment, remission rates or numbers dropping out (all moderate‐quality estimates). The sample size was small (n = 72 for this comparison) and there was imprecision in the estimates, therefore our confidence in these findings is limited.
Evidence for the effectiveness of augmenting current antidepressant treatment with another antidepressant was mixed. There was moderate‐quality evidence from one study that augmenting with mianserin was associated with a greater reduction in depression severity scores from baseline, an increased response to treatment and increased remission of depressive symptoms. There was no evidence of a difference in numbers dropping out of treatment (moderate‐quality) but there was considerable imprecision in this estimate. A second study, of augmentation of current antidepressant treatment with mirtazapine, found no evidence of a difference in depressive symptoms or other outcomes (remission, quality of life, numbers dropping out of treatment; all estimates high‐quality) at any time point, although there was weak evidence of an increased response to treatment at 12 weeks (but not at later follow‐up). There were also no differences in resource use costs between treatment groups or evidence that augmenting current treatment with mirtazapine was cost‐effective compared with continuing on antidepressant monotherapy.
The single study (n = 102) that focused on augmentation of current antidepressant therapy with the anxiolytic busiprone found no evidence of a difference in depressive symptoms (low‐quality) or numbers dropping out (moderate‐quality). No data were reported for response to treatment or remission rates. There was considerable imprecision (very wide confidence intervals) in the estimates of effect for the depression outcomes and therefore our confidence in these findings is limited.
Seven studies examined the effectiveness of augmenting current antidepressant therapy with an antipsychotic (cariprazine, olanzapine, quetiapine or ziprasidone). All found evidence of a greater reduction in depression severity scores from baseline in the augmentation group compared with the control group. For cariprazine (moderate‐quality), quetiapine (high‐quality) and ziprasidone (moderate‐quality), this difference was modest, corresponding to an average difference of between 1.5 and 2.7 points on the depression severity scale used that equate to an effect size of approximately 0.18 to 0.42 SD. The NICE guidelines group (NICE 2004) previously suggested that a difference of around 0.33 SD equated to a minimum clinically important difference (MCID) and, therefore, the effects seen here may be clinically relevant. However, there is some evidence that patients with TRD require larger improvements in their symptoms to report feeling better (Button 2015). A larger effect was observed in the study of augmentation with olanzapine (n = 20) but certainty in this estimate was judged to be low due to imprecision.
We also found evidence to support a beneficial effect of adjunctive treatment with an antipsychotic for the dichotomous outcome of response to treatment. The effect was largest for olanzapine, but this estimate was judged to be of low quality due to considerable imprecision.The evidence for all other antipsychotics was judged to be of moderate quality.
Findings for the more stringent outcome of remission of depressive symptoms (where measured) were mixed. There was moderate‐quality evidence that augmentation with quetiapine was associated with an increased likelihood of remission of depressive symptoms compared with continuing on an existing antidepressant treatment alone. There was no evidence of a difference for cariprazine (moderate‐quality). For ziprasidone, there was no evidence of a difference when remission was determined using an observer‐rated measure (HAM‐D), but increased likelihood of remission when depressive symptoms were self‐reported (QIDS‐SR) (both estimates, moderate‐quality). Remission rates were not reported for olanzapine.
There was moderate‐quality evidence of a differential dropout rate when current antidepressant therapy was augmented with cariprazine, quetiapine or ziprasidone, with those who received an antipsychotic drug being more likely to drop out. There was no evidence of a difference in dropout rates when current therapy was augmented with olanzapine but the quality of this estimate was low. The most common reason for dropping out was an adverse event. Definitions of adverse events varied between included studies but reports of adverse events were higher in the groups who received augmented treatment with an antipsychotic compared with those who continued antidepressant therapy alone or augmented with a placebo.
Data for all other outcomes were limited. Two trials found no evidence of a difference in quality of life for participants whose current treatment was augmented with quetiapine compared with placebo (moderate‐quality). One trial found no evidence of a difference in resource use costs (to health and social services, patients and carers) when current antidepressant treatment was augmented with mirtazapine compared with placebo.
None of the studies we identified reported data on social adjustment or functioning.
Due to the small number of studies identified, we were unable to conduct any subgroup analyses or explore the robustness of the findings through sensitivity analyses. Our confidence in the majority of the effect estimates obtained in this review was lowered by the imprecision of these estimates.
In summary, there is a small body of evidence on the effectiveness of augmenting current antidepressant therapy with a second pharmacological treatment. There is currently insufficient evidence to support the addition of busiprone to current antidepressant treatment but further trials may change this conclusion. There is evidence of a benefit for augmenting antidepressant therapy with the antidepressant mianserin (based on a single study) or with an antipsychotic, but this needs to be balanced against the increased likelihood of dropping out of treatment or experiencing an adverse event as a side effect of treatment. There is currently insufficient evidence to support switching existing antidepressant treatment to another antidepressant but further trials may change this conclusion.
Overall completeness and applicability of evidence
Our search was comprehensive; we utilised the CMDCTR's trial reference and studies registers, which were collated from searches (from inception) of multiple databases, and also included assessment of unpublished literature accessed by contacting study authors. In addition, we screened reference lists of included studies and contacted study authors for any unpublished or ongoing studies.
Despite this, we identified only a small body of evidence. Individual comparisons were supported by only one, two or three studies. Importantly, we identified a number of gaps in the literature. There were no trials of increasing the dose of existing antidepressant medication and only one of switching to another antidepressant treatment that were eligible for inclusion in our review. Only two trials assessed efficacy outcomes (depressive symptoms, response to treatment and remission) using a self‐report tool. Only three studies reported quality of life data, none reported on social adjustment/functioning and only one study reported economic outcomes. Only one study (Kessler 2018) assessed outcomes beyond the short term (duration of studies ranged between six and eight weeks) so long‐term outcomes of treatment of most treatment strategies are unknown.
Findings from this review are applicable to adults with TRD defined as "depression (meeting diagnostic criteria) that has not responded to at least 4 weeks treatment with a therapeutic dose of antidepressant medication". Definitions of TRD varied between studies ranging from between one and three previous failed treatments. Duration of treatment in most trials was six to eight weeks which is comparable with other pharmacological intervention trials in depressed patients. The age and gender distribution of participants recruited in the included studies reflect the average age/gender profile of depressed patients ‐ women in their 40s. In terms of the severity of depression, mean depression scores on the HAM‐D/MADRS equated to moderate/severe depression. Nine trials recruited outpatients from hospital (secondary care) outpatient clinics. Only one trial recruited participants from primary care. Whilst we know that many patients with TRD will be seen in primary care settings (Thomas 2013), similarities in terms of age, gender and severity of depression increase the generalisability of review findings. Four studies were conducted in the US, one each in Canada, England, Finland and France, and two in multiple countries across Europe, North America and Australia. As such, findings may have limited generalisability to other countries where the system for delivering mental health care is substantially different from those described in the included studies.
Quality of the evidence
We assessed the quality of the evidence using the GRADE approach for each of the outcomes presented in the summary of findings tables (Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8).
Study limitations
All studies were judged to be at a low or unclear (due to insufficient reporting) risk of selection bias. Outcome assessors were blinded in all included studies and a placebo intervention was administered in the comparator arm of all but one study to protect against performance and detection bias. We had no concerns in relation to completeness of outcome data. Although we had some concerns with regards to the possibility of selective outcome reporting in some of the studies, we did not consider that these would reduce the confidence in the estimates of effect as none of the issues identified directly related to the outcomes of interest. Overall, we did not consider any of the included studies to be at a sufficient risk of bias to warrant downgrading the evidence for any outcome.
Consistency of effect
For each of the majority of comparisons made in this review, only one study provided data. For two of the treatment strategies considered (augmenting with quetiapine and augmenting with ziprasidone), data were provided from two or more studies. No significant heterogeneity was observed for any outcome for augmentation of current treatment with ziprasidone. An I2 value of 42% was observed for the numbers of participants dropping out when current antidepressant treatment was augmented with quetiapine compared to placebo. The confidence intervals for three studies contributing to this estimate overlapped and a statistical test for heterogeneity was not significant (P = 0.18). No significant heterogeneity was observed for any other outcomes for these comparisons. We did not therefore downgrade any of the evidence included in the review for inconsistency.
Imprecision
Imprecision is the key issue affecting our confidence in the body of evidence identified in this review. For each of many of the comparisons examined, only one study contributed data. Some of the included studies had very small sample sizes (e.g. Dunner 2007; Shelton 2001) and were arguably underpowered to detect a difference between treatment groups. Many of the effect estimates obtained had wide confidence intervals. For augmentation with the anxiolytic buspirone, the estimates of treatment effects (mean change in depression severity, response to treatment and numbers dropping out) were judged to have very serious imprecision and are therefore of low quality. Estimates of treatment effectiveness and numbers dropping out for augmentation with olanzapine also exhibited very serious imprecision, as did those for switching to or augmenting current treatment with mianserin. For each of these treatment strategies, the true effect may be substantially different from the estimate of the effect obtained in this review.
Indirectness
The studies included in this review were free from indirectness in terms of the comparisons of interest (all direct comparisons) as well as the target population, types of intervention, comparator and methods of outcome determination.
Publication bias
We minimised the likelihood of publication bias by searching for and including unpublished and ongoing studies. However, due to the small numbers of studies available for each comparison of interest, we were unable to perform a formal test of small‐study bias (Eggers test) and are therefore unable to reach any formal conclusion regarding the absence of publication bias for this review.
Potential biases in the review process
We attempted to contact authors of included studies in order to resolve any queries related to their study and/or to obtain missing information relevant to this review. This, combined with a very comprehensive search strategy, including searching trial registers for unpublished literature along with the inclusion of non‐English language papers, minimised the likelihood of publication bias, although the small number of included studies precluded the use of a funnel plot to formally test for such bias. Moreover, our attempts to obtain missing information addressed the potential for selective outcome reporting.
We judged two studies to be at high risk of bias for selective outcome reporting although these issues did not relate directly to the outcomes of interest and therefore we judged these as unlikely to bias the findings of this review. All but one study included a placebo group, minimising the potential for detection and performance bias. All but one (Kessler 2018) of the included studies were industry sponsored and, as shown by previous reviews (e.g. Lundh 2017), may be more likely to give a favourable view of the drug trialled.
The small number of included studies precluded us from undertaking prespecified subgroup analyses and we were therefore unable to determine whether severity of depression and length of the acute treatment phase were potential effect modifiers.
Agreements and disagreements with other studies or reviews
We identified seven previous systematic reviews of pharmacological interventions for treatment‐resistant depression. The majority restricted their inclusion to randomised controlled trials. Most looked at the effectiveness of augmentation strategies (with an antipsychotic or other pharmacological agent, Edwards 2013; Liu 2015; Zhou 2015; Zhou 2015a). Two reviews examined the effectiveness of switching to another antidepressant (Bschor 2010; Ruhe 2006) and one looked at increasing the dose of existing antidepressant treatment (Ruhe 2006a). The definition of treatment‐resistant depression was not always clear within the reviews’ eligibility criteria and some also considered non‐standard pharmacological treatments. Consequently, other reviews have included a somewhat different set of trials.
Zhou 2015 conducted a systematic review and network meta‐analysis of augmenting antidepressant treatment with an atypical antipsychotic. Whilst their definition of TRD was similar to ours (current episode of major depressive disorder diagnosed according to standard diagnostic interview and an inadequate response to at least one course of conventional antidepressant treatment), they did not specify the length of the initial treatment trial. RCTs of augmentation with an atypical antipsychotic compared with either augmentation with another atypical antipsychotic or with placebo were suitable for inclusion. Several of their included studies were excluded from our review as formal diagnostic criteria were not applied at the point of randomisation or because the length of prior antidepressant treatment was less than four weeks. The review included studies examining the atypical antipsychotics risperidone and aripiprazole, whereas these studies did not meet our inclusion criteria. Conversely, Zhou 2015 did not include any evidence for ziprasidone or cariprazine (the one study of cariprazine included in our review was published beyond the search dates of Zhou 2015). Nonetheless, Zhou's findings are broadly consistent with ours ‐ atypical antipsychotics were more efficacious than placebo (SMDs ranged from ‐0.27 to ‐0.43). All, apart from risperidone, had more side effect discontinuations than placebo (ORs ranged from 2.72 to 6.40), but only quetiapine (mean 250 mg to 350 mg daily) had more all‐cause dropouts than placebo (OR = 1.89).
Zhou 2015a also published a systematic review and network meta‐analysis of all pharmacological augmentation agents (evidence was identified for aripiprazole, bupropion, buspirone, lamotrigine, lithium, methylephenidate, olanzpine, pindolol, quetiapine, risperidone, thyroid hormone). TRD was defined as in Zhou 2015. The authors identified one small trial of the anxiolytic buspirone (Appelberg 2001, also included in our review). None of the nine trials of augmentation with lithium compared with placebo met the inclusion criteria for this review (as formal diagnostic criteria were not applied at the point of randomisation or because the length of prior antidepressant treatment was less than four weeks). Four included RCTs of augmentation with pindolol compared with placebo also failed to meet our inclusion criteria.
Edwards 2013 examined the effectiveness of augmenting an SSRI with either lithium or an antipsychotic. TRD was defined as failure to respond to at least two previous antidepressants in the current episode of depression. No continuous outcome of depression severity was reported. Edwards identified evidence for aripiprazole, olanzapine, quetiapine and lithium. We excluded all three aripiprazole studies included in the Edwards review as none applied formal diagnostic criteria at the point of randomisation. Five of six olanzapine studies were excluded from our review for the same reason (Shelton 2005a being the exception). The review found evidence that augmenting current antidepressant therapy with olanzapine increased response rates (OR = 1.60, 95% CI 1.01 to 2.53). This estimate, based on five studies, was more precise than ours. None of the quetiapine studies included in our review (Bauer 2009 (ONYX); El‐Khalili 2010 (PEARL); McIntyre 2007) met the inclusion criteria for Edwards 2013 as only one treatment failure was required for entry in these trials and that treatment did not have to be an SSRI. No studies of ziprasidone were included. Dunner 2007 was excluded by Edwards 2013 as the dosages were high and not used in the UK. Papakostas 2015 was published beyond the dates of their search as was the cariprazine study included in our review (Durgam 2016).
Liu 2015 carried out a systematic review of the effectiveness of augmenting current antidepressant therapy with the beta‐blocker pindolol in participants with a primary diagnosis of unipolar TRD who failed to respond to at least one SSRI compared with continuing on antidepressant therapy (with or without placebo). None of the five trials identified met the inclusion criteria for our review (three did not meet our criteria for TRD, one included bipolar patients, and the mean age of participants was greater than 74 years in the fifth study).
Two systematic reviews examined the effectiveness of switching to another antidepressant. Bschor 2010 defined TRD as depression determined using formal diagnostic criteria and a failure to respond to four weeks or more of antidepressant treatment at standard or higher dose. In addition to the one study included in our review (Ferreri 2001), the authors included two further studies that did not meet our inclusion criteria (both examined a switch to fluoxetine and neither employed formal diagnostic criteria to diagnose depression at the point of randomisation to treatment). Bschor 2010 found no evidence a difference in response to treatment or remission from depression when switching to another antidepressant compared with continuing on current antidepressant treatment (no continuous measure of depression symptoms was considered in the review). Ruhe 2006 examined switching antidepressants after an insufficient response to an SSRI and included six RCTs in their review. One of these was the STAR*D study, from which three levels met the criteria for inclusion in the review. Thus, there were eight randomised comparisons in total. Only one (Ferreri 2001) met our inclusion criteria (seven did not include a comparator arm that continued on current antidepressant therapy alone or with placebo). The review authors' definition of TRD was not described in the paper.
Finally, Ruhe 2006a examined the effectiveness of dose escalating in participants who had failed to respond to a minimum of three weeks of treatment with an SSRI. They identified seven randomised studies, none which met the inclusion criteria for our review (four did not meet our definition of TRD as participants had only had three weeks of prior SSRI treatment, two had no comparator group that continued on the same dose of the SSRI, and one did not apply diagnostic criteria at point of randomisation).
Authors' conclusions
Implications for practice.
This review found moderate‐ to high‐quality evidence that augmenting antidepressant treatment with an antipsychotic – cariprazine (moderate), quetiapine (high) or ziprasidone (moderate) ‐ for individuals with treatment‐resistant depression reduced depressive symptoms over the short term (8 to 12 weeks). Findings from a small study of the antipsychotic olanzapine also suggested augmenting current antidepressant treatment with this drug may reduce symptoms of depression but this study was of low quality. However, those who received adjunctive antipsychotic treatment were more likely to experience adverse events or not tolerate treatment compared with those who received placebo. As such, adjunctive treatment with an antipsychotic may be less acceptable.
There was no evidence that augmenting antidepressant treatment with an anxiolytic, buspirone, reduced symptoms of depression compared with placebo. However, this evidence was judged to be low‐quality.
There was some evidence that augmenting existing antidepressant treatment with a second antidepressant may be beneficial but this was not consistent across different drugs. There was moderate‐quality evidence that adjunctive treatment with mianserin reduced depressive symptoms over the short term (six weeks). However, there was no evidence of improvements in depressive symptoms when existing antidepressant treatment was augmented with mirtazapine compared with placebo.
There was no evidence that switching to a different antidepressant treatment (mianserin) was beneficial compared with placebo. This evidence was judged to be moderate quality due to imprecision in the results.
No evidence was identified for other treatment strategies, such as increasing the dose of existing antidepressant treatment or augmenting with other medications (e.g. lithium or beta‐blockers).
Implications for research.
Only a small number of studies were included in our review and most of these examined the effectiveness of a single treatment strategy ‐ augmenting antidepressant treatment with a non‐antidepressant medication ‐ most commonly, an antipsychotic medication. Evidence is therefore needed on the effectiveness of other pharmacological approaches to the management of treatment‐resistant depression in adults in order to improve outcomes for the large group of patients who do not respond to antidepressant treatment. Future trials need to examine strategies such as switching to a different antidepressant, increasing the dose of antidepressant monotherapy, combining two antidepressant treatments, as well as evidence on other augmentation strategies.
Future trials in this area need to give careful consideration to their design. Many of the trials that were excluded from this review did not apply diagnostic criteria for depression at the point of randomisation. We also excluded a large number of trials as they lacked a suitable comparator. In order to make a meaningful comparison that will inform clinical practice, studies need to include a 'control' group that continues on existing antidepressant monotherapy. In addition, it is important that future trials are independent of the pharmaceutical industry to minimise the potential for bias.
Future studies also need to ensure that they include outcomes that are important to patients (and caregivers), such as quality of life, which were rarely measured in studies included this review. Most of the existing evidence only relates to outcomes over the short term. It is important that future research evaluates longer‐term outcomes and gathers data about resource use in order to inform discussions about allocation of resources based on evidence of both clinical and cost‐effectiveness. Moreover, it is important that adverse effects are consistently recorded in future studies in order to enable patients and clinicians to have an informed discussion about the potential benefits of treatment in light of possible adverse events.
As the evidence base in this area increases, it will be important that researchers compare the effectiveness of different pharmacological interventions with each other, and with psychological interventions. Advances in the development of methods for network meta‐analysis that enable multiple treatment options to be compared simultaneously will provide evidence to further inform policy and practice.
Acknowledgements
We would like to thank Michelle Taylor for her help in assessing full papers for this review.
This research was supported by the National Institute for Health Research (NIHR) Biomedical Research Centre and by the Applied Research Collaboration West (NIHR ARC West) at the University Hospitals Bristol NHS Foundation Trust and the University of Bristol, UK.
The authors and the Cochrane Common Mental Disorders Editorial Team, are grateful to the following peer reviewers for their time and comments: Stephana J. Cherak, Sumeet Gupta, Nuala Livingstone and Gill Worthy. They would also like to thank copy editor, Anne Lethaby.
CRG funding acknowledgement: the National Institute for Health Research (NIHR) is the largest single funder of the CCMD Group.
Disclaimer: the views and opinions expressed therein are those of the review authors and do not necessarily reflect those of the NIHR, the NHS or the Department of Health and Social Care.
Appendices
Appendix 1. MEDLINE (Ovid): CCMD's core search strategy used to inform the Group's specialised register
A weekly search alert based on condition + RCT filter only 1. [MeSH Headings]: eating disorders/ or anorexia nervosa/ or binge‐eating disorder/ or bulimia nervosa/ or female athlete triad syndrome/ or pica/ or hyperphagia/ or bulimia/ or self‐injurious behavior/ or self mutilation/ or suicide/ or suicidal ideation/ or suicide, attempted/ or mood disorders/ or affective disorders, psychotic/ or bipolar disorder/ or cyclothymic disorder/ or depressive disorder/ or depression, postpartum/ or depressive disorder, major/ or depressive disorder, treatment‐resistant/ or dysthymic disorder/ or seasonal affective disorder/ or neurotic disorders/ or depression/ or adjustment disorders/ or exp antidepressive agents/ or anxiety disorders/ or agoraphobia/ or neurocirculatory asthenia/ or obsessive‐compulsive disorder/ or obsessive hoarding/ or panic disorder/ or phobic disorders/ or stress disorders, traumatic/ or combat disorders/ or stress disorders, post‐traumatic/ or stress disorders, traumatic, acute/ or anxiety/ or anxiety, castration/ or koro/ or anxiety, separation/ or panic/ or exp anti‐anxiety agents/ or somatoform disorders/ or body dysmorphic disorders/ or conversion disorder/ or hypochondriasis/ or neurasthenia/ or hysteria/ or munchausen syndrome by proxy/ or munchausen syndrome/ or fatigue syndrome, chronic/ or obsessive behavior/ or compulsive behavior/ or behavior, addictive/ or impulse control disorders/ or firesetting behavior/ or gambling/ or trichotillomania/ or stress, psychological/ or burnout, professional/ or sexual dysfunctions, psychological/ or vaginismus/ or Anhedonia/ or Affective Symptoms/ or *Mental Disorders/
2. [Title/ Author Keywords]: (eating disorder* or anorexia nervosa or bulimi* or binge eat* or (self adj (injur* or mutilat*)) or suicide* or suicidal or parasuicid* or mood disorder* or affective disorder* or bipolar i or bipolar ii or (bipolar and (affective or disorder*)) or mania or manic or cyclothymic* or depression or depressive or dysthymi* or neurotic or neurosis or adjustment disorder* or antidepress* or anxiety disorder* or agoraphobia or obsess* or compulsi* or panic or phobi* or ptsd or posttrauma* or post trauma* or combat or somatoform or somati#ation or medical* unexplained or body dysmorphi* or conversion disorder or hypochondria* or neurastheni* or hysteria or munchausen or chronic fatigue* or gambling or trichotillomania or vaginismus or anhedoni* or affective symptoms or mental disorder* or mental health).ti,kf.
3. [RCT filter]: (controlled clinical trial.pt. or randomized controlled trial.pt. or (randomi#ed or randomi#ation).ab,ti. or randomly.ab. or (random* adj3 (administ* or allocat* or assign* or class* or control* or determine* or divide* or distribut* or expose* or fashion or number* or place* or recruit* or subsitut* or treat*)).ab. or placebo*.ab,ti. or drug therapy.fs. or trial.ab,ti. or groups.ab. or (control* adj3 (trial* or study or studies)).ab,ti. or ((singl* or doubl* or tripl* or trebl*) adj3 (blind* or mask* or dummy*)).mp. or clinical trial, phase ii/ or clinical trial, phase iii/ or clinical trial, phase iv/ or randomized controlled trial/ or pragmatic clinical trial/ or (quasi adj (experimental or random*)).ti,ab. or ((waitlist* or wait* list* or treatment as usual or TAU) adj3 (control or group)).ab.)
4. (1 and 2 and 3)
Records were screened for reports of RCTs within the scope of the Cochrane Common Mental Disorders Group. Secondary reports of RCTs were tagged to the appropriate study record.
