

SUMMARY
Long non-coding RNAs (lncRNAs) are critical for regulating HOX genes, aberration of which is a dominant mechanism for leukemic transformation. How HOX gene-associated lncRNAs regulate hematopoietic stem cell (HSC) function and contribute to leukemogenesis remains elusive. We found that HOTTIP is aberrantly activated in acute myeloid leukemia (AML) to alter HOXA-driven topologically associated domain (TAD) and gene expression. HOTTIP loss attenuates leukemogenesis of transplanted mice, while reactivation of HOTTIP restores leukemic TADs, transcription, and leukemogenesis in the CTCF-boundary-attenuated AML cells. Hottip aberration in mice abnormally promotes HSC self-renewal leading to AML-like disease by altering the homeotic/hematopoietic gene-associated chromatin signature and transcription program. Hottip aberration acts as an oncogenic event to perturb HSC function by reprogramming leukemic-associated chromatin and gene transcription.
In Brief
Luo et al. find that the lncRNA HOTTIP is overexpressed in acute myeloid leukemia (AML). They show that HOTTIP coordinates topologically associated domain organization in the AML genome, including the posterior HOXA genes and various key hematopoietic regulator loci, and is important for AML growth.
Graphical Abstract
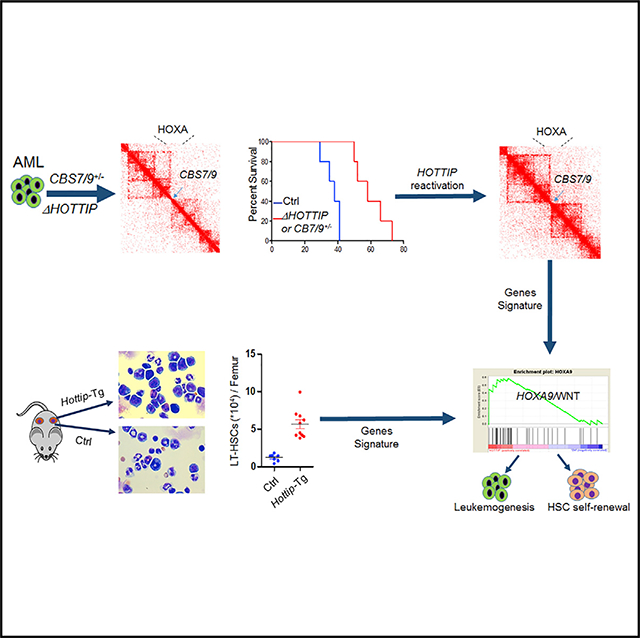
INTRODUCTION
HOX genes, especially HOXA and HOXB families, are critical for hematopoietic lineage development (Deng et al., 2013, 2016; Dou et al., 2016). Activation of HOX genes is a dominant mechanism of leukemic transformation, perhaps by altering self-renewal and differentiation properties of hematopoietic stem and progenitor cells (HS/PCs) (Andreeff et al., 2008; Drabkin et al., 2002). Although overexpression of HOX genes in acute myeloid leukemia (AML) has been attributed to specific chromosomal rearrangements involved in the mixed lineage leukemia (MLL) gene (KMT2A) or abnormalities such as mutations in NPM1 (Meyer et al., 2009; Rice and Licht, 2007), the molecular mechanisms that drive HOX genes activation are not fully understood.
HOX genes are critical for embryonic development, and their expression patterns are temporally and spatially restricted (Deng et al., 2016; Deschamps and van Nes, 2005; Forlani et al., 2003). The lineage-restricted expression pattern of HOX genes during hematopoiesis resembles their expression in early development. Generally, anterior HOX genes are highly activated in most primitive hematopoietic stem cells (HSCs) and downregulated upon lineage commitment, while posterior HOX genes are expressed in committed lineages (Sauvageau et al., 1994; Spencer et al., 2015). The different HOX genes clusters also exhibit specific patterns of lineage-specific expression. For example, HOXA genes are expressed in immature myeloid cells that are believed to play an important role in myeloid progenitor proliferation (Crooks et al., 1999; Fuller et al., 1999; So et al., 2004; Thorsteinsdottir et al., 2002).
Furthermore, the overexpression of certain HOX genes, such as HOXA9, is a strong marker of poor prognosis in leukemia patients (Collins and Hess, 2016; Golub et al., 1999), while lower expression of HOXA9 and HOXB4 are favorable predictors for AML patient outcome (Andreeff et al., 2008; Zangenberg et al., 2009), suggesting that targeting posterior HOXA genes may provide insight into AML therapy. Recently, we identified a CTCF boundary located between HOXA7 and HOXA9 (CBS7/9) that plays a critical role in maintaining posterior HOXA genes’ topologically associated domain (TAD) allowing for the aberrant HOXA gene expression (Luo et al., 2018). However, the molecular mechanism by which CBS7/9 initiates the aberrant TAD and transcription of posterior HOXA genes remain elusive.
Several HOX gene loci-associated long non-coding RNAs (lncRNAs) regulate transcription of HOX genes through their influence on the epigenetic landscape (Deng et al., 2016; Wang et al., 2011). In particular, the HOXA locus associated lncRNA HOTTIP acts as an epigenetic regulator that recruits WDR5/MLL complex to coordinate active chromatin modifications and HOXA gene expression (Wang et al., 2011). Although during limb development, expression of HOTTIP was suggested to act in cis and positively correlates with the formation of posterior HOXA gene TAD, whether HOTTIP directly binds to and regulates its chromatin targets including the HOXA locus remains unknown. It has shown that knockout (KO) of HOTTIP strongly inhibits the 5′ tip of HOXA genes (e.g., HOXA13 and HOXA11), but the inhibitory effect is gradually diminished when genes move toward the anterior end (e.g., HOXA10–HOXA7) (Wang et al., 2011). Interestingly, HOX genes, especially posterior HOXA9 and HOXA10, are frequently activated in AML, which predicts poor prognosis and treatment responses. However, the role of HOTTIP in HSC function and myeloid malignancies, and the mechanism by which HOTTIP regulates its chromatin targets in leukemogenesis, remain completely unknown.
RESULTS
HOTTIP Loss Results in Inhibition of Genes Critical for Hematopoiesis and AML Leukemogenesis
To unbiasedly uncover non-coding sequences involved in HOX gene regulation in AML, we screened all CTCF sites and lncRNAs important for HOXA9 expression within four HOX gene loci in MLL-AF9 rearranged MOLM13 AML cells using a CRISPR/Cas9 lentivirus screening library. Besides the CBS7/9 boundary, HOTTIP lncRNA was also identified as critical for aberrant HOXA9 expression (Luo et al., 2018). HOTTIP is downregulated in the CBS7/9-disrupted (CBS7/9+/−) MOLM13 cells (Figure 1A), suggesting that HOTTIP acts downstream of the CBS7/9 boundary to regulate posterior HOXA genes. To test this, we specifically deleted HOTTIP (HOTTIP−/−) by CRISPR/Cas9 in MOLM13 cells (Figure S1A). We compared transcriptomes between wild-type (WT) and HOTTIP−/− MOLM13 cells by performing RNA-sequencing (RNA-seq) analysis. A total of 706 genes exhibited greater than 2-fold decreases whereas 513 genes had increased expression upon HOTTIP−/− (Figure 1B). HOTTIP−/− impaired the transcription of not only HOXA13-HOXA9 genes but also many genes important for hematopoiesis and leukemogenesis (Figures 1B and 1C), suggesting that HOTTIP may directly regulate hematopoietic genes in AML besides the posterior HOXA genes. Gene ontology (GO) analysis revealed that many pathways were affected by both CBS7/9−/− and HOTTIP−/− (Figure 1D), including cell cycle, apoptosis, myeloid/leukocyte cell differentiation, JAK-STAT signaling, and regulation of cell development. In addition, pathways regulating hematopoietic cell lineage and myeloid differentiation were specifically affected by HOTTIP−/− (Figure 1D). Furthermore, when we subjected the RNA-seq data to gene set enrichment analysis (GSEA), the top ranked pathways affected by the HOTTIP−/− were those involved in JAK-STAT, NOTCH, cell adhesion, and progression of AML (Figures 1E and S1B).
Figure 1. HOTTIP−/− Perturbs HOXA Gene-Mediated Oncogenic Transcription Program.

(A) HOTTIP levels in WT versus CBS7/9+/− MOLM13 cells by RNA-seq. FPKM, fragments per kilobase of transcript per million mapped reads.
(B) Heatmap of >2-fold up-and downregulated genes upon HOTTIP KO by RNA-seq.
(C) mRNA levels of HOXA genes in WT and HOTTIP KO MOLM13 cells.
(D) GO analysis of genes whose expression was altered by HOTTIP KO.
(E) Enrichment of decreased genes involved in JAK-STAT (top) and AML (bottom) pathways upon HOTTIP KO by GSEA.
(F) Hi-C interacting maps in part of the human chromosome 7p15 region containing the HOXA locus comparing WT and HOTTIP KO MOLM13 cells.
See also Figure S1.
Further comparison of the expression profiles between CBS7/9+/− and HOTTIP−/− MOLM13 cells revealed that 33% of differentially regulated genes were co-regulated by both CBS7/9 boundary and HOTTIP (Figure S1C, top). Among them, 33% of genes downregulated and 24% of genes upregulated overlapped between HOTTIP−/− and CBS7/9+/− (Figure S1C, bottom). The significant overlapping of co-regulated genes by the HOTTIP−/− and CBS7/9−/− in MOLM13 cells indicated that HOTTIP may act downstream of the CBS7/9 boundary to coordinate active chromatin domain and gene transcription in AML cells. The critically downregulated genes were validated by qRT-PCR (Figure S1D).
To test the role of HOTTIP in AML genome organization, we then carried out high-throughput chromosome conformation capture (Hi-C) analysis to assess whether HOTTIP is required to organize the HOXA locus TAD in the AML genome by comparing WT and HOTTIP−/− MOLM13 cells (Figure 1F). KO of HOTTIP disrupted the posterior HOXA locus TAD, but did not affect anterior HOXA locus TAD that is demarcated by the CBS7/9 boundary (Figure 1F). Thus, the data revealed that HOTTIP is involved in organization of TAD in the AML genome to drive aberrant posterior HOXA gene expression.
HOTTIP lncRNA Is Aberrantly Expressed in a Subset of AML Patients and Cells
We then analyzed database: TCGA-LAML and database: TARGET-AML RNA-seq datasets to examine HOTTIP expression patterns. Compared with the NPM1 WT (NPM1C−) and MLL WT (MLLr−) cases (n = 245), NPM1-mutated (NPM1C+) or MLL-rearranged (MLLr+) AML cases (n = 76) exhibited elevated levels of HOTTIP expression (Figure 2A). Overall survival was significantly longer in patients having AML with low HOTTIP expression (bottom 30th percentile) than those having AML with higher HOTTIP expression (top 30th percentile, Figure 2B). Given that HOTTIP is aberrantly expressed in the MLLr+ and NPM1C+ AML (Figures S2A and S2B), we further analyzed RNA-seq data obtained from the database: TCGA-LAML and database: TARGET-AML for the correlations between expression levels of HOTTIP and of posterior HOXA genes and leukemogenic genes. HOTTIP expression positively correlated with expression of posterior HOXA genes, especially 5′ posterior tip of HOXA genes, as well as TWIST1, MEIS1 and PBX3 in AML, although the Pearson R value is relatively weak for non-HOXA genes (Figure 2C). Thus, HOTTIP plays an important role in the pathogenesis and prognosis of AML patients.
Figure 2. HOTTIP lncRNA Is Aberrantly Expressed in a Specific Subset of AML.

(A) The HOTTIP levels In NPM1C− and MLLr− AML cases and in NPM1C+ and MLLr+ AML cases obtained from the TCGA-LAML and TARGET-AML datasets. Violin plots show mean, Interquartile, and 1.5× Interquartile. The width shows the probability density.
(B) Kaplan-Meier curve of overall survival probabilities of the patients having AML with high or low HOTTIP levels from the TCGA-LAML and TARGET-AML datasets.
(C) Significant correlation between the expression of HOTTIP and posterior HOXA genes, MEIS1, TWIST1, and PBX3 in the TCGA-LAML and TARGET-AML datasets. Pearson correlation and corresponding p value are calculated by the cor.test of R.
See also Figure S2.
