

Summary
Chromatin domains and their associated structures must be faithfully inherited through cellular division to maintain cellular identity. Yet, accessing the localized strategies preserving chromatin domain inheritance, specifically the transfer of parental, pre-existing nucleosomes with their associated post-translational modifications (PTMs) during DNA replication is challenging in living cells. We devised an inducible, proximity-dependent labeling system to irreversibly mark replication-dependent H3.1 and H3.2 histone-containing nucleosomes at desired loci in mouse embryonic stem cells such that their fate after DNA replication could be followed. Strikingly, repressed chromatin domains are preserved through the local re-deposition of parental nucleosomes. In contrast, nucleosomes decorating active chromatin domains do not exhibit such preservation. Notably, altering cell fate leads to an adjustment in the positional inheritance of parental nucleosomes that reflects the corresponding changes in chromatin structure. These findings point to important mechanisms that contribute to parental nucleosome segregation to preserve cellular identity.
Keywords: post-translational histone modifications, epigenetic, repressed chromatin domains, gene silencing, gene expression, polycomb, parental nucleosome segregation, DNA replication
Graphical Abstract
Introduction
Genome function and cellular identity are maintained through the structural organization of chromatin domains that either facilitate or impede transcription. The presence of specific histone post-translational modifications (PTMs) within these chromatin domains not only correlates with a given transcription status, but also facilitates the formation of repressive chromatin structures that impact gene expression (Reinberg and Vales, 2018). In order to maintain the integrity of these gene expression programs through cell division, the molecular features defining these chromatin structures must be transmitted, rather than established anew. Important determinants to establishing different types of chromatin states are the histone PTMs decorating primarily the tails of histones H3 and H4 (Stillman, 2018). Thus, in addition to DNA replication, chromosome duplication should entail the accurate reassembly/segregation of parental nucleosomes onto the appropriate locale of each daughter DNA molecule. This latter process involves a tightly coupled deposition of histones to the replication machinery behind the replication fork (Cusick et al., 1981; Herman et al., 1981). The founding studies on the structure of replicated chromatin establishes that parental histones are deposited onto newly synthesized DNA relatively quickly and that both replicated DNA strands capture equal amounts of parental histones (Annunziato, 2013; Petryk et al., 2018; Yu et al., 2018). It is now accepted that parental canonical histones, i.e. replication-dependent histones, rapidly re-assemble behind the replication fork starting with the H3-H4 tetrameric core followed by H2A-H2B dimer deposition (Campos and Reinberg, 2009; Xu et al., 2010; Alabert and Groth, 2012; Campos et al., 2014). Moreover, clear evidence that specific histone PTMs have the potential to be transmitted across mitosis (epigenetically inherited) was derived from studies of two distinct methylated states of histone H3 within the (H3-H4)2 core: tri-methylated H3K9 (H3K9me3) and di- or tri-methylated H3K27 (H3K27me2/me3) (Grewal and Moazed, 2003; Margueron et al., 2009; Margueron and Reinberg, 2011) and a model to account for this inheritance was recently put forward (Reinberg and Vales, 2018). Notably, both of these histone modifications are associated with repressed chromatin domains.
Nucleosomes vary in the composition of their histone components. While only one histone H4 isoform has been identified, there are several histone H3 variants including H3.1 and H3.2 that differ at one amino acid and are considered the canonical replication-coupled versions (Tagami et al., 2004). The observation that parental H3.1- and H3.2-containing nucleosomes are re-deposited as intact (H3.1-H4)2 or (H3.2-H4)2 tetramers upon DNA replication (Xu et al., 2010), supports a model for the local inheritance of histone PTMs. However, direct testing for the local re-deposition of parental canonical tetramers at a particular locus has not been achieved. In particular, the in vivo re-deposition of parental histones within the general vicinity of their original genomic position has not yet been examined through direct methods, but instead through proteomics, marking of newly replicated DNA and ChIP-sequencing techniques (Zee et al., 2012; Alabert et al., 2014; Clement et al., 2018; Reveron-Gomez et al., 2018). Although, the combination of these approaches provides insights into the “bulk” re-deposition of parental nucleosomes, these studies cannot determine the fidelity of such re-deposition at a given chromatin domain, key to tackling the mechanisms of epigenetic inheritance.