Similar weekly search alerts were also be conducted on OVID Embase and PsycINFO, using relevant subject headings (controlled vocabularies) and search syntax, appropriate to each resource.
Appendix 2. Review search strategies
1 MEDLINE (Ovid)
Database: Ovid MEDLINE(R) Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations, Ovid MEDLINE(R) Daily and Ovid MEDLINE(R) <(1 January 2016 to 31 December 2018) >
1 Depressive Disorder, Treatment‐Resistant/ 2 (depress* and ((antidepress* or SSRI* or SNRI* or (serotonin adj3 (uptake or reuptake or re‐uptake)) or medication* or psychotropic or treatment* or respon*) adj2 fail*)).ti,ab,kf. 3 (depress* and ((antidepress* or SSRI* or SNRI* or (serotonin adj3 (uptake or reuptake or re‐uptake)) or psychotropic medication* or treatment*) adj2 ("no respon*" or "not respon*" or nonrespon* or non‐respon* or unrespon*))).ti,ab,kf. 4 (depress* adj3 (refractor* or resistan* or chronic* or persist*)).ti,ab,kf. 5 (depress* adj3 (relaps* or recurr*)).ti,kf. 6 (depress* and (augment* or potentiat*)).mp. 7 or/1‐6 8 randomized controlled trial.pt. or exp randomized controlled trial/ or exp Randomized Controlled Trials as Topic/ 9 controlled clinical trial.pt. 10 (RCT or randomi#ed or at random or (random* adj3 (assign* or allocat* or divide* or division or number*))).ti,ab,kf. 11 ((placebo or sham or mock or fake or dummy) and (control* or group?)).ti,ab,kf. 12 double‐blind*.ti,ab,kf,hw. 13 trial.ti. 14 ((cluster or crossover* or cross‐over*) adj3 (random* or trial or study or control* or group?)).ti,ab,kf. 15 or/8‐14 16 7 and 15 17 letter/ 18 editorial/ 19 news/ 20 exp historical article/ 21 Anecdotes as topic/ 22 comment/ 23 case report/ 24 (letter or comment*).ti. 25 exp animals/ not humans/ 26 exp Animals, Laboratory/ 27 exp Animal Experimentation/ not (exp human experimentation/ or humans/) 28 exp Models, Animal/ 29 exp rodentia/ 30 (rat or rats or mouse or mice or rodent*).ti. 31 or/17‐30 32 16 not 31 33 (2016* or 2017*).yr,dc,ed,ep. 34 (in‐data‐review or in‐process or publisher).st. 35 33 or 34 36 32 and 35
2 Embase (Ovid)
(1 January 2016 to 31 December 2018)
1 treatment resistant depression/ 2 (depress* and ((antidepress* or SSRI* or SNRI* or (serotonin adj3 (uptake or reuptake or re‐uptake)) or medication* or psychotropic or treatment* or respon*) adj2 fail*)).ti,ab,kf. 3 (depress* and ((antidepress* or SSRI* or SNRI* or (serotonin adj3 (uptake or reuptake or re‐uptake)) or psychotropic medication* or treatment*) adj2 ("no respon*" or "not respon*" or nonrespon* or non‐respon* or unrespon*))).ti,ab,kf. 4 (depress* adj3 (refractor* or resistan* or chronic* or persist*)).ti,ab,kf. 5 (depress* adj3 (relaps* or recurr*)).ti,kf. 6 (depress* and (augment* or potentiat*)).mp. 7 or/1‐6 8 randomized controlled trial/ or "randomized controlled trial (topic)"/ 9 crossover procedure/ 10 "double blind procedure"/ 11 "single‐blind procedure"/ 12 (RCT or randomi#ed or at random or (random* adj3 (assign* or allocat* or divide* or division or number*))).ti,ab,kf. 13 trial.ti. 14 ((cluster or crossover* or cross‐over*) adj3 (random* or trial or study or control* or group?)).ti,ab,kf. 15 double‐blind*.ti,ab. 16 ((placebo or sham or mock or fake or dummy) and (control* or group?)).ti,ab,kf. 17 or/8‐16 18 7 and 17 19 letter.pt. or letter/ 20 note.pt. 21 editorial.pt. 22 case report/ or case study/ 23 (letter or comment*).ti. 24 exp animal/ not human/ 25 nonhuman/ 26 exp experimental animal/ 27 exp animal experiment/ 28 exp animal model/ 29 exp rodent/ 30 (rat or rats or mouse or mice or rodent*).ti. 31 or/19‐30 32 18 not 31
3 PsycINFO (Ovid)
(1 January 2016 to 31 December 2018)
1 (depress* and ((antidepress* or SSRI* or SNRI* or (serotonin adj3 (uptake or reuptake or re‐uptake)) or medication* or psychotropic or treatment* or respon*) adj2 fail*)).ti,ab,id. 2 (depress* and ((antidepress* or SSRI* or SNRI* or (serotonin adj3 (uptake or reuptake or re‐uptake)) or psychotropic medication* or treatment*) adj2 ("no respon*" or "not respon*" or nonrespon* or non‐respon* or unrespon*))).ti,ab,id. 3 (depress* adj3 (refractor* or resistan* or chronic* or persist*)).ti,ab,id. 4 (depress* and (augment* or potentiat*)).mp. 5 treatment resistant depression/ 6 (depress* adj3 (relaps* or recurr*)).ti,id. 7 or/1‐6 8 clinical trials/ 9 (RCT or randomi#ed or at random or (random* adj3 (assign* or allocat* or divide* or division or number*))).ti,ab,id. 10 double‐blind*.ti,ab,id,hw. 11 ((placebo or sham or mock or fake or dummy) and (control* or group?)).ti,ab,id. 12 trial.ti. 13 ((cluster or crossover* or cross‐over*) adj3 (random* or trial or study or control* or group?)).ti,ab,id. 14 or/8‐13 15 7 and 14 16 (authored book or book or edited book).pt. 17 scientific communication/ 18 case report/ 19 (letter or comment*).ti. 20 exp animals/ or animal models/ 21 (rat or rats or mouse or mice or rodent*).ti. 22 or/16‐21 23 15 not 22
4 Cochrane Central Register of Controlled Trials (CENTRAL), Issue 12, 2018
(1 January 2016 to 31 December 2018) #1MeSH descriptor: [Depressive Disorder, Treatment‐Resistant] explode all trees #2(depress* and ((antidepress* or SSRI* or SNRI* or (serotonin near/3 (uptake or reuptake or re‐uptake)) or medication* or psychotropic or treatment* or respon*) near/2 fail*)):ti,ab,kw #3(depress* and ((antidepress* or SSRI* or SNRI* or (serotonin near/3 (uptake or reuptake or re‐uptake)) or "psychotropic medication" or "psychotropic medications" or treatment*) near/2 ("no respon*" or "not respon*" or nonrespon* or non‐respon* or unrespon*))):ti,ab,kw #4(depress* near/3 (refractor* or resistan* or chronic* or persist*)):ti,ab,kw #5(depress* near/3 (relaps* or recurr*)):ti,kw #6(depress* and (augment* or potentiat*)):ti,ab,kw #7#1 or #2 or #3 or #4 or #5 or #6
5 Web of Science (Clarivate Analytics)
(1 January 2016 to 31 December 2018)
# 15 #11 not #14 # 14 #13 OR #12 # 13 TS=((animal* near/2 experiment*) or (animal* near/2 model*) or (animal* near/2 laborator*)) # 12 TI= (rat or rats or mouse or mice or rodent* or animal* or comment* or letter or "case study" or "case report" or anecdote* or editorial* or news ) # 11 #10 AND #6 # 10 #9 OR #8 OR #7 # 9 TI= trial # 8 TS= (RCT or randomized or randomised or "at random" or (random* near/3 (assign* or allocat* or divide* or division or number*))) # 7 TS= ((controlled near/2 "clinical trial") or double‐blind* or ((placebo or sham or mock or fake or dummy) and (control* or group?)) or ((cluster or crossover* or cross‐over*) near/3 (random* or trial or study or control* or group?))) # 6 #5 OR #4 OR #3 OR #2 OR #1 # 5 TS=(depress* and (augment* or potentiat*)) # 4 TS=(depress* near/3 (relaps* or recurr*)) # 3 TS=(depress* near/3 (refractor* or resistan* or chronic* or persist*)) # 2 TS=(depress* and ((antidepress* or SSRI* or SNRI* or (serotonin near/3 (uptake or reuptake or re‐uptake)) or "psychotropic medication" or "psychotropic medications" or treatment*) near/2 ("no respon*" or "not respon*" or nonrespon* or non‐respon* or unrespon*))) # 1 TS=((depress* and ((antidepress* or SSRI* or SNRI* or (serotonin near/3 (uptake or reuptake or re‐uptake)) or medication* or psychotropic or treatment* or respon*) near/2 fail*)))
6 International trials registers search (all years)
Types of Study = Interventional
Condition 1 = treatment resistant depression
Condition 2 = refractory depression
Condition 3 = recurrent depression
Condition 4 = chronic depression
Data and analyses
Comparison 1. Switch to mianserin.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Depressive symptoms (HAM‐D) | 1 | 71 | Mean Difference (IV, Fixed, 95% CI) | ‐1.80 [‐5.23, 1.63] |
| 2 Dropouts | 1 | 72 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.08 [0.94, 4.59] |
| 3 Response (≥ 50% improvement in HAM‐D score) | 1 | 71 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.32 [0.76, 2.27] |
| 4 Remission (HAM‐D total score ≤ 8) | 1 | 71 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.97 [0.88, 4.42] |
Comparison 2. Augment current antidepressant with mianserin.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Depressive symptoms (HAM‐D) | 1 | 70 | Mean Difference (IV, Fixed, 95% CI) | ‐4.80 [‐8.18, ‐1.42] |
| 2 Dropouts | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.38, 2.72] |
| 3 Response (≥ 50% reduction in HAM‐D score) | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.70 [1.03, 2.78] |
| 4 Remission (HAM‐D total score ≤ 7) | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.38 [1.09, 5.16] |
Comparison 3. Augment current antidepressant with mirtazapine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Depressive symptoms (BDI‐II) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1 12 weeks | 1 | 431 | Mean Difference (IV, Fixed, 95% CI) | ‐1.70 [‐4.03, 0.63] |
| 1.2 24 weeks | 1 | 402 | Mean Difference (IV, Fixed, 95% CI) | ‐0.90 [‐3.39, 1.59] |
| 1.3 52 weeks | 1 | 388 | Mean Difference (IV, Fixed, 95% CI) | 0.10 [‐2.38, 2.58] |
| 2 Depressive symptoms (PHQ‐9, 12 weeks) | 1 | 429 | Mean Difference (IV, Fixed, 95% CI) | ‐0.89 [‐2.08, 0.30] |
| 3 Dropouts | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 12 weeks | 1 | 480 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.50 [0.15, 1.62] |
| 3.2 24 weeks | 1 | 480 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.50 [0.19, 1.30] |
| 3.3 52 weeks | 1 | 480 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.18, 1.18] |
| 4 Response (≥ 50% improvement in BDI‐II score) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 12 weeks | 1 | 431 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [0.97, 1.54] |
| 4.2 24 weeks | 1 | 402 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.83, 1.23] |
| 4.3 52 weeks | 1 | 388 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.82, 1.22] |
| 5 Remission (BDI‐II total score ≤ 9) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 12 weeks | 1 | 431 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.21 [0.88, 1.65] |
| 5.2 24 weeks | 1 | 402 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.86, 1.55] |
| 5.3 52 weeks | 1 | 388 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.74, 1.30] |
| 6 Quality of life (EQ‐5D‐5L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.1 12 weeks | 1 | 447 | Mean Difference (IV, Fixed, 95% CI) | ‐0.01 [‐0.06, 0.04] |
| 6.2 24 weeks | 1 | 403 | Mean Difference (IV, Fixed, 95% CI) | ‐0.02 [‐0.07, 0.03] |
| 6.3 52 weeks | 1 | 388 | Mean Difference (IV, Fixed, 95% CI) | ‐0.03 [‐0.08, 0.02] |
| 7 Quality of life (SF‐12 ‐ aggregate mental functioning) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 7.1 12 weeks | 1 | 418 | Mean Difference (IV, Fixed, 95% CI) | 3.61 [1.23, 5.99] |
| 7.2 24 weeks | 1 | 392 | Mean Difference (IV, Fixed, 95% CI) | 1.98 [‐0.64, 4.60] |
| 7.3 52 weeks | 1 | 373 | Mean Difference (IV, Fixed, 95% CI) | 1.29 [‐1.44, 4.02] |
| 8 Quality of life (SF‐12 ‐ aggregate physical functioning) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 8.1 12 weeks | 1 | 418 | Mean Difference (IV, Fixed, 95% CI) | ‐1.76 [‐4.20, 0.68] |
| 8.2 24 weeks | 1 | 392 | Mean Difference (IV, Fixed, 95% CI) | ‐2.49 [‐5.04, 0.06] |
| 8.3 52 weeks | 1 | 373 | Mean Difference (IV, Fixed, 95% CI) | ‐0.98 [‐3.61, 1.65] |
3.5. Analysis.
Comparison 3 Augment current antidepressant with mirtazapine, Outcome 5 Remission (BDI‐II total score ≤ 9).
Comparison 4. Augment current antidepressant with buspirone (anxiolytic).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Depressive symptoms (MADRS) | 1 | 102 | Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐9.48, 8.88] |
| 2 Dropouts | 1 | 108 | Risk Ratio (M‐H, Random, 95% CI) | 0.6 [0.23, 1.53] |
Comparison 5. Augment current antidepressant with cariprazine (antipsychotic).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Depressive symptoms (MADRS) | 1 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 1.1 1‐2 mg/d | 1 | 537 | Mean Difference (Fixed, 95% CI) | ‐0.9 [‐2.29, 0.49] |
| 1.2 2‐4.5mg/d | 1 | 535 | Mean Difference (Fixed, 95% CI) | ‐2.1 [‐3.63, ‐0.57] |
| 1.3 Any dose | 1 | 808 | Mean Difference (Fixed, 95% CI) | ‐1.50 [‐2.74, ‐0.25] |
| 2 Dropouts | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 1‐2 mg/d | 1 | 545 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.43 [0.94, 2.17] |
| 2.2 2‐4.5mg/d | 1 | 545 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.92 [1.30, 2.84] |
| 2.3 Pooled dose | 1 | 821 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.68 [1.16, 2.41] |
| 3 Response (≥ 50% improvement in MADRS score) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 1‐2 mg/d | 1 | 537 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [1.03, 1.53] |
| 3.2 2‐4.5mg/d | 1 | 535 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [1.06, 1.57] |
| 3.3 Pooled dose | 1 | 808 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [1.07, 1.52] |
| 4 Remission (MADRS total score ≤ 10) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 1‐2mg/d | 1 | 537 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.83, 1.37] |
| 4.2 2‐4.5mg/d | 1 | 535 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.83, 1.38] |
| 4.3 Pooled data | 1 | 808 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.86, 1.33] |
Comparison 6. Augment current antidepressant with olanzapine (antipsychotic).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Depressive symptoms (MADRS) | 1 | 20 | Mean Difference (IV, Fixed, 95% CI) | ‐12.4 [‐22.44, ‐2.36] |
| 2 Depressive symptoms (HAM‐D) | 1 | 20 | Mean Difference (IV, Random, 95% CI) | ‐7.90 [‐16.76, 0.96] |
| 3 Dropouts | 1 | 20 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.04, 2.69] |
| 4 Response (≥ 50% reduction in MADRS score) | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 6.0 [0.87, 41.21] |
Comparison 7. Augment current antidepressant with quetiapine (antipsychotic).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Depressive symptoms (MADRS or HAM‐D) | 3 | Std. Mean Difference (Random, 95% CI) | Subtotals only | |
| 1.1 150 ‐200 mg/d | 3 | 670 | Std. Mean Difference (Random, 95% CI) | ‐0.34 [‐0.53, ‐0.14] |
| 1.2 300mg/d | 2 | 610 | Std. Mean Difference (Random, 95% CI) | ‐0.33 [‐0.49, ‐0.17] |
| 1.3 Any dose | 3 | 977 | Std. Mean Difference (Random, 95% CI) | ‐0.32 [‐0.46, ‐0.18] |
| 2 Dropouts | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 150 ‐ 200 mg/d | 3 | 684 | Risk Ratio (M‐H, Random, 95% CI) | 1.18 [0.86, 1.63] |
| 2.2 300 mg/d | 2 | 624 | Risk Ratio (M‐H, Random, 95% CI) | 1.82 [1.29, 2.57] |
| 2.3 Any dose | 3 | 997 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.90, 1.95] |
| 3 Response (≥ 50% reduction in MADRS or HAM‐D score) | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 150 ‐200 mg/d | 3 | 673 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [1.02, 1.40] |
| 3.2 300 mg/d | 2 | 610 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [1.10, 1.50] |
| 3.3 Any dose | 3 | 977 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [1.09, 1.43] |
| 4 Quality of life (% max score of Q‐LES‐Q‐SF) | 2 | Mean Difference (Random, 95% CI) | Subtotals only | |
| 4.1 150 mg/d | 2 | 591 | Mean Difference (Random, 95% CI) | 0.70 [‐2.31, 3.71] |
| 4.2 300 mg/d | 2 | 589 | Mean Difference (Random, 95% CI) | 0.35 [‐2.06, 2.77] |
| 4.3 Any dose | 2 | 884 | Mean Difference (Random, 95% CI) | 0.57 [‐1.52, 2.65] |
| 5 Remission (MADRS score ≤ 8/HAM‐D score ≤ 7) | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 150 ‐ 200 mg/d | 3 | 673 | Risk Ratio (M‐H, Random, 95% CI) | 1.51 [1.18, 1.92] |
| 5.2 300 mg/d | 2 | 610 | Risk Ratio (M‐H, Random, 95% CI) | 1.53 [1.13, 2.07] |
| 5.3 Any dose | 3 | 977 | Risk Ratio (M‐H, Random, 95% CI) | 1.53 [1.22, 1.90] |
Comparison 8. Augment current antidepressant with ziprasidone (antipsychotic).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Depressive symptoms (HAM‐D) | 2 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 1.1 80 mg/d | 1 | 41 | Mean Difference (Fixed, 95% CI) | ‐0.91 [‐4.84, 3.02] |
| 1.2 40 ‐ 160 mg/d (mean 98 mg/d) | 1 | 139 | Mean Difference (Fixed, 95% CI) | ‐3.1 [‐5.19, ‐1.01] |
| 1.3 160 mg/d | 1 | 39 | Mean Difference (Fixed, 95% CI) | ‐2.56 [‐6.72, 1.60] |
| 1.4 Any dose | 2 | 199 | Mean Difference (Fixed, 95% CI) | ‐2.73 [‐4.53, ‐0.93] |
| 2 Depressive symptoms (MADRS) | 1 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 2.1 80 mg/d | 1 | 41 | Mean Difference (Fixed, 95% CI) | ‐1.53 [‐6.94, 3.88] |
| 2.2 160 mg/d | 1 | 39 | Mean Difference (Fixed, 95% CI) | ‐3.82 [‐9.64, 2.00] |
| 2.3 Any dose | 1 | 60 | Mean Difference (Fixed, 95% CI) | ‐2.62 [‐7.47, 2.23] |
| 3 Depressive symptoms (QIDS‐SR) | 1 | 139 | Mean Difference (IV, Fixed, 95% CI) | ‐2.5 [‐3.83, ‐1.17] |
| 3.1 40 ‐ 160 mg/d (mean 98 mg/d) | 1 | 139 | Mean Difference (IV, Fixed, 95% CI) | ‐2.5 [‐3.83, ‐1.17] |
| 4 Dropouts | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 80 mg/d | 1 | 41 | Risk Ratio (M‐H, Random, 95% CI) | 2.10 [0.89, 4.96] |
| 4.2 160 mg/d | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 2.11 [0.88, 5.03] |
| 4.3 40 ‐ 160 mg/d (mean 98 mg/d) | 1 | 139 | Risk Ratio (M‐H, Random, 95% CI) | 1.40 [0.80, 2.47] |
| 4.4 Any dose | 2 | 199 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [1.01, 2.55] |
| 5 Response (≥ 50% reduction in MADRS/HAM‐D score) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 80 mg/d (MADRS) | 1 | 41 | Risk Ratio (M‐H, Random, 95% CI) | 1.90 [0.39, 9.28] |
| 5.2 160 mg/d (MADRS) | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 3.16 [0.72, 13.76] |
| 5.3 40 ‐ 160 mg/d (mean 98 mg/d) (HAM‐D) | 1 | 139 | Risk Ratio (M‐H, Random, 95% CI) | 1.71 [0.97, 3.00] |
| 5.4 Any dose | 2 | 199 | Risk Ratio (M‐H, Random, 95% CI) | 1.80 [1.07, 3.04] |
| 6 Response (≥ 50% reduction in QIDS‐SR) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 40 ‐ 160 mg/d (mean 98 mg/d) | 1 | 139 | Risk Ratio (M‐H, Random, 95% CI) | 2.34 [1.16, 4.72] |
| 7 Remission (MADRS/HAM‐D) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 80 mg/d | 1 | 41 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.06, 14.22] |
| 7.2 160 mg/d | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 4.21 [0.52, 34.36] |
| 7.3 40 ‐ 160 mg/d (mean 98 mg/d) | 1 | 139 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [0.77, 1.96] |
| 7.4 Any dose | 2 | 199 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [0.81, 2.00] |
| 8 Remission (QIDS‐SR) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 40 ‐ 160 mg/d (mean 98 mg/d) | 1 | 139 | Risk Ratio (M‐H, Random, 95% CI) | 2.33 [1.03, 5.25] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Appelberg 2001.
| Methods | Parallel group RCT | |
| Participants |
Country: Finland Setting: Outpatient care N randomised: 108 (n = 64 (62.7%) female (of 102 participants included in analyses)) Age: Mean 44 years (SD not reported; range 18 ‐ 61) Inclusion: Patients being treated or seeking treatment for depression, 18 years of age or older, meeting DSM‐IV criteria for a major depressive episode Exclusion: Psychotic or bipolar depression, being regarded by the clinician in charge of treatment to be seriously suicidal, severe neurologic or somatic disease, mental disorder due to a general medical condition, substance‐induced disorders, and other psychiatric disorders (except generalised anxiety disorder and specific phobias) |
|
| Interventions | Group 1: Augmentation of current SSRI with buspirone (20 mg/d ‐ 60 mg/d) for six weeks Group 2: Augmentation of current SSRI with placebo for six weeks |
|
| Outcomes | Post‐treatment assessment (6 weeks): 1. Depressive symptoms, continuous (MADRS) 2. Dropouts |
|
| Definition of TRD | Had received fluoxetine or citalopram for at least 6 weeks without showing an antidepressant response according to the psychiatrist in charge of treatment. For at least the last 4 weeks before inclusion, their daily fluoxetine dose was at least 30 mg or their daily citalopram dose at least 40 mg | |
| Notes | Funding: Supported by a grant from Bristol‐Myers Quibb, Finland. Respone to treatment (defined as an improvement in CGI‐S score ≥ 2 points; data not included in this review as the CGI‐S is a generic measure of illness severity rather than specifically measuring depression symptomology) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised but no information given about how the sequence was generated |
| Allocation concealment (selection bias) | Unclear risk | No information about how allocation was concealed. "After a single‐blind placebo wash‐in period of 2 weeks while continuing their SSRI, the patients were randomly assigned to adjunctive treatment with buspirone … or placebo". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The study was designed as a placebo‐controlled, double‐blind ….. Buspirone and placebo were administered as tablets that were identical in appearance". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The investigators, but not the patients, were aware of the placebo wash‐in period. The double‐blind phase began immediately after baseline and lasted for 6 weeks … Buspirone and placebo were administered as tablets that were identical in appearance". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | LOCF used to account for missing data. Six participants (3 from each arm) were excluded from the analyses as no observations available. "The primary assessment was based on last observation carried forward (LOCF) according to the principle of intention to treat .... One hundred and thirteen patients were enrolled in the study … Five patients were removed from the study during the placebo wash‐in phase ... Six more patients (3 in the buspirone and 3 in the placebo group) were removed from the study because of a protocol violation before week 1 ... One hundred and two patients had at least 1 evaluation after placebo wash‐in phase and were included in the final analysis." |
| Selective reporting (reporting bias) | High risk | No protocol available. MADRS scores reported as percentage mean change and reported for weeks 1 and 6 only. Mean scores for all time points are shown on a graph (with no measure of precision). |
| Other bias | Low risk | No other concerns identified |
Bauer 2009 (ONYX).