HOTTIP Establishes Aberrant Chromatin Signature to Drive AML-Specific Transcription Profile
To further investigate how HOTTIP regulates aberrant posterior HOXA locus TAD and gene expression in AML, we carried out chromatin isolation by RNA purification (ChIRP-seq) to examine global HOTTIP binding in the MLLr+ AML genome of WT and HOTTIP−/− MOLM13 cells. HOTTIP lncRNA bound to HOXA9-HOXA13 in cis, but not to the anterior HOXA genes or the HOXB locus (Figures 3A and S3A–S3C). HOTTIP−/− greatly reduced the binding of HOTTIP to the posterior HOXA genes (Figures 3A and S3B), supporting that HOTTIP is a regulator of posterior HOXA genes. The global binding site distribution of HOTTIP in AML genome revealed that HOTTIP mainly binds to non-coding regions (Figure 3B). Although HOTTIP bound to 3,767 genomic sites, it only directly bound to the promoters of 259 annotated genes that are mainly involved in hematopoiesis, myeloid cell differentiation, cell-cycle progression, and JAK-STAT and WNT signaling pathways (Figure 3C), concomitant with GO enriched pathways obtained from the changed transcriptomic profiles upon HOTTIP−/− (Figure 1D). In addition, the GO analysis of HOTTIP-bound intergenic regions also revealed that HOTTIP targets are consistently involved in chromatin organization, myeloid cell differentiation, and hematopoiesis (Figure S3D). Interestingly, HOTTIP also bound in trans in PBX3, MYC, KIT, CD33, MEIS2, and RUNX1 promoters (Figures 3D and S3E). To examine whether HOTTIP is indeed regulating the hematopoietic transcription program, we performed de novo motif analysis of the HOTTIP binding sites from ChlRP-seq (Figure 3E and Table S1). Consistently, the top transcription factor (TF) motifs bound by HOTTIP are those involved in HS/PC function, such as RUNX1, MYC, E-box, and STAT5 motifs (Figure 3E, top), suggesting that these factors may interact with HOTTIP to mediate its function in hematopoiesis. To confirm this notion, we carried out RNA immunoprecipitation (RIP) and showed that HOTTIP physically interacts with phosphorylated STAT5A, MYC, RUNX1, DOT1L, and MLL1 complex, but not HDAC1 and IKAROS controls (Figures 3F and S3F). Thus, HOTTIP regulates the hematopoietic chromatin landscape and transcription program by interacting with hematopoietic-specific TFs and epigenetic regulators.
Figure 3. HOTTIP Reprograms AML Chromatin and Regulates Leukemic Specific Transcription Networks.

(A) ChIRP-seq analysis of HOTTIP binding in WT and HOTTIP KO MOLM13 cells.
(B) Pie chart shows global HOTTIP binding distribution in the human AML genome.
(C) GO analysis of HOTTIP regulated transcription and signal pathways.
(D) Changes in HOTTIP binding (ChIRP-seq), chromatin accessibility (ATAC-seq), and MLL1 recruitment and H3K4me3 enrichment (ChIP-seq) upon HOTTIP KO.
(E) Shared top TF binding motifs enriched by the HOTTIP ChIRP-seq (top) and the ATAC-seq altered peaks (bottom) upon HOTTIP−/−by the de novo motif analysis.
(F) HOTTIP and protein interactions detected by RIP.
(G) ChIP-seq analysis of changes in H3K4me3, H3K27me3, and H3K79me2 modification levels upon HOTTIP−/− in MOLM13 cells.
(H) Altered ATAC-seq accessibility of genomic regions upon HOTTIP−/−. Box plots show horizontal line (zero Z score, mean), box indicating the median with upper and lower quartiles, and whiskers indicating the highest and lowest values. p value is calculated by Kolmogorov-Smirnov test.
(I) ATAC-seq analysis of altered chromatin accessibility upon HOTTIP−/− in MOLM13 cells.
To test whether HOTTIP controls the chromatin signature of its targets, we performed chromatin immunoprecipitation sequencing (ChIP-seq) and ATAC-seq (assay for transposase-accessible chromatin using sequencing) in WT and HOTTIP−/− MOLM13 cells. Consistent with transcriptional changes of the posterior HOXA genes, HOTTIP−/− resulted in marked decreases in H3K4me3 and H3K79me2, while expanding and elevating H3K27me3 levels in the posterior HOXA gene domain (Figure 3G). In contrast, HOTTIP−/− affected neither the anterior HOXA gene domain nor the HOXB gene locus (Figures 3G and S3G). In HOTTIP−/− cells, significantly gained or lost chromatin accessibility in the subset of genomic regulatory regions was observed (Figure 3H). HOTTIP−/− led to a significant decrease in chromatin accessibility only in the posterior HOXA gene domain, but not anterior HOXA genes or HOXB gene clusters (Figures 3I and S3H; Table S2). In addition, the MLL1 recruitment and chromatin modification/accessibility of a subset of non-HOXA genes were impaired upon loss of HOTTIP binding, consistent with the binding of HOTTIP to these genes (Figures 3D and S3E; Table S2). The de novo motif analysis of ATAC-seq altered peaks confirmed that the top HOTTIP-bound motifs also exhibit significant chromatin accessibility alteration (Figure 3E, bottom; Table S1). Thus, HOTTIP establishes an aberrant HOXA gene-associated chromatin signature to drive ectopic transcription profile in the MLLr+ AML.
HOTTIP Loss Perturbs AML Cell Proliferation and Prolongs Survival of the Transplanted AML Mouse Models
We next assessed the effects of HOTTIP−/− on leukemic cell growth and viability. Compared with WT MOLM13 cells, HOTTIP−/− showed consistent inhibitions of the posterior HOXA genes (Figure 4A) and cell proliferation (Figure 4B). Cell-cycle analysis revealed that HOTTIP−/− blocked MOLM13 cells in the G1 phase and significantly reduced the G2/M phases (Figure 4C), suggesting that HOTTIP controls AML cell proliferation by regulating cell-cycle progression consistent with RNA-seq analysis (Figure 1D). To exclude any effect of possible regulatory elements presented in the genomic HOTTIP region, we created CRISPR/dCas9-KRAB-mediated HOTTIP epigenetic silenced clones using the KRAB-repressive domain that recruits H3K9 methyltransferase Suv39H1 (Gilbert et al., 2013) to specifically target the promoter region of HOTTIP gene in MLLr+ MOLM13 and NPM1C+ OCI-AML3 cells. dCas9-KRAB targeted at the HOTTIP promoter elevated H3K9me2 levels while inhibiting H3K4me3 levels across HOXA9-HOXA13 genes (Figures S4A and S4B). Inhibition of HOTTIP significantly reduced HOTTIP binding to posterior HOXA and non-HOXA genes in both cell lines (Figures S4C–S4E). As a result HOTTIP target gene expression, cell proliferation, and cell-cycle progression were specifically inhibited by HOTTIP inhibition (Figures S4F–S4K), consistent to the phenotypes of HOTTIP−/−.
Figure 4. HOTTIP−/− Perturbs Cell Proliferation and Prolongs Survival of the Transplanted AML Mouse Models.

(A) qRT-PCR analysis of HOXA gene expression in WT and HOTTIP−/− MOLM13 clones.
(B) Proliferation curves of WT and HOTTIP−/− MOLM13 cells.
(C) Cell-cycle analysis of WT and HOTTIP−/− MOLM13 clones.
(D) Kaplan-Meier curves of NSG mice transplanted with WT and HOTTIP−/− MOLM13 cells.
(E) hCD45+ cell chimerism in BM, spleen (SP), and PB of NSG mice receiving WT (n = 4) or HOTTIP−/− (n = 4) MOLM13 cells.
(F) Hematoxylin and eosin (H&E) and anti-hCD45 immunostaining (brown) of femur sections from mice transplanted with WT or HOTTIP−/− MOLM13 cells for 16 days.
(G) Kaplan-Meier curves of NSG mice transplanted with WT or HOTTIP−/− primary AML patient BM cells carrying MLLr+ (LPP4), NPM1C+Flt3-ITD+ (974), or NPM1C−FLT3-ITD+ (886) mutations.
(H) hCD45+ cell chimerism in BM, SP, and PB of NSG mice receiving WT (n = 4) or HOTTIP−/− (n = 4) primary AML cells.
Data in (A), (B), (C), (E), and (H) are presented as mean ± SD; *p < 0.05; **p < 0.01; ***p < 0.001 by Student’s t test. See also Figure S4.
To test whether HOTTIP loss affects AML leukemogenesis; in vivo, we transplanted 5 × 105 WT or HOTTIP−/− MOLM13 cells into irradiated NSG mice. All mice transplanted with WT MOLM13 cells died around 40 days after transplantation while mice receiving HOTTIP−/− cells survived more than 70 days (Figure 4D). Indeed, fluorescence-activated cell sorting (FACS) analysis of recipients at 35 days after transplantation revealed that the human CD45+ (hCD45+) cell chimerism was significantly reduced in mice receiving HOTTIP−/− MOLM13 cells (Figure 4E). Consistently, immunostaining of femur sections showed that mice transplanted with HOTTIP−/− cells had decreased infiltration of hCD45+ AML blasts in bone marrow (BM) (Figure 4F). Thus, deletion of HOTTIP reduces the AML leukemic burden in vivo.
Furthermore, HOTTIP was deleted in primary AML cells with MLLr+ (#LPP4), NPM1C+;FLT3-ITD+ (#974), or FLT3-ITD+ (#886) obtained from patients by CRISPR/Cas9 editing. Both #LPP4 and #974 exhibited elevated expression of HOTTIP and posterior HOXA9-A13 genes, while #886 had low HOTTIP and posterior HOXA gene expression (Figure S4L). We then transplanted 2 × 105 control or HOTTIP−/− primary AML cells into NSG mice. Interestingly, mice receiving control MLLr+ (#LPP4) or NPM1C+/FLT3-ITD+ (#974) AML cells all died around 31 days after transplantation, while mice transplanted with corresponding HOTTIP−/− AML cells survived for up to 45 days (Figure 4G). FACS analysis revealed that HOTTIP−/− dramatically decreased the hCD45+ cell chimerism in BM, spleen, and peripheral blood (PB) of recipients (Figure 4H). In contrast, HOTTIP−/− neither prolonged the survival nor decreased hCD45+ cell chimerism in mice transplanted with FLT3-ITD+ (#886) (Figures 4G and 4H). Thus, loss of HOTTIP decreases tumor burden and attenuates leukemic progression in vivo specific for HOTTIP-activated AML patients carrying MLLr+ or NPM1C+ mutations.
HOTTIP Activation Rescues AML-Associated Chromatin Signature and Transcription Profile in CBS7/9+/− AML Cells
Since CBS7/9+/− strongly suppressed HOTTIP expression (Figure 1A), we next sought to test whether HOTTIP acts downstream of CBS7/9 to organize the AML-associated TAD and transcription profile using the dCAS9-VP160 mediated promoter activation of the endogenous HOTTIP gene in CBS7/9+/− MOLM13 cells. Reactivation of HOTTIP in the CBS7/9+/− MOLM13 cells largely restored the expression of the posterior HOXA9-HOXA13 genes (Figure 5A). HOTTIP reactivation partially rescued the defective cellular proliferation in the CBS7/9+/− MOLM13 cells by escaping G1-phase blockage and restoring the G2/M phase (Figures 5B and 5C).
Figure 5. Activation of HOTTIP Rescues the HOXA Gene Chromatin Defects in the CBS7/9+/− AML Cells.

(A) qRT-PCR analysis of HOXA gene expression in WT, CBS7/9+/−, and the dCas9-VP-160-mediated HOTTIP-activated MOLM13 clones.
(B) Proliferation curves of the WT, CBS7/9+/−, and the dCas9-VP-160-mediated HOTTIP-activated MOLM13 cells.
(C) Cell-cycle analysis of the WT, CBS7/9+/−, and dCas9-VP-160-mediated HOTTIP-activated MOLM13 clones.
(D) Heatmap of RNA-seq analysis shows up-and downregulated genes of WT and the dCas9-VP-160-mediated HOTTIP-activated clones compared with the CBS7/9+/− clone.
(E) qRT-PCR validation of the key altered genes identified by RNA-seq comparing WT, CBS7/9+/−, and the dCas9-VP-160-mediated HOTTIP-activated clones.
(F) Enrichment of upregulated genes involved in HOXA9 (top) and AML (bottom) pathways upon HOTTIP activation in the CBS7/9+/− MOLM13 by GSEA.
(G) ATAC-seq analysis of chromatin accessibility in WT, CBS7/9+/−, and the dCas9-VP-160-mediated HOTTIP-activated MOLM13 cells.
(H) ChIP-seq analysis of MLL1 recruitment in WT, CBS7/9+/−, and the dCas9-VP-160-mediated HOTTIP-activated MOLM13 cells.
(I) Hi-C interacting maps in part of the human chromosome 7p15 region containing the HOXA locus comparing WT, CBS7/9+/−, and the dCas9-VP-160-mediated HOTTIP-activated MOLM13 cells.
(J) Kaplan-Meier curve of NSG mice transplanted with WT, CBS7/9+/−, or the dCas9-VP-160-mediated HOTTIP-activated MOLM13 cells.
(K) FACS analysis of hCD45+ cell chimerism in BM, spleen (SP), and PB of NSG mice 14 days after transplantation of WT (n = 4), CBS7/9+/− (n = 4), or the dCas9-VP-160-mediated HOTTIP-activated MOLM13 cells (n = 4).
Data in (A), (B), (C), (E), and (K) are presented as mean ± SD; *p < 0.05; **p < 0.01; ***p < 0.001; ns: not significant by Student’s t-test. See also Figure S5.