In the case of embryonic stem cells (ESCs), their pluripotent state depends on both the active transcription of genes encoding the ‘pluripotent factors’, i.e. Pou5f1, Nanog and Sox2 (among others), and the repression of genes encoding lineage-specifying developmental regulators (Boyer et al., 2006; Ng and Surani, 2011; Dowen et al., 2014). The Polycomb group (PcG) of proteins maintain the status of such gene repression in part through: 1) the catalysis of H3K27me2/me3 by PRC2 and 2) H3K27me2/me3 providing a platform for the recruitment of PRC1 and chromatin compaction (Boyer et al., 2006; Margueron and Reinberg, 2011; Pengelly et al., 2013; Simon and Kingston, 2013), although alternative pathways have been proposed (Cooper et al., 2016; Almeida et al., 2017). Indeed, perturbations of PcG proteins can aberrantly affect differentiation and foster a loss in cellular identity (Boyer et al., 2006; Margueron and Reinberg, 2011). Thus, the integrity of chromatin domains must be conveyed in a precise spatial and temporal manner to ensure proper development and to establish the maintenance of cellular identity.
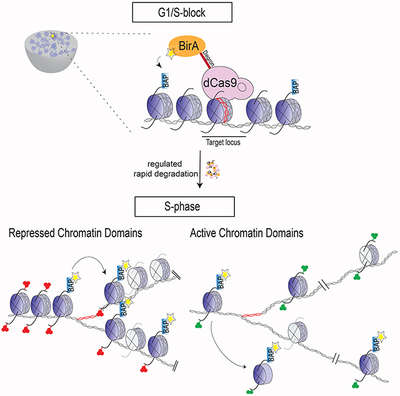
To gain insights into how parental nucleosome re-deposition might contribute to epigenetic inheritance, we devised a method that permanently labels H3.1- and H3.2-containing nucleosomes at desired actively transcribed or repressed genes in ESCs. This system allows us to follow the fate of the “marked” parental nucleosome upon DNA replication when ESCs are in a pluripotent state or enter into a specific differentiation program. The findings reveal that nucleosomes from repressed (but not active) chromatin domains are re-deposited within the same chromatin domain after DNA replication, whereas parental nucleosomes from actively transcribed genes are dispersed. Moreover, by inducing an altered cell fate from ESCs, the local re-deposition of parental nucleosomes during DNA replication at defined loci now reflects their conversion from a repressed to an active chromatin domain. These findings point to the inheritance of repressed versus active chromatin domains.
Results
Marking nucleosomes at a specific locus to follow their segregation upon DNA replication.
To investigate the segregation of parental nucleosomes, we developed a bio-orthogonal system to irreversibly mark H3.1 and H3.2 in vivo at candidate loci to follow their segregation at the nucleosome level across cellular division in mouse ESCs (Figure 1A). First, we introduced a Biotin Acceptor Peptide (BAP) motif sequence into the N-terminus of the endogenous H3.1 and H3.2 loci to biotinylate H3 chromatin using the Escherichia coli Biotin Ligase (BirA) (Kulyyassov et al., 2011; Shoaib et al., 2013). To recruit BirA to biotinylate nucleosomes at specific loci, BirA was fused to the catalytically inactive dCas9. To ensure the exclusive labeling of parental histones at unique selected genes, we restricted biotinylation to a tight temporal window by expressing dCas9-BirA from an integrated cassette under the control of a doxycycline-inducible promoter and incorporating an FKBP degradation domain (DD) (Banaszynski et al., 2006). This latter feature was essential in order to ensure that dCas9-DD-BirA expression before S-phase would be short-lived and then absent at entry into S-phase, yet nucleosome labelling would still be detectable. With this system in hand, the expression of chosen gRNAs allowed us to control spatial (gene) specificity of biotinylation (Chen et al., 2013), resulting in the desired deposition of the biotin tag, exclusively at the chosen locus of interest (Figure 1A). We then created clonal dCas9-DD-BirA-expressing ESCs containing Flag-BAP knock-ins to the N-terminus of 13 endogenous copies of replication-dependent H3.1 and H3.2 in the Hist1h3 cluster (Figures 1A and S1A–B). Chromatin immunoprecipitation (ChIP) followed by western blots of Flag-BAP-H3 showed that the tagged-histones were incorporated into both active and repressive chromatin as defined by the presence of H3K4me3 and H3K27me3, respectively (Figure S1C).