| Methods | Parallel group RCT | |
| Participants |
Country: Australia, Canada, Europe, South Africa Setting: Outpatient care N randomised: (n = 329 (67.6%) female (of 487 included in analyses)) Age: Mean 45.44 years (SD 10.53). Inclusion: Patients aged 18 to 65 years with a documented DSM‐IV‐TR diagnosis of MDD (single episode [296.2x] or recurrent [296.3x]). Diagnoses confirmed by the Mini‐International Neuropsychiatric Interview. HAM‐D‐17 total score > 20 and HAM‐D item 1 (depressed mood) score ≥ 2 at enrolment and randomisation. Exclusion: Any DSM‐IV Axis I disorder other than MDD within 6 months prior to enrolment; any DSM‐IV Axis II disorder significantly impacting the patient’s current psychiatric status; duration of current MDD episode > 12 months or < 4 weeks from enrolment; substance or alcohol abuse or dependence, as defined by DSM‐IV criteria, within 6 months prior to enrolment; any clinically significant medical illness, such as renal or hepatic impairment, or coronary artery disease; conditions that could affect absorption or metabolism of study medication risk of suicide or homicide; HAM‐D item 3 score of ≥ 3; or suicide attempt within the past 6 months. Patients requiring psychotherapy (other than supportive therapy), unless psychotherapy had been ongoing for ≥ 3 months before randomisation. Drugs that induce or inhibit the hepatic metabolising cytochrome P450 3A4 enzymes within 2 weeks prior to randomisation. Quetiapine > 25 mg/d for insomnia within 7 days before randomisation, known lack of response following 4 weeks’ treatment with quetiapine ≥ 50 mg/d for depression or were receiving quetiapine ≥ 50 mg/d at enrolment |
|
| Interventions | Group 1: Augmentation of current antidepressant with quetiapine XR (150 mg/d) for six weeks Group 2: Augmentation of current antidepressant with quetiapine XR (300 mg/d) for six weeks Group 3: Augmentation of current antidepressant with placebo for six weeks |
|
| Outcomes | Post‐treatment assessment (6 weeks): 1. Depressive symptoms, continuous (MADRS, HAM‐D) 2. Dropouts 3. Response, dichotomous (≥ 50% reduction in MADRS total score from randomisation) 4. Remission, dichotomous (MADRAS total score ≤ 8) 5. Quality of life, continuous (Q‐LES‐Q‐SF) 6. Adverse events |
|
| Definition of TRD | Inadequate response during the current episode to amitriptyline, bupropion, citalopram, duloxetine, escitalopram, fluoxetine, paroxetine, sertraline or venlafaxine given for ≥ 6 weeks at adequate doses (minimum effective dose according to label and including at least 1 dose increase as permitted by label) | |
| Notes | Funding: Study funded by AstraZeneca | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised but no information given about how the randomisation sequence was generated. "eligible patients ... were randomly assigned (1:1:1 ratio) to receive 6 weeks of double‐blind treatment with 1 of 3 treatment regimens". |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to enable a judgment ''eligible patients ... were randomly assigned ..". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind and placebo given to control group so unlikely that either participants or study personnel would have been aware of treatment allocation. "This was a 6‐week, randomized, double‐blind, parallel‐group, placebo‐controlled, phase III, double‐dummy study." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Not specifically stated that outcome assessors were blind to treatment allocation but study described as double‐blind and interventions that were identical in appearance."'This was a 6‐week, randomized, double‐blind, parallel‐group, placebo‐controlled, phase III, double‐dummy study." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | LOCF used to account for missing data. MITT analysis carried out. Six randomised participants could not be included in it. Missing data balanced across groups. "Efficacy analyses were based on the MITT [modified intention to treat] population; a last‐observation‐carried‐forward (LOCF) approach was used for missing data ... modified intent‐to‐treat (MITT) population (all patients assigned to randomized treatment who took study medication and who had a MADRS assessment at randomization and at least 1 valid MADRS assessment after randomization)." |
| Selective reporting (reporting bias) | Unclear risk | Very limited information in the protocol about which outcomes will be assessed but all those mentioned were reported in the paper. Additional outcomes reported that did not appear in the protocol (e.g. adverse events) |
| Other bias | Low risk | No other concerns identified |
Dunner 2007.
| Methods | Parallel group RCT | |
| Participants |
Country: US Setting: Outpatients N randomised: 64 (n = 31 (50.8%) female (of 61 included in analyses)) Age: Mean 44 years (SD 11) Inclusion: Adult outpatients aged 21 to 65 years meeting DSM‐IV criteria for MDD and MADRS total score ≥ 20 Exclusion: Current DSM‐IV diagnosis of any psychotic disorder, post‐traumatic stress disorder, panic disorder, or obsessive‐compulsive disorder; DSM‐IV substance abuse or dependence disorder in the past 3 months; history of treatment with an atypical antipsychotic agent; treatment with fluoxetine, a monoamine oxidase inhibitor, or electroconvulsive therapy during the 6 weeks prior to study entry; any clinically significant abnormality on electrocardiogram (ECG); current therapy with medications known to prolong the corrected QT interval; any acute or unstable medical illness; pregnancy or breast feeding |
|
| Interventions | Group 1: Augmentation of sertraline with ziprasidone (160 mg) for eight weeks Group 2: Augmentation of sertraline with ziprasidone (80 mg) for eight weeks Group 3: Sertraline for eight weeks |
|
| Outcomes | Post‐treatment assessment (8 weeks): 1. Depressive symptoms, continuous (MADRS, HAM‐D) 2. Dropouts 3. Response, dichotomous (≥ 50% decrease from baseline MADRS score) 4. Remission, dichotomous (MADRS score ≤ 10) 5. Adverse events |
|
| Definition of TRD | Nonresponse to at least 1 course of treatment of at least 4 weeks’ duration with a clinically appropriate dose of an SSRI or non‐SSRI antidepressant. Also confirmed prospectively by a failure to achieve at least a 30% decrease in MADRS score and a CGI‐S score ≥ 4 following a 6‐week open‐label sertraline (100 to 200 mg/d) treatment period | |
| Notes | Funding: Study funded by Pfizer Inc | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised but no information given about how the randomisation sequence was generated. "Subjects who continued to meet TRD criteria at the conclusion of the 6‐week sertraline lead‐in period were randomly assigned, in a 1:1:1 ratio". |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to make a judgment. "Subjects who continued to meet TRD criteria at the conclusion of the 6‐week sertraline lead‐in period were randomly assigned, in a 1:1:1 ratio". |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial so likely that participants and those delivering the intervention knew their treatment allocation. "After completion of the 6‐week lead‐in study, nonresponders were randomized to open‐label treatment with 8 weeks of …" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors were blinded to participants' treatment assignment. ''The clinician who rated efficacy was blind to the subject's treatment assignment, and, whenever possible, the rater who conducted a subject's baseline assessments rated that subject at end point." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Three randomised participants discontinued prior to taking the study drug and were not included in the analyses. LOCF used to account for missing data for the remaining participants. Minor discrepancy between the text and the table with regards to numbers of participants (text stated 61 but table reported data for 60 participants) |
| Selective reporting (reporting bias) | Unclear risk | No study protocol available. All outcomes detailed in the methods are reported in the study results. |
| Other bias | Low risk | No other concerns identified |
Durgam 2016.
| Methods | Parallel group RCT | |
| Participants |
Country: USA, Estonia, Finland, Slovakia, Sweden, Ukraine Setting: Outpatients N randomised: 819 (n = 578 (71.2%) female (of 812 included in the safety population)) Age: 45.7 years (mean 11.6) Inclusion: Outpatients, aged 18‐65 years, meeting DSM‐IV‐TR criteria for MDD without psychotic features and had a current depressive episode (duration ≥ 8 weeks to ≤ 24 months). MADRS total score ≥ 22, BMI between 18 and 40, normal results from physical examinations, laboratory tests and electrocardiogram or abnormal results that were not considered clinically significant Exclusion: Principal Axis I disorder other than MDD, or any Axis I disorder other than MDD that was the primary focus of treatment within 6 months of screening; and Axis II disorder that might interfere with participation and judged by the investigator; alcohol/substance abuse or dependence within 6 months of screening; or lifetime history of depressive episodes with psychotic or catatonic features, bipolar disorder, schizophrenia or other psychotic disorder, panic disorder, obsessive‐compulsive disorder, anorexia or bulimia, and dementia or other cognitive disorder, suicide risk. Refractory depression (inadequate response ≥ 3 antidepressants at sufficient dosages and duration for the current episode); antidepressant augmentation with any medication within 1 week of baseline (up to 4 weeks for some medications) or 5 half‐lives of the medication concomitant use of antipsychotics, anxiolytics, or stimulants; or psychotherapy for depression within 3 months of screening. |
|
| Interventions | Group 1: Augmentation of current antidepressant with cariprazine (1 mg/d to 2 mg/d) for eight weeks Group 2: Augmentation of current antidepressant with cariprazine (2 mg/d to 4.5 mg/d) for eight weeks Group 3: Augmentation of current antidepressant with placebo for eight weeks |
|
| Outcomes | Post‐treatment assessment (8 weeks): 1. Depressive symptoms, continuous (MADRS) 2. Dropouts 3. Response, dichotomous (≥ 50% decrease from baseline MADRS score) 4. Remission, dichotomous (MADRS score ≤ 10) 5. Adverse events |
|
| Definition of TRD | Ongoing inadequate response during the current episode to antidepressant treatment for ≥ 6 weeks at recommended doses per label guidelines | |
| Notes | Funding: Supported by funding from Forest Laboratories, LLC (Jersey City, New Jersey), an Allergan affiliate and Gedeon Richter Plc (Budapest, Hungary) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Sequence generated by web based software. "Patient identification and randomization codes were generated by an interactive voice/web system". |
| Allocation concealment (selection bias) | Low risk | Centralised allocation using a web‐based system. "Patient identification and randomization codes were generated by an interactive voice/web system, and study medication was identical in appearance". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Placebo‐administered. Study participants reported to be blind to treatment allocation. "Patients and study staff were blinded throughout treatment … and study medication was identical in appearance. Breaking the blind resulted in discontinuation from the study." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Placebo‐administered. Study staff reported to be blind to treatment allocation. "Patients and study staff were blinded throughout treatment … and study medication was identical in appearance. Breaking the blind resulted in discontinuation from the study". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT population included all those with at least one post‐baseline assessment. LOCF used. 11 randomised participants not included in the ITT population. 1 and 5 from the cariprazine 1 mg/d to 2 mg/d and 2 mg/d to 4.5 mg/d groups, and 5 from the placebo group. No significant imbalance between arms. "The safety population included all patients who received ≥ 1 dose of double‐blind study drug. The intent‐to‐treat population included all patients in the safety population who had ≥ 1 post‐baseline MADRS total score assessment." "The safety population included 812 patients 4 of whom (2 each for placebo and cariprazine 2 mg/d to 4.5 mg/d) did not have a post‐baseline MADRS assessment and were excluded from efficacy analyses". |
| Selective reporting (reporting bias) | Unclear risk | Very limited information in the protocol about which outcomes would be assessed but all those mentioned were reported in the paper. Additional outcomes reported that did not appear in the protocol (e.g. adverse events, response/remission) |
| Other bias | Low risk | No other concerns identified |
El‐Khalili 2010 (PEARL).
| Methods | Parallel group RCT | |
| Participants |
Country: US Setting: Outpatients N randomised: 446 (n = 313 (72.5%) female (of 432 included in the analyses)) Age: Mean 44.5 years (SD 12.8) Inclusion: Patients aged 18 to 65 years with a documented DSM‐IV‐TR diagnosis of MDD (single episode [296.2x] or recurrent [296.3x], confirmed by the Mini‐International Neuropsychiatric Interview). HAM‐D‐17 total score > 20 and HAM‐D item 1 (depressed mood) score ≥ 2 at enrolment and randomisation Exclusion: Any DSM‐IV Axis I disorder other than MDD within 6 months prior to enrolment; any DSM‐IV Axis II disorder significantly impacting the patient’s current psychiatric status; duration of current MDD episode > 12 months or < 4 weeks from enrolment; history of substance or alcohol abuse or dependence, as defined by DSM‐IV criteria, within 6 months prior to enrolment; clinically significant medical illness; risk of suicide or homicide; HAM‐D item 3 score of ≥ 3; or suicide attempt within the past 6 months; requiring psychotherapy (other than supportive therapy), unless psychotherapy had been ongoing for ≥ 3 months before randomisation |
|
| Interventions | Group 1: Augmentation of current antidepressant with quetiapine XR (150 mg/d) for six weeks Group 2: Augmentation of current antidepressant with quetiapine XR (300 mg/d) for six weeks Group 3: Augmentation of current antidepressant with placebo for six weeks |
|
| Outcomes | Post‐treatment assessment (6 weeks): 1. Depressive symptoms, continuous (MADRS, HAM‐D) 2. Dropouts 3. Response, dichotomous (≥ 50% reduction in MADRS total score from randomisation) 4. Remission, dichotomous (MADRS total score <= 8) 5. Quality of life, continuous (Q‐LES‐Q‐SF) 6. Adverse events |
|
| Definition of TRD | Inadequate response during the current episode to amitriptyline, bupropion, citalopram, duloxetine, escitalopram, fluoxetine, paroxetine, sertraline or venlafaxine given for ≥ 6 weeks at adequate doses (minimum effective dose according to label and including at least 1 dose increase as permitted by label) | |
| Notes | Funding: Study funded by AstraZeneca | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation sequence used. "Patients were randomized (in a 1:1:1 ratio) in a non‐centre‐specific manner using a computer‐based system". |
| Allocation concealment (selection bias) | Low risk | Sequence generation done by computer so would not have been possible to anticipate. Study treatments administered in identical packaging. "Patients were randomized (in a 1:1:1 ratio) in a non‐centre‐specific manner using a computer‐based system … Quetiapine XR 50 mg and 300 mg tablets were identical in appearance, smell, and taste to their respective placebo tablets". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study described as double‐blind. Identical interventions administered to participants. Not specifically stated that study personnel were blind to treatment but likely due to the use of interventions that were identical in appearance. "Quetiapine XR 50 mg and 300 mg tablets were identical in appearance, smell, and taste to their respective placebo tablets". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Study described as double‐blind. Not specifically stated that outcome assessors were blind to treatment but likely due to the use of interventions that were identical in appearance. "Quetiapine XR 50 mg and 300 mg tablets were identical in appearance, smell, and taste to their respective placebo tablets". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | LOCF used to account for missing data for all but 14 participants. Slightly larger number missing from the quetiapine 300 mg/d group. "The modified intent‐to‐treat (MITT) population (randomized patients who received study drug, and had randomization and ≥ 1 post‐randomization MADRS total scores) was used for analysis of primary and secondary variables. For all efficacy analyses, missing data were handled using a last observation carried forward (LOCF) approach. 446 patients were randomised .... and 432 were analysed for efficacy (MITT population) after 13 patients were excluded due to missing/invalid randomization or post‐randomization MADRS scores." |
| Selective reporting (reporting bias) | Low risk | All outcomes listed in the protocol were reported. Missing standard deviations for mean change from baseline in depression symptoms (but these could be calculated from other information provided) |
| Other bias | Low risk | No other concerns identified |
Ferreri 2001.
| Methods | Parallel group RCT | |
| Participants |
Country: France Setting: Inpatients and outpatients N randomised: 104 Age: Mean 46.6 years (SD 12.6) Inclusion: Diagnosis of major depression according to DSM‐III‐R and have scores ≥ 25 on the Hamilton Depression Rating Scale 17 items (HAM‐D) and scores ≥ 28 on the HARD diagram scale [Mood‐Anxiety‐Retardation‐Danger (Humeur‐Angoisse‐Ralentissement‐Danger)], after at least 6 weeks of treatment with 20 mg/day of fluoxetine. Both in‐ and outpatients were included. Exclusion: Pregnant or lactating women, women without contraception at childbearing age, somatic disease condition that could interfere with everyday life activities, psychiatric disorders with psychotic features (with the exception of psychotic mood disorder), unreliable patients, in particular drug and alcohol abuse and dependence, treatment with psychotropic drugs other than fluoxetine, with the exception of benzodiazepines if benzodiazepine equivalent dosage was equal to or lower than 15 mg/day of diazepam |
|
| Interventions | Group 1: Augmentation of current antidepressant (fluoxetine 20 mg/d) with mianserin (60 mg/d) for six weeks Group 2: Switched treatment to Mianserin (60 mg/d) given with placebo for six weeks Group 3: Augmentation of current antidepressant (fluoxetine 20 mg/d) with placebo for six weeks |
|
| Outcomes | Post‐treatment assessment (6 weeks): 1. Depressive symptoms, continuous (HAM‐D‐17) 2. Dropouts 3. Response, dichotomous (≥ 50% reduction in HAM‐D score from baseline) 4. Remission, dichotomous (HAM‐D total score ≤ 8) 5. Adverse events |
|
| Definition of TRD | Inadequate response after at least 6 weeks of treatment with 20 mg/d of fluoxetine | |
| Notes | Funding: Study funded by Organon Pharmaceuticals, France | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Limited information about how sequence was generated but likely random. "Patients were randomized according to a randomization schedule by blocks of four up to 120." |
| Allocation concealment (selection bias) | Unclear risk | No information about how allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind and placebo given to control group so unlikely that either participants or study personnel would have been aware of treatment allocation. "Placebo of fluoxetine was provided in the form of identical capsules and, similarly, placebo of mianserin and mianserin were also indistinguishable." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Not specifically stated that outcome assessors were blind to treatment allocation but study described as double‐blind and interventions were identical in appearance. "Placebo of fluoxetine was provided in the form of identical capsules and, similarly, placebo of mianserin and mianserin were also indistinguishable." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT population included all participants who had taken at least one dose of the study drug. LOCF used to account for missing data. "For missing data the method of Last Observation Carried Forward was used … The intent‐to‐treat population consisted of all randomized patients who took at least one dose of a study drug … One patient was lost at day 0; thus, the intent‐to‐treat population consisted of 103 patients treated." |
| Selective reporting (reporting bias) | Unclear risk | No study protocol available. All outcomes detailed in the methods were reported in the study results. |
| Other bias | Low risk | No other concerns identified |
Kessler 2018.
| Methods | Parallel group RCT | |
| Participants |
Country: UK Setting: Primary care N randomised: 480 Age: 50.15 years (SD 13.16) Inclusion: Aged 18‐75 years. Treated for depression with any of the following SSRI or SNRI antidepressants for at least 6 weeks at recommended (British National Formulary, BNF) doses: fluoxetine, sertraline, citalopram, escitalopram, fluvoxamine, paroxetine, duloxetine or venlafaxine; scored 14 or more on the Beck Depression Inventory (BDI) and have adhered to their medication and meet ICD‐10 criteria for depression (assessed using the Computerised Interview Schedule – Revised version (CIS‐R). Exclusion: Currently taking combined or augmented antidepressant treatment, having their medication managed by a psychiatrist, dementia (formal diagnosis), bipolar disorder, psychosis or alcohol or substance abuse/dependence, pregnant, planning a pregnancy or breastfeeding, unable to complete the study questionnaires, previous adverse reaction to mirtazapine, currently being treated with a monoamine oxidase inhibitor (MAOI), including moclobemide, or with other medical contraindications to mirtazapine |
|
| Interventions | Group 1: Augmentation of current antidepressant with mirtazapine (30 mg/d) for up to 52 weeks Group 2: Augmentation of current antidepressant with placebo for up to 52 weeks |
|
| Outcomes | Assessed at 12, 24 and 52 weeks (unless specified): 1. Depressive symptoms, continuous (BDI‐II, PHQ‐9) 2. Dropouts 3. Response, dichotomous (≥ 50% reduction in BDI‐II total score from baseline) 4. Remission, dichotomous (BDI‐II < 10) 5. Social functioning (SF‐12 aggregate mental functioning, SF‐12 aggregate physical functioning) 6. Quality of life, continuous (EQ‐5D‐5L) 7. Economic outcomes (health and social care use, costs to patients/carers, time off work) 8. Adverse events (12 and 52 weeks) |
|
| Definition of TRD | Inadequate response to fluoxetine, sertraline, citalopram, escitalopram, fluvoxamine, paroxetine, duloxetine or venlafaxine given for at least six weeks at BNF recommended dose and adherent to antidepressant treatment | |
| Notes | Funding: National Institute for Health Research (NIHR) Health Technology Assessment programme, UK | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation sequence generated by a computer. "Participants were randomised using the automated randomisation service provided by the Bristol Randomised Trials Collaboration (BRTC). Randomisation was carried out by means of a computer‐generated code to ensure concealment of allocation." |
| Allocation concealment (selection bias) | Low risk | Centralised allocation provided by an independent organisation using a computer‐generated code. "Participants were randomised using the automated randomisation service provided by the Bristol Randomised Trials Collaboration (BRTC). Randomisation was carried out by means of a computer‐generated code to ensure concealment of allocation". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Placebo‐administered. Study participants reported to be blind to treatment allocation. "Participants were randomly assigned to one of two treatments: (1) 1 × 15 mg encapsulated mirtazapine tablet daily for 2 weeks followed by 2 × 15 mg encapsulated mirtazapine tablets for up to 50 weeks or (2) identical placebo tablets…. Participants, clinicians, outcome assessors and the research team were blinded to allocation." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Placebo‐administered. Study staff reported to be blind to treatment allocation. "Participants were randomly assigned to one of two treatments: (1) 1 × 15 mg encapsulated mirtazapine tablet daily for 2 weeks followed by 2 × 15 mg encapsulated mirtazapine tablets for up to 50 weeks or (2) identical placebo tablets…. Participants, clinicians, outcome assessors and the research team were blinded to allocation." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analyses carried out. "The primary comparative analyses of clinical effectiveness were conducted according to the principle of intention to treat (ITT) …. A number of sensitivity analyses were conducted to assess the impact of missing primary outcome data on the main findings." |
| Selective reporting (reporting bias) | Unclear risk | PHQ‐9 measured at 12, 24 and 52 weeks but only data for 12 weeks was reported. |
| Other bias | Low risk | No other concerns identified |
McIntyre 2007.