Next, we carried out RNA-seq, ATAC-seq, ChIP-seq, and Hi-C analysis using WT, CBS7/9+/−, and the HOTTIP-activated CBS7/9+/− MOLM13 cells. CBS7/9+/− downregulated 865 genes involved in myeloid differentiation and cell-cycle controls (Luo et al., 2018). HOTTIP activation largely reversed the transcription profiles of the gene sets affected by the CBS7/9+/−, making them closely resemble WT MOLM13 cells (Figure 5D). These genes are involved in hematopoiesis, myeloid differentiation, and cell-cycle controls (Figures 5D and 5E). GSEA revealed that the pathways involved in HOXA9 regulation, AML progression, JAK-STAT signaling, and NOTCH signaling were enriched in HOTTIP-activated cells as compared with CBS7/9+/− MOLM13 cells (Figures 5F and S5A). In addition, chromatin accessibility and MLL1 recruitment was largely rescued in the posterior HOXA gene domain (Figures 5G and 5H; Table S2) and the promoters of RUNX1, TWIST1, STAT5A, and MYC (Figures S5B and S5C; Table S2), but not HOXB genes (Figure S5D). Consistently, the posterior HOXA locus TAD that was largely disrupted by the CBS7/9+/− was rescued by HOTTIP reactivation (Figure 5I). Thus, HOTTIP plays a critical role in establishing and maintaining leukemic specific HOXA locus TAD and gene-expression profiles.
To evaluate whether HOTTIP reactivation functionally rescues the CBS7/9+/−-mediated anti-leukemia effect, we again transplanted 2 × 105 WT, CBS7/9+/−, or HOTTIP-activated CBS7/9+/− MOLM13 cells into irradiated NSG mice. All mice transplanted with CBS7/9+/− cells died between 29 and 39 days after transplantation, while the mice receiving WT or HOTTIP-activated CBS7/9+/− MOLM13 cells survived only 14–25 days (Figure 5J). FACS analysis of recipients at 14 days after transplantation revealed that the hCD45+ cell chimerism in the mice receiving HOTTIP-reactivated CBS7/9+/− MOLM13 cells was restored to compatible levels of mice transplanted with WT cells (Figure 5K). Thus, reactivation of HOTTIP in the CBS7/9+/− MOLM13 cells rescues leukemic HOXA locus TAD, chromatin signature, and transcription profile to reverse the CBS7/9+/−-mediated antileukemic effects.
Hottip Transgenic Expression in the Hematopoietic Compartment Perturbs HSC Pools and Leads to AML-like Disease in Mice
Hottip expression is high in HSCs and early progenitors but decreases upon terminal differentiation (Figure S6A). Since HOTTIP is aberrantly expressed in the MLLr+ and NPM1C+ AMLs (Figure 2A) resulting in dysregulation of HOX gene-associated chromatin domain and gene expression (Figures 1B and 1F), enforced expression of HOTTIP in HS/PC may perturb HSC function in vivo. We then generated transgenic mice that express Hottip lncRNA under the control of the hematopoietic-specific Vav1 enhancer and promoter (Figure S6B). The transgene was integrated in chromosome 10qE4 (Chr10:60117191) (Figure S6C). Two transgenic mouse lines were obtained, which exhibited ~5- and ~11-fold increase, respectively, in Hottip expression compared with the endogenous Hottip levels in BM cells (Figure S6D). The Hoxa9-Hoxa13 genes were also aberrantly elevated upon Hottip transgenic expression (Figure S6E). When a cohort of WT and Hottip-Tg mice from both lines (6–18 months old) were analyzed, the Hottip-Tg mice exhibited increased white blood cell and neutrophil counts (Figure 6A) and most of them developed splenomegaly (Figure S6F), indicating that enforced expression of Hottip led to perturbation of hematopoiesis. FACS analysis of BM cells revealed that the c-Kit+ cell population was significantly increased in Hottip-Tg mice (Figure 6B). Importantly, ~32% of Hottip-Tg mice died by 18 months of age (Figure 6C). May-Giemsa staining of PB smears and BM cytospins revealed increases in immature myeloid cells in these Hottip-Tg mice (Figure 6D). Further histological analysis identified myeloid cell infiltration in the liver of these mice (Figure 6D). However, whole-exome sequencing of bulk tumor (BM) and non-tumor (skin) cells from five diseased Hottip-Tg mice did not reveal any recurrent mutation in genes implicated in HSC regulation or leukemogenesis in the BM of Hottip-Tg mice (Table S3). The only recurrently mutated gene in Hottip-Tg BM cells is Mroh2a, which has unknown cellular function. Thus, Hottip overexpression plays an intrinsic oncogenic role in the pathogenesis of myeloid malignancies in vivo.
Figure 6. Hottip Transgenic Expression in Hematopoiesis Perturbs HSC Pools and Leads to AML-like Disease.

(A) Parameters of blood counts were summarized from 6- to 20-month-old Hottip-Tg (n = 15) and age-matched WT (n = 8) mice. WBC, white blood cells; NE, neutrophils; RBC, red blood cells. Data are presented as mean ± SD; *p < 0.05; **p < 0.01 by Student’s t test.
(B) FACS analysis and quantitation of c-Kit (CD117+) cells within total BM cells of 6- to 20-month-old WT (n = 7) and Hottip-Tg (n = 15) mice. Data show all dots as mean ± SD by Student’s t test. Horizontal bars represent mean. Data are presented as mean ± SD; ***p < 0.001 by Student’s t test.
(C) Kaplan-Meier curve of WT (n = 21) and Hottip-Tg (n = 31) mice over 20 months.
(D) Images of PB smears, BM cytospins, and liver sections prepared from representative WT and Hottip-Tg mice. Scale bar, 20 μm.
(E) FACS analysis of LSK and LK populations in the BM Lin− cells (top) as well as LT-HSC, ST-HSC, and MPP populations in the BM LSK cells (bottom) of representative young (8–16 weeks old) WT and age-matched Hottip-Tg mice.
(F) Quantitation of the total LSK, LK, LT-HSC, ST-HSC, and MPP populations per femur of young WT (n = 7) and Hottip-Tg (n = 10) mice. Quantitation data are presented as mean ± SD; ***p < 0.001 by Student’s t test.
FACS analyses of the BM HSCs of young WT and Hottip-Tg mice (8–16 weeks old) revealed that overexpression of Hottip increased the frequencies and pools of lineage−Sca-1+c-Kit+ (LSK) cell population in mice (Figures 6E and 6F). Furthermore, both the frequencies and total numbers of long-term (LT) and short-term (ST) HSCs were dramatically increased in Hottip-Tg mice (Figure 6F). Of note, alteration of proportions of common myeloid progenitor (CMP), megakaryocyte-erythroid progenitor (MEP), granulocyte-macrophage progenitor (GMP), and mature lineage cell populations in the BM were not evident in young Hottip-Tg mice (Figures S6G and S6H), but these abnormalities developed as mice were aging.
To determine the impact of the Hottip transgene levels on the hematological phenotypes, we generated the homozygous Hottip-Tg (HottipHomo-Tg) mice with 1-fold increase in the levels of Hottip expression as compared with the Hottip-Tg mice (Figure S6I, left), with a shorter disease latency than Hottip-Tg mice. Half of the HottipHomo-Tg mice (5 of 10) developed AML-like disease within 8 months of age. These mice exhibited severe anemia, splenomegaly, >20% of blast in PB or BM, and increased CD117+CD11b+ immature myeloid populations in their BM (Figures S6I–S6L). When young HottipHomo-Tg were analyzed for composition of hematopoietic populations, the HottipHomo-Tg mice had higher frequencies and pools of LSK and lineage−Sca-1−c-Kit+ (LK) cell populations than Hottip-Tg mice (Figure S6M). Consistently, the frequencies and total numbers of LT- and ST-HSCs, and multipotent progenitor cells (MPPs) were significantly higher in the BM of HottipHomo-Tg mice than those of WT or Hottip-Tg mice (Figure S6N). In addition, the frequencies of GMP were increased, and MEP frequencies were decreased in the BM of HottipHomo-Tg mice as compared with WT or Hottip-Tg mice, which did not exhibit such alterations at such a young age (Figure S6O). These data suggest that the Hottip overexpression alters HSC pool and homeostasis in vivo in a gene-dosage-dependent manner.
Hottip Regulates the Balance of Self-Renewal and Differentiation of HSCs
Next, we sought to investigate the role of Hottip in HSC function by assessing the frequencies of colony-forming unit cells (CFU-C) in the BM and spleen of WT and Hottip-Tg mice (Figure 7A). The frequencies of each type of CFU-C including CFU-GM (granulocyte/monocyte), BFU-E (burst-forming unit-erythrocyte), and CFU-GEMM (granulocyte/erythrocyte/monocyte/megakaryocyte) were significantly higher in both BM and spleen of Hottip-Tg mice (Figure 7A). When replating assays were performed on WT and Hottip-Tg LSK cells purified from the BM of mice, a higher replating potential was observed in Hottip-Tg LSK cells (Figure 7B).
Figure 7. Hottip Transgenic Expression Perturbs HSC Function, Leading to AML-like Disease.

(A) Frequencies of CFU-Cs in the BM and spleen cells from WT and Hottip-Tg mice. GM, granulocytes/macrophages; BFU-E, burst-forming unit-erythrocytes; GEMM, granulocytes/erythrocytes/monocytes/megakaryocytes.
(B) Frequencies of colonies per 100 LSK cells in WT and Hottip-Tg BM cells are shown (1st). Colonies were replated every 7 days for four times (2nd-5th).
(C) Paired-daughter cell assays were performed on CD34− LSK cells from WT, Hottip-Tg mice, Asxl2−/− mice, and AML-ETO mice.
(D) FACS analyses of CD45.2 (donor) chimerisms in the PB of recipients (CD45.1) receiving WT or Hottip-Tg BM cells in first transplantation (top) and second transplantation (bottom).
(E) Kaplan-Meier curve of second transplantation receiving WT (n = 5) and Hottip-Tg (n = 5) BM cells (top) and appearance of spleens and femur of representative WT and moribund Hottip-Tg mice receiving second transplantation (bottom).
(F) Images of PB smears (top) and BM cytospins (bottom) prepared from representative WT and moribund Hottip-Tg mice receiving second transplantation. Scale bar, 20 μm.
(G) FACS analyses showing CD45.2 versus CD45.1 chimerism as well as their respective lineage distribution and LSK/LK cell populations (within Lin− cells) in the BM of representative WT or Hottip-Tg mice receiving second transplantation.
Data in (A), (B), and (D) are presented as mean ± SD; *p < 0.05; **p < 0.01; ***p < 0.001 by Student’s t test. See also Figure S7.
Both symmetric and asymmetric cell divisions are required for the preservation of normal HSC pool and continuous production of blood cells. To test whether Hottip aberration alters cell fates of HS/PCs, we performed paired-daughter cell assays to assess the proportions of symmetric self-renewal, symmetric differentiation, and asymmetric divisions using primitive CD34− LSK cells isolated from WT and Hottip-Tg BM. Overexpression of Hottip increased in proportion to CD34− LSK cells with symmetric self-renewal capacity (Figure 7C). In contrast, the proportion of cells that underwent symmetric differentiation were decreased, while the asymmetric division of CD34− LSK cells was not affected (Figure 7C). The abnormal behavior of CD34− LSK caused by Hottip-Tg is similar to ASXL2−/− or AML-ETO expression, which enhances HSC self-renewal and blocks myeloid differentiation.
To further interrogate the effects of the ectopic Hottip expression on HS/PC proliferation and differentiation, we performed liquid cultures and analyzed weekly the frequencies and total numbers of c-Kit+ cells and CFU-Cs in the progenies. FACS analysis and colony assays revealed that Hottip-Tg LK cells gave rise to a greater number of c-Kit+ progenies and a higher number of CFU-Cs than WT LK cells at each time point assayed (Figures S7A–S7C). Thus, the ectopic expression of Hottip in HS/PCs leads to an increased expansion of c-Kit+ cells and is likely accompanied by increased proliferation and impaired differentiation, two hallmarks of leukemogenesis.
Cell-Autonomous Effect of Hottip Overexpression on HS/PC Functions
Next, we carried out competitive transplantation assays to examine the repopulating capacity of Hottip-Tg and WT BM cells in comparison with WT competitor BM cells in recipient mice. When the donor cell chimerism was analyzed kinetically in the PB of recipient mice for 6 months, the CD45.2 (donor) cell population remained ~50% in mice receiving WT BM cells, whereas the CD45.2 chimerism in mice receiving Hottip-Tg BM cells steadily increased, reaching ~80% 6 months after transplantation (Figure 7D, top). Strikingly, Hottip-Tg BM cells generated higher proportions and numbers of LSKs in recipient animals as compared with WT BM and competitor BM cells (Figure S7D). The proportions of each mature lineage population produced by Hottip-Tg BM cells were comparable with that produced by WT and competitor BM cells up to 6 months after transplantation (Figure S7D). However, when we transplanted HottipHomo-Tg BM cells into recipients (Figure S7E), 40% of the HottipHomo-Tg recipients developed AML-like disease within 6 months post transplantation (Figure S7F). The HottipHomo-Tg BM cells exhibited a significantly enhanced repopulation capacity and generated higher proportions and numbers of LSK, LK, LT-HSC, CMP, and immature myeloid cells (CD117+CD11b+ and CD117+Gr1+) as compared with WT and competitor BM cells (Figure S7G), indicating that Hottip transgene perturbs HSC function and hematopoiesis in a gene-dosage-dependent manner.