Figure 1. Precise labeling of H3.1 and H3.2 histones in living cells.

(A) Overview of the system to assess in vivo chromatin domain inheritance in mESCs. A master cell line containing endogenous Flag-BAP tagged versions of H3.1 and H3.2, a stably integrated doxycycline (Dox)-inducible dCas9-DD-BirA, and transducible gRNAs spanning 5 kb of a candidate locus is arrested in G1 phase. Following a 6 hr pulse of minimal doxycycline (Dox) and exogenous biotin, nearby tagged parental nucleosomes are biotinylated (blue histones and yellow asterisks). Wash-off of media releases cells into S-phase wherein the re-distribution of biotin-H3 at a mononucleosomal level is assayed in newly synthesized chromatin. (B) Cas9 immunofluorescence and DAPI staining of mESC nuclei with dCas9-DD-BirA targeted to the Gatat6 locus in G1/S-blocked cells ±Dox (top and middle panels), and in early S-phase cells (1 hr release after G1/S-block) following Dox and thymidine wash-off (bottom panel). Data is representative of two sets of biological experiments in which n=3 in each experiment. (C) Quantitation of Cas9 in (B), where the mean fluorescence intensity is compared to G1/S-block - Dox and early S-phase nuclei (top) or within the nucleus of G1/S-block +Dox (bottom). Statistical significance was determined using Student’s t-test (***p<0.001 and **** p<0.0001) and data are represented as mean ±SEM. (D) ChIP-seq analysis of G1/S-blocked cells at the Gata6 locus following dCas9-DD-BirA recruitment. Native Flag (top panel) and biotin and IgG (lower panels). Recruitment significance was determined by a Mann-Whitney test (p<0.0001). For ChIP-seq tracks, the data were normalized using Drosophila chromatin spike-in and the tracks show a scaling factor processed with the bioconductor package ChIPSeqSpike (see STAR Methods).
To spatially recruit dCas9-DD-BirA and biotinylate local parental H3 incorporated into chromatin at a unique desired gene, we stably expressed an array of ~29-35 guide RNAs (gRNAs) spanning 5 kb of sequence from a candidate locus (Figure 1A). To test the specificity and extent of temporal control on dCas9-DD-BirA expression, we introduced 33 gRNAs targeting BirA to 5 kb upstream of the Gata6 gene. We found that during the last step of a double thymidine G1-block synchronization (Figure S1D), a 6 hr pulse with a minimal amount of doxycycline was sufficient to observe an individual focus of dCas9-DD-BirA using immunofluorescence (Figure 1B, ±Dox). As BirA is targeted for proteasome degradation through its fusion with a degradation domain (Figure S1E), a wash-off step to release cells into early S-phase resulted in undetectable BirA expression (Figure 1B, lower panel). Quantitation of the mean fluorescence intensity from the Cas9 focus, showed a specific and transient enrichment of dCas9 at the G1/S-block (Figure 1C). In accordance, ChIP-quantitative PCR (ChIP-qPCR) of dCas9 at the Gata6 locus shows its transient enrichment and subsequent elimination in early S-phase and the next cell-cycle (Figure S2A).
Having established conditions for treating cells at the G1-phase with a minimal dose of doxycycline and exogenous biotin for 6 hr, chromatin at the G1/S transition was isolated and digested with MNase to enrich for mononucleosomes. Biotinylated and Flag-BAP-H3.1 and -H3.2 chromatin were then isolated by immunoprecipitation using Flag, biotin or IgG antibodies and in this case, ChIP-sequencing (seq) was performed (Figure 1D). We observed Flag-BAP-H3 incorporation throughout the genome (Figure 1D, top panel), and a significant enrichment of doxycycline-dependent biotin at the Gata6 locus through ChIP-seq (Mann-Whitney test, p<0.0001; Figure 1D, middle panels). However, we can detect signals elsewhere in the genome and such signals were present in IgG controls as well. We reasoned that the high sensitivity of ChIP-seq coupled with the intended, low temporal expression of dCas9-DD-BirA results in an uninterpretable signal-to-noise ratio in the case of ChIP-seq. Thus, ChIP-qPCR was used hereafter to quantitate parental biotin-H3.1 and -H3.2 nucleosome re-deposition.