| Methods | Parallel group RCT | |
| Participants |
Country: Canada Setting: Outpatient N randomised: 58 Age: 44.5 years (SD 11) Inclusion: Adults (18–65 years of age) with a DSM‐IV diagnosis of major depression; a 17‐item Hamilton Depression Scale (HAM‐D) score of ≥ 18; a Clinical Global Impression of Severity (CGI‐S) score of ≥ 4 (moderately ill); and a 14‐item Hamilton Anxiety Scale (HAM‐A) score of ≥ 14. These criteria had to be met both at screening and baseline. In addition, all patients had been treated for their current episode of major depressive disorder with a single SSRI/venlafaxine tablet at a therapeutic dose for at least 6 weeks. Exclusion: Exclusion criteria included: a DSM‐IV diagnosis of substance abuse or dependence within 6 months of screening, and patients who received an antipsychotic or benzodiazepine 7 days prior to entering the study, or a potent cytochrome P450 inhibitor or inducer 14 days prior to entering the study. Patients who were pregnant, breastfeeding, or at risk of suicide in the investigator’s opinion were also excluded. |
|
| Interventions | Group 1: Augmentation of SSRI/venlafaxine with quetiapine (mean 182 mg/d) for 8 weeks Group 2: Augmentation of SSRI/venlafaxine with placebo for 8 weeks |
|
| Outcomes | Post‐treatment assessment (8 weeks): 1. Depressive symptoms, continuous (HAM‐D‐17) 2. Dropouts 3. Response, dichotomous (≥ 50% reduction in HAM‐D score from baseline) 4. Remission, dichotomous (HAM‐D total score ≤ 7) 5. Adverse events |
|
| Definition of TRD | Inadequate response to at least six weeks of a single SSRI or venlafaxine at an acceptable therapeutic dose | |
| Notes | Funding: Study funded by AstraZeneca | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised but no information given about how the sequence was generated |
| Allocation concealment (selection bias) | Unclear risk | No information about how allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind and placebo given to control group so unlikely that either participants or study personnel would have been aware of treatment allocation |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Not specifically stated that outcome assessors were blind to treatment allocation but study described as double‐blind and placebo given to control group |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All randomised participants were included in the analyses. "The intent‐to‐treat (ITT) population (any randomized patient who received at least one dose of study medication) was used in all statistical analyses... LOCF analyses are reported for the above measures". |
| Selective reporting (reporting bias) | Unclear risk | Study protocol did not provide details of the outcomes to be assessed. All outcomes and time points described in the methods were reported in the results. |
| Other bias | Low risk | No other concerns identified |
Papakostas 2015.
| Methods | Parallel group RCT | |
| Participants |
Country: US Setting: Outpatient N randomised: 139 (n = 98 (70.5%) female)) Age: Mean 44.5 years (SD 12.8) Inclusion: men and women 18–65 years of age with a primary diagnosis of current major depressive disorder according to DSM‐IV criteria and confirmed with the Structured Clinical Interview for DSM‐IV and a score >= 10 on the 16‐item Quick Inventory of Depressive Symptomatology–Self‐Rated (QIDS‐SR) at screening Exclusion: Patients were considered ineligible for participation in the study if they were pregnant, breastfeeding, or lactating or if they were women of childbearing potential who were not using a medically accepted means of contraception; high risk of suicide or homicide or other reasons the investigator might deem a patient unsafe to enrol in the study; an unstable medical illness or an uncontrolled seizure disorder; a history of multiple adverse reactions or an allergy to the study drug; and a DSM‐IV diagnosis of a substance use disorder (including alcohol) active within the past 6 months, bipolar disorder (current or past), or any psychotic disorder or psychotic symptoms (current or past). Patients who had received an adequate trial of escitalopram during the current major depressive episode (prior to study entry) or any lifetime trial of ziprasidone were also excluded, as were patients who had received an investigational psychotropic drug within 3 months of their screening visit. In addition, patients who had failed to benefit after more than three antidepressant trials of adequate dose and duration during the current major depressive episode were excluded. |
|
| Interventions | Group 1: Augmentation of escitalopram with ziprasidone (40 mg/d to 160 mg/d) for eight weeks Group 2: Augmentation of escitalopram with placebo for eight weeks |
|
| Outcomes | Post‐treatment assessment (8 weeks): 1. Depressive symptoms, continuous (HAM‐D, QIDS‐SR) 2. Dropouts 3. Response, dichotomous (≥ 50% reduction in MADRS total score from randomisation; ≥ 50% reduction in QIDS‐SR score from randomisation) 4. Remission, dichotomous (HAM‐D score ≤ 7; QIDS‐SR score ≤ 5) 5. Adverse events |
|
| Definition of TRD | Confirmed prospectively by failure to respond to an 8‐week open‐label trial of escitalopram (10 mg/d to 30 mg/d). Continuing to meet DSM‐IV criteria for MDD and had a QIDS‐SR score ≥10 | |
| Notes | Funding: Supported by a National Institute of Mental Health (NIMH) grant, Pfizer (which supplied blinded ziprasidone and placebo pills), and Forest Laboratories (which supplied escitalopram) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation sequence. "Patients were randomly assigned to receive adjunctive ziprasidone or placebo, in a 1:1 ratio, throughout the remainder of the study. A central randomization center used a computer‐generated list of random numbers to allocate treatments." |
| Allocation concealment (selection bias) | Low risk | Centralised allocation, adequately concealed. "A central randomization center used a computer‐generated list of random numbers to allocate treatments. None of the investigators, study clinicians, clinical raters, or participants at any of the study sites had access to the randomization list". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and study personnel were blind to treatment allocation due to the use of a placebo. "Ziprasidone (20 mg per capsule) and placebo were in capsule form and identical in appearance. Independent pharmacists prepared ziprasidone and placebo capsules. Study drugs were prepackaged in bottles and consecutively numbered for each patient according to the randomization schedule." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors were kept blind to treatment allocation. "None of the investigators, study clinicians, clinical raters, or participants at any of the study sites had access to the randomization list. Ziprasidone (20 mg per capsule) and placebo were in capsule form and identical in appearance. Independent pharmacists prepared ziprasidone and placebo capsules. Study drugs were prepackaged in bottles and consecutively numbered for each patient according to the randomization schedule." |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 37 participants reported as having dropped out. ITT analyses carried out (including all 139 randomised participants). No information about how any missing data was handled. "All analyses used the intent‐to‐treat data set (including all randomized patients)." |
| Selective reporting (reporting bias) | Unclear risk | All outcomes listed in the clinical trial registry entry were reported in the paper. Authors reported carrying out additional analyses; they reported an additional measure QIDS‐SR for secondary outcomes compared with what was listed in the protocol. |
| Other bias | Low risk | No other concerns identified |
Shelton 2001.
| Methods | Parallel group RCT | |
| Participants |
Country: US Setting: Outpatients N randomised: 28 (8 of whom randomised to a treatment arm not included in this review) (n = 21 (75%) female (of 28 included in whole study)) Age: Mean 42 years (SD 11) Inclusion: Outpatients who met DSM‐IV criteria for recurrent major depression without psychotic features and were resistant to conventional antidepressant pharmacotherapy Exclusion: History of psychosis, dysthymic disorder, or bipolar disorder |
|
| Interventions | Group 1: Augmentation of fluoxetine with olanzapine (5 mg/d to 20 mg/d) for eight weeks Group 2: Augmentation of fluoxetine with placebo for eight weeks (Other arms not relevant to this review: olanzapine (5 mg/d to 20 mg/d) plus placebo for eight weeks) |
|
| Outcomes | Post‐treatment assessment (8 weeks): 1. Depressive symptoms, continuous (MADRS, HAM‐D) 2. Dropouts 3. Response, dichotomous (≥ 50% reduction in MADRS total score from randomisation) 4. Adverse events |
|
| Definition of TRD | History of failure to respond to antidepressants of two different classes, one of which was not an SSRI, after at least 4 weeks of therapy at an acceptable therapeutic dose. Failure to respond was confirmed prospectively during a six‐week screening period in which fluoxetine (20 mg/d to 60 mg/d) was given. At entry, patients were required to score ≥ 20 on the 21‐item Hamilton Depression Rating Scale and still meet DSM‐IV criteria (confirmed by study author). | |
| Notes | Funding: Supported in part by Eli Lilly and Company | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stated randomised but sequence generation not described. "Non‐responders to fluoxetine were randomly assigned in a 1:1:1 ratio". |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to enable a judgment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind. Placebo used in the arm that did not receive augmentation with antipsychotic. "The second phase was an 8‐week double‐blind trial in which non‐responders to fluoxetine were randomly assigned in a 1:1:1 ratio to receive … fluoxetine plus placebo ('fluoxetine') or olanzapine plus fluoxetine ('combination')". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Described as double‐blind. Placebo used in the arm that did not receive augmentation with antipsychotic |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants were analysed in the group to which they were randomised with LOCF used to account for missing data. 'Analyses were performed on an intent‐to‐treat basis…. The last observation carried forward was used to analyse weekly visits". |
| Selective reporting (reporting bias) | High risk | HAM‐D reported to be the primary outcome in the published paper. Dichotomous data not given for HAM‐D but were for MADRS. FDA documentation suggested that MADRS was a post hoc analysis and that the ‘response’ outcome was originally based on reduction in HAM‐D score. |
| Other bias | Low risk | No other concerns identified |
BDI and BDI‐II: Beck Depression Inventory BMI: body mass index BNF: British National Formulary BRTC: Bristol Randomised Trials Collaboration CGI‐S: Clinical Global Impressions ‐ Severity DSM‐III‐R: Diagnostic and Statistical Manual of Mental Disorders (version 3, revised). DSM‐IV and DSM‐IV‐TR: Diagnostic and Statistical Manual of Mental Disorders (version 4, text revision). ECG: electrocardiogram EQ‐5D‐5L: EuroQoL 5 level version FDA: Food and Drug Administration HAM‐A: Hamilton Anxiety Rating Scale HAM‐D and HAM‐D‐17: Hamilton Depression Rating Scale HARD: Humeur‐Angoisse‐Ralentissement‐Danger diagramme [Mood‐Anxiety‐Retardation‐Danger depression scale] ICD‐10: International Classification of Diseases ITT: intention‐to‐treat LOCF: last observation carried forward MADRS: Montgomery Asberg Depression Rating Scale MAOI: monoamine oxidase inhibitor MDD: major depressive disorder MITT: modified intention‐to treat PHQ‐9: Patient Health Questionnaire QIDS‐SR: Quick Inventory of Depressive Symptomatology ‐ Self Report Q‐LES‐Q‐SF: Quality of Life Enjoyment and Satisfaction Questionnaire ‐ Short Form RCT: randomised controlled trial SD: standard deviation SF‐12: Short‐Form SNRI: serotonin‐norepinephrine reuptake inhibitor SSRI: selective serotonin reuptake inhibitor
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Aarnell 2002 | Not TRD (no duration criteria defined for prior antidepressant treatment) |
| ADAPT‐A 2012 | Diagnostic criteria not applied at point of randomisation |
| Baca 2005 | Not TRD (included participants that were intolerant to AD) |
| Barbee 2011 | Diagnostic criteria not applied at point of randomisation |
| Bares 2013 | Compared two switching strategies. No control comparison of continuing on initial antidepressant monotherapy |
| Bauer 2013 (RUBY) | Compared switching to an antipsychotic (not a treatment strategy covered by the review) with augmenting with quetiapine or augmenting with lithium. No control comparison of continuing on initial antidepressant monotherapy |
| Bauer 2018 | Diagnostic criteria not applied at point of randomisation |
| Benkert 1997 | Not TRD (prior antidepressant therapy for 3 weeks only). Not MDD (included patients with minor depression) |
| Berman 2007 | Diagnostic criteria not applied at point of randomisation |
| Berman 2009 | Diagnostic criteria not applied at point of randomisation |
| Birkenhager 2004 | Compared two switching strategies. No control comparison of continuing on initial antidepressant monotherapy. |
| Bose 2012 | Compared dose increase with switch. No control comparison of continuing on initial antidepressant monotherapy |
| Brecht 2011 | Not TRD (MDD only were randomised, non‐responders during trial all got a dose increase) |
| Chaput 2004 | Not a comparison of interest. Compared switch to cognitive behavioural therapy plus quetiapine augmentation to CBT plus placebo. No control comparison of continuing on initial antidepressant monotherapy |
| Cheon 2017 | Compared two augmentation strategies. No control comparison of continuing on initial antidepressant monotherapy |
| Cipriani 2013 | Not TRD (dose and duration of prior antidepressant treatment not specified) |
| Corya 2006 | Diagnostic criteria not applied at point of randomisation |
| Doree 2007 | Compared augmentation with lithium with augmentation with quetiapine. No control comparison of continuing on initial antidepressant monotherapy |
| e‐Therapeutics 2013 | Compared two switching strategies. No control comparison of continuing on initial antidepressant monotherapy. |
| Earley 2018 | Diagnostic criteria not applied at point of randomisation |
| Edwards 1998 | Not TRD (dose and duration of prior antidepressant not defined) |
| Fang 2010 | Diagnostic criteria not applied at point of randomisation |
| Fang 2011 | Not TRD (medical history‐based definition of TRD, no dose or duration for prior AD defined) |
| Fava 1994 | Three‐arm trial that compared increasing dose of current antidepressant with two augmentation strategies (desipramine or lithium). No control comparison of continuing on initial antidepressant monotherapy |
| Fava 2001 | Not TRD (included patients who were intolerant as well as resistant) |
| Fava 2002 | Three‐arm trial that compared increasing dose of current antidepressant with two augmentation strategies (desipramine or lithium). No control comparison of continuing on initial antidepressant monotherapy |
| Fava 2018 | Diagnostic criteria not applied at point of randomisation |
| Fornaro 2014 | Compared two switching strategies. No control comparison of continuing on initial antidepressant monotherapy |
| Franco‐Chaves 2013 | Compared two switching strategies and combination treatment. No control comparison of continuing on initial antidepressant monotherapy |
| Furukawa 2011 | Not TRD (3 weeks duration of prior AD treatment) |
| Garry 1963 | Not TRD (all treatments for depression were discontinued for two weeks prior to randomisation into the trial) |
| Gonul 1999 | Compared two augmentation strategies. No control comparison of continuing on initial antidepressant monotherapy |
| Grant 2017 | Compared dose escalation with a switching strategy. No control comparison of continuing on initial antidepressant monotherapy |
| Han 2015 | Compared augmenting current antidepressant treatment with an antipsychotic and switching to another antidepressant. No control comparison of continuing on initial antidepressant monotherapy |
| Hides 2006 | Not TRD (participants had major depression which had not responded to CBT, participants had no prior antidepressant treatment) |
| Hobart 2018 | Diagnostic criteria not applied at point of randomisation |
| Horikoshi 2017 | Compared two augmentation strategies. No control comparison of continuing on initial antidepressant monotherapy |
| Joffe 1993 | Diagnostic criteria not applied at point of randomisation |
| Joffe 2006 | Not TRD (range of doses reported for previous antidepressant use included subtherapeutic dose) |
| Kamijima 2013 | Diagnostic criteria not applied at point of randomisation |
| Kamijima 2018 | Diagnostic criteria not applied at point of randomisation |
| Keitner 2009 | Not TRD (only three weeks of prior antidepressant treatment) |
| Kennedy 2003 | Compared cognitive therapy and lithium augmentation of current antidepressant treatment. No control comparison of continuing on initial antidepressant monotherapy |
| Kocsis 1988 | Not TRD (participants had chronic depression and had failed to respond to two weeks treatment with imipramine) |
| Lejoyeux 2015 | Not a comparison of interest. Compared three tapering strategies. No control comparison of continuing on initial antidepressant monotherapy |
| Lenox‐Smith 2008 | Compared two switching strategies. No control comparison of continuing on initial antidepressant monotherapy |
| Lenze 2015 (IRL GREY) | Diagnostic criteria not applied at point of randomisation |
| Li 2006 | Compared augmentation of current antidepressant therapy with lithium and switching to another antidepressant. No control comparison of continuing on initial antidepressant monotherapy |
| Li 2013 | Compared two switching strategies. No control comparison of continuing on antidepressant monotherapy |
| Licht 2002 | Diagnostic criteria not applied at point of randomisation |
| Maes 1996 | Not TRD at the point of randomisation. Participants who had not responded to previous antidepressant medication underwent a ten‐day washout followed by a week of trazadone. Participants who then had HAM‐D ≥ 14 were randomised to augment with pindolol, fluoxetine or placebo. |
| Maes 1999 | Not TRD (no dose or duration criteria defined) |
| Marcus 2008 | Diagnostic criteria not applied at point of randomisation |
| Marwood 2017 | Compared two augmentation strategies. No control comparison of continuing on initial antidepressant monotherapy |
| Mattingly 2006 | Not TRD (dosage criteria for prior antidepressant treatment not defined) |
| Medhus 1994 | Not TRD (no dosage criteria defined, "presumed adequate dosage") |
| Mohamed 2017 (VAST‐D) | Compared switching to another antidepressant with augmenting with an antidepressant. No control comparison of continuing on initial antidepressant monotherapy |
| Montgomery 2014 (REVIVE) | Compared two switching strategies. No control comparison of continuing on initial antidepressant monotherapy |
| Moreno 1997 | Not all participants had received an antidepressant at the manufacturer's minimum recommended therapeutic dose. |
| Nakajima 2011 | Not TRD (run‐in phase with antidepressant was two weeks duration at which point non‐responders were randomised) |
| NCT00273624 | Study terminated early. No results available. TRD defined as two courses of antidepressants from different classes for at least three weeks at an adequate dose |
| NCT00296517 | Not a comparison of interest. Nonresponders to paroxetine had their dose tapered down and augmented with either bupropion or placebo. |
| NCT01052077 | Diagnostic criteria not applied at point of randomisation |
| NCT02272517 | Compared two switching strategies. No control comparison of continuing on initial antidepressant monotherapy |
| NCT02960763 | Compared an augmentation strategy with switching to another antidepressant. No control comparison of continuing on initial antidepressant monotherapy |
| NCT02977299 | Compared augmentation with aripiprazole with augmentation with rTMS. No control comparison of continuing on initial antidepressant monotherapy |
| Nierenberg 2003 | Diagnostic criteria not applied at point of randomisation |
| Nolen 1993 | Not TRD (only three weeks of prior antidepressant treatment) |
| Perahia 2008 | Compared two switching strategies (immediate switch versus immediate switch with tapering down of original antidepressant). No control comparison of continuing on initial antidepressant monotherapy |
| Perez 1999 | TRD defined as failure to respond to a minimum of six weeks of antidepressant treatment, however dose was only required to have been fixed at a therapeutic level for the final two weeks. |
| Petrescu 2014 | Compared dose escalation with switching to another antidepressant. No control comparison of continuing on initial antidepressant monotherapy |
| Poirier 1999 | Compared two switching strategies. No control comparison of continuing on initial antidepressant monotherapy |
| Rapaport 2006 (ARISe‐RD) | Diagnostic criteria not applied at point of randomisation |
| Ravindran 2006 | Compared adjunctive olanzapine with adjunctive risperidone. No control comparison of continuing on initial antidepressant monotherapy |
| Rele 2015 | Compared three switching strategies. No control comparison of continuing on initial antidepressant monotherapy |
| Romera 2010 | Compared two switching strategies (early or delayed switch). No control comparison of continuing on initial antidepressant monotherapy |
| Ruhe 2009 | Diagnostic criteria not applied at point of randomisation |
| Russell 1995 | Not treatment‐resistant depression. Participants with chronic depression (with or without dysthymia, 'double depression') were randomised to sertraline or imipramine (i.e. not TRD). In a second part of the study, those who did not respond were crossed over to the other treatment (i.e. not randomised). Responders continued in a maintenance study. |
| Rybakowski 1999 | Compared augmentation with lithium to augmentation with carbamazine. No control comparison of continuing on initial antidepressant monotherapy |
| Santos 2008 | Participants had a history of TRD. Study did not state that treatment resistance had to be in the current episode. TRD defined as failure to respond to a minimum of two antidepressants of different classes at the maximum‐tolerated dose for at least six weeks |
| Schindler 2007 | Compared two augmentation strategies. No control comparison of continuing on initial antidepressant monotherapy |
| Schweizer 1990 | TRD defined as failure to respond to fluoxetine 20 mg/day for 3 weeks |
| Schweizer 2001 | TRD defined as failure to respond to three weeks of antidepressant therapy |
| Shelton 2005a | Compared two augmentation strategies. No control comparison of continuing on initial antidepressant monotherapy |
| Shelton 2005b | Diagnostic criteria not applied at point of randomisation |
| Small 1981 | Not TRD. Included participants who had received lower than recommended tricyclic antidepressant dose who had been unable to tolerate side effects |
| Song 2007 | Compared two switching strategies. No control comparison of continuing on initial antidepressant monotherapy |
| Souche 1991 | Diagnostic criteria not applied at point of randomisation |
| Souery 2011 | Phase 1: compared two switching strategies (no control group). Phase 2: not randomised |
| STAR*D 2004 | Evaluated a sequence of different treatment options for depression. Participants who failed to respond to treatment were randomised into the next phase of the study. Continuation of current antidepressant therapy was not a treatment option at any phase of the study. There was, therefore, no control comparison of continuing on initial antidepressant monotherapy. |
| Stein 1993 | Not TRD (only three weeks of prior antidepressant treatment) |
| Sun 2004 | Described as 'refractory depression' but participants had not received prior treatment for their depression |
| Sunderland 1994 | Not TRD (patients were drug‐free for three weeks before randomisation) |
| Tanghe 1997 | Not TRD (no dose or duration criteria for prior AD treatment) |
| Thase 2000 | Compared switching to mirtazapine with switching to sertraline. No control comparison of continuing on initial antidepressant monotherapy |
| Thase 2006 | Not TRD (included participants intolerant to medication) |
| Thase 2007 | Diagnostic criteria not applied at point of randomisation |
| Thase 2011 | Diagnostic criteria not applied at point of randomisation |
| Thase 2015 (PYXIS) | Diagnostic criteria not applied at point of randomisation |
| Thase 2015a (POLARIS) | Diagnostic criteria not applied at point of randomisation |
| Yang 2005 | Compared two switching therapies. No control comparison of continuing on initial antidepressant monotherapy |
| Yoshimura 2012 | Compared two switching strategies. No control comparison of continuing on initial antidepressant monotherapy |
| Yoshimura 2014 | Compared three augmentation strategies. No control comparison of continuing on initial antidepressant monotherapy |
| Zhao 2003 | Compared two switching strategies. No control comparison of continuing on initial antidepressant monotherapy |
| Zusky 1988 | Not TRD. Included participants unable to tolerate manufacturer's recommended dose |
AD: Antidepressant CBT: Cognitive behavioural therapy MDD: Major depressive disorder rTMS: Repetitive transcranial magnetic stimulation TRD: Treatment‐resistant depression
Characteristics of studies awaiting assessment [ordered by study ID]
Cao 2005.
| Methods | Randomised controlled trial |
| Participants | Chinese depression diagnosis. Failure to respond to two antidepressants for at least six weeks |
| Interventions | 1. Venlafaxine 2. Venlafaxine and quetiapine |
| Outcomes | Measured at 1, 2, 4 and 6 weeks. 1. Depression severity HAM‐D 2. Treatment‐emergent symptom scales (TESS) 3. Treatment efficacy (dichotomous outcome) |
| Notes | No information about the dose of prior antidepressants reported and no response received from study authors. Unclear whether there was a control group that continued on current antidepressant therapy |
Clunie 2001.
| Methods | Randomised controlled trial |
| Participants | Depressed patients (no information about method of diagnosis) who failed to respond to clomipramine |
| Interventions | 1. Phenelzine 2. Moclobemide |
| Outcomes | Not reported |
| Notes | Trial protocol from National Institute for Health Research Research Registry. No publications relating to the study identified. No information about method of diagnosis or duration and dose of prior treatment. Unclear whether there was a control group that continued on current antidepressant therapy. No response from contact person listed. |
Euctr‐002130‐11‐Es 2007.
| Methods | Randomised controlled trial |
| Participants | Diagnosis of depression according to DSM‐IV‐TR criteria, failure to respond to SSRI at correct dose for a minimum of 6 weeks, score of >= 14 of HAM‐D‐17 |
| Interventions | 1. Venlafaxine 75 mg to 150 mg 2. Lithium carbonate 400 mg 3. Nortriptyline 25 mg 4. Control group |
| Outcomes | Primary outcome measure: HAM‐D‐17 |
| Notes | Trial registry entry. No publications relating to the study identified. Study authors could not be identified to enable requests for further information. Unclear whether interventions were given as monotherapy or in addition to current antidepressant treatment. Details of control condition also unclear |
Gulrez 2012.
| Methods | Randomised controlled trial |
| Participants | Diagnosis of MDD according to DSM‐IV‐TR criteria, HAM‐D score of >= 16 after 4 weeks of treatment with an SSRI |
| Interventions | 1. Bupropion, starting at 150 mg/d and increasing to 300 mg/d after 1 week, in addition to SSRI 2. Placebo in addition to SSRI |
| Outcomes | Change in HAM‐D and MADRS scores Remission: HAM‐D score <= 7 or MADRS score <= 12 Spontaneously reported adverse events |
| Notes | Dose of previous SSRI treatment could not be determined from the paper and no response received from corresponding study author. |
Moica 2018.
| Methods | Randomised controlled trial |
| Participants | Major depressive disorder, single or recurrent, without psychotic symptoms and an inadequate response to the antidepressant therapy (the use of minimal doses accepted as effective for a period of at least 4 to 6 weeks). At the time of the enrolment, all participants were treated with duloxetine 60 mg per day. |
| Interventions | 1. Current antidepressant treatment (duloxetine 60 mg/d) augmented with quetiapine (150 mg/d) 2. Continue on duloxetine (60 mg/d) monotherapy |
| Outcomes | Change in HAM‐D score |
| Notes | Unclear how long participants had been receiving current antidepressant (duloxetine) treatment. Also unclear if/when formal diagnostic criteria applied. No response received from corresponding study author |
Zhu 2003.
| Methods | Randomised controlled trial |
| Participants | Diagnosis of depression according to CCMD‐3 (Chinese Classification of Mental Disorders). Failure to respond to 6 weeks of treatment with an SSRI. HAM‐D score >= 18 |
| Interventions | 1. Mirtazapine (30 mg/d to 45 mg/d) for 6 weeks 2. Continue on original antidepressant therapy for 6 weeks |
| Outcomes | Measured at 1, 2, 4 and 6 weeks 1. Depression severity (HAM‐D) 2. Treatment‐emergent symptom scales (TESS) |
| Notes | Details of treatment resistance could not be determined. Unable to contact study author |
CCMD‐3: Chinese Classification of Mental Disorders version 3 DSM‐IV‐TR: Diagnostic and Statistical Manual of Mental Disorders HAM‐D: Hamilton Depression Rating Scale HAM‐D‐17: Hamilton Depression Rating Scale‐17 MADRS: Montgomery Asberg Depression Rating Scale SSRI: selective serotonin reuptake inhibitor TESS: Treatment Emergent Symptoms Scale
Differences between protocol and review
In the protocol, we defined the comparator intervention as 'antidepressant medication ‐ either continuing on the initial antidepressant monotherapy or another antidepressant.'. Hence, switching to another antidepressant was listed in the protocol as both an intervention and comparator of interest. In order to make a meaningful comparison, studies need to include a group that continue on existing antidepressant monotherapy. We therefore decided not to consider switching to another antidepressant a suitable control condition and amended our inclusion criteria accordingly.