To enforce LT-HSC self-renewal and disease development, we carried out secondary transplantation by transplanting WT or Hottip-Tg BM cells from primary recipients (1 × 106 cells) into second recipients. In this context, 80% of the Hottip-Tg second transplanted recipients developed AML-like diseases as evident by splenomegaly and significant increases of immature myeloid cells in the PB or BM (Figures 7E–7G). The donor cell chimerism in the PB of the second recipients transplanted with Hottip-Tg BM cells continuously increased, from 80% to 93% 6 months after the second transplantation (Figure 7D, bottom). Hottip-Tg BM cells generated much higher proportions and numbers of LSKs and immature myeloid cells in second-recipient mice when compared with WT BM cells and competitor BM cells (Figure 7G). Thus, ectopic expression of Hottip in hematopoiesis increases the repopulating capacity of HS/PCs and promotes HSC self-renewal, leading to AML-like disease.
Transgenic Expression of Hottip Remodels Chromatin Accessibility and Alters Hematopoietic Transcription Programs
It is conceivable that transgenic expression of Hottip remodels the HOX gene-associated chromatin domain and drives the hematopoietic transcription program to perturb HS/PC function. To examine this, we performed ATAC-seq and RNA-seq using BM LT- and ST-HSCs purified from WT and Hottip-Tg mice. RNA-seq revealed that a total of 535 genes exhibited >2-fold increases, whereas 275 genes had decreased expression in LT-HSCs upon Hottip transgenic expression (Figure 8A). Upregulated genes included Hoxa9-a13, Nanog, Sox2, Myc, Meis1, Runx1, Kit, Slamf1, Gata2, and Pbx3 (Figure 8B), many of which were directly bound by Hottip (Figure S8A) suggesting that Hottip regulates posterior Hoxa genes and hematopoietic transcription networks. Furthermore, GO analysis revealed that an Hottip-Tg altered transcription program is involved in pathways associated with the pluripotency of stem cells, cell-fate commitment, LT potentials, HSC proliferation, myeloid progenitor differentiation, and NOTCH, JAK-STAT, and WNT signaling (Figure 8C). Similar hematopoietic pathways and genes were also altered in ST-HSCs upon Hottip transgenic expression (Figures S8B and S8C). Concomitantly, ATAC-seq revealed that Hottip aberration enhanced chromatin accessibility in the endogenous posterior Hoxa gene cluster and non-Hoxa gene targets (Figures 8D and S8D–S8F; Table S2). As a control, Hottip expression did not alter chromatin accessibility in the anterior Hoxb gene domain (Figure S8G), which also plays an important role in HSC function. We further grouped ATAC-seq promoter gained or lost peaks and carried out GO analysis to examine the pathways associated with gain or loss of promoter accessibility upon Hottip aberration in BM HSCs. Gained peaks were associated with pathways involved in the pluripotency of stem cells, cell-fate commitment, LT potentials, HSC proliferation, and JAK-STAT and WNT signaling, while lost peaks were associated with hematopoietic cell lineage, the cellular differentiation program, and myeloid progenitor cell differentiation (Figures 8E and S8E). Thus, Hottip acts as an epigenetic regulator directing hematopoietic transcription programs.
Figure 8. Transgenic Expression of Hottip Alters HSC Chromatin Signature and Hematopoietic Transcription Programs.

(A) Scatterplot of RNA-seq analysis of >2-fold differentially expressed genes upon overexpression of Hottip in BM LT-HSCs.
(B) Heatmap showing changed expression of representative genes upon Hottip overexpression.
(C) GO analysis of the HOTTIP affected genes.
(D) ATAC-seq analysis of chromatin accessibility in WT and Hottip-Tg BM LT-HSCs.
(E) ATAC-seq promoter density map of LT-HSCs sorted from WT and Hottip-Tg BM. Upregulated (top) or downregulated (bottom) ATAC-seq promoter peaks correlate with GO enriched pathways annotated by GREAT (gemomic regions enrichment of annotations tool) analysis.
(F) Primary MLLr+ AML patient samples with elevated HOTTIP expression were treated with DMSO or ICG-001 (500 nM) and the LT culture-initiating cells frequency of each group was determined. The black bars represent the mean frequency of each group. Data are presented as mean ± SD.
(G) Primary MLLr+ AML patient samples MLL7 (2.5 million cells) were transplanted into NSG mice. The mice were treated with vehicle (n = 4) or ICG-001 (50 mg/kg; n = 5) and sacrificed when they showed signs of illness.
See also Figure S8.
In both LT- and ST-HSCs, WNT signaling pathway was upregulated by Hottip transgenic expression (Figures 8C, 8E, and S8E), suggesting that the canonical WNT signaling pathway may play an important role in Hottip-driven leukemia. WNT signaling is required for HSC homeostasis and leukemia development (Lento et al., 2013; Reya et al., 2003). To test the clinical implication of Hottip-driven leukemogenesis, we treated primary AML patient samples carrying MLLr+ and exhibiting elevated HOTTIP expression with DMSO (control) or 500 nM ICG-001, a canonical WNT inhibitor (Emami et al., 2004) (Figures S8H–S8J). As a result, LT culture initiating cell frequency of the HOTTIP-expressed AML samples were significantly inhibited by ICG-001 while HOTTIP silenced/or weakly expressed MLLr− AML samples were resistant to the ICG-001 (Figures 8F, S8I, and S8J). Furthermore, the primary MLLr+ AML patient samples MLL7 that expressed high levels of HOTTIP were transplanted into NSG mice and treated with vehicle (control) or ICG-001 (50 mg/kg). Treatment of ICG-001 significantly prolonged the survival of leukemic mice (Figure 8G) by decreased CD45/CD33 human leukemic blast in BM and eradicated the human leukemic blast in spleen and liver (Figure S8K). Thus, MLLr+ AML expressing a high level of HOTTIP is indeed sensitive to WNT inhibitor, and the WNT signaling pathway facilitates Hottip-driven leukemogenesis.
DISCUSSION
HOTTIP is known to coordinate transcription of the 5′ tip of HOXA genes (Wang et al., 2011). We characterized a function of HOTTIP in regulating the balance of HSC self-renewal and differentiation. The HOTTIP action is, in part, dependent on its ability to directly bind to and regulate genes and pathways that are required for HS/PC regulation. It is particularly interesting that the WNT pathway is required for self-renewal of leukemia stem cells (LSCs) in AML that are driven by MLLr+ or its targets, MEIS1 and HOXA9 (Wang et al., 2010; Yeung et al., 2010). HOTTIP is highly expressed in MLLr+ or NPM1C+ mutated AML patients and the HOTTIP-expressed AML is sensitive to the WNT inhibitor. Thus, HOTTIP may be also involved in regulation of LSCs carrying MLLr+ or NPM1C+ mutations and represents a therapeutic opportunity for eradication of LSCs, in part through manipulating the WNT pathway.
MLLr+ AML is one of the most devastating subtypes of AML, being associated with poor prognosis and chemoresistance. Although NPM1C+ mutation alone is generally associated with favorable prognosis, co-existence of FLT3-ITD and/or DNMT3A mutations predicts an increased risk of relapse and poorer outcome (Gale et al., 2015; Grimwade et al., 2016). HOXA9 is a strong predictor of poor prognosis in AML (Collins and Hess, 2016). MEIS1 and PBX3 are oncogenes that co-express with many HOX genes, especially HOXA9, to stimulate the proliferation of HSCs (Li et al., 2016; Takeda et al., 2006). They are highly expressed in AML cases carrying MLLr+ or NPM1C+ mutation and depend on HOTTIP lncRNA. Interestingly, AMLs harboring NPM1C+ with aberrant HOXA9/A10 genes and homeotic oncogenes, MEIS1 and PBX3, are synergistically required for the maintenance of NPM1C+-driven AML (Brunetti et al., 2018; Dovey et al., 2017; Kuhn et al., 2016). The reports are consistent with Hottip overexpression resulting in activation of Hoxa9, Meis1, and Pbx3 in the HSC population, promoting HSC self-renewal and leukemogenesis.
Although TADs are mostly conserved across cell types and species, TADs are indeed structural and functional chromosomal units that constrain enhancer/promoter communication for specific transcription programs (Valton and Dekker, 2016). Altered TAD might result in inappropriate promoter/enhancer interactions to alter transcription of oncogenes or tumor suppressors (Groschel et al., 2014; Taberlay et al., 2016). Chromatin boundaries, CTCF binding sites in many cases, play a critical role in defining TADs and chromatin signature within the TAD (Luo et al., 2018; Narendra et al., 2015). Given that expression of HOTTIP restores CBS7/9-mediated posterior HOXA locus TAD and leukemogenesis, it is likely that stratification of CTCF boundary and oncogenic TAD in the HOXA locus by HOTTIP lncRNA may be exploited by MLLr+ or NPM1C+ AML cells to promote leukemogenesis.
Apart from the HOXA genes, HOTTIP lncRNA also bound and regulated a subset of non-HOX genes. RUNX1 is required for definitive hematopoiesis and could act to promote the survival of MLLr+ leukemia cells (Goyama et al., 2013). Intriguingly, epithelial-to-mesenchymal transition genes were recently shown to control AML blast migration and invasion, and to be linked to aggressiveness and poor prognostic outcomes of MLL-AF9-mediated AML (Stavropoulou et al., 2016). The question remains as to whether HOTTIP directly regulates these non-HOXA genes or modulates them through an indirect mechanism. Although KO of HOTTIP decreased more than 80% of HOTTIP transcript levels, it is interesting that there is only approximately 50% reduction in HOTTIP binding to its putative targets, posterior HOXA genes. In contrast, the binding of HOTTIP in the trans-regulated non-HOXA genes was almost completely eliminated. Of note, transgenic expression of Hottip from chromosome 10 is able to activate Hoxa and non-Hox genes. It is likely that HOTTIP mainly acts in cis to regulate the posterior HOXA genes in normal development due to its low expression levels. However, when HOTTIP is overexpressed, such as in AML or in transgenic mice, the overexpressed HOTTIP may go to other chromatin sites besides the posterior HOXA genes. Thus, HOTTIP could act in cis and/or in trans in a context-dependent manner. The mechanism of HOTTIP lncRNA in gene regulation warrants further investigation.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Suming Huang (shuang4@pennstatehealth.psu.edu).
All unique/stable reagents including plasmids and transgenic Hottip mice generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement. The plasmids generated in this study have been deposited to Addgene (pLKO5-SgHottip-GFP-CRISPRi, Addgene ID # 134989; pLKO5-SgHottip-GFP-CRISPRa, Addgene ID # 134990). At the meantime, we are in the process to deposit the transgenic Hottip mouse lines to the Jackson Laboratory.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
AML Patient Samples
The primary MLL patients were carried MLLr+. MLL1 is female and MLL2-MLL7 are male. Non-MLL patients are female. The primary AML patient samples, #886, FLT3-ITD+/NPM1 WT, female; #974, MLLr−/NPM1C+/FLT3-ITD+, male; #LPP4, MLLr+/NPM1 WT, male, were obtained via approval of the Institutional Review Board of the University of Florida in accordance with the Declaration of Helsinki.
Cell Lines
HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% bovine serum (FBS). MOLM13 with carrying MLL-AF9 rearrangement and OCI-AML3 cells carrying NPM1C+ and DNMT3AR882C mutations were cultured in RPMI 1640 medium with 10% FBS and alpha-MEM with 15–20% FBS, respectively.
Transgenic (Tg) Mouse (Mus musculus) Model and Animal Experiments
The complete coding region sequence of mouse Hottip was cloned into downstream of a Vav1 promoter (HS321/45-vav vector) followed by Vav1 enhancer to ensure transgene expression solely in hematopoiesis (Ogilvy et al., 1999; Yang et al., 2018). The plasmid DNA was digested with SacII to remove the pBlueScript II SK backbone and was used for injection into pronuclei of eggs from C57BL/6 mice. Two Hottip-Tg founder mice were obtained by PCR screening of the tail genomic DNAs with primers (Table S4). Transgenic founder mice (line1 founder is male, line2 founder is female) were crossed with WT C57BL/6 mice and the siblings of the Hottip negative and Hottip-Tg mice (both male and female, 2–20 month of age) were used throughout the study. The levels of transgenic expression of Hottip were confirmed by RNA-seq or RT-qPCR analysis.
For in vivo xenotransplantation study procedures, non-obese diabetic (NOD)/LtSz-severe combined immunodeficiency (SCID) IL2Rγcnull (NSG) mice containing both male and female at age of 6–10 weeks old were injected with primary AML patient cells and AML cell lines, as detailed below. All animal studies were conducted in accordance with the regulatory guidelines by the Institutional Animal Care and Use Committee (IACUC) at Penn State Hershey Medical Center and University of Texas Health Science Center at San Antonio.