Additional testing of the system demonstrated doxycycline-inducible and transient dCas9 recruitment to the Hoxc6 gene (Figure S2B) and accordingly, enrichment of biotinylated chromatin at the Hoxc6 locus (Figure S2C), as also evidenced by ChIP-qPCR (Figure S2D). Comparable results were obtained when dCas9 was recruited to the Ebf1 locus (Figure S2E–F). Therefore, our system allows for permanent histone labeling in vivo with notable spatial resolution (Figure 1B D) and tight temporal control of dCas9-DD-BirA recruitment to chromatin (Figure 1B C and S2A–B), resulting in the labeling of parental H3.1 and H3.2 at a single nucleosome level at a desired gene.
Inheritance of parental nucleosomes from repressive chromatin domains
A rigorous and sustainable repression of key developmental regulators through cell division is required to maintain the ESC pluripotent state (Boyer et al., 2006; Margueron and Reinberg, 2011; Dowen et al., 2014). To determine the localized strategies for the re-deposition of parental nucleosomes following DNA replication, we first assayed the local re-distribution of biotinylated Flag-BAP-H3.1 and -H3.2 at repressed chromatin domains through a timeline of 12, 24, and 48 hr after releasing ESCs from a G1/S-block (Figures 2A and S3A). Among the best studied and developmentally consequential repressed domains in ESCs are the Hox clusters (Figure S3B) (Boyer et al., 2006). Therefore, we tested parental nucleosome re-deposition by targeting a 5 kb area within the Hoxc6 gene in dCas9-DD-BirA expressing cells (Figure S2C). These cells were G1/S-blocked and given a 6 hr doxycycline and biotin pulse and subsequent wash-off to label parental nucleosomes. The cells were then released and followed for 12, 24, and 48 hr. Chromatin from these cells was collected, processed through native biotin ChIP, and a 35 kb window spanning 15 kb upstream and downstream from and including the 5 kb recruitment area was assayed for the presence of biotin at a high resolution of 500 bp through qPCR. To quantitatively analyze the parental biotin ChIP-qPCR, we used spike-in Drosophila melanogaster chromatin and normalized the data to input, spike-in, and minus-doxycycline native chromatin. Hoxc6 ChIP-qPCR interrogation of chromatin from cells arrested at the G1/S transition showed a robust biotin peak upstream of the dCas9-DD-BirA recruitment site towards the Hoxc6 transcriptional start site (TSS), as well as a less intense peak towards the 3′ end of the transcriptional unit (Figure 2B, 0 hr). This unexpected pattern of nucleosome biotinylation likely depends on the differences in accessibility of BirA to nucleosomes within repressed condensed chromatin, as a different chromatin marking was observed when analyzing euchromatic open chromatin (see below). Nonetheless, the labeling was very specific at the gene level (see Figure 1), allowing us to draw conclusions regarding nucleosome re-deposition at different genes. Subsequent release of the G1/S-blocked ESCs and analysis of the segregation of labeled parental nucleosomes revealed that the biotinylated nucleosomes segregated within the vicinity of the Hoxc6 locus (Figure 2B, time 12-24 hr), until the signal became undetectable at 48 hr (Figure 2B). Furthermore, quantitative analysis of the highest peak showed a drop in parental biotinylated chromatin enrichment from 1.00 to 0.40 through the first cell division (Figure 2C, Table 1). These findings suggest the positional inheritance of parental nucleosomes to the repressed Hoxc6 gene in ESCs.
Figure 2. Repressed parental H3 domains are re-distributed locally.

(A) Experimental timeline whereby ESCs are synchronized at G1/S phase (0 hr) and released to continue the cell cycle. Cells were harvested at 12 hr, 24 hr, and 48 hr intervals. (B-G) Native mononucleosomal biotin ChIP-qPCR in G1/S-blocked mESCs that were released following a 6 hr pulse of Dox and exogenous biotin, with BirA targeted to the Hoxc6 (B, C), Ebf1 (D, E), and Meis2 (F, G) loci. Data shows the average of 3 biological replicates spanning a 35 kb area at a resolution of 500 bp. For (C), (E), and (G), the graph highlights the biotin enrichment taken from the highest peak of the corresponding assays: Hoxc6 primer set 22 (B), Ebf1 primer set 22 (D), and Meis2 primer set 23 (F). All biotin enrichment levels are relative to input, normalized to Drosophila chromatin spike-in, followed by subtraction of the minus-Dox (-Dox) control. Error bars represent standard error. For panels (C), (E), and (G), the dataset for 0 hr was set to 1.