We also stated that missing data would be addressed in additional analyses assuming best (all who dropped out had positive outcomes) and worst (all who dropped out had negative outcomes) case scenarios. However, we later agreed that this was not necessary because study level data were more robust than participant level data imputations determined by review authors.
Contributions of authors
NW drafted the protocol, which was finalised following comments from all review authors. PD, SI and NW screened abstracts. PD, SI, NW, CW, DK and GL assessed full papers for eligibility. PD and SI extracted data from included studies. PD carried out the analyses and wrote the first draft of the review, which all review authors commented on. CW died in April 2018. Update searches were run, screening and data extraction were carried out after this date and substantive changes to the review made.
Sources of support
Internal sources
Cochrane Common Mental Disorders Review Group, UK.
External sources
-
NIHR ARC West, UK.
This research was supported by the National Institute for Health Research Applied Research Collaboration West (NIHR ARC West). The views expressed in this article are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health and Social Care.
-
NIHR Biomedical Research Centre at the University Hospitals Bristol NHS Foundation Trust and the University of Bristol, UK, UK.
This research was also supported by the NIHR Biomedical Research Centre at University Hospitals Bristol NHS Foundation Trust and the University of Bristol. The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care.
Declarations of interest
GL has received payments for lectures from pharmaceutical companies.
PD, SI, DK, and NW have no conflicts to declare.
DK was the chief investigator for the MIR study (Kessler 2018); GL and NW were co‐investigators.
Author CW deceased: declarations of interest published in the protocol (Williams 2013)."CW has no conflicts to declare".
Deceased
New
References
References to studies included in this review
Appelberg 2001 {published data only}
- Appelberg BG, Syvalahti E, Muhonen T, Koskinen T, Naukkarinen H, Mehtonen OP. Patients with severe depression may benefit from buspirone augmentation of selective SSRIs. 153rd Annual Meeting of the American Psychiatric Association, 2000 May 13‐18; Chicago (IL). 2000:NR470. [DOI] [PubMed]
- Appelberg BG, Syvalahti EK, Koskinen TE, Mehtonen OP, Muhonen TT, Naukkarinen HH. Patients with severe depression may benefit from buspirone augmentation of selective serotonin reuptake inhibitors: results from a placebo‐controlled, randomized, double‐blind, placebo wash‐in study. Journal of Clinical Psychiatry 2001;62(6):448‐52. [DOI] [PubMed] [Google Scholar]
Bauer 2009 (ONYX) {published data only}
- Bauer M, Pretorius HW, Constant EL, Earley WR, Szamosi J, Brecher M. Extended‐release quetiapine as adjunct to an antidepressant in patients with major depressive disorder: results of a randomized, placebo‐controlled, double‐blind study. Journal of Clinical Psychiatry 2009;70:540‐9. [DOI] [PubMed] [Google Scholar]
- NCT00351910. A multi‐centre, double‐blind, randomised, parallel‐group, placebo‐controlled phase III study of the efficacy and safety of quetiapine fumarate sustained release (Seroquel SRTM) in combination with an antidepressant in the treatment of patients with major depressive disorder with inadequate response to an antidepressant treatment. clinicaltrials.gov/show/NCT00351910 (first received 13 July 2006).
Dunner 2007 {published data only}
- Dunner D, Amsterdam JD, Shelton RC, Loebel A. Adjunctive ziprasidone for resistant depression: 8‐week pilot study. 45th Annual NCDEU (New Clinical Drug Evaluation Unit) Meeting; 2005 June 6 ‐ 9; Boca Raton (FL). 2005:123.
- Dunner DL, Amsterdam JD, Shelton RC, Hassman HA, Rosenthal M, Romano SJ. Adjunctive ziprasidone in treatment‐resistant depression: a pilot study. 156th Annual Meeting of the American Psychiatric Association; 2003 May 17‐22; San Francisco (CA). 2003:NR238.
- Dunner DL, Amsterdam JD, Shelton RC, Loebel A, Romano SJ. Efficacy and tolerability of adjunctive ziprasidone in treatment‐resistant depression: a randomized, open‐label, pilot study. Journal of Clinical Psychiatry 2007;68(7):1071‐7. [DOI] [PubMed] [Google Scholar]
- Dunner DL, Amsterdam JD, Shelton RC, Romano SJ, Loebel A. Adjunctive ziprasidone in treatment‐resistant depression: randomized, double‐blind, 8‐week pilot study. Neuropsychopharmacology 2004;29 Suppl 1:S99. [Google Scholar]
- Romano SJ, Amsterdam JD, Shelton RC, Hassman H, Rosenthal MH, Dunner DL. Adjunctive ziprasidone in treatment‐resistant depression: pilot study. 156th Annual Meeting of the American Psychiatric Association; 2003 May 17‐22 San Francisco (CA). 2003:170.
Durgam 2016 {published data only}
- Durgam S, Earley W, Guo H, Li D, Nemeth G, Laszlovszky I, et al. Efficacy and safety of adjunctive cariprazine in inadequate responders to antidepressants: a randomized, double‐blind, placebo‐controlled study in adult patients with major depressive disorder. Journal of Clinical Psychiatry 2016;77(3):371. [DOI] [PubMed] [Google Scholar]
- Fava M, Durgam S, Mergel V, Earley W, Nemeth G, Laszlovszky I. Efficacy and safety of cariprazine as adjunctive therapy in major depressive disorder: a double‐blind, placebo‐controlled study. Neuropsychopharmacology 2014;39(Suppl 1):S352‐3. [Google Scholar]
El‐Khalili 2010 (PEARL) {published data only}
- El‐Khalili N, Joyce M, Atkinson S, Buynak R. Adjunctive extended‐release quetiapine fumarate (quetiapine XR) in patients with major depressive disorder and inadequate antidepressant response. 161st Annual Meeting of the American Psychiatric Association; 2008 May 3‐8; Washington (DC). 2008:129.
- El‐Khalili N, Joyce M, Atkinson S, Buynak RJ, Datto C, Lindgren P, et al. Extended‐release quetiapine fumarate (quetiapine XR) as adjunctive therapy in major depressive disorder (MDD) in patients with an inadequate response to ongoing antidepressant treatment: a multicentre, randomized, double‐blind, placebo‐controlled study. International Journal of Neuropsychopharmacology 2010;13:917‐32. [DOI] [PubMed] [Google Scholar]
- NCT00326105. A multicenter, double‐blind, randomized, parallel‐group, placebo‐controlled phase III study of the efficacy and safety of quetiapine fumarate sustained release (Seroquel SRTM) in combination with an antidepressant in the treatment of patients with major depressive disorder with inadequate response to an antidepressant treatment (PEARL STUDY). clinicaltrials.gov/show/NCT00326105 (first received 16 May 2006).
Ferreri 2001 {published data only}
Kessler 2018 {published data only}
- ISRCTN06653773. Mirtazapine for treatment resistant depression. isrctn.com/ISRCTN06653773 (first received 20 September 2012).
- Kessler D, Burns A, Tallon D, Lewis G, MacNeill S, Round J, et al. Combining mirtazapine with SSRIs or SNRIs for treatment‐resistant depression: the MIR RCT. Health Technology Assessment 2018; Vol. 22, issue 63:1‐136. [DOI] [PMC free article] [PubMed]
- Kessler DS, MacNeill SJ, Tallon D, Lewis G, Peters TJ, Hollingwoth W, et al. Mirtazapine added to SSRIs or SNRIs for treatment resistant depression in primary care: phase III randomised placebo controlled trial (MIR). BMJ 2018; Vol. 363:k4218. [DOI] [PMC free article] [PubMed]
- Tallon D, Wiles N, Campbell J, Chew‐Graham C, Dickens C, Macleod U, et al. Mirtazapine added to selective serotonin reuptake inhibitors for treatment‐resistant depression in primary care (MIR trial): study protocol for a randomised controlled trial. Trials 2016;17:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
McIntyre 2007 {published data only}
- McIntyre A, Gendron A, McIntyre A. Quetiapine adjunct to selective serotonin reuptake inhibitors or venlafaxine in patients with major depression, comorbid anxiety, and residual depressive symptoms: a randomized, placebo‐controlled pilot study. Depression & Anxiety 2007; Vol. 24, issue 7:487‐94. [DOI] [PubMed]
Papakostas 2015 {published data only}
- Ionescu D, Baer L, Shelton R, Papakostas G. Ziprasidone vs. placebo augmentation of escitalopram for patients with vs. without anxious depression. Neuropsychopharmacology 2015;40(Suppl 1):S172‐3. [Google Scholar]
- Ionescu DF, Shelton RC, Baer L, Meade KH, Swee MB, Fava M, et al. Ziprasidone augmentation for anxious depression. International Clinical Psychopharmacology 2016;31(6):341‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iovieno N, Shelton RC, Petrie SR, Cusin C, Fava M, Papakostas GI. Efficacy of ziprasidone augmentation of escitalopram for cognitive symptoms of major depressive disorder. Journal of Clinical Psychiatry 2018;79(1):55‐9. [DOI] [PubMed] [Google Scholar]
- Mischoulon D, Shelton RC, Baer L, Bobo WV, Curren L, Fava M, et al. Ziprasidone augmentation of escitalopram for major depressive disorder: cardiac, endocrine, metabolic, and motoric effects in a randomized, double‐blind, placebo‐controlled study. Journal of Clinical Psychiatry 2017;78(4):449‐55. [DOI] [PubMed] [Google Scholar]
- NCT00633399. Ziprasidone augmentation of SSRIs for patients with major depressive disorder (MDD) that do not sufficiently respond to treatment with SSRIs. clinicaltrials.gov/show/NCT00633399 (first received 12 March 2008).
- Papakostas G, Fava M, Mischoulon D, Shelton R. Ziprasidone augmentation of escitalopram for major depressive disorder: Safety and tolerability. Neuropsychopharmacology 2015;40(Suppl 1):S146‐7. [Google Scholar]
- Papakostas G, Swee MB, Baer L, Shelton RC. An 8‐week, randomized, double‐blind, placebo‐controlled trial of adjunctive ziprasidone in patients with major depressive disorder receiving treatment with escitalopram. Neuropsychopharmacology 2014;39(Suppl 1):S344‐5. [Google Scholar]
- Papakostas GI, Fava M, Baer L, Swee MB, Jaeger A, Bobo WV, et al. Ziprasidone augmentation of escitalopram for major depressive disorder: efficacy results from a randomized, double‐blind, placebo‐controlled Study. American Journal of Psychiatry 2015;172:1251‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Shelton 2001 {published and unpublished data}
- Shelton RC, Tohen M, Stahl S, Jacobs T, Gannon KS, Tollefson GD. The study of olanzapine plus fluoxetine in treatment‐resistant major depressive disorder without psychotic features. Schizophrenia Research. 2000; Vol. Number 1 ‐ Special Issue:193.
- Shelton RC, Tollefson GD, Tohen M, Stahl S, Gannon KS, Jacobs TG, et al. A novel augmentation strategy for treating resistant major depression. American Journal of Psychiatry 2001;158:131‐4. [DOI] [PubMed] [Google Scholar]
- Tohen M, Shelton R, Tollefson GD, Stahl S, Jacobs T, Gannon KS. Olanzapine plus fluoxetine: double‐blind and open‐label results in treatment‐resistant major depressive disorder. European Neuropsychopharmacology 1999;9(Suppl 5):246. [Google Scholar]
- Tollefson G, Gannon K, Jacobs T, Shelton R, Tohen M, Stahl S. The study of olanzapine plus fluoxetine in treatment‐resistant major depressive disorder without psychotic features. XI World Congress of Psychiatry ; 1999 April 6‐11; Hamburg (Germany) 1999;Abstracts Volume II:133. [Google Scholar]
- Tollefson GD, Shelton R, Tohen M, Stahl S, Jacobs T, Buras W, et al. Efficacy of olanzapine, fluoxetine and combination therapy in treatment‐resistant major depressive disorder without psychotic features. European Neuropsychopharmacology 1998;8(Suppl 2):S178. [Google Scholar]
- Tollefson GD, Shelton RC, Tohen MF, Stahl SM, Jacobs L, Gannon KS. The study of olanzapine plus fluoxetine in treatment‐resistant MDD without psychotic features. 152nd Annual Meeting of the American Psychiatric Association; 1999 May 15‐20; Washington (DC). 1999:NR455.
References to studies excluded from this review
Aarnell 2002 {published data only}
- Aarnell G, Borjesson A, Eder DN, Olausson B, Hetta J. Treatment resistant major depression; a comparison of fluoxetine/olanzapine combination therapy with venlafaxine. International Journal of Neuropsychopharmacology 2002;5(Suppl 1):141. [Google Scholar]
- Aarnell G, Borjesson AM, Eder DN, Olausson B, Hetta J. Treatment resistant major depression. XIIth World Congress of Psychiatry; 2002 August 24‐29; Yokohama, Japan. 2002.
ADAPT‐A 2012 {published data only}
- Dording C, Cassiello C, King IVF, Pencina M, Fava M, Mischoulon D. The effects of aripiprazole on the subscales of the Kellner Symptom Questionnaire in treatment resistant depression. International Clinical Psychopharmacology 2013;28:238‐44. [DOI] [PubMed] [Google Scholar]
- Fava M, Mischoulon D, Iosifescu D, Witte J, Pencina M, Flynn M, et al. A double‐blind, placebo‐controlled study of aripiprazole adjunctive to antidepressant therapy among depressed outpatients with inadequate response to prior antidepressant therapy (ADAPT‐A Study). Psychotherapy and Psychosomatics 2012;81:87‐97. [DOI] [PubMed] [Google Scholar]
- Mischoulon D, Witte J, Levy M, Papakostas GI, Pet LR, Hsieh WH, et al. Efficacy of dose increase among nonresponders to low‐dose aripiprazole augmentation in patients with inadequate response to antidepressant treatment: randomized, double‐blind, placebo‐controlled, efficacy trial. Journal of Clinical Psychiatry 2012;73:353‐7. [DOI] [PubMed] [Google Scholar]
- NCT00683852. A double‐blind, placebo‐controlled study of aripiprazole adjunctive to antidepressant therapy (ADT) among outpatients with major depressive disorder who have responded inadequately to prior ADT. clinicaltrials.gov/show/NCT00683852 (first received 26 May 2008).
Baca 2005 {published data only}
- Baca BE, Giner UJ, Leal CC, Vallejo RJ, Garcia CC, Prieto LR. Venlafaxine extended release versus conventional antidepressants in the remission of depressive disorders after previous antidepressant failure: ARGOS study. Depression and Anxiety 2005;22(2):68‐76. [DOI] [PubMed] [Google Scholar]
Barbee 2011 {published data only}
- Barbee JG, Thompson TR, Jamhour NJ, Stewart JW, Conrad EJ, Reimherr FW, et al. A double‐blind placebo‐controlled trial of lamotrigine as an antidepressant augmentation agent in treatment‐refractory unipolar depression. Journal of Clinical Psychiatry 2011;72(10):1405‐12. [DOI] [PubMed] [Google Scholar]
- NCT00901407. Lamotrigine augmentation in resistant depression. https://clinicaltrials.gov/show/NCT00901407 (first received 13 May 2009).
Bares 2013 {published data only}
- Bares M, Novak T, Kopecek M, Stopkova P, Cermak J, Kozeny J, et al. Antidepressant combinations and monotherapy in the treatment of resistant depression: a randomised, open‐label study. 24th Congress of the European College of Neuropsychopharmacology; 2011 September 3‐7; Paris (France). 2011; Vol. 21:S356.
- Bares M, Novak T, Kopecek M, Stopkova P, Cermak J, Kozeny J, et al. Antidepressant monotherapy compared with combinations of antidepressants in the treatment of resistant depressive patients: a randomized, open‐label study. International Journal of Psychiatry in Clinical Practice 2013;17:35‐43. [DOI] [PubMed] [Google Scholar]
Bauer 2013 (RUBY) {published data only}
- Bauer M, Dell'osso L, Kasper S, Pitchot W, Dencker Vansvik E, Kohler J, et al. Extended‐release quetiapine fumarate (quetiapine XR) monotherapy and quetiapine XR or lithium as add‐on to antidepressants in patients with treatment‐resistant major depressive disorder. Journal of Affective Disorders 2013;151:209‐19. [DOI] [PubMed] [Google Scholar]
- Euctr2008‐000908‐91. A randomised, 6‐week, multicentre, open‐label, rater‐blinded parallel group study comparing quetiapine extended release monotherapy and augmentation with Lithium augmentation in patients with treatment resistant depression ‐ RUBY. clinicaltrialsregister.eu/ctr‐search/search?query=eudract_number:2008‐000908‐91 (first received 16 October 2008).
- Franco MM, Figueira ML, Petrov P, Rihmer Z, Vavrusov L, Dencker‐Vansvik E, et al. Quetiapine XR monotherapy and quetiapine XR + ongoing antidepressant versus lithium + ongoing AD for stage II treatment‐resistant major depressive disorder. 23rd European College of Neuropsychopharmacology Congress; 2010 August 28‐September 1; Amsterdam, Netherlands. 2010; Vol. 20, issue Suppl 3:S347.
- NCT00789854. Comparing quetiapine XR monotherapy and augmentation with lithium augmentation in TRD patients. clinicaltrials.gov/show/NCT00789854 (first received 13 November 2008).
Bauer 2018 {published data only}
- Bauer M, Hefting N, Lindsten A, Josiassen MK, Hobart M. A randomised, placebo‐controlled 24‐week study evaluating adjunctive brexpiprazole in patients with major depressive disorder. Acta Neuropsychiatrica 2018;31(1):27‐35. [DOI] [PubMed] [Google Scholar]
Benkert 1997 {published data only}
- Anghelescu I, Szegedi A, Philipp M, Staab HJ, Benkert O. Paroxetine vs. maprotiline: effects of dose escalation in non‐responding depressed outpatients after 3 weeks of treatment. Pharmacopsychiatry 1995;28:159. [Google Scholar]
- Benkert O, Szegedi A, Wetzel H, Staab HJ, Meister W, Philipp M. Dose escalation vs. continued doses of paroxetine and maprotiline: a prospective study in depressed out‐patients with inadequate treatment response. Acta Psychiatrica Scandinavica 1997;95:288‐96. [DOI] [PubMed] [Google Scholar]
- Szegedi A, Wetzel H, Angersbach D, Dunbar GC, Schwarze H, Philipp M, et al. A double‐blind study comparing paroxetine and maprotiline in depressed outpatients. Pharmacopsychiatry 1997;30(3):97‐105. [DOI] [PubMed] [Google Scholar]
Berman 2007 {published data only}
- Berman RM, Marcus RN, Swanink R, McQuade RD, Carson WH, Corey‐Lisle PK, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a multicenter, randomized, double‐blind, placebo‐controlled study. Journal of Clinical Psychiatry 2007;68(6):843‐53. [DOI] [PubMed] [Google Scholar]
Berman 2009 {published data only}
- Berman RM, Fava M, Thase ME, Trivedi MH, Swanink R, McQuade RD, et al. Aripiprazole augmentation in major depressive disorder: a double‐blind, placebo‐controlled study in patients with inadequate response to antidepressants. CNS Spectrums 2009;14(4):197‐206. [DOI] [PubMed] [Google Scholar]
Birkenhager 2004 {published data only}
- Birkenhager TK, Broek WW, Mulder PG, Bruijn JA, Moleman P. Efficacy and tolerability of tranylcypromine versus phenelzine: a double‐blind study in antidepressant‐refractory depressed inpatients. Journal of Clinical Psychiatry 2004;65(11):1505‐10. [DOI] [PubMed] [Google Scholar]
Bose 2012 {published data only}
- Bose A, Tsai J, Li D. Early non‐response in patients with severe depression: escitalopram up‐titration versus switch to duloxetine. Clinical Drug Investigation 2012;32:373‐85. [DOI] [PubMed] [Google Scholar]
Brecht 2011 {published data only}
- Brecht S, Desaiah D, Marechal ES, Santini AM, Podhorna J, Guelfi JD. Efficacy and safety of duloxetine 60 mg and 120 mg daily in patients hospitalized for severe depression: a double‐blind randomized trial. Journal of Clinical Psychiatry 2011;72:1086‐94. [DOI] [PubMed] [Google Scholar]
Chaput 2004 {published data only}
- Chaput Y, Magnan A, Gendron A. The co‐administration of quetiapine or placebo to cognitive‐behavior therapy in treatment refractory depression: a preliminary trial. BMC Psychiatry 2008;8:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaput Y, Magnan A, Lebel MJ. Adjunctive quetiapine administration to cognitive‐behavior therapy. 157th Annual Meeting of the American Psychiatric Association; 2004 May 1‐6; New York (NY). 2004:NR511.