METHOD DETAILS
SsecCRISPR-Cas9 Mediated HOTTIP lncRNA Knock-Out and Lentivirus Production
HOTTIP knockout (KO) MOLM13 leukemia cell was generated according to the Neon Transfection User Guide and the Alt-R CRISPR-Cas9 System User Guide (2014) Neon® Transfection System for transfecting mammalian cells, including primary and stem cells, with high transfection efficiency (Integrated DNA Technologies, available at https://tools.thermofisher.com/content/sfs/manuals/neon_device_man.pdf). Briefly, CRISPR-RNA (crRNA) and tracrRNA were mixed and annealed in 95°C for 5 min and then cooled down to room temperature. The crRNA:tracrRNA duplex and S.p. Cas9 Nuclease components were combined together and then mixed with 500,000 AML cells for electroporation with Neon® System. After 24 hr or 96 hr, 100 μL of cell suspension was used for DNA extraction using Qiagen quick Extract kit and the mutation was verified through Sanger sequence. HOTTIP−/−-#1 targeted region is Chr7: 27241953–27241985; HOTTIP−/−-#2 targeted region is Chr7: 27240098–27240123.
dCas9-Mediated Inactivation and Overexpression of HOTTIP in AML Cells
Two guide RNAs plasmid targeting the promoter regions of HOTTIP (Table S4) were designed using the Zhang laboratory web tool (http://crispr.mit.edu), and cloned into the pLKO5.sgRNA.EFS.tRFP vector (Addgene #57824). The gRNA plasmids encoding mCherry and puromycin resistance were co-transfected with a plasmid encoding dCas9-KRAB (pHR-SFFV-dCas9-BFP-KRAB, addgene plasmid #46911) or dCas9-VP160 (pAC94-pmax-dCas9VP160–2A-puro, addgene plasmid number #48226) in MOLM13 and OCI-AML3 cells. After 24 hr post-transfection, MOLM13 or OCI-AML3 cells were selected with 2 μg/mL puromycin for another 48 hr, and then FACS sorted for RFP+ cells. RNA was extracted from RFP+ cells, and RT-qPCR was performed according to the primers list (Table S4).
Transgenic Integrating Location Identification
The PCR-based method-TAIL-PCR (Thermal Asymmetric Interlaced PCR) which relies on a series of PCR amplifications with gene specific and degenerate primers to reliably amplify the integration sites was performed according to previous report (Pillai et al., 2008). In briefly, the primary PCR reaction was performed by mixing 50–100 ng genomic DNA, 2.5 mM dNTPs, 10 uM SP1 primer, 10 mM AD primer, and 1 U Taq polymerase in 20 μL 1X reaction buffer. In the secondary or tertiary PCR primers amplification, 1 μL first or secondary 10–20 fold diluted PCR products were used as templates in the reaction, respectively. Finally, the tertiary PCR products were verified through Sanger sequencing.
Cell Cycle Analysis
WT and modified MOLM13 cells were harvested and washed with phosphate buffered saline (PBS). The washed cells were fixed by adding 70% ethanol drop wise to the pellet with vortexing and incubated overnight at 4°C. After fixation, cells were washed with PBS twice. The PBS washed cells were treated with the staining buffer (RNase A, Triton X-100, propidium iodide) and then incubated at 37°C for 30 min in the dark. Stained samples were proceeded on the BD Accuri™ C6 plus flow cytometry (BD Biosciences), and cell cycle data analysis was performed using FlowJo program. Triplicate experiments were performed for each sample.
Hematopoietic Stem/Progenitor Cells (HS/PCs) Sorting, Analysis and Colony Assay
Flow cytometric analysis of Hottip transgenic mice was performed as previously described (Wang et al., 2014) using a BD LSRII flow cytometer. All data were analyzed by FlowJo-V10 software. Briefly, LK (Lin− Kit+), LSK (Lin− Sca1+ Kit+), Long Term (LT) and Short-Term (ST)-HSC cells were derived from total bone marrow (BM) cells, and Lin+ BM cells was pre-depleted by Miltenyi Biotec magnetic beads (130–110-470), then the leftover Lin− BM cells were stained with Lin, Sca-1, c-kit, CD34, CD135 and CD16/32 antibody and sorted by BD FUSION flow cytometer. The purity of selected cells were over 95%.
Total white blood cells were obtained after lysis of Peripheral blood (PB) with red cell lysis buffer (Thermo fisher). Single-cell suspensions from bone marrow (BM), spleen and PB were stained with panels of fluorochrome-conjugated antibodies. Flow cytometric analysis of hematopoietic stem / progenitor cells (HS/PCs) was performed as previously described (Li et al., 2011). The analyses were performed using the FACS Canto II or LSR Fortessa flow cytometer (BD LSRFortessa™). All data was analyzed by FlowJo.V10 software. For colony and replating assays, bone marrow (BM) cells from the femurs and tibias of 6–8 week-old mice stained with antimurine cKit-APC, Sca1-PE antibodies and a panel of antibody-conjugated goat anti-Rat IgG BioMag beads (Qiagen) for lineages (Lin), then cells were sorted on FACS ARIA II (Becton Dickinson), thus the enriched Lin− Sca1+ Kit+ population (LSK cells) were obtained. For colony-forming unit (CFU) assays, BM (1×104 cells/plate) or spleen cells (5×104 cells/plate) were plated in triplicate in methylcellulose medium (Methocult M3231) supplemented with mIL-3 (mouse interleukin 3, 10 ng/mL), hIL-6 (human interleukin 6, 100 ng/mL), hEpo (human erythropoietin, 4 U/mL) and mSCF (mouse stem cell factor, 100 ng/mL), and scored in 8–10 days. For replating assays, CFU assays were performed with LSK cells in methylcellulose medium supplemented with the same cytokine cocktails. Colonies were passaged every 7 days for 4 sequential plating.
Competitive Repopulation Assay
1 ×106 BM cells (CD45.2+) from Ctrl or Hottip-Tg mice were mixed with 1 ×106 competitor BM cells (CD45.1) from B6.SJL mice and then transplanted into lethally irradiated (950 centigray) recipients (B6.SJL) by tail vein injection. Transplanted mice were monitored daily for signs of disease development.
Suspension Culture Assay
The LK cells were incubated in suspension culture containing 30% FBS, 2% BSA, and a combination of cytokines (mouse interleukin-3, human interkeukin-6, human erythropoietin, and mouse stem cell factor). At weekly intervals, cultures were mixed by pipetting and half of the culture media were removed, which was then replaced by newly prepared medium with the same combinations of cytokines. Cells in the collected media were counted and used for flow cytometric analysis. Total CFUs generated at each time point in the suspension culture were evaluated by culturing a fraction of the expanded cells in the colony assay as described above.
RNA Immunoprecipitation (RIP) Assay
The RNA-IP protocol was modified from previous reported (Deng et al., 2016; Tsai et al., 2010). The MOLM13 AML cells were collected and washed with PBS (e.g. 107 cells in 2 mL PBS), resuspended in freshly prepared nuclear isolation buffer (1.28 M sucrose, 40 mM Tris-HCl pH 7.5, 20 mM MgCl2, 4% Triton X-100), and then kept on ice for 20 min (with frequent mixing). Nuclei were precipitated by centrifugation at 2,500 g for 15 min, and then resuspended in freshly prepared lysis buffer (10 mM HEPES-KOH pH7, 150 mM KCl, 5 mM MgCl2, 5 mM EDTA, 0.5% IGEPAL-CA-630, 0.5 mM dithiothreitol, 0.2 mg/mL Heparin, 100 U/mL RNase OUT, 100 U/mL Superase IN, protease inhibitor tablet). Nuclei were sonicated with the Bioruptor™ UCD200. The suspension was centrifuged three times at 14,000 g at 4°C for 10 min, and supernatant was collected and precipitated with antibody (2–10 μg) overnight at 4°C with rotation. The precipitant was captured by the equilibrated Protein A/G magnetic beads followed by washing four times in ice-cold NT2 buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM MgCl2, 0.05% IGEPAL-CA-630) supplemented with 0.02 mg/mL heparin. The RNA-protein complexes were eluted twice with 500 μL SDS-EDTA (50 mM Tris pH 8.0, 100 mM NaCl, 10 mM EDTA, 1% SDS) for 10 min at 65°C. Coprecipitated HOTTIP RNA was isolated by resuspending beads in TRIzol RNA extraction reagent, eluted with nuclease-free water, treated with TURBO DNase, and then detected by RT-qPCR.
RNA Isolation, Quantitative RT-PCR, as well as RNA-Sequencing and Data Analysis
Total RNAs from MOLM13 cells, OCI-AML3 AML cells, or primary Hottip-Tg mice long-term (LT) and short-term (ST)-HSCs were purified with the RNeasy mini-isolation kit according to manufacturer’s instructions (Qiagen, MD, USA). A total of 2 μg RNA was subjected to reverse-transcription with Superscript II reverse Transcriptase (Invitrogen) and analyzed by a real-time polymerase chain reaction (PCR) Detection System (Bio-Rad). Primer sequences are listed in the supplemental Information (Table S4).
Paired end RNA-Seq was performed by Pennsylvania State University College of Medicine Genome Science Facility according to standard protocols. All of sequencing reads were processed and aligned to the mouse or human genome assembly (mm9 or hg19) using TopHat (version 2.0) and Bowtie2 (Langmead et al., 2009; Trapnell et al., 2009, 2012). To prevent false positives, a stringent approach was taken to identify differentially expressed genes. First, FPKM (paired-end fragments per kilobase of exon model per million mapped reads) was calculated for each gene and further normalized (RMS-FPKM). Second, to prevent false positives due to the fluctuation of detection among genes with low expression levels, only genes with 50 or more reads in one of the conditions (WT control or HOTTIP manipulations) were included in the analysis. Differential expression was determined according to abundance estimations (FPKMs) processed with Cufflinks v2.2.1 and Cuffdiff (Trapnell et al., 2010). Differentially expressed genes were identified if the ratio of RMS-FPKM in the two conditions was greater than 2.0 fold, or undetectable in one condition but detectable by more than 50 reads in the other. The heatmaps and scatter plots were based on log2 transformation of the RMS-FPKM values. Expression level increased or decreased genes were marked with red or blue, respectively. The GO mapping of differentially expressed genes were performed with Gorilla (Eden et al., 2009). The normalized expression data was uploaded to Integrated Genomic Viewer (IGV) for visulization. The sequence reads have been deposited in the NCBI GEO under accession number (GSE114981).
Chromatin Immunoprecipitation (ChIP) Assay
ChIP were performed as described previously (Deng et al., 2013). Briefly, Nuclei were sonicated with the Bioruptor™ UCD200. Chromatin samples prepared from 5×106 cells of MOLM13 cells were immunoprecipitated with antibodies against MLL1, H3K4me3, H3K9me2, H3K27me3 and H3K79me2, separately. The immunoprecipitates were subjected to a series of washing steps to remove non-specific binding materials. After reverse-crosslinking, the DNA samples were purified and then analyzed by real-time quantitative PCR. Final results represent percentage of input chromatin and error bars indicate standard deviations (S.D.) through triplicate experiments. The MLL1, H3K4me3, H3K79me2 and H3K27me3 ChIP-DNA libraries were prepared using Illumina’s TruSeq ChIP Sample Preparation Kit according to the manufacturer’s instructions (Cat #IP-202–1012). The quality of the library was checked with Agilent TapeStation. Final libraries were submitted to paired-end sequencing of 100 bp length on an Illumina HiSeq 3000.
Chromatin Isolation by RNA Immunoprecipitation (CHIRP) Assay
The Chromatin Isolation by RNA Immunoprecipitation (CHIRP) assay was carried out based on the protocol described in a previous studies (Chu et al., 2011) with some modifications. Briefly, 20 million cells were collected and cross-linked in 20 mL of PBS buffer containing 1% formaldehyde at room temperature for 10 mins. Cross-linked cells were washed by chilled PBS 2 times, lysed in 1mL per 100 mg of cell pellet in cell lysis buffer (50 mM Tris-Cl pH 7.0,10 mM EDTA, 1% SDS, supplemented with PMSF, DTT, proteinase inhibitors (P.I.) and Superase-in in fresh), and sonicated using a Bioruptor™ UCD200 (Diagenode) to prepare chromatin. Chromatin was diluted 2 times using hybridization buffer (750 mM NaCl, 1% SDS, 50 mM Tris 7.0, 1 mM EDTA, 15% formamide, add DTT, PMSF, P.I, and Superase-in fresh) and hybridized with 100 pmole of biotinylated DNA probes targeting HOTTIP or LacZ containing 100 μL of Streptavidin-magnetic C1 beads (Invitrogen). RNA and DNA hybrids were purified, washed 5 times with washing buffer (2x SSC, 0.5% SDS), and subjected to analysis by RT-qPCR. RNA binding proteins were subjected to analysis by western blotting with antibodies. Probes and primers are listed in the Table S4. CHIRP libraries were prepared using Illumina’s TruSeq ChIP Sample Preparation Kit according to the manufacturer’s instructions (Catalog: #IP-202–1012). The quality of the library was checked with Agilent TapeStation. Final libraries were submitted to paired-end sequencing of 100 bp length on an Illumina HiSeq 2500. All genomics datasets were deposited in the NCBI GEO under accession number (GSE114981).