Table 1.
Quantitative analysis of biotinylated nucleosomes remaining across DNA duplication
| Gene Name | Chromatin state | % Biotin Remaining | |
|---|---|---|---|
| Δt 0-12 hr | Δt 0-24 hr | ||
| Hoxc6 | Repressed | 40% | 31% |
| Ebf1 | Repressed | 50% | 39% |
| Meis2 | Repressed | 63% | 32% |
| Gata2 (−RA) | Repressed | 34% | 10% |
| Gata6 (−RA) | Repressed | 57% | 20% |
| Ccna2 | Active | 12% | 4% |
| Pou5f1 | Active | 7% | 1% |
| Nanog | Active | 20% | 1% |
| Gata2 (+RA) | Active | 0% | 0% |
| Gata6 (+RA) | Active | 8% | 1% |
Candidate loci (left column) analyzed for the percentage of biotin retention 12 and 24 hr after release from the G1/S-block and within S-phase of the cell-cycle (right columns). The status of gene expression for each gene is indicated (middle column). The Gata loci in which Retinoid Acid (RA) was added to induce gene expression are indicated. The expression of the other genes was analyzed under steady-state conditions.
To further analyze whether the observed positional inheritance of the repressed Hoxc6 domain was a general phenomenon across repressed chromatin domains, dCas9-DD-BirA was similarly recruited to two other transcriptionally inactive domains on separate chromosomes (Figure S3C and S3D). We selected gRNAs targeting a 5 kb region in the Ebf1 or the Meis2 gene and observed an analogous biotin enrichment upstream and downstream from the recruitment areas in G1/S-blocked ESCs (Figure 2D and 2F, respectively, 0 hr). Similar to the Hoxc6 locus, DNA replication resulted in the dilution of the parental biotin signals within their respective regions (Figure 2D and 2F, 12-48 hr), and quantitative analysis of the most intense peak at each locus showed a drop in parental biotinylated chromatin enrichment from 1.00 to either 0.50 or 0.63 through the first cell division for Ebf1 and Meis2, respectively (Figure 2E and 2G, Table 1). These observations from three independent repressed chromatin domains point to parental histones being re-deposited locally.
Lack of inheritance of parental nucleosomes from active chromatin domains
That parental histones were retained locally during replication of repressed chromatin domains warranted the comparable analysis of active chromatin domains. We employed gRNAs tiling to a 5 kb region upstream of the Ccna2 gene and during the final step of double thymidine synchronization, induced the transient expression of dCas9-DD-BirA and subsequent biotinylation of Flag-BAP-H3.1 and -H3.2 at Ccna2 chromatin, as performed above. We then assayed the local re-distribution of biotinylated Flag-BAP-H3.1- and -H3.2-labeled nucleosomes and the results showed a lower level of nucleosome labeling spread across a much broader area from the Ccna2 5 kb recruitment site (Figure 3A, 0 hr), likely reflecting that the chromatin is “open” and more accessible to the dCas9-DD-BirA. Subsequent to the release of the G1/S-block, the biotin enriched parental histones could not be detected after DNA replication (Figure 3A, 12 hr). Quantitative analysis of the highest biotin peak surrounding the Ccna2 5 kb recruitment area showed a drastic drop in parental biotinylated chromatin enrichment from 1.00 to 0.12 through the first cell division (Figure 3B, 12 hr and Table 1). These data point to the non-local re-distribution of parental H3.1 and H3.2 histones in the Ccna2 active domain.
Figure 3. Dispersed segregation of active parental H3 domains.