Cheon 2017 {published data only}
- Cheon EJ, Lee KH, Park YW, Lee JH, Koo BH, Lee SJ, et al. Comparison of the efficacy and safety of aripiprazole versus bupropion augmentation in patients with major depressive disorder unresponsive to selective serotonin reuptake inhibitors: a randomized, prospective, open‐label study. Journal of Clinical Psychopharmacology 2017;37(2):193‐9. [DOI] [PubMed] [Google Scholar]
- Koo BH, Cheon EJ, Lee KH, Park YW, Lee JH, Lee SJ, et al. Effects of aripiprazole versus bupropion adjunctive to selective serotonin reuptake inhibitor on the specific symptoms of depression: a randomised prospective open‐label multi‐center study. 29th European College of Neuropsychopharmacology Congress; 2016 September 17‐20; Vienna, Austria. 2017; Vol. 26:S461.
- Lee K, Lee JB, Park YW, Lee JH, Lee SJ, Koo BH, et al. Comparison of aripiprazole augmentation versus bupropion combination for patient with major depressive disorder. 27th European College of Neuropsychopharmacology Congress; 2014 October 18‐21; Berlin, Germany. 2014:S452‐3.
Cipriani 2013 {published data only}
- Cipriani A, Girlanda F, Agrimi E, Barichello A, Beneduce R, Bighelli I, et al. Effectiveness of lithium in subjects with treatment‐resistant depression and suicide risk: a protocol for a randomised, independent, pragmatic, multicentre, parallel‐group, superiority clinical trial. BMC Psychiatry 2013;13:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girlanda F, Cipriani A, Agrimi E, Appino MG, Barichello A, Beneduce R, et al. Effectiveness of lithium in subjects with treatment‐resistant depression and suicide risk: results and lessons of an underpowered randomised clinical trial. BMC Research Notes 2014;7:731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT00927550. Lithium and standard therapy in resistant depression. clinicaltrials.gov/show/NCT00927550 (first received 25 June 2009).
Corya 2006 {published data only}
- Corya SA, Williamson D, Sanger TM, Briggs SD, Case M, Tollefson G. A randomized, double‐blind comparison of olanzapine/fluoxetine combination, olanzapine, fluoxetine, and venlafaxine in treatment‐resistant depression. Depression and Anxiety 2006;23(6):364‐72. [DOI] [PubMed] [Google Scholar]
Doree 2007 {published data only}
- Doree JP, Rosiers J, Lew V, Gendron A, Elie R, Stip E, et al. Quetiapine augmentation of treatment‐resistant depression: a comparison with lithium. Current Medical Research and Opinion 2007;23(2):333‐41. [DOI] [PubMed] [Google Scholar]
- Doree JP, Tourjman SV, Kunicki S, Desrosiers J, Vanier C, Elie R, et al. Comparison of quetiapine versus lithium treatment of resistant depression. 157th Annual Meeting of the American Psychiatric Association; 2004 May 1‐6; New York (NY). 2004:NR725.
Earley 2018 {published data only}
- Earley WR, Guo H, Nemeth G, Harsanyi J, Thase ME. Cariprazine augmentation to antidepressant therapy in major depressive disorder: results of a randomized, double‐blind, placebo‐controlled trial. Psychopharmacology Bulletin 2018;48(4):62‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Edwards 1998 {published data only}
- Edwards DR. A double‐blind, placebo‐controlled trial of lithium augmentation of antidepressants in treatment‐resistant depression in elderly patients. National Research Register 1998. [Google Scholar]
e‐Therapeutics 2013 {published data only}
- Euctr2013‐000719‐26. A single centre, double blind, non‐inferiority study to evaluate the antidepressant activity of Viotra TM compared with amitriptyline in the treatment of major depressive disorder (MDD) in patients who have an unsatisfactory response. clinicaltrialsregister.eu/ctr‐search/search?query=eudract_number:2013‐000719‐26 (first received 12 April 2013).
- NCT02014363. Double blind, non‐inferiority study to evaluate the antidepressant activity of ETS6103 compared with amitriptyline in the treatment of major depressive disorder (MDD) in patients who have an unsatisfactory response to selective serotonin re‐uptake inhibitors (SSRIs). clinicaltrials.gov/show/NCT02014363 (first received 18 December 2013).
Fang 2010 {published data only}
- Fang Y, Yuan C, Xu Y, Chen J, Wu Z, Cao L, et al. Comparisons of the efficacy and tolerability of extended‐release venlafaxine, mirtazapine, and paroxetine in treatment‐resistant depression: a double‐blind, randomized pilot study in a Chinese population. Journal of Clinical Psychopharmacology 2010;30(4):357‐64. [DOI] [PubMed] [Google Scholar]
Fang 2011 {published data only}
- Fang Y, Yuan C, Xu Y, Chen J, Wu Z, Cao L, et al. A pilot study of the efficacy and safety of paroxetine augmented with risperidone, valproate, buspirone, trazodone, or thyroid hormone in adult Chinese patients with treatment‐resistant major depression. Journal of Clinical Psychopharmacology 2011;31:638‐42. [DOI] [PubMed] [Google Scholar]
Fava 1994 {published data only}
- Fava M, Rosenbaum JF, McGrath PJ, Stewart JW, Amsterdam JD, Quitkin FM. Lithium and tricyclic augmentation of fluoxetine treatment for resistant major depression: a double‐blind, controlled study. American Journal of Psychiatry 1994;151(9):1372‐4. [DOI] [PubMed] [Google Scholar]
Fava 2001 {published data only}
- Fava M, Dunner DL, Greist JH, Preskorn SH, Trivedi MH, Zajecka J, et al. Efficacy and safety of mirtazapine in major depressive disorder patients after SSRI treatment failure: an open‐label trial. Journal of Clinical Psychiatry 2001;62(6):413‐20. [DOI] [PubMed] [Google Scholar]
- Fava M, Greist JH, Preskorn SH, Trivedi MH, Zajecka JM, Cohen M, et al. An open‐label study with mirtazapine in depressed patients who are SSRI treatment failures. 152nd Annual Meeting of the American Psychiatric Association; 1999 May 15‐20; Washington (DC). 1999.
Fava 2002 {published data only}
- Fava M, Alpert J, Nierenberg A, Lagomasino I, Sonawalla S, Tedlow J, et al. Double‐blind study of high‐dose fluoxetine versus lithium or desipramine augmentation of fluoxetine in partial responders and nonresponders to fluoxetine. Journal of Clinical Psychopharmacology 2002;22(4):379‐87. [DOI] [PubMed] [Google Scholar]
Fava 2018 {published data only}
- Fava M, Durgam S, Earley W, Lu K, Hayes R, Laszlovszky I, et al. Efficacy of adjunctive low‐dose cariprazine in major depressive disorder: a randomized, double‐blind, placebo‐controlled trial. International Clinical Psychopharmacology 2018;33(6):312‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT00854100. A double‐blind, placebo‐controlled study of caripazine (RGH‐188) as adjunctive therapy In major depressive disorder. clinicaltrials.gov/show/NCT00854100 (first received 2 March 2009).
Fornaro 2014 {published data only}
- Fornaro M, Martino M, Mattei C, Prestia D, Vinciguerra V, Berardis D, et al. Duloxetine‐bupropion combination for treatment‐resistant atypical depression: a double‐blind, randomized, placebo‐controlled trial. European Neuropsychopharmacology 2014;24:1269‐78. [DOI] [PubMed] [Google Scholar]
Franco‐Chaves 2013 {published data only}
- Franco‐Chaves JA, Mateus CF, Luckenbaugh DA, Martinez PE, Mallinger AG, Zarate CA Jr. Combining a dopamine agonist and selective serotonin reuptake inhibitor for the treatment of depression: a double‐blind, randomized pilot study. Journal of Affective Disorders 2013;149:319‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT00086307. Combining a dopamine agonist and selective serotonin reuptake inhibitor for treatment of depression: a double‐blind, randomized study. clinicaltrials.gov/show/NCT00086307 (first received 30 June 2004).
Furukawa 2011 {published data only}
- Furukawa TA, Akechi T, Shimodera S, Yamada M, Miki K, Watanabe N, et al. Strategic use of new generation antidepressants for depression: SUN‐D study protocol. Trials 2011;12:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemoto N, Tanaka S, Furukawa TA, Kato T, Mantani A, Ogawa Y, et al. Strategic use of new generation antidepressants for depression: SUN‐D protocol update and statistical analysis plan. Trials 2015;16:459. [DOI] [PMC free article] [PubMed] [Google Scholar]
Garry 1963 {published data only}
- Garry JW, Leonard TJ. Trial of amitriptyline in chronic depression. British Journal of Psychiatry 1963;109:54‐5. [DOI] [PubMed] [Google Scholar]
Gonul 1999 {published data only}
- Gonul AS, Oguz A, Yabanoglu I, Aslan SS, Turan T. Buspiron and pindolol in augmentation therapy of treatment‐resistant depression. European Neuropsychopharmacology 1999;9(Suppl 5):215. [Google Scholar]
Grant 2017 {published data only}
- Grant JE, Redden SA, Leppink EW. Double‐blind switch study of vilazodone in the treatment of major depressive disorder. International Clinical Psychopharmacology 2017;32(3):121‐6. [DOI] [PubMed] [Google Scholar]
Han 2015 {published data only}
- Han C, Wang SM, Kwak KP, Won WY, Lee HY, Chang CM, et al. Aripiprazole augmentation versus antidepressant switching for patients with major depressive disorder: a 6‐week, randomized, rater‐blinded, prospective study. Journal of Psychiatric Research 2015;66‐67:84‐94. [DOI] [PubMed] [Google Scholar]
- NCT01488266. Comparison of aripiprazole augmentation vs switching to different class of antidepressants for patients with MDD who are partially/minimally responsive to current antidepressants: randomized, rater‐blinded, prospective study. clinicaltrials.gov/show/NCT01488266 (first received 8 December 8 2011).
Hides 2006 {published data only}
- Hides L, Lubman D, Wong L, Carroll S, Kay‐Lambkin F, Allen N, et al. Treating coexisting depression and alcohol/other drug misuse in young people. 29th Australian Association for Cognitive and Behaviour Therapy Annual Conference; 2006 October 18 ‐ 23; Sydney, Australia. 2006:50.
Hobart 2018 {published data only}
- Hobart M, Skuban A, Zhang P, Augustine C, Brewer C, Hefting N, et al. A randomized, placebo‐controlled study of the efficacy and safety of fixed‐dose brexpiprazole 2 mg/d as adjunctive treatment of adults with major depressive disorder. Journal of Clinical Psychiatry 2018;79(4):7. [DOI] [PubMed] [Google Scholar]
Horikoshi 2017 {published data only}
- Horikoshi SH, Miura IM, Yamamoto SY, Abe OA, Ito MI, Watanabe KW, et al. Low and high dose aripiprazole augmentation in major depressive disorder: a randomised, open‐label study. 30th European College of Neuropsychopharmacology Congress; 2017 September 2‐5; Paris, France. 2017; Vol. 27 Suppl 4:S861.
Joffe 1993 {published data only}
- Joffe RT, Singer W, Levitt AJ, MacDonald C. A placebo‐controlled comparison of lithium and triiodothyronine augmentation of tricyclic antidepressants in unipolar refractory depression. Archives of General Psychiatry 1993;50(5):387‐93. [DOI] [PubMed] [Google Scholar]
Joffe 2006 {published data only}
- Joffe RT, Sokolov ST, Levitt AJ. Lithium and triiodothyronine augmentation of antidepressants. Canadian Journal of Psychiatry 2006;51(12):791‐3. [PUBMED: 17168254] [DOI] [PubMed] [Google Scholar]
Kamijima 2013 {published data only}
- Kamijima K, Higuchi T, Ishigooka J, Ohmori T, Ozaki N, Kanba S, et al. Aripiprazole augmentation to antidepressant therapy in Japanese patients with major depressive disorder: a randomized, double‐blind, placebo‐controlled study (ADMIRE study). Journal of Affective Disorders 2013;151:899‐905. [DOI] [PubMed] [Google Scholar]
Kamijima 2018 {published data only}
- Kamijima K, Kimura M, Kuwahara K, Kitayama Y, Tadori Y. Randomized, double‐blind comparison of aripiprazole/sertraline combination and placebo/sertraline combination in patients with major depressive disorder. Psychiatry and Clinical Neurosciences 2018;72(8):591‐601. [DOI] [PubMed] [Google Scholar]
Keitner 2009 {published data only}
- Keitner G, Ninan P, Ryan C, Garlow S, Solomon D, Nemeroff C, et al. A randomized, placebo‐controlled trial of risperidone augmentation for patients with difficult‐to‐treat unipolar, nonpsychotic major depression. 46th Annual New Clinical Drug Evaluation Unit Meeting; 2006 June 12 ‐ 15; Boca Raton (FL). 2006:142.
- Keitner GI, Garlow SJ, Ryan CE, Ninan PT, Solomon DA, Nemeroff CB, et al. A randomized, placebo‐controlled trial of risperidone augmentation for patients with difficult‐to‐treat unipolar, non‐psychotic major depression. Journal of Psychiatric Research 2009;43(3):205‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keitner GI, Ninan PT, Ryan CE, Garlow S, Solomon DA, Nemeroff CB, et al. A randomized, placebo‐controlled trial of risperidone augmentation for patients with difficult‐to‐treat unipolar, nonpsychotic major depression. Neuropsychopharmacology 2005;30 Suppl 1:S175. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kennedy 2003 {published data only}
- Kennedy SH, Segal ZV, Cohen NL, Levitan RD, Gemar M, Bagby RM. Lithium carbonate versus cognitive therapy as sequential combination treatment strategies in partial responders to antidepressant medication: an exploratory trial. Journal of Clinical Psychiatry 2003;64(4):439‐44. [DOI] [PubMed] [Google Scholar]
Kocsis 1988 {published data only}
- Kocsis JH, Frances AJ, Voss C, Mann JJ, Mason BJ, Sweeney J. Imipramine treatment for chronic depression. Archives of General Psychiatry 1988;45(3):253‐7. [DOI] [PubMed] [Google Scholar]
- Kocsis JH, Sutton BM, Frances AJ. Long‐term follow‐up of chronic depression treated with imipramine. Journal of Clinical Psychiatry 1991;52(2):56‐9. [PubMed] [Google Scholar]
Lejoyeux 2015 {published data only}
- Lejoyeux M, Matharan S, Bodinat C. How to switch to agomelatine after an unsuccessful try with paroxetine or venlafaxine. CNS Spectrums 2015;20:29‐38. [DOI] [PubMed] [Google Scholar]
- Lejoyeux M, Picarel‐Blanchot F, Bodinat C. How to switch to agomelatine after an initial treatment failure of paroxetine or venlafaxine?. 26th European College of Neuropsychopharmacology Congress; 2013 October 5‐10; Barcelona, Spain. 2013:S341‐2.
Lenox‐Smith 2008 {published data only}
- Lenox‐Smith AJ, Jiang Q. Venlafaxine extended release versus citalopram in patients with depression unresponsive to a selective serotonin reuptake inhibitor. International Clinical Psychopharmacology 2008;23(3):113‐9. [DOI] [PubMed] [Google Scholar]
Lenze 2015 (IRL GREY) {published data only}
- Hsu JH, Mulsant BH, Lenze EJ, Karp JF, Lavretsky H, Roose SP, et al. Impact of prior treatment on remission of late‐life depression with venlafaxine and subsequent aripiprazole or placebo augmentation. American Journal of Geriatric Psychiatry 2016;24(10):918‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JH, Mulsant BH, Lenze EJ, Sanches M, Karp JF, Reynolds CF, et al. Clinical predictors of extrapyramidal symptoms associated with aripiprazole augmentation for the treatment of late‐life depression in a randomized controlled trial. Journal of Clinical Psychiatry 2018;79(4):no pagination. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneriya SH, Robbins‐Welty GA, Smagula SF, Karp JF, Butters MA, Lenze EJ, et al. Predictors and moderators of remission with aripiprazole augmentation in treatment‐resistant late‐life depression: an analysis of the IRL‐grey randomized clinical trial. JAMA Psychiatry 2016;73(4):329‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenze EJ, Mulsant BH, Blumberger DM, Karp JF, Newcomer JW, Anderson SJ, et al. Efficacy, safety, and tolerability of augmentation pharmacotherapy with aripiprazole for treatment‐resistant depression in late life: a randomised, double‐blind, placebo‐controlled trial. Lancet 2015;386(10011):2404‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT00892047. Incomplete response in late life depression: getting to remission. clinicaltrials.gov/show/NCT00892047 (first received 4 May 4 2009).
- Smagula SF, Wallace ML, Anderson SJ, Karp JF, Lenze EJ, Mulsant BH, et al. Combining moderators to identify clinical profiles of patients who will, and will not, benefit from aripiprazole augmentation for treatment resistant late‐life major depressive disorder. Journal of Psychiatric Research 2016;81:112‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2006 {published data only}
- Li HZ, Zhang YL, Mu JL, Zhang YL. Venlafaxine extended release and lithium salt for major depressive disorder in nonresponders to selective serotonin reuptake inhibitors. Chinese Journal of Clinical Rehabilitation 2006;10(30):4‐6. [Google Scholar]
Li 2013 {published data only}
- Li X, Xing B, Yu E, Chen W, Wu H. The combined treatment of venlafaxine and quetiapine for treatment‐resistant depression: a clinical study. Journal of Neuropsychiatry and Clinical Neurosciences 2013;25:157‐60. [DOI] [PubMed] [Google Scholar]
Licht 2002 {published data only}
- Licht RW, Qvitzau S. Mianserin augmentation, sertraline dose increase or extended duration of treatment in major depression insufficiently responding to 6 weeks of sertraline treatment. International Journal of Neuropsychopharmacology 2000;3(Suppl 1):236. [Google Scholar]
- Licht RW, Qvitzau S. Treatment strategies in patients with major depression not responding to first‐line sertraline treatment. A randomised study of extended duration of treatment, dose increase or mianserin augmentation. Psychopharmacology 2002;161(2):143‐51. [DOI] [PubMed] [Google Scholar]
- Licht RW, Qvitzav S. Mianserin augmentation, sertraline dose increase or extended duration of treatment in major depression insufficiently responding to 6 weeks of sertraline treatment: a randomized, double‐blind study. 39th Annual Meeting of the American College of Neuropsychopharmacology; 2000 December 10‐14; San Juan, Puerto Rico. 2000:150.
- Licht RW, Qvitzav S. Mianserin augmentation, sertraline dose increase, or extended duration of treatment in patients with major depression nonresponding after six weeks of sertraline treatment: a randomized, double‐blind study. 154th Annual Meeting of the American Psychiatric Association; 2001 May 5‐10; New Orleans (LA). 2001:NR369.
- Licht RW, Qvitzav S. Mianserin augmentation, sertraline dose increase, or extended duration of treatment in patients with major depression nonresponding after six weeks of sertraline treatment: a randomized, double‐blind study. 155th Annual Meeting of the American Psychiatric Association; 2002 May 18‐23; Philadelphia (PA). 2002.
Maes 1996 {published data only}
- Maes M, Vandoolaeghe E, Desnyder R. Efficacy of treatment with trazodone in combination with pindolol or fluoxetine in major depression. Journal of Affective Disorders 1996;41(3):201‐10. [DOI] [PubMed] [Google Scholar]
Maes 1999 {published data only}
- Maes M, Libbrecht I, Hunsel F, Campens D, Meltzer H Y. Pindolol and mianserin augment the antidepressant activity of fluoxetine in hospitalized major depressed patients, including those with treatment resistance. Journal of Clinical Psychopharmacology 1999;19(2):177‐82. [DOI] [PubMed] [Google Scholar]
Marcus 2008 {published data only}
- Marcus RN, McQuade RD, Carson WH, Hennicken D, Fava M, Simon JS, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a second multicenter, randomized, double‐blind, placebo‐controlled study. Journal of Clinical Psychopharmacology 2008;28(2):156‐65. [DOI] [PubMed] [Google Scholar]
Marwood 2017 {published data only}
- Marwood L, Taylor R, Goldsmith K, Romeo R, Holland R, Pickles A, et al. Study protocol for a randomised pragmatic trial comparing the clinical and cost effectiveness of lithium and quetiapine augmentation in treatment resistant depression (the LQD study). BMC Psychiatry 2017;17(1):231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT03004521. Lithium versus quetiapine in treatment resistant depression. clinicaltrials.gov/show/NCT03004521 (first received 29 December 2016).
Mattingly 2006 {published data only}
- Mattingly G, Ilivicky H, Canale J, Anderson R. Quetiapine augmentation for treatment‐resistant depression. 46th Annual New Clinical Drug Evaluation Unit Meeting; 2006 June 12‐15; Boca Raton (FL). 2006:154.
Medhus 1994 {published data only}
- Medhus A, Heskestad S, Tjemsland L. Mianserin added to tricyclic antidepressants in depressed patients not responding to a tricyclic antidepressant alone: a randomized, placebo‐controlled, double‐blind study. Nordic Journal of Psychiatry 1994;48(5):355‐8. [Google Scholar]
Mohamed 2017 (VAST‐D) {published data only}
- Mohamed S. Effect of antidepressant switching vs augmentation on remission among patients with major depressive disorder unresponsive to antidepressant treatment: the VAST‐D randomized clinical trial. JAMA 2017;318:132‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed S, Johnson GR, Vertrees JE, Guarino PD, Weingart K, Young IT, et al. The VA augmentation and switching treatments for improving depression outcomes (Vast‐D) study: rationale and design considerations. Psychiatry Research 2015;229:760‐70. [DOI] [PubMed] [Google Scholar]
- NCT01421342. CSP #576 ‐ VA augmentation and switching treatments for improving depression outcomes (VAST‐D). clinicaltrials.gov/show/NCT01421342 (first received 22 August 2011).
- Yoon J, Zisook S, Park A, Johnson GR, Scrymgeour A, Mohamed S. Comparing cost‐effectiveness of aripiprazole augmentation with other "next‐step" depression treatment strategies: a randomized clinical trial. Journal of Clinical Psychiatry 2018;80(1):e1‐8. [DOI] [PubMed] [Google Scholar]
- Zisook S, Tal I, Weingart K, Hicks P, Davis LL, Chen P, et al. Characteristics of U.S. veteran patients with major depressive disorder who require "next‐step" treatments: a VAST‐D report. Journal of Affective Disorders 2016;206:232‐40. [DOI] [PubMed] [Google Scholar]
Montgomery 2014 (REVIVE) {published data only}
- Haggstrom L, Nielsen RZ, Danchenko N, Poulsen L. A randomised, double‐blind, study of vortioxetine versus agomelatine in adults with major depressive disorder (MDD) with inadequate response to SSRI/SNRI treatment. 26th European College of Neuropsychopharmacology Congress; 2013 October 5‐9; Barcelona, Spain. 2013; Vol. 23:S412.
- Haggstrom L, Nielsen RZ, Dragheim M. Randomized, double‐blind, study of vortioxetine versus agomelatine in adults with MDD after inadequate response to SSRI or SNRI treatment. 21st European Congress of Psychiatry; 2013 April 6‐9; Nice, France. 2013; Vol. 28:3009.
- Montgomery SA, Nielsen RZ, Poulsen LH, Haggstrom L. A randomised, double‐blind study in adults with major depressive disorder with an inadequate response to a single course of selective serotonin reuptake inhibitor or serotonin‐noradrenaline reuptake inhibitor treatment switched to vortioxetine or agomelatine. Human Psychopharmacology: Clinical and Experimental 2014;29:470‐82; Erratum in: Human Psychopharmacology: Clinical and Experimental 2015; 30:327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01488071. A randomised, double‐blind, parallel‐group, active‐controlled, flexible dose study evaluating the effects of Lu AA21004 versus agomelatine in adult patients suffering from major depressive disorder with inadequate response to antidepressant treatment. clinicaltrials.gov/show/NCT01488071 (first received 8 December 2011).