ChIP-seq and ChIRP-seq Data Analysis
The ChIP-seq or ChIRP-seq raw data were processed through cutadapt (http://cutadapt.readthedocs.io, version 1.2.0) to remove adaptors and low quality reads (Martin, 2011). Cutadapt-filtered reads aligned to human reference genome (hg19) using Bowtie2 with default parameters (Langmead et al., 2009), and the quality of these trimmed data was evaluated by FastQC program (Wingett and Andrews, 2018). After alignment, SAM files were converted to BAM files and sorted using Samtools (Li et al., 2009). Peak calling was performed using peak calling algorithm MACS2 (Zhang et al., 2008). The bedGraphToBigWig program was employed to generate the bigWig file of fragment or read coverages, including control and experimental datasets (https://www.encodeproject.org/software/bedgraphtobigwig/). All sequencing tracks were viewed using the Integrated Genomic Viewer (Robinson et al., 2011). Peaks annotation was carried out with the command “annotatePeaks.pl” from HOMER package (Heinz et al., 2010). For ChIRP-seq motif analysis, the de novo motif analysis was performed by the “findmotifsgenome.pl” from the HOMER motif discovery algorithm (Heinz et al., 2010). The genes and pathways regulated by the HOTTIP bound promoters or intergenic regions were analyzed and annotated by the Gene Ontology analysis with the Database for Annotation, Visualization and Integrated Discovery (DAVID) tool (https://david.ncifcrf.gov/, Version 6.8) (Huang da et al., 2009a; Huang da et al., 2009b). Each GO term with a p value more than 1 × 10^−3 is used for cutoff (threshold: 10^−3). All genomics datasets were deposited in the NCBI GEO under accession number (GSE114981).
Assay for Transposase-Accessible Chromatin Using Sequencing (ATAC-seq)
ATAC-seq was performed using the Nextera DNA library preparation kit as described previously (Buenrostro et al., 2015). In Brief, 5 ×104 cells in single cell suspension were used for library preparation. Washed cells were re-suspended in lysis buffer containing 10 mM Tris-HCL (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 0.1 % NP-40. After washing with cold 1x phosphate buffered saline (PBS) buffer, cells were treated with Tn5 Transposes for transposition reaction at 37°C for 30 min. DNA was purified using the MinElute Kit (QIAGEN). Library fragments were amplified using 1x NEB next PCR master mix and 1.25 μM indexed Nextra PCR primers (Ad1_noMX and Ad2.1–2.4 barcoded primers) with following PCR conditions: 72°C for 5 min, 98°C for 30 s, followed by thermocycling at 98°C for 10 s, 63°C for 30 s and 72°C for 1 min. The eluted DNA was used in a quantitative PCR (qPCR) reaction to estimate the optimum number of amplification cycles. Libraries were quantified using qPCR (Kapa Library Quantification Kit for Illumina, Roche), and libraries were purified with AMPure beads (Beckman Coulter), and the quality of the DNA library was examined by Agilent Bioanalyzer 2100 prior to sequencing with 2×100 bp paired-end reads on an Illumina NextSeq 500. Each sample includes two replicates for statistical analysis.
ATAC-seq Analysis
We normally carried out two biological replicates for all of the ATAC-seq experiments (Luo et al., 2018). For quality control, first, each replicate should have 50 million reads for paired-end sequencing. Second, the alignment rate of each replicate is more than 95%. Third, we also removed the mitochondrial related reads from total reads after alignment and PCR duplicates were also removed. Finally, non-uniquely aligned reads were filtered based on MAPQ scores with samtools (MAPQ > 30), and plotPCA from BiocGenerics package in R package (R/3.6.1) was carried out to identify the variance between control and treatment groups. Moreover, fragSizeDist from ATACseqQC package in R package was carried out to show the fragment size distribution for control and treatment groups. In additional, the library complexity was analyzed including nucleosome free region signals (NFRs), mono-nucleosome, di-nucleosome and tri-nucleosome signals according to previously report (Tarbell and Liu, 2019).
Briefly, all of the raw fastq files were processed through cutadapt (http://cutadapt.readthedocs.io, version 1.2.0) to remove adaptors and low quality reads (Martin, 2011). Cutadapt-filtered reads aligned to human or mouse genome (hg19 or mm9) using Bowtie2 with default parameters (version Bowtie 2/2.2.6) (Langmead et al., 2009), and the quality of these trimmed data was evaluated by FastQC program (version 0.11.8) (Wingett and Andrews, 2018). After alignment, SAM files were converted to BAM files and sorted using Samtools (version 1.8.0) (Li et al., 2009). PCR duplicates were removed using Picard MarkDuplicates (version 2.0.1), and mitochondrial reads were removed with Samtools (Corces et al., 2017). ENCODE blacklist regions were filtered (https://sites.google.com/site/anshulkundaje/projects/blacklists). Peak calling was performed using peak calling algorithm MACS2 with parameters (“-gmm -p 1e-9–nolambda -f BAMPE–nomodel –shiftsize=100-extsize200”) (Zhang et al., 2008). bedGraphToBigWig program was employed to generate the bigWig file of fragment or read coverages, including control and experimental datasets (https://www.encodeproject.org/software/bedgraphtobigwig/). All sequencing tracks were viewed using the Integrated Genomic Viewer (IGV/2.4.19) (Robinson et al., 2011). Peaks annotation was carried out with the command “annotatePeaks.pl” from HOMER package (version 4.8) (Heinz et al., 2010) and GREAT (McLean et al., 2010). DEseq2 (Benjamini-Hochberg adjusted p<0.05; FoldChange≥2) were also performed to find the differential binding sites between two peak files, including control and treatment groups with C+G normalized and “reads in peaks” normalized data (Ross-Innes et al., 2012). The de novo motif analysis was performed by the “findmotifsgenome.pl” from the HOMER package (Heinz et al., 2010). For each genomic feature (peaks or chromVAR annotation), we calculated the chromatin accessibility median deviation z-score (for chromVAR features) or fragment counts (for peaks) in control and treatment groups with chromVAR package in R language (Rubin et al., 2019; Schep et al., 2017). Pearson’s correlation coefficient and Pearson’s χ2-test were carried out to calculate overall similarity between the replicates of ATAC-seq global open chromatin signatures. All genomics datasets were deposited in the NCBI GEO under accession number (GSE114981).
Mouse Exome Sequencing Assay
The whole exome sequencing (WES) was carried out to identify candidate mutations in the exomes of genes. Genomic DNA was isolated from mice ear and BM cells including wildtype and the diseased Hottip-Tg mice, and genomic exome library was captured and constructed according to SureSelectXT Mouse All Exon kit (Agilent, Part Number:5190–4641), and then 100 bp paired-end sequencing was performed using an Illumina NovaSeq S1. Raw sequencing reads were processed through cutadapt (http://cutadapt.readthedocs.io, version 1.2.0) to remove adaptors and low quality reads. These clean reads were mapped to the whole mouse genome (mm10) using BWA with the default settings (bwa/0.7.4) (Liang et al., 2009). The PCR duplicates were removed with Picard with default parameters (version 1.88) (http://broadinstitute.github.io/picard/), recalibrated with GATK with default setting (version 3.7) (McKenna et al., 2010), and compared the variance between wildtype and Hottip-Tg group with Strelka (version 2.9.2, default setting)(Kim et al., 2018), and then variant bases were annotated with SnpEff (latest version) (http://snpeff.sourceforge.net/SnpEff_manual.html) (Cingolani et al., 2012). All genomics datasets were deposited in the NCBI GEO under accession number (GSE114981).
Xenotransplantation of Human Leukemic Cells and Patient-Derived Xenografts (PDX)
Non-obese diabetic (NOD)/LtSz-severe combined immunodeficiency (SCID) IL2Rγcnull (NSG) mice were housed in sterile conditions using high-efficiency particulate arrestance filtered micro-isolators and fed with irradiated food and acidified water. Adult mice (6–10 weeks old) were sublethally irradiated with 280 cGy of total body irradiation before injection of leukemic cells. WT control, CBS7/9+/− cells, HOTTIP−/− cells, and HOTTIP-VP-CBS7/9+/− MOLM13 cells (in 300 μL of PBS) were injected into the NSG mice by tail-vein injection at a dose of 0.5 × 106 cells/mouse. For Patient-Derived Xenografts (PDX) assay, control or HOTTIP−/− primary AML patients (#886, FLT3-ITD+/NPM1C−; # 974, NPM1C+/FLT3-ITD+; LPP4, MLLr+/NPM1C−) were injected into the NSG mice at 1.8 × 105 cells/mouse. Daily monitoring of mice for symptoms of disease (ruffled coat, hunched back, weakness and reduced motility) determined the time of killing for injected animals with signs of distress. NSG mice were humanely killed at the time of moribund. Peripheral blood was collected by retro-orbital bleeding, bone (tibias, femurs and pelvis) and spleen were dissected. BM cells were isolated by flushing the bones. Spleens were mashed through a 70-pm mesh filter and made into single cell suspensions. PB was prepared for flow cytometry by ammonium chloride treatment to remove red cells. Human CD45 chimerism in these hematopoietic tissues was analyzed by flow cytometry (FACS LSR II–BD Biosciences, San Jose, CA, USA). All data were analyzed by FlowJo7.6 software.
For histopathology analyses, femurs were fixed in formaldehyde, decalcified, and paraffin embedded. Spleens were treated similarly except for the step of decalcification. Sections (4.5 μm) were stained with hematoxylin/eosin (H&E) or immunohistochemistry staining with anti-hCD45 antibody (Abcam, ab10559) and detected using an HRP conjugated compact polymer system with DAB as the chromogen. The slides were then observed with a conventional microscope.
Hi-C Assay
Hi-C assay was performed to generate a genome-wide interaction as described previously with Arima-HiC Kit (Cat: A410030) (https://arimagenomics.com/) with minor modifications. In brief, 5 million cells were collected and cross-linked in 10 mL of PBS buffer containing 1 % formaldehyde at room temperature for 10 min. The reaction was quenched by 0.125 M glycine solution. Cross-linked cell pellet were washed in 1x PBS buffer and collected. Cross-linked cell pellet was treated with lysis buffer and incubate at 4°C for 15 min, and then conditioning solution was added to continue incubate at 62°C for 10 min. Reaction was stopped by adding stop solution and incubating at 37°C for 15 min. Cell pellet was digested with reaction buffer and restriction enzyme cocktail (Arima-HiC Kit) overnight at 37°C with rotation. Digested DNA was purified with DNA purification beads (AMPure XP Beads), and the concentration of DNA was measured with Qubit. 750 ng of DNA per sample was sheared through sonication (Bioruptor) with default parameters (30 seconds ON, 30 seconds OFF pulse intervals). Fragmented DNA was then size-selected to have a size distribution between 200–600 bp. 250 ng of size-selected DNA was used to generate sequence library with KAPA Hyper Prep Kit (Catalog # KK8500, KK4824 and KK8502). Final libraries were submitted to paired-end sequencing of 100 bp length on an Illumina HiSeq 2500.
Hi-C Sequence Data Analysis
Raw sequence reads were first cleaned to remove adapter and low quality reads with bbmap and bbduk.sh (https://jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/bbduk-guide/). The paired-end sequencing data was trimmed from the 3′ end of enzymatic sequences with homerTools (homerTools trim −3 Arima -mis 0 -matchStart 20 -min 20) from Homer software (version 4.8). Trimmed reads were aligned to human reference genome (hg19) using Bowtie2 with parameters (“-n 1 -m 1 -p 8”) (Langmead et al., 2009). The remainder of the analysis was performed using juicer (version 1.5.5) (Durand et al., 2016b) and Homer (Heinz et al., 2010). Paired-end sequencing was used to make a tag directory with makeTagDirectory package from Homer software. A normalized interaction matrix was generated with the analyzeHiC program with parameters (1 Mb resolution for all chromosomes, and 100 kb resolution for specific chromosome), and the intra-chromatin interactions within specific loci were also generated with the analyzeHiC program in Homer software via parameters (-res 10,000 -superRes 20,000 -pos chromosome location). In depth explanations of normalization, generation of Hi-C correlation matrices, principal component analysis (PCA) and identifying significant interactions were performed as previously described (Lin et al., 2012). These interaction matrices for Hi-C heatmap were visualized with Juicebox (Durand et al., 2016a) and Java Treeview (Saldanha, 2004). All genomics datasets were deposited in the NCBI GEO under accession number (GSE114981).
Long Term Culture-Initiating Cells (LTC-IC) Assay
Primary MLL rearranged or non-MLL-AML primary samples were seeded into 96 wells plate with MS5 stroma cells in 200 μL medium (IMEM + 10 % FBS + 20 ng/mL human IL-3, IL-6, TPO, SCF and FLT3 ligand). Different cell doses were put for 10 or 20 wells per cell dose. Cells were either vehicle DMSO treated or treated with 500 nM ICG001 for one week. After one week, all medium were replenished with fresh medium without drugs and further cultured for other two weeks with medium replenished every week. Cell clusters (cobberstone) were scored from each well and the initiating frequency were calculated using online resources-WEHI Extreme Limiting Dilution Analysis (http://bioinf.wehi.edu.au/software/elda/).