- Papakostas G, Dragheim M, Nielsen RZ. Efficacy and tolerability of vortioxetine is independent of previous treatment in MDD patients switched after an inadequate response. 27th European College of Neuropsychopharmacology Congress; 2014 October 18‐21; Berlin, Germany. 2014; Vol. 24:S466.
- Papakostas GI, Nielsen RZ, Dragheim M, Tonnoir B. Efficacy and tolerability of vortioxetine versus agomelatine, categorized by previous treatment, in patients with major depressive disorder switched after an inadequate response. Journal of Psychiatric Research 2018;101:72‐9. [DOI] [PubMed] [Google Scholar]
Moreno 1997 {published data only}
- Moreno FA, Gelenberg AJ, Bachar K, Delgado PL. Pindolol augmentation of treatment‐resistant depressed patients. Journal of Clinical Psychiatry 1997;58(10):437‐9. [DOI] [PubMed] [Google Scholar]
Nakajima 2011 {published data only}
- Nakajima S, Uchida H, Suzuki T, Watanabe K, Hirano J, Yagihashi T, et al. Is switching antidepressants following early nonresponse more beneficial in acute‐phase treatment of depression? A randomized open‐label trial. Progress in Neuro‐Psychopharmacology & Biological Psychiatry 2011;35:1983‐9. [DOI] [PubMed] [Google Scholar]
NCT00273624 {published data only}
- NCT00273624. Olanzapine augmentation therapy in treatment‐resistant depression: a double‐blind placebo‐controlled trial. clinicaltrials.gov/show/NCT00273624 (first received 9 January 2006).
NCT00296517 {published data only}
- NCT00296517. Clinical evaluation of 323U66 SR in patients with depression ‐ placebo‐controlled, double‐blind, comparative study in patients with depression who did not respond sufficiently to selective serotonin re‐uptake inhibitors. clinicaltrials.gov/show/NCT00296517 (first received 27 February 2006).
NCT01052077 {published data only}
- NCT01052077. A phase 2, multicenter, randomized, double‐blind, placebo‐controlled study of the safety and efficacy of OPDC‐34712 (1 to 3 mg/Day) as adjunctive therapy in the treatment of adults with major depressive disorder STEP‐D222. clinicaltrials.gov/show/NCT01052077 (first received 20 January 2010).
NCT02272517 {published data only}
- NCT02272517. An interventional, randomised, double‐blind, parallel‐group, active‐comparator, flexible‐dose study on the efficacy of vortioxetine versus escitalopram on cognitive dysfunction in patients with inadequate response to current antidepressant treatment of major depressive disorder. clinicaltrials.gov/show/NCT02272517 (first received 23 October 2014).
NCT02960763 {published data only}
- NCT02960763. Optimizing outcomes of treatment‐resistant depression in older adults (OPTIMUM). clinicaltrials.gov/show/NCT02960763 (first received 10 November 2016).
NCT02977299 {published data only}
- NCT02977299. Comparative effectiveness research trial for antidepressant incomplete and non‐responders with TRD (ASCERTAINTRD). clinicaltrials.gov/show/NCT02977299 (first received 30 November 2016).
Nierenberg 2003 {published data only}
- Nierenberg AA, Papakostas GI, Petersen T, Montoya HD, Worthington JJ, Tedlow J, et al. Lithium augmentation of nortriptyline for subjects resistant to multiple antidepressants. Journal of Clinical Psychopharmacology 2003;23(1):92‐5. [DOI] [PubMed] [Google Scholar]
Nolen 1993 {published data only}
- Nolen WA, Haffmans PM, Bouvy PF, Duivenvoorden HJ. Monoamine oxidase inhibitors in resistant major depression: a double‐blind comparison of brofaromine and tranylcypromine in patients resistant to tricyclic antidepressants. Journal of Affective Disorders 1993;28(3):189‐97. [DOI] [PubMed] [Google Scholar]
Perahia 2008 {published data only}
- Perahia DG, Quail D, Desaiah D, Corruble E, Fava M. Switching to duloxetine from selective serotonin reuptake inhibitor antidepressants: a multicenter trial comparing 2 switching techniques. Journal of Clinical Psychiatry 2008;69(1):95‐105. [DOI] [PubMed] [Google Scholar]
- Perahia DG, Quail D, Desaiah D, Montejo AL, Schatzberg AF. Switching to duloxetine in selective serotonin reuptake inhibitor non‐ and partial‐responders: effects on painful physical symptoms of depression. Journal of Psychiatric Research 2009;43:512‐8. [DOI] [PubMed] [Google Scholar]
Perez 1999 {published data only}
- Perez V, Soler J, Puigdemont D, Alvarez E, Artigas F. A double‐blind, randomized, placebo‐controlled trial of pindolol augmentation in depressive patients resistant to serotonin reuptake inhibitors. Archives of General Psychiatry 1999;56(4):375‐9. [DOI] [PubMed] [Google Scholar]
Petrescu 2014 {published data only}
- Petrescu B, Vasile D, Vasiliu O, Tudor C, Mangalagiu A, Ungureanu D. SSRI dose escalation versus duloxetine in treatment of major depressive disorder not responding to initial SSRI. 27th European College of Neuropsychopharmacology Congress; 2014 October 18‐21; Berlin, Germany. 2014:S455‐6.
Poirier 1999 {published data only}
- Poirier MF. The concept of resistant depression and therapeutic strategies, particularly with venlafaxine. Encephale 1999;25 Spec No 2:55‐7. [PubMed] [Google Scholar]
- Poirier MF. Venlafaxine versus paroxetine in the treatment of resistant depression. Sixth World Congress of Biological Psychiatry; 1997 June 22‐27; Nice, France. 1997.
- Poirier MF, Boyer P. Venlafaxine and paroxetine in treatment‐resistant depression. Double‐blind, randomised comparison. British Journal of Psychiatry 1999;174:12‐6. [DOI] [PubMed] [Google Scholar]
- Poirier MF, Boyer P. Venlafaxine versus paroxetine for treatment resistant depression. XXIst Collegium Internationale Neuro Psychopharmacologicum; 1998 July 12‐16; Glasgow, Scotland. 1998.
- Schaeffer P, Poirier FM, Boyer P. Double‐blind trial of venlafaxine and paroxetine for treatment‐resistant depression. 11th European College of Neuropsychopharmacology Congress; 1998 October 31‐ November 4; Paris, France. 1998.
Rapaport 2006 (ARISe‐RD) {published data only}
- Mahmoud RA, Pandina GJ, Turkoz I, Kosik‐Gonzalez C, Canuso CM, Kujawa MJ, et al. Risperidone for treatment‐refractory major depressive disorder: a randomized trial. Annals of Internal Medicine 2007;147(9):593‐602. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB, Canuso CM, Mahmoud R, Loescher A, Turkoz I, Rapaport MH, et al. Augmentation with risperidone in chronic resistant depression: a double‐blind placebo‐controlled maintenance trial. Neuropsychopharmacology 2004;29 Suppl 1:S159. [Google Scholar]
- Rapaport MH, Canuso CM, Loescher A, Lasser R, Gharabawi G. Preliminary results from the ARISe‐RD (risperidone augmentation in resistant depression) trial. 156th Annual Meeting of the American Psychiatric Association; 2003 May 17‐22, San Francisco (CA). 2003:NR179.
- Rapaport MH, Canuso CM, Rouillon F, Leblanc J, Young AH, Loescher A, et al. Results from the augmentation with risperidone in resistant depression trial. 157th Annual Meeting of the American Psychiatric Association; 2004 May 1‐6; New York (NY). 2004:NR799.
- Rapaport MH, Gharabawi GM, Canuso CM, Mahmoud RA, Keller MB, Bossie CA, et al. Effects of risperidone augmentation in patients with treatment‐resistant depression: results of open‐label treatment followed by double‐blind continuation. Neuropsychopharmacology 2006;31(11):2505‐13. [DOI] [PubMed] [Google Scholar]
Ravindran 2006 {published data only}
- NCT01282632. Risperidone vs. olanzapine as add‐on treatment in treatment resistant depression. clinicaltrials.gov/show/NCT01282632 (first received 25 January 2011).
- Ravindran L, Lam R, Chaput Y, Enns M, Levitt A. Impact of low vs. high dose olanzapine or risperidone on outcome and side effects in non‐psychotic treatment resistant depression. 46th Annual NCDEU (New Clinical Drug Evaluation Unit) Meeting; 2006 June 12 ‐ 15; Boca Raton (FL). 2006:251.
- Ravindran L, Lam R, Chaput Y, Enns M, Levitt A. Impact of low vs. high dose olanzapine or risperidone on outcome and side effects in non‐psychotic treatment resistant depression (TRD). World Psychiatric Association International Congress; 2006 July 12 ‐ 16; Istanbul, Turkey. 2006:325.
Rele 2015 {published data only}
- Clayton AH, Gommoll C, Chen D, Nunez R, Mathews M. Sexual dysfunction during treatment of major depressive disorder with vilazodone, citalopram, or placebo: results from a phase IV clinical trial. International Clinical Psychopharmacology 2015;30:216‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rele S, Millet R, Paik JW, Kim S, Masand PS, Patkar AA. An 8‐week randomized, double‐blind trial comparing efficacy, safety, and tolerability of 3 vilazodone dose‐initiation strategies following switch from SSRIS and SNRIs in major depressive disorder. Primary Care Companion to CNS Disorders 2015;17(4):no pagination. [DOI] [PMC free article] [PubMed] [Google Scholar]
Romera 2010 {published data only}
- Romera I, Gilaberte I, Perez V, Menchon JM, Papen R, Schacht A, et al. Early antidepressant switch in MDD with pain symptoms: functional improvement. 11th International Forum on Mood and Anxiety Disorders; 2011 November 9‐11; Budapest, Hungary. 2011.
- Romera I, Perez V, Manuel Menchon J, Schacht A, Papen R, Neuhauser D, et al. Early vs. conventional switching of antidepressants in patients with MDD and moderate to severe pain: a double‐blind randomized study. Journal of Affective Disorders 2012;143:47‐55. [DOI] [PubMed] [Google Scholar]
- Romera I, Perez V, Menchon JM, Schacht A, Papen R, Neuhauser D, et al. Early switch strategy in patients with major depression disorder: a double‐blind, randomized study. Journal of Clinical Psychopharmacology 2012;32:479‐86. [DOI] [PubMed] [Google Scholar]
- Romera I, Perez V, Menchon JM, Schacht A, Papen R, Neuhauser D, et al. Early vs. delayed switch to duloxetine in patients with major depressive disorder not exhibiting early improvement with escitalopram: double‐blind randomized study. International Journal of Psychiatry in Clinical Practice 2010;14:34. [Google Scholar]
Ruhe 2009 {published data only}
- Mocking RJT, Verburg HF, Westerink AM, Assies J, Vaz FM, Koeter MWJ, et al. Fatty acid metabolism and its longitudinal relationship with the hypothalamic‐pituitary‐adrenal axis in major depression: associations with prospective antidepressant response. Psychoneuroendocrinology 2015;59:1‐13. [DOI] [PubMed] [Google Scholar]
- Mocking RJT, Verburg HF, Westerink AM, Assies J, Vaz FM, Koeter MWJ, et al. Longitudinal interplay between paroxetine response, cortisol and fatty acid metabolism in major depressive disorder abstract. 27th European College of Neuropsychopharmacology Congress; 2014 October 18‐21; Berlin, Germany. 2014:S396‐7.
- Ruhe HG, Booij J, Weert HC, Reitsma JB, Franssen EJ, Michel MC, et al. Evidence why paroxetine dose escalation is not effective in major depressive disorder: a randomized controlled trial with assessment of serotonin transporter occupancy. Neuropsychopharmacology 2009;34(4):999‐1010. [DOI] [PubMed] [Google Scholar]
- Ruhe HG, Booij J, Veltman DJ, Michel MC, Schene AH. Successful pharmacologic treatment of major depressive disorder attenuates amygdala activation to negative facial expressions: a functional magnetic resonance imaging study. Journal of Clinical Psychiatry 2012;73:451‐9. [DOI] [PubMed] [Google Scholar]
- Ruhe HG, Khoenkhoen SJ, Ottenhof KW, Koeter MW, Mocking RJT, Schene AH. Longitudinal effects of the SSRI paroxetine on salivary cortisol in major depressive disorder. Psychoneuroendocrinology 2015;52:261‐71. [DOI] [PubMed] [Google Scholar]
- Ruhe HG, Rooijen G, Veltman DJ, Schene AH. Twelve weeks of paroxetine treatment improves prefrontal brain activity. An fMRI study using the Tower of London. 23rd European College of Neuropsychopharmacology (ECNP) Congress; 2010 August 28‐September 01; Amsterdam, The Netherlands. 2010; Vol. 20 Suppl 3:S380.
Russell 1995 {published data only}
- Finkelstein SN, Berndt ER, Greenberg PE, Parsley RA, Russell JM, Keller MB, Chronic Depression Study group. Improvement in subjective work performance after treatment of chronic depression: some preliminary results. Psychopharmacolgy Bulletin 1996;32(1):33‐40. [PubMed] [Google Scholar]
- Miller IW, Keitner GI, Schatzberg AF, Klein DN, Thase ME, Rush AJ, et al. The treatment of chronic depression, part 3: psychosocial functioning before and after treatment with sertraline or imipramine. Journal of Clinical Psychiatry 1998;59(11):608‐19. [DOI] [PubMed] [Google Scholar]
- Rush AJ, Koran LM, Keller MB, Markowitz JC, Harrison WM, Miceli RJ, et al. The treatment of chronic depression, part 1: study design and rationale for evaluating the comparative efficacy of sertraline and imipramine as acute, crossover, continuation, and maintenance phase therapies. Journal of Clinical Psychiatry 1998;59(11):589‐97. [PubMed] [Google Scholar]
- Russell JM, Kornstein SG, Shea MT, McCullough JP, Harrison WM, Hirschfeld RM, et al. Chronic depression and comorbid personality disorders: response to sertraline versus imipramine. Journal of Clinical Psychiatry 2003;64(5):554‐61. [DOI] [PubMed] [Google Scholar]
- Russell JM, McCullough JP, Trapp G, McGee MD, Hirschfeld RM. Response to antidepressant treatment in chronically depressed patients with comorbid personality disorder. Psychopharmacology Bulletin 1995;31(3):612. [Google Scholar]
- Thase ME, Rush AJ, Howland RH, Kornstein SG, Kocsis JH, Gelenberg AJ, et al. Double‐blind switch study of imipramine or sertraline treatment of antidepressant‐resistant chronic depression. Archives of General Psychiatry 2002;59(3):233‐9. [DOI] [PubMed] [Google Scholar]
Rybakowski 1999 {published data only}
- Rybakowski JK, Suwalska A, Chlopocka‐Wozniak M. Potentiation of antidepressants with lithium or carbamazepine in treatment‐resistant depression. Neuropsychobiology 1999;40(3):134‐9. [DOI] [PubMed] [Google Scholar]
- Suwalska A, Rybakowski J. Potentiation of antidepressants with lithium or carbamazepine. 9th European College of Neuropsychopharmacology Congress; 1996 September 21‐26; Amsterdam, The Netherlands. 1996.
- Suwalska A, Rybakowski J. Potentiation of antidepressants with lithium or carbamazepine: factors associated with therapeutic efficacy. 11th European College of Neuropsychopharmacology Congress; 1998 October 31‐November 4: Paris, France. 1998.
Santos 2008 {published data only}
- NCT00652171. Efficacy and safety of antidepressant augmentation with lamotrigine. clinicaltrials.gov/show/NCT00652171 (first received 3 April 2008).
- Santos MA, Rocha FL, Hara C. Efficacy and safety of antidepressant augmentation with lamotrigine in patients with treatment‐resistant depression: a randomized, placebo‐controlled, double‐blind study. Primary Care Companion to the Journal of Clinical Psychiatry 2008;10(3):187‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schindler 2007 {published data only}
- Schindler F, Anghelescu IG. Lithium versus lamotrigine augmentation in treatment resistant unipolar depression: a randomized, open‐label study. International Clinical Psychopharmacology 2007;22(3):179‐82. [DOI] [PubMed] [Google Scholar]
Schweizer 1990 {published data only}
- Schweizer E, Rickels K, Amsterdam JD, Fox I, Puzzuoli G, Weise C. What constitutes an adequate antidepressant trial for fluoxetine?. Journal of Clinical Psychiatry 1990;51(1):8‐11. [PubMed] [Google Scholar]
Schweizer 2001 {published data only}
- Rickels K, DiMartinis N, Rynn MA, Schweizer EE. Therapeutic equivalence of 50 mg and 150 mg sertraline in outpatient major depression: results from a clinical trial. 152nd Annual Meeting of the American Psychiatric Association; 1999 May 15‐20; Washington (DC). 1999.
- Rickels K, Schweizer E, Rynn M, DiMartinis N. Sertraline 50 mg is as effective as 150 mg in MDD. XI World Congress of Psychiatry; 1999 August 6‐11; Hamburg, Germany. 1999; Vol. II:128.
- Schweizer E, Rynn M, Mandos LA, DeMartinis N, Garcia‐Espana F, Rickels K. The antidepressant effect of sertraline is not enhanced by dose titration: results from an outpatient clinical trial. International Clinical Psychopharmacology. 2001;16(3):137‐43. [DOI] [PubMed] [Google Scholar]
Shelton 2005a {published data only}
- NCT00179244. Risperidone vs. bupropion ER augmentation of SSRIs in treatment‐resistant depression. clinicaltrials.gov/show/NCT00179244 (first received 15 September 2005).
- Shelton RC, Addington S, Thakkar V. Risperidone vs. bupropion combined with SSRIs in treatment resistant unipolar major depression. Neuropsychopharmacology 2005;30 Suppl 1:S238. [Google Scholar]
Shelton 2005b {published data only}
- Shelton RC. Atypical antipsychotics: treatment of depression. 152nd Annual Meeting of the American Psychiatric Association; 1999 May 15‐20; Washington (DC). 1999.
- Shelton RC, Williamson DJ, Corya SA, Sanger TM, Campen LE, Case M, et al. Olanzapine/fluoxetine combination for treatment‐resistant depression: a controlled study of SSRI and nortriptyline resistance. Journal of Clinical Psychiatry 2005;66(10):1289‐97. [DOI] [PubMed] [Google Scholar]
Small 1981 {published data only}
- Small JG, Kellams JJ, Dennis JL, Milstein V. Comparison of molindone and tranylcypromine in the treatment of refractory depression. Journal of Clinical Pharmacology 1981;21:351‐8. [DOI] [PubMed] [Google Scholar]
Song 2007 {published data only}
- Song ZW, Liu XB, Li YD. Venlafaxine combined with low‐dose risperidone for treatment‐resistant depression. Journal of Clinical Rehabilitative Tissue Engineering Research 2007;11(52):10486‐8. [Google Scholar]
Souche 1991 {published data only}
- Baumann P, Nil R, Souche A, Montaldi S, Baettig D, Lambert S, et al. A double‐blind, placebo‐controlled study of citalopram with and without lithium in the treatment of therapy‐resistant depressive patients: a clinical, pharmacokinetic, and pharmacogenetic investigation. Journal of Clinical Psychopharmacology 1996;16(4):307‐14. [DOI] [PubMed] [Google Scholar]
- Souche A, Montaldi S, Uehlinger C, Kasas A, Reymond MJ, Reymond P, et al. Treatment of resistant depression with the citalopram‐lithium combination: methodology of a double‐blind multicenter study and preliminary results. Encephale 1991;17(3):213‐9. [PubMed] [Google Scholar]
- Souche A, Montaldi S, Uehlinger C, Kasas A, Reymond MJ, Reymond PBP, et al. Treatment of resistant depression by association citalopram‐lithium; methodology of a multicenter double‐blind study, and preliminary results. Encephale 1990;16(6):465‐6. [PubMed] [Google Scholar]
Souery 2011 {published data only}
- ACTRN12610000564055. Patterns of treatment resistance and switching strategies in unipolar affective disorder. www.anzctr.org.au/ACTRN12610000564055.aspx (first received 9 July 2010).
- Souery D, Serretti A, Calati R, Oswald P, Massat I, Konstantinidis A, et al. Citalopram versus desipramine in treatment resistant depression: effect of continuation or switching strategies: a randomized open study. World Journal of Biological Psychiatry 2011;12:364‐75. [DOI] [PubMed] [Google Scholar]
- Souery D, Serretti A, Montgomery S, Kasper S, Zohar J, Mendlewicz J. Advances on the treatment of resistant depression. International Journal of Psychiatry in Clinical Practice 2010;14:13. [Google Scholar]
STAR*D 2004 {published data only}
- Bech P, Fava M, Trivedi MH, Wisniewski SR, Rush AJ. Outcomes on the pharmacopsychometric triangle in bupropion‐SR vs. buspirone augmentation of citalopram in the STAR*D TRIAL. Acta Psychiatrica Scandinavica 2012;125:342‐8. [DOI] [PubMed] [Google Scholar]
- Fava M, Rush AJ, Wisniewski SR, Nierenberg AA, Alpert JE, McGrath PJ, et al. A comparison of mirtazapine and nortriptyline following two consecutive failed medication treatments for depressed outpatients: a STAR*D report. American Journal of Psychiatry 2006;163(7):1161‐72. [DOI] [PubMed] [Google Scholar]
- Gaynes BN, Dusetzina SB, Ellis AR, Hansen RA, Farley JF, Miller WC, et al. Treating depression after initial treatment failure: directly comparing switch and augmenting strategies in STAR*D. Journal of Clinical Psychopharmacology 2012;32:114‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen RA, Dusetzina SB, Ellis AR, Stürmer T, Farley JF, Gaynes BN. Risk of adverse events in treatment‐resistant depression: propensity‐score‐matched comparison of antidepressant augment and switch strategies. General Hospital Psychiatry 2012;34:192‐200. [DOI] [PubMed] [Google Scholar]
- McGrath PJ, Stewart JW, Fava M, Trivedi MH, Wisniewski SR, Nierenberg AA, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. American Journal of Psychiatry 2006;163(9):1531‐41. [DOI] [PubMed] [Google Scholar]
- Nierenberg AA, Fava M, Trivedi MH, Wisniewski SR, Thase ME, McGrath PJ, et al. A comparison of lithium and T3 augmentation following two failed medication treatments for depression: a STAR*D report. American Journal of Psychiatry 2006;163(9):1519‐30. [DOI] [PubMed] [Google Scholar]
- Rush AJ, Fava M, Wisniewski SR, Lavori PW, Trivedi MH, Sackeim HA, et al. Sequenced treatment alternatives to relieve depression (STAR*D): rationale and design. Controlled Clinical Trials 2004;25(1):119‐42. [DOI] [PubMed] [Google Scholar]
- Rush AJ, Trivedi M, Wisniewski SR, Fava M. An update on the STAR*D trial. 158th Annual Meeting of the American Psychiatric Association; 2005 May 21‐26; Atlanta (GA). 2005:9C.