In Vivo and In Vitro Drug Treatment
For in vitro experiment, primary MLLr+ AML primary samples were either mock-treated or treated 500 nM ICG-001 for 5 days in liquid medium. Cells were then harvested and stained with 0.1% Nitro Blue Tetrazolium chloride (NBT) which is converted into a dark deposit in myeloid differentiated cells and were scored out of the total cell counted. For in vivo experiment, mice were distributed into their respective groups randomly. MLLr+ AML cells were transplanted into 6–12 weeks old sub-leathally irradiated (250 cGy) NSG mice (male or female) via intra-femoral route. Two weeks after transplantation, the mice were control-treated or treated with ICG-001 (50 mg/kg; 5 days a week for 2 weeks) in PEG300/D5W (3:1) via intra-peritoneal route. When animals showed signs of sickness, the mice were suffocated in the CO2 chamber and confirmed dead by cervical dislocation. The leukemic mouse is defined by >20% human AML engraftment with CD45 and CD33 positive cells in the bone marrow.
QUANTIFICATION AND STATISTICAL ANALYSIS
Differences between experimental groups were determined by the Student’s t-test or analysis of variance (ANOVA) followed by New-man-Keuls multiple comparison tests. p value <0.05 is considered significant (*), p value <0.01 is considered highly significant (**), p value <0.001 is considered extremely significant (***). TCGA datasets were obtained from The Cancer Genome Atlas (TCGA) database (https://gdac.broadinstitute.org/). Pearson’s χ2-test also was applied to determining significance of the enrichment of prognostic data from published TCGA human de novo AML datasets (Mukaka, 2012). For in vivo experiment, sample size chosen was based on the generalized linear model with Bonferroni multiple comparison adjustments; with the proposed sample size of at least five mice/ group/genotype. Animals were randomly assigned to each study. For all in vitro experiments, at least three independent experiments with more than three biological replicates for each condition/genotype were performed to ensure adequate statistical power.
DATA AND CODE AVAILABILITY
The accession number for the RNA-seq, ATAC-seq, ChIP-seq, ChIRP-seq, whole exome-seq and HiC-seq reported in this paper are GSE114981 and GSE113191.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-H3K4me3 antibody, rabbit monoclonal | Millipore | Cat#04–745; RRID:AB_1163444 |
| Anti-H3K27me3 antibody, rabbit polyclonal | Millipore | Cat#07–449; RRID:AB_310624 |
| Anti-H3K9me2 antibody, rabbit polyclonal | Millipore | Cat#17–681, RRID:AB_1977531 |
| Anti-H3K79me2 antibody, rabbit polyclonal | Abcam | Cat#ab3594; RRID:AB_303937 |
| Anti-CD45 antibody, rabbit polyclonal | Abcam | Cat#ab10559, RRID:AB_442811 |
| Anti-Ly-6A/E (Sca-1) antibody, mouse monoclonal | BioLegend | Cat#108111, RRID:AB_313348 |
| Anti-CD117 (c-kit) antibody, mouse monoclonal | BioLegend | Cat#135135, RRID:AB_2632808 |
| Anti-CD34 antibody, mouse monoclonal | BD Biosciences | Cat#553733, RRID:AB_395017 |
| Anti-CD135 antibody, mouse monoclonal | BD Biosciences | Cat#558995, RRID:AB_397174 |
| Anti-CD16 antibody, mouse monoclonal | BD Biosciences | Cat#555405, RRID:AB_395805 |
| Anti-KMT2A/MLL antibody, rabbit polyclonal | Novus Biologicals | Cat#NB600–248, RRID:AB_2145479 |
| Anti-WDR5 antibody, mouse monoclonal | Abcam | Cat#ab56919, RRID:AB_946146 |
| Anti-DOT1L antibody, rabbit polyclonal | Novus Biologicals | Cat# NB100–40845, RRID:AB_789636 |
| Anti-p-STAT5A, rabbit polyclonal | Cell Signaling Technology | Cat# 9351, RRID:AB_2315225 |
| Anti-STAT5A, rabbit monoclonal | Cell Signaling Technology | Cat# 94205, RRID:AB_2737403 |
| Anti-HDAC1, rabbit polyclonal | Abcam | Cat# ab7028, RRID:AB_305705 |
| Anti-RUNX1, rabbit polyclonal | Abcam | Cat# ab23980, RRID:AB_2184205 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipofectamine 3000 reagent | Thermo Fisher Scientific | Cat#L3000–008 |
| Proteinase K | Thermo Fisher Scientific | Cat#25530049 |
| Alt-R® S.p. Cas9 Nuclease 3NLS | Integrated DNA Technologies | Cat#1074181 |
| Alt-R® CRISPR-Cas9 tracrRNA | Integrated DNA Technologies | Cat#1072532 |
| Alt-R® Cas9 Electroporation Enhancer | Integrated DNA Technologies | Cat#1075915 |
| Protease inhibitor Cocktail | Abcam | Cat#ab65621 |
| Dynabeads™ Protein G | Thermo Fisher Scientific | Cat#10003D |
| Dynabeads™ Protein A | Thermo Fisher Scientific | Cat#10001D |
| Dynabeads™ MyOne™ Streptavidin C1 | Thermo Fisher Scientific | Cat#65001 |
| SUPERase• In™ RNase Inhibitor | Thermo Fisher Scientific | Cat#AM2694 |
| RNaseOUT™ Ribonuclease Inhibitor | Thermo Fisher Scientific | Cat#10777019 |
| Pierce Protease Inhibitor Tablets | Thermo Fisher Scientific | Cat#A32963 |
| TURBO™ DNase | Thermo Fisher Scientific | Cat#AM2238 |
| AMPure XP beads | Beckman Coulter | Cat#A63881 |
| Critical Commercial Assays | ||
| RNeasy mini-isolation kit | QIAGEN | Cat#74106 |
| Neon™ Transfection System Kit | Thermo Fisher Scientific | Cat#MPK1025 |
| QIAquick Gel Extract kit | QIAGEN | Cat#28706 |
| Alt-R® CRISPR-Cas9 Control Kit | Integrated DNA Technologies | Cat#1072554 |
| mirVana PARIS kit | Thermo Fisher Scientific | Cat#AM1556 |
| Superscript II reverse Transcriptase | Thermo Fisher Scientific | Cat#18064014 |
| QIAquick PCR purification kit | QIAGEN | Cat#28106 |
| QIAprep Spin Miniprep Kit | QIAGEN | Cat#27106 |
| QIAGEN Plasmid Plus Maxi Kit | QIAGEN | Cat#12965 |
| Nextera DNA Library Preparation Kit | illumina | Cat#FC-121–1030 |
| Arima-HiC Kit | Arima | Cat#A410030 |
| KAPA Hyper Prep Kit | KAPA | Cat # KK8500, KK4824 and KK8502 |
| SureSelectXT Mouse All Exon | Agilent | Cat # 5190–4641 |
| pGEM®-T Easy Vector Systems | Promega | Cat#A137A |
| SingleShot™ SYBR® Green One-Step Kit | Bio-Rad Laboratories | Cat#1725095 |
| Deposited Data | ||
| RNA-seq in WT vs HOTTIP-KO MOLM13 | This paper | GEO: GSE114981 |
| ATAC-seq in WT vs HOTTIP-KO MOLM13 | This paper | GEO: GSE114981 |
| RNA-seq in WT vs CBS7/9-KO MOLM13 | (Luo et al., 2018) | GEO: GSE113191 |
| CHIRP-seq of WT vs HOTTIP-KO MOLM13 | This paper | GEO: GSE114981 |
| ChIP-seq of WT vs HOTTIP-KO MOLM13 | This paper | GEO: GSE114981 |
| LSK RNA-seq of WT vs Hottip-Tg mice | This paper | GEO: GSE114981 |
| LSK ATAC-seq of WT vs Hottip-Tg mice | This paper | GEO: GSE114981 |
| LT-HSC RNA-seq of WT vs Hottip-Tg mice | This paper | GEO: GSE114981 |
| ST-HSC RNA-seq of WT vs Hottip-Tg mice | This paper | GEO: GSE114981 |
| LT-HSC ATAC-seq of WT vs Hottip-Tg mice | This paper | GEO: GSE114981 |
| ST-HSC ATAC-seq of WT vs Hottip-Tg mice | This paper | GEO: GSE114981 |
| HiC-seq of WT vs HOTTIP-KO MOLM13 | This paper | GEO: GSE114981 |
| HiC-seq of HOTTIP-KO MOLM13 | This paper | GEO: GSE114981 |
| HiC-seq of CBS7/9-KO MOLM13 | This paper | GEO: GSE114981 |
| HiC-seq of CBS7/9-KO-VP-HT MOLM13 | This paper | GEO: GSE114981 |
| Mouse whole exome sequencing | This paper | GEO: GSE114981 |
| Experimental Models: Cell Lines | ||
| MOLM-13 | DSMZ | Cat# ACC-554, RRID:CVCL_2119 |
| HEK293T | ATCC | Cat# CRL-3216, RRID:CVCL_0063 |
| OCI-AML3 | DSMZ | Cat# ACC-582, RRID:CVCL_1844 |
| Experimental Models: Organisms/Strains | ||
| Hottip-Transgenic mouse | This paper | N/A |
| Xenograft AML mouse model | This paper | N/A |
| Oligonucleotides | ||
| sgRNAs | This paper | see Table S4 |
| RT-qPCR primers | This paper | see Table S4 |
| ChIP-PCR primers | This paper | see Table S4 |
| crRNAs | This paper | see Table S4 |
| ATAC primers | This paper | see Table S4 |
| CHIRP Probes | This paper | see Table S4 |
| Recombinant DNA | ||
| pL-CRISPR.EFS.GFP | Heckl et al., 2014 | Addgene Plasmid #57818; RRID:Addgene_57818 |
| pLKO5.sgRNA.EFS.tRFP | Heckl et al., 2014 | Addgene Plasmid #57824; RRID:Addgene_57824 |
| lentiCRISPR v2 | Sanjana et al., 2014 | Addgene Plasmid # 52961; RRID:Addgene_52961 |
| pHR-SFFV-dCas9-BFP-KRAB | Gilbert et al., 2013 | Addgene Plasmid #46911; RRID:Addgene_46911 |
| pAC94-pmax-dCas9VP160–2A-puro | Cheng et al., 2013 | Addgene Plasmid #48226; RRID:Addgene_48226 |
| pMD2.G | Will et al., 2015 | Addgene Plasmid # 12259; RRID:Addgene_12259 |
| psPAX2 | Will et al., 2015 | Addgene Plasmid #12260; RRID:Addgene_12260 |
| pLKO5-SgHottip-GFP-CRISPRi | This paper | Addgene Plasmid # 134989 |
| pLKO5-SgHottip-GFP-CRISPRa | This paper | Addgene Plasmid # 134990 |
| Software and Algorithms | ||
| TopHat | (Trapnell et al., 2012) | https://ccb.jhu.edu/software/tophat/ |
| Bowtie2 | (Langmead and Salzberg, 2012) | http://bowtiebio.sourceforge.net/bowtie2/ |
| R | N/A | https://www.r-project.org/ |
| Cufflinks | (Trapnell et al., 2010) | http://cole-trapnell-lab.github.io/cufflinks/ |
| Cuffdiff | (Trapnell et al., 2010) | http://cole-trapnell-lab.github.io/cufflinks/ |
| Integrated Genomic Viewer | (Robinson et al., 2011) | http://software.broadinstitute.org/ |
| Deeptools | (Ramirez et al., 2014) | https://deeptools.github.io/ |
| Gene Set Enrichment Analysis (GSEA) | (Subramanian et al., 2005) | http://software.broadinstitute.org/gsea/ |
| chromVAR | (Schep et al., 2017) |
https://bioconductor.org/packages/release/bioc/ html/chromVAR.html |
| Homer | (Heinz S et al., 2010) | http://homer.ucsd.edu/homer/index.html |
| Juicer | (Neva C. Durand, 2016) | https://github.com/aidenlab/juicer/ |
| Juicebox | (Neva C. Durand, 2016) | http://aidenlab.org/juicebox/ |
Highlights.
HOTTIP reprograms 3D AML genome and drives leukemic transcription profile in AML
HOTTIP binds and regulates genes important for hematopoiesis and leukemogenesis
HOTTIP KO attenuates AML progression by impairing leukemic transcription program
Hottip aberration perturbs HSC self-renewal leading to AML-like disease in mice
Significance.
The initiation and progression of acute myeloid leukemia (AML) have so far been studied mainly within the realm of mutations and/or dysregulation of key protein-coding genes. Whether and how long non-coding RNA (lncRNA) misregulation can lead to oncogenesis remains to be explored in AML. Dysregulation of HOXA genes (e.g., HOXA9) is a dominant mechanism for hematopoietic deregulation and leukemogenesis. Our study demonstrates that HOTTIP lncRNA coordinates topologically associated domain organization of AML genome including the posterior HOXA genes and various key hematopoietic regulator loci. Expression of HOTTIP is required for AML driven by mixed lineage leukemia fusions/NPM1 mutation and is sufficient to initiate leukemic transformation of hematopoietic stem cells. These findings provide a framework for developing targeted therapeutics for AML.
ACKNOWLEDGMENTS
We thank Penn State College of Medicine Genome Science Facility. This work was supported by grants from National Institutes of Health (S.H., R01DK110108, R01CA204044, HL141950; M.X., R01CA172408, R01HL145883, R01HL141950; Y.Q., R01HL144712; W.L., R01HG007538, R01CA228140), Program grants by Cancer Research UK and Bloodwise (C.W.E.S.), San Antonio Cancer Council (X.M.) and the Four Diamonds Fund (S.H.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.ccell.2019.10.011.