- Rush AJ, Trivedi MH, Wisniewski SR, Stewart JW, Nierenberg AA, Thase ME, et al. Bupropion‐SR, sertraline, or venlafaxine‐XR after failure of SSRIs for depression. New England Journal of Medicine 2006;354(12):1231‐42. [DOI] [PubMed] [Google Scholar]
- Thase ME, Friedman ES, Biggs MM, Wisniewski SR, Trivedi MH, Luther JF, et al. Cognitive therapy versus medication in augmentation and switch strategies as second‐step treatments: a STAR*D report. American Journal of Psychiatry 2007;164(5):739‐52. [DOI] [PubMed] [Google Scholar]
- Trivedi MH, Fava M, Wisniewski SR, Thase ME, Quitkin F, Warden D, et al. Medication augmentation after the failure of SSRIs for depression. New England Journal of Medicine 2006;354(12):1243‐52. [DOI] [PubMed] [Google Scholar]
Stein 1993 {published data only}
- Stein G, Bernadt M. Double‐blind trial of lithium carbonate in tricyclic resistant depression. Birch NJ (ed). Lithium: Inorganic Pharmacology and Psychiatric Use. Oxford, Washington DC: IRL Press Ltd, 1988:35‐6. [Google Scholar]
- Stein G, Bernadt M. Lithium augmentation therapy in tricyclic‐resistant depression. A controlled trial using lithium in low and normal doses. British Journal of Psychiatry 1993;162:634‐40. [DOI] [PubMed] [Google Scholar]
Sun 2004 {published data only}
- Sun Q, Zen D, Luo S. Comparative study of citalopram combined with buspirone for treatment of refractory depression. Journal of Clinical Psychological Medicine 2004;14(4):221‐2. [Google Scholar]
Sunderland 1994 {published data only}
- Sunderland T, Cohen RM, Molchan S, Lawlor BA, Mellow AM, Newhouse PA, et al. High‐dose selegiline in treatment‐resistant older depressive patients. Archives of General Psychiatry 1994;51(8):607‐15. [DOI] [PubMed] [Google Scholar]
Tanghe 1997 {published data only}
- Tanghe A, Steenan J, Bollen E, Van Reynghe, Voxvrie G, Dendooven M, et al. Moclobemide and amitriptyline alone or in combination in therapy resistant depression. Human Psychopharmacology 1997;12(5):509‐10. [Google Scholar]
Thase 2000 {published data only}
- Thase ME, Kremer C, Rodrigues H. Mirtazapine versus sertraline after SSRI non‐response. 39th Annual Meeting of the American College of Neuropsychopharmacology; 2000 December 10‐14; San Juan, Puerto Rico. 2000:260.
- Thase ME, Kremer C, Rodrigues H. Mirtazapine versus sertraline after SSRI nonresponse. 52nd Institute on Psychiatric Services; 2000 October 25‐29th; Philadelphia, PA. 2000.
Thase 2006 {published data only}
- Thase ME, Shelton RC, Khan A. Treatment with venlafaxine extended release after SSRI nonresponse or intolerance: a randomized comparison of standard‐ and higher‐dosing strategies. Journal of Clinical Psychopharmacology 2006;26(3):250‐8. [DOI] [PubMed] [Google Scholar]
Thase 2007 {published data only}
- Houston JP, Lau K, Aris V, Liu W, Fijal BA, Heinloth AN, et al. Association of common variations in the norepinephrine transporter gene with response to olanzapine‐fluoxetine combination versus continued‐fluoxetine treatment in patients with treatment‐resistant depression: a candidate gene analysis. Journal of Clinical Psychiatry 2012;73:878‐85. [DOI] [PubMed] [Google Scholar]
- NCT00035321. The study of olanzapine plus fluoxetine in combination for treatment‐resistant depression without psychotic features. clinicaltrials.gov/ct2/show/NCT00035321 (first received 3 May 2002).
- Thase ME, Corya SA, Osuntokun O, Case M, Henley DB, Sanger TM, et al. A randomized, double‐blind comparison of olanzapine/fluoxetine combination, olanzapine, and fluoxetine in treatment‐resistant major depressive disorder. Journal of Clinical Psychiatry 2007;68(2):224‐36. [DOI] [PubMed] [Google Scholar]
Thase 2011 {published data only}
- NCT00797966. Study of the safety and efficacy of OPC‐34712 as adjunctive therapy in the treatment of patients with major depressive disorder. clinicaltrials.gov/show/NCT00797966 (first received 25 November 2008).
- Thase M, Fava M, Hobart M, Skuban A, Zhang P, McQuade RD, et al. Efficacy of adjunctive OPC‐34712 across multiple outcome measures in major depressive disorder: phase II, randomized, placebo‐controlled study. 50th Annual Meeting of the American College of Neuropsychopharmacology; 2011 December 4‐8; Waikoloa (HI). 2011; Vol. 36:S302‐4.
Thase 2015 (PYXIS) {published data only}
- NCT01360645. A phase 3, multicenter, randomized, double‐blind, placebo‐controlled trial of the safety and efficacy of fixed dose OPC‐34712 as adjunctive therapy in the treatment of adults with major depressive disorder, the Pyxis trial. clinicaltrials.gov/show/NCT01360645 (first received 25 May 2011).
- Thase ME, Youakim JM, Skuban A, Hobart M, Augustine C, Zhang P, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo‐controlled study in patients with inadequate response to antidepressants. Journal of Clinical Psychiatry 2015;76:1224‐31. [DOI] [PubMed] [Google Scholar]
Thase 2015a (POLARIS) {published data only}
- NCT01360632. A phase 3, multicenter, randomized, double‐blind, placebo‐controlled trial of the safety and efficacy of two fixed doses of OPDC‐34712 as adjunctive therapy in the treatment of adults with major depressive disorder, the Polaris trial. clinicaltrials.gov/show/NCT01360632 (first received 25 May 2011).
- Thase ME, Youakim JM, Skuban A, Hobart M, Zhang P, McQuade RD, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double‐blind study. Journal of Clinical Psychiatry 2015;76:1232‐40. [DOI] [PubMed] [Google Scholar]
Yang 2005 {published data only}
- Yang C, Wen Q, Wang X, Liu X. Comparative study of citalopram combined with amitriptyline for treatment of refractory depression. International Medicine and Health Guidance News 2005;11(4):69‐70. [Google Scholar]
Yoshimura 2012 {published data only}
- Yoshimura R, Kishi T, Hori H, Ikenouchi‐Sugita A, Katsuki A, Umene‐Nakano W, et al. Comparison of the efficacy between paroxetine and sertraline augmented with aripiprazole in patients with refractory major depressive disorder. Progress in Neuro‐Psychopharmacology & Biological Psychiatry 2012;39:355‐7. [DOI] [PubMed] [Google Scholar]
- Yoshimura R, Kishi T, Hori H, Ikenouchi‐Sugita A, Umene‐Nakano W, Katsuki A, et al. Comparison the efficacy between paroxetine and sertraline augmented with aripiprazole in patients with refractory major depressive disorder. 28th CINP World Congress of Neuropsychopharmacology; 2012 June 3‐7; Stockholm, Sweden. 2012:199. [DOI] [PubMed]
Yoshimura 2014 {published data only}
- Yoshimura R, Hori H, Umene‐Nakano W, Ikenouchi‐Sugita A, Katsuki A, Atake K, et al. Comparison of lithium, aripiprazole and olanzapine as augmentation to paroxetine for inpatients with major depressive disorder. Therapeutic Advances in Psychopharmacology 2014;4:123‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhao 2003 {published data only}
- Zhao Z, Xian Y, Zhou H. A controlled trial of venlafaxine and paroxetine in treatment‐refractory depression. Journal of Clinical Psychological Medicine 2003;13(1):26‐7. [Google Scholar]
Zusky 1988 {published data only}
- Zusky PM, Biederman J, Rosenbaum JF, Manschreck TC, Gross CC, Weilberg JB, et al. Adjunct low‐dose lithium carbonate in treatment‐resistant depression: a placebo‐controlled study. Journal of Clinical Psychopharmacology 1988;8(2):120‐4. [PubMed] [Google Scholar]
References to studies awaiting assessment
Cao 2005 {published data only}
- Cao Y, Song L, Wang Z. Controlled study of venlafaxine combined with quetiapine in refractory depression. Journal of Clinical Psychosomatic Diseases 2005;11(2):129‐30. [Google Scholar]
Clunie 2001 {published data only}
- Clunie F. Double blind randomised controlled trial comparing moclobemide and phenelzine in treatment resistant depression. National Research Register 2001. [Google Scholar]
Euctr‐002130‐11‐Es 2007 {published data only}
- Euctr‐002130‐11‐Es. Therapeutic strategies in major depressive disorder resistant to treatment with SSRIs. Pragmatic, parallel group, randomised trial with blinded evaluation [Estrategias terapéuticas en Trastorno Depresivo Mayor resistente a tratamiento con Inhibidores Selectivos de la Recaptación de la Serotonina. Ensayo clínico pragmático, paralelo, aleatorizado con evaluación enmascarada]. clinicaltrialsregister.eu/ctr‐search/trial/2007‐002130‐11/ES (first received 4 January 2008).
Gulrez 2012 {published data only}
- Gulrez G, Badyal DK, Deswal RS, Sharma A. Bupropion as an augmenting agent in patients of depression with partial response. Basic & Clinical Pharmacology & Toxicology 2012;110:227‐30. [DOI] [PubMed] [Google Scholar]
Moica 2018 {published data only}
- Moica T, Grecu IG, Sabau D, Szasz S, Grecu GM, Moica S, et al. The efficacy of quetiapine XR as an adjunctive therapy to duloxetine in depressed patients with inadequate response to monotherapy. Farmacia 2018;66(2):354‐7. [Google Scholar]
Zhu 2003 {published data only}
- Zhu H, Yu J, Zheng H. A study of switching to mirtazapine for treatment‐resistant depression. Shanghai Archives of Psychiatry 2003;15:355‐7. [Google Scholar]
Additional references
Anderson 2008
- Anderson IM, Ferrier IN, Baldwin RC, Cowen PJ, Howard L, Lewis G, et al. Evidence‐based guidelines for treating depressive disorders with antidepressants: a revision of the 2000 British Association for Psychopharmacology guidelines. Journal of Psychopharmacology 2008;22(4):343‐96. [DOI] [PubMed] [Google Scholar]
Anttila 2001
- Anttila SA, Leinonen EV. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Reviews 2001;7(3):249‐64. [PUBMED: 11607047] [DOI] [PMC free article] [PubMed] [Google Scholar]
APA 2000
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Text Revision (DSM‐IV‐TR). 4th Edition. Washington, DC: American Psychiatric Association, 2000. [Google Scholar]
APA 2010
- American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder. psychiatryonline.org/guidelines.aspx (accessed 6 April 2013).
Bauer 1999
- Bauer M, Dopfmer S. Lithium augmentation in treatment‐resistant depression: meta‐analysis of placebo‐controlled studies. Journal of Clinical Psychopharmacology 1999;19(5):427‐34. [DOI] [PubMed] [Google Scholar]
Beck 1961
- Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Archives of General Psychiatry 1961;4:561‐71. [DOI] [PubMed] [Google Scholar]
Beck 1996
- Beck AT, Steer RA, Brown GK. Beck Depression Inventory: Manual. 2nd Edition. San Antonio: The Psychological Corporation, 1996. [Google Scholar]
Bschor 2010
- Bschor T, Baethge C. No evidence for switching the antidepressant: systematic review and meta‐analysis of RCTs of a common therapeutic strategy. Acta Psychiatrica Scandinavica 2010;121(3):174‐9. [PUBMED: 19703121] [DOI] [PubMed] [Google Scholar]
Button 2015
- Button KS, Kounali D, Thomas L, Wiles NJ, Peters TJ, Welton NJ, et al. Minimal clinically important difference on the Beck Depression Inventory‐II according to the patient's perspective. Psychological Medicine 2015;45(15):3269‐79. [PUBMED: 26165748] [DOI] [PMC free article] [PubMed] [Google Scholar]
Carvalho 2007
- Carvalho AF, Cavalcante JL, Castelo MS, Lima MCO. Augmentation strategies for treatment‐resistant depression: a literature review. Journal of Clinical Pharmacy and Therapeutics 2007;32:415‐28. [DOI] [PubMed] [Google Scholar]
Carvalho 2008
- Carvalho AF, Machado JR, Cavalcante JL. Augmentation strategies for treatment‐resistant depression. Current Opinion in Psychiatry 2008;22(1):7‐12. [DOI] [PubMed] [Google Scholar]
Cohen 1988
- Cohen J. Statistical Power Analysis in the Behavioural Sciences. Hillsdale, NJ: Lawrence Erlbaum Associates, Inc, 1988. [Google Scholar]
Cooper 2011
- Cooper C, Katona C, Lyketsos K, Blazer D, Brodaty H, Rabins P, et al. A systematic review of treatments for refractory depression in older people. American Journal of Psychiatry 2011;168:681‐8. [DOI] [PubMed] [Google Scholar]
Edwards 2013
- Edwards SJ, Hamilton V, Nherera L, Trevor N. Lithium or an atypical antipsychotic drug in the management of treatment‐resistant depression: a systematic review and economic evaluation. Health Technology Assessment (Winchester, England) 2013;17(54):1‐190. [PUBMED: 24284258] [DOI] [PMC free article] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Smith GD, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fava 2005
- Fava M. Diagnosis and definition of treatment‐resistant depression. Biological Psychiatry 2005;53:649‐59. [DOI] [PubMed] [Google Scholar]
Feighner 1972
- Feighner JP, Robins E, Guze SB, Woodruff RA, Winokur G, Munoz R. Diagnostic criteria for use in psychiatric research. Archives of General Psychiatry 1972;26:57‐63. [DOI] [PubMed] [Google Scholar]
Fekadu 2009
- Fekadu A, Wooderson S, Donaldson C, Markopoulou K, Masterson B, Poon L, et al. A multidimensional tool to quantify treatment resistance in depression: the Maudsley staging method. Journal of Clinical Psychiatry 2009;70(2):177‐84. [DOI] [PubMed] [Google Scholar]
Ferrari 2013
- Ferrari AJ, Somerville AJ, Baxter AJ, Norman R, Patten SB, Vos T, et al. Global variation in the prevalence and incidence of major depressive disorder: a systematic review of the epidemiological literature. Psychological Medicine 2013;43(3):471‐81. [PUBMED: 22831756] [DOI] [PubMed] [Google Scholar]
Friedman 2004
- Friedman MA, Detweiler‐Bedell JB, Leventhal HE, Horne R, Keitner GI, Miller IW. Combined psychotherapy and pharmacotherapy for the treatment of major depressive disorder. Clinical Psychology: Science and Practice 2004;11:47‐68. [Google Scholar]
Gibson 2010
- Gibson TB, Jing Y, Smith Carls G, Kim E, Bagalman JE, Burton WN, et al. Cost burden of treatment resistance in patients with depression. American Journal of Managed Care 2010;16(5):370‐7. [PUBMED: 20469957] [PubMed] [Google Scholar]
Hamilton 1960
- Hamilton M. A rating scale for depression. Journal of Neurology, Neurosurgery and Psychiatry 1960;23:56‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011a
- Higgins JPT, Altman DG, Cochrane Statistical Methods Group and the Cochrane Bias Methods Group. Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Ijaz 2018
- Ijaz S, Davies P, Williams CJ, Kessler D, Lewis G, Wiles N. Psychological therapies for treatment‐resistant depression in adults. Cochrane Database of Systematic Reviews 2018, Issue 5. [DOI: 10.1002/14651858.CD010558.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kessler 2013
- Kessler RC, Bromet EJ. The epidemiology of depression across cultures. Annual Review of Public Health 2013;34:119‐38. [PUBMED: 23514317] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lingjaerde 1987
- Lingjaerde O, Ahlfors UG, Bech P, Dencker SJ, Elgen K. The UKU side effect rating scale. A new comprehensive rating scale for psychotropic drugs and a cross‐sectional study of side effects in neuroleptic‐treated patients. Acta Psychiatrica Scandinavica 1987;334(Supplementum):1‐100. [PUBMED: 2887090] [DOI] [PubMed] [Google Scholar]
Liu 2015
- Liu Y, Zhou X, Zhu D, Chen J, Qin B, Zhang Y, et al. Is pindolol augmentation effective in depressed patients resistant to selective serotonin reuptake inhibitors? A systematic review and meta‐analysis. Human Psychopharmacology 2015;30(3):132‐42. [PUBMED: 25689398] [DOI] [PubMed] [Google Scholar]
Luborsky 1962
- Luborsky L. Clinician's judgments of mental health. Archives of General Psychiatry 1962;7:407‐17. [DOI] [PubMed] [Google Scholar]
Lundh 2017
- Lundh A, Lexchin J, Mintzes B, Schroll JB, Bero L. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2017, Issue 2. [DOI: 10.1002/14651858.MR000033.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mahlich 2018
- Mahlich J, Tsukazawa S, Wiegand F. Estimating prevalence and healthcare utilization for treatment‐resistant depression in Japan: a retrospective claims database study. Drugs ‐ Real World Outcomes 2018;5(1):35‐43. [PUBMED: 29185233] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mathers 2005
- Mathers CD, Loncar D. Updated Projections of Global Mortality and Burden of Disease, 2002‐2030: Data Sources, Methods and Results (Working Paper). www.who.int/healthinfo/statistics/bod_projections2030_paper.pdf (accessed 11 December 2019).
McManus 2000
- McManus P, Mant A, Mitchell PB, Montgomery WS, Marley J, Auland ME. Recent trends in the use of antidepressant drugs in Australia. Medical Journal of Australia 2000;173:458‐61. [DOI] [PubMed] [Google Scholar]
Middleton 2001
- Middleton N, Gunnell D, Whitley E, Dorling D, Frankel S. Secular trends in antidepressant prescribing in the UK, 1975‐1998. Journal of Public Health Medicine 2001;23:262‐7. [DOI] [PubMed] [Google Scholar]
Montgomery 1979
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. British Journal of Psychiatry 1979;134:382‐9. [DOI] [PubMed] [Google Scholar]
NHS Digital 2015
- NHS Digital. Prescriptions dispensed in the community. England 2004‐14. digital.nhs.uk/catalogue/PUB17644 (accessed 9th October 2018).
NHS Information Centre 2011
- NHS Information Centre. Prescription cost analysis England 2010. www.hscic.gov.uk/pubs/prescostanalysis2010 (accessed 6 April 2013).
NICE 2004
- National Institute for Health and Clinical Excellence. Depression: the treatment and management of depression in adults. National Clinical Practice Guideline 23. www.nice.org.uk/CG23 (accessed prior to 5 December 2019).
NICE 2009
- National Institute for Health and Clinical Excellence. Depression: the treatment and management of depression in adults (update). National Clinical Practice Guideline 90. www.nice.org.uk/CG90 (accessed 6 April 2013).
Nierenberg 2007
- Nierenberg AA, Katz J, Fava M. A critical overview of the pharmacologic management of treatment‐resistant depression. Psychiatric Clinics of North America 2007;30:13‐29. [DOI] [PubMed] [Google Scholar]
Pampallona 2004
- Pampallona S, Bollini P, Tibaldi G, Kupelnick B, Munizza C. Combined pharmacotherapy and psychological treatment for depression. Archives of General Psychiatry 2004;61:714‐9. [DOI] [PubMed] [Google Scholar]
Papakostas 2008
- Papakostas GI, Fava M, Thase ME. Treatment of SSRI‐resistant depression: a meta‐analysis comparing within‐versus across‐class switches. Biological Psychiatry 2008;63:699‐704. [DOI] [PubMed] [Google Scholar]
Papakostas 2009
- Papakostas GI. Managing partial response or nonresponse: switching, augmentation, and combination strategies for major depressive disorder. Journal of Clinical Psychiatry 2009;70(Suppl 6):16‐25. [DOI] [PubMed] [Google Scholar]
Pincus 1998
- Pincus HA, Tanielian TL, Marcus SC, Olfon M, Zarin DA, Thompson J, et al. Prescribing trends in psychotropic medications. JAMA 1998;279:526‐31. [DOI] [PubMed] [Google Scholar]
Ruhe 2006
- Ruhe HG, Huyser J, Swinkels JA, Schene AH. Switching antidepressants after a first selective serotonin reuptake inhibitor in major depressive disorder: a systematic review. Journal of Clinical Psychiatry 2006;67(12):1836‐55. [PUBMED: 17194261] [DOI] [PubMed] [Google Scholar]
Ruhe 2006a
- Ruhe HG, Huyser J, Swinkels JA, Schene AH. Dose escalation for insufficient response to standard‐dose selective serotonin reuptake inhibitors in major depressive disorder: systematic review. British Journal of Psychiatry 2006;189:309‐16. [PUBMED: 17012653] [DOI] [PubMed] [Google Scholar]
Shelton 2008
- Shelton RC, Papakostas GI. Augmentation of antidepressants with atypical antipsychotics for treatment‐resistant major depressive disorder. Acta Psychiatrica Scandinavica 2008;117:253‐9. [DOI] [PubMed] [Google Scholar]
Spitzer 1978
- Spitzer RL, Robins E. Research diagnostic criteria: rationale and reliability. Archives of General Psychiatry 1978;35(6):773‐82. [DOI] [PubMed] [Google Scholar]
Stimpson 2002
- Stimpson N, Agrawal N, Lewis G. Randomised controlled trials investigating pharmacological and psychological interventions for treatment‐refractory depression. Systematic review. British Journal of Psychiatry 2002;181:284‐94. [DOI] [PubMed] [Google Scholar]
Thase 1997
- Thase ME, Rush AJ. When at first you don't succeed: sequential strategies for antidepressant nonresponders. Journal of Clinical Psychiatry 1997;58 Suppl 13:23‐9. [PubMed] [Google Scholar]
Thomas 2013
- Thomas L, Kessler D, Campbell J, Morrison J, Peters TJ, Williams C, et al. Prevalence of treatment‐resistant depression in primary care: cross‐sectional data. British Journal of General Practice 2013;63(617):e852‐8. [PUBMED: 24351501] [DOI] [PMC free article] [PubMed] [Google Scholar]
Trivedi 2006
- Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, et al. Evaluation of outcomes with citalopram for depression using measurement‐based care in STAR*D: implications for clinical practice. American Journal of Psychiatry 2006;163:28‐40. [DOI] [PubMed] [Google Scholar]
Wang 2003
- Wang PS, Simon G, Kessler RC. The economic burden of depression and the cost‐effectiveness of treatment. International Journal of Methods in Psychiatric Research 2003;12(1):22‐33. [PUBMED: 12830307] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ware 1993
- Ware JE, Snow KK, Kosinski M, Gandek B. SF‐36 Health Survey: Manual and Interpretation Guide. Boston, Massachusetts: The Health Institute, New England Medical Center, 1993. [Google Scholar]
WHO 1992
- World Health Organization. International Statistical Classification of Diseases and Related Health Problems. 10th Edition. Geneva: World Health Organization, 1992. [Google Scholar]
WHOQOL 1998
- WHOQOL Group. The World Health Organisation Quality of Life Assessment (WHOQOL): development and general psychometric properties. Social Science and Medicine 1998;46:1569‐85. [DOI] [PubMed] [Google Scholar]
Wing 1994
- Wing J. Measuring Mental Health Outcomes: a Perspective from the Royal College of Psychiatrists. Outcomes into Clinical Practice. London: BMJ Publishing, 1994. [Google Scholar]
WPA 1974
- World Psychiatric Association. Symposium on therapy resistant depression. Pharmacopsychiatry 1974;7:69‐74. [Google Scholar]
Zhou 2015
- Zhou X, Keitner GI, Qin B, Ravindran AV, Bauer M, Giovane C, et al. Atypical antipsychotic augmentation for treatment‐resistant depression: a systematic review and network meta‐analysis. International Journal of Neuropsychopharmacology 2015;18(11):pyv060. [DOI: 10.1093/ijnp/pyv060] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhou 2015a
- Zhou X, Ravindran AV, Qin B, Giovane C, Li Q, Bauer M, et al. Comparative efficacy, acceptability, and tolerability of augmentation agents in treatment‐resistant depression: systematic review and network meta‐analysis. Journal of Clinical Psychiatry 2015;76(4):e487‐98. [PUBMED: 25919841] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Williams 2013
- Williams CJ, Taylor M, Kessler D, Lewis G, Wiles N. Pharmacological interventions for treatment‐resistant depression in adults. Cochrane Database of Systematic Reviews 2013, Issue 6. [DOI: 10.1002/14651858.CD010557] [DOI] [PMC free article] [PubMed] [Google Scholar]