REFERENCES
- Andreeff M, Ruvolo V, Gadgil S, Zeng C, Coombes K, Chen W, Kornblau S, Baron AE, and Drabkin HA (2008). HOX expression patterns identify a common signature for favorable AML. Leukemia 22, 2041–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunetti L, Gundry MC, Sorcini D, Guzman AG, Huang YH, Ramabadran R, Gionfriddo I, Mezzasoma F, Milano F, Nabet B, et al. (2018). Mutant NPM1 maintains the leukemic state through HOX expression. Cancer Cell 34, 499–512.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 109,21.29.21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AW, Wang H, Yang H, Shi L, Katz Y, Theunissen TW, Rangarajan S, Shivalila CS, Dadon DB, and Jaenisch R (2013). Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 23, 1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C, Qu K, Zhong FL, Artandi SE, and Chang HY (2011). Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 44, 667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, and Ruden DM (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CT, and Hess JL (2016). Role of HOXA9 in leukemia: dysregulation, cofactors and essential targets. Oncogene 35, 1090–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GM, Fuller J, Petersen D, Izadi P, Malik P, Pattengale PK, Kohn DB, and Gasson JC (1999). Constitutive HOXA5 expression inhibits erythropoiesis and increases myelopoiesis from human hematopoietic progenitors. Blood 94, 519–528. [PubMed] [Google Scholar]
- Deng C, Li Y, Liang S, Cui K, Salz T, Yang H, Tang Z, Gallagher PG, Qiu Y, Roeder R, et al. (2013). USF1 and hSET1A mediated epigenetic modifications regulate lineage differentiation and HoxB4 transcription. PLoS Genet. 9, e1003524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C, Li Y, Zhou L, Cho J, Patel B, Terada N, Li Y, Bungert J, Qiu Y, and Huang S (2016). HoxBlinc RNA recruits Set1/MLL complexes to activate Hox gene expression patterns and mesoderm lineage development. Cell Rep. 14, 103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschamps J, and van Nes J (2005). Developmental regulation of the Hox genes during axial morphogenesis in the mouse. Development 132, 2931–2942. [DOI] [PubMed] [Google Scholar]
- Dou DR, Calvanese V, Sierra MI, Nguyen AT, Minasian A, Saarikoski P, Sasidharan R, Ramirez CM, Zack JA, Crooks GM, et al. (2016). Medial HOXA genes demarcate haematopoietic stem cell fate during human development. Nat. Cell Biol 18, 595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dovey OM, Cooper JL, Mupo A, Grove CS, Lynn C, Conte N, Andrews RM, Pacharne S, Tzelepis K, Vijayabaskar MS, et al. (2017). Molecular synergy underlies the co-occurrence patterns and phenotype of NPM1-mutant acute myeloid leukemia. Blood 130, 1911–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drabkin HA, Parsy C, Ferguson K, Guilhot F, Lacotte L, Roy L, Zeng C, Baron A, Hunger SP, Varella-Garcia M, et al. (2002). Quantitative HOX expression in chromosomally defined subsets of acute myelogenous leukemia. Leukemia 16, 186–195. [DOI] [PubMed] [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, and Aiden EL (2016a). Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES, and Aiden EL (2016b). Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, and Yakhini Z (2009). GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, Moon RT, Teo JL, Kim HY, Moon SH, et al. (2004).A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected]. Proc. Natl. Acad. Sci. U S A 101, 12682–12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forlani S, Lawson KA, and Deschamps J (2003). Acquisition of Hox codes during gastrulation and axial elongation in the mouse embryo. Development 130,3807–3819. [DOI] [PubMed] [Google Scholar]
- Fuller JF, McAdara J, Yaron Y, Sakaguchi M, Fraser JK, and Gasson JC (1999). Characterization of HOX gene expression during myelopoiesis: role of HOX A5 in lineage commitment and maturation. Blood 93, 3391–3400. [PubMed] [Google Scholar]
- Gale RE, Lamb K, Allen C, El-Sharkawi D, Stowe C, Jenkinson S, Tinsley S, Dickson G, Burnett AK, Hills RK, and Linch DC (2015). Simpson’s paradox and the impact of different DNMT3A mutations on outcome in younger adults with acute myeloid leukemia. J. Clin. Oncol 33, 2072–2083. [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. (2013). CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, et al. (1999). Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 286, 531–537. [DOI] [PubMed] [Google Scholar]
- Goyama S, Schibler J, Cunningham L, Zhang Y, Rao Y, Nishimoto N, Nakagawa M, Olsson A, Wunderlich M, Link KA, et al. (2013). Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J. Clin. Invest 123, 3876–3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimwade D, Ivey A, and Huntly BJ (2016). Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood 127, 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groschel S, Sanders MA, Hoogenboezem R, de Wit E, Bouwman BAM, Erpelinck C, van der Velden VHJ, Havermans M, Avellino R, van Lom K, et al. (2014). A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157, 369–381. [DOI] [PubMed] [Google Scholar]
- Heckl D, Kowalczyk MS, Yudovich D, Belizaire R, Puram RV, McConkey ME, Thielke A, Aster JC, Regev A, and Ebert BL (2014). Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat. Biotechnol 32, 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineagedetermining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Kallberg M, Chen X, Kim Y, Beyter D, Krusche P, and Saunders CT (2018). Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594. [DOI] [PubMed] [Google Scholar]
- Kuhn MW, Song E, Feng Z, Sinha A, Chen CW, Deshpande AJ, Cusan M, Farnoud N, Mupo A, Grove C, et al. (2016). Targeting chromatin regulators inhibits leukemogenic gene expression in NPM1 mutant leukemia. Cancer Discov. 6, 1166–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lento W, Congdon K, Voermans C, Kritzik M, and Reya T (2013). Wnt signaling in normal and malignant hematopoiesis. Cold Spring Harb. Perspect. Biol 5, 10.1101/cshperspect.a008011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, Yang FC, and Xu M (2011). Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 118, 4509–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Chen P, Su R, Hu C, Li Y, Elkahloun AG, Zuo Z, Gurbuxani S, Arnovitz S, Weng H, et al. (2016). PBX3 and MEIS1 cooperate in hematopoietic cells to drive acute myeloid leukemias characterized by a core transcrip-tome of the MLL-rearranged disease. Cancer Res. 76, 619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang SY, Moghimi B, Crusselle-Davis VJ, Lin IJ, Rosenberg MH, Li X, Strouboulis J, Huang S, and Bungert J (2009). Defective erythropoiesis in transgenic mice expressing dominant-negative upstream stimulatory factor. Mol. Cell. Biol 29, 5900–5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Benner C, Mansson R, Heinz S, Miyazaki K, Miyazaki M, Chandra V, Bossen C, Glass CK, and Murre C (2012). Global changes in the nuclear positioning of genes and intra- and interdomain genomic interactions that orchestrate B cell fate. Nat. Immunol 13, 1196–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Wang F, Zha J, Li H, Yan B, Du Q, Yang F, Sobh A, Vulpe C, Drusbosky L, et al. (2018). CTCF boundary remodels chromatin domain and drives aberrant HOX gene transcription in acute myeloid leukemia. Blood 132, 837–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, and DePristo MA (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer C, Kowarz E, Hofmann J, Renneville A, Zuna J, Trka J, Ben Abdelali R, Macintyre E, De Braekeleer E, De Braekeleer M, et al. (2009). New insights to the MLL recombinome of acute leukemias. Leukemia 23, 1490–1499. [DOI] [PubMed] [Google Scholar]
- Mukaka MM (2012). Statistics corner: a guide to appropriate use of correlation coefficient in medical research. Malawi Med. J 24, 69–71. [PMC free article] [PubMed] [Google Scholar]
- Narendra V, Rocha PP, An D, Raviram R, Skok JA, Mazzoni EO, and Reinberg D (2015). CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347, 1017–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogilvy S, Metcalf D, Gibson L, Bath ML, Harris AW, and Adams JM (1999). Promoter elements of vav drive transgene expression in vivo throughout the hematopoietic compartment. Blood 94, 1855–1863. [PubMed] [Google Scholar]
- Pillai MM, Venkataraman GM, Kosak S, and Torok-Storb B (2008). Integration site analysis in transgenic mice by thermal asymmetric interlaced (TAIL)-PCR: segregating multiple-integrant founder lines and determining zygosity. Transgenic Res. 17, 749–754. [DOI] [PubMed] [Google Scholar]
- Ramirez F, Dundar F, Diehl S, Gruning BA, and Manke T (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, and Weissman IL (2003). A role for Wnt signalling in selfrenewal of haematopoietic stem cells. Nature 423, 409–414. [DOI] [PubMed] [Google Scholar]
- Rice KL, and Licht JD (2007). HOX deregulation in acute myeloid leukemia. J. Clin. Invest 117, 865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin AJ, Parker KR, Satpathy AT, Qi Y, Wu B, Ong AJ, Mumbach MR, Ji AL, Kim DS, Cho SW, et al. (2019). Coupled single-cell CRISPR screening and epigenomic profiling reveals causal gene regulatory networks. Cell 176, 361–376.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ (2004). Java Treeview—extensible visualization of microarray data. Bioinformatics 20, 3246–3248. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvageau G, Lansdorp PM, Eaves CJ, Hogge DE, Dragowska WH, Reid DS, Largman C, Lawrence HJ, and Humphries RK (1994). Differential expression of homeobox genes in functionally distinct CD34+ subpopulations of human bone marrow cells. Proc. Natl. Acad. Sci. U S A 91, 12223–12227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So CW, Karsunky H, Wong P, Weissman IL, and Cleary ML (2004). Leukemic transformation of hematopoietic progenitors by MLL-GAS7 in the absence of Hoxa7 or Hoxa9. Blood 103, 3192–3199. [DOI] [PubMed] [Google Scholar]
- Spencer DH, Young MA, Lamprecht TL, Helton NM, Fulton R, O’Laughlin M, Fronick C, Magrini V, Demeter RT, Miller CA, et al. (2015). Epigenomic analysis of the HOX gene loci reveals mechanisms that may control canonical expression patterns in AML and normal hematopoietic cells. Leukemia 29, 1279–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavropoulou V, Kaspar S, Brault L, Sanders MA, Juge S, Morettini S, Tzankov A, Iacovino M, Lau IJ, Milne TA, et al. (2016). MLL-AF9 expression in hematopoietic stem cells drives a highly invasive AML expressing EMT-related genes linked to poor outcome. Cancer Cell 30, 43–58. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberlay PC, Achinger-Kawecka J, Lun AT, Buske FA, Sabir K, Gould CM, Zotenko E, Bert SA, Giles KA, Bauer DC, et al. (2016). Threedimensional disorganization of the cancer genome occurs coincident with long-range genetic and epigenetic alterations. Genome Res. 26, 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda A, Goolsby C, and Yaseen NR (2006). NUP98-HOXA9 induces long-term proliferation and blocks differentiation of primary human CD34+ hematopoietic cells. Cancer Res. 66, 6628–6637. [DOI] [PubMed] [Google Scholar]
- Tarbell ED, and Liu T (2019). HMMRATAC: a Hidden Markov ModeleR for ATAC-seq. Nucleic Acids Res. 47, e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsteinsdottir U, Mamo A, Kroon E, Jerome L, Bijl J, Lawrence HJ, Humphries K, and Sauvageau G (2002). Overexpression of the myeloid leukemia-associated Hoxa9 gene in bone marrow cells induces stem cell expansion. Blood 99, 121–129. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, and Chang HY (2010). Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valton AL, and Dekker J (2016). TAD disruption as oncogenic driver. Curr. Opin. Genet. Dev 36, 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Li Z, He Y, Pan F, Chen S, Rhodes S, Nguyen L, Yuan J, Jiang L, Yang X, et al. (2014). Loss of Asxl1 leads to myelodysplastic syndrome-like disease in mice. Blood 123, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, et al. (2011). A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 472, 120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, Zon LI, and Armstrong SA (2010). The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 327, 1650–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will B, Vogler TO, Narayanagari S, Bartholdy B, Todorova TI, da Silva Ferreira M, Chen J, Yu Y, Mayer J, Barreyro L, et al. (2015). Minimal PU.1 reduction induces a preleukemic state and promotes development of acute myeloid leukemia. Nat. Med 21, 1172–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingett SW, and Andrews S (2018). FastQ Screen: a tool for multi-genome mapping and quality control. F1000Res 7, 1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Kurtenbach S, Guo Y, Lohse I, Durante MA, Li J, Li Z, Al-Ali H, Li L, Chen Z, et al. (2018). Gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood 131, 328–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung J, Esposito MT, Gandillet A, Zeisig BB, Griessinger E, Bonnet D, and So CW (2010). beta-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 18, 606–618. [DOI] [PubMed] [Google Scholar]
- Zangenberg M, Grubach L, Aggerholm A, Silkjaer T, Juhl-Christensen C, Nyvold CG, Kjeldsen E, Ommen HB, and Hokland P (2009). The combined expression of HOXA4 and MEIS1 is an independent prognostic factor in patients with AML. Eur. J. Haematol 83, 439–448. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq, ATAC-seq, ChIP-seq, ChIRP-seq, whole exome-seq and HiC-seq reported in this paper are GSE114981 and GSE113191.