Native mononucleosomal biotin ChIP-qPCR from G1/S-blocked mESCs that were released following a 6 hr Dox and exogenous biotin pulse, with BirA targeted to the Ccna2 (A, B), Pou5f1 (C, D), and Nanog (E, F) loci. Data shown is the average of 3 biological replicates spanning a 25 kb area at a resolution of 500 bp. For (B) (D), and (F), the graph highlights the biotin enrichment taken from the highest peak of the corresponding assays: Ccna2 primer set 10 (A), Pou5f1 primer set 9 (C), and Nanog primer set 40 (E). All biotin enrichment levels are relative to input, normalized to Drosophila chromatin spike-in, followed by subtraction of the -Dox control. Error bars represent standard error. For panels (B), (D), and (F), the dataset for 0 hr was set to 1.
As active domains replicate earlier in S-phase (Rhind and Gilbert, 2013), we examined whether the loss of Ccna2 biotinylated chromatin occurs as early as 6 hr, a time at which mouse ESCs (mESCs) are in mid S-phase prior to cell division (Figure S4A). Indeed, native biotin ChIP-qPCR of Ccna2 chromatin showed a drastic decrease in the biotin signal at this time point and a more pronounced loss at 12 hr (Figure S4B, 6 hr and 12 hr). To determine whether loss of biotin-H3.1 and -H3.2 was the result of histone turnover in transcriptionally active Ccna2, we inhibited transcription by the addition of triptolide during S-phase progression in our system. The addition of triptolide significantly reduced global mRNA levels and drastically inhibited Ccna2 mRNA expression without altering its protein expression within the time frame of the experiment (Figure S4C–E). Importantly, this transcription inhibition was ineffectual in decreasing the biotin signal (Figure S4B, 6hr + triptolide), suggesting that the loss in parental H3.1 and H3.2 in the Ccna2 active domain is not due to transcriptional turnover. Thus, biotinylated parental histones, at least within the Ccna2 locus, distribute non-locally upon DNA replication, revealing an unexpected, but biologically sound difference in the spatial inheritance of nucleosomes at active versus repressed loci (see Discussion), which could not be appreciated with previously published genome-wide approaches.
To interrogate whether this non-local distribution of parental histones is specific to the Ccna2 locus or represents a wider phenomenon of nucleosome inheritance associated with active domains, we expanded our studies and analyzed the loci of pluripotent factors Pou5f1 and Nanog, which are highly expressed in ESCs and encode transcription factors critical for ESC pluripotency and self-renewal capacity (Takahashi and Yamanaka, 2006). Targeting dCas9-DD-BirA to either of these loci in G1/S-blocked cells again resulted in a broader biotin enrichment surrounding the 5 kb gRNA recruitment area (Figure 3C and 3E, respectively). Furthermore, and similar to the case of chromatin associated with the Ccna2 locus, release of the G1/S-block led to a loss in biotin signal upon the first cell division (Figure 3C and 3E, respectively). We quantitatively assessed the reduction in the biotin signal from the peaks of Pou5f1 and Nanog, which again revealed a drastic drop in biotin enrichment from 1.00 to 0.07 for Pou5f1 (Figure 3D, 12 hr) and from 1.00 to 0.20 for Nanog (Figure 3F, 12 hr) within the first cell division (Table 1). The loss in biotin signal following DNA replication in active chromatin domains suggests that parental H3.1- and H3.2-containing nucleosomes are randomly re-deposited within active genes. Alternatively, the loss in biotin signal could be due to a trivial loss in detection. The latter is not likely given the results obtained with two other active domains, as shown below. These results point to the distinction between nucleosome re-deposition in active versus repressed chromatin, with biotinylated parental histones being dispersed in the case of active domains and positionally inherited in the case of repressed ones.
The segregation of parental nucleosomes requires DNA replication
To assess whether DNA replication is required for the local and non-local segregation of parental nucleosomes associated with repressed and active chromatin domains, respectively, we examined biotin signal dilution when DNA replication is blocked. To this end, shortly after cells were released from the G1/S-block, biotinylated nucleosomes present on the Hoxc6 and Pou5f1 cells were kept in S-phase within a 12 or 24 hr timeline of aphidicolin treatment (Figures 4A and S5A). The biotin-enriched peaks at the repressed Hoxc6 and active Pou5f1 loci (Figures 2C and 3D), significantly sustained biotin enrichment in the presence of aphidicolin (Figure 4B E), which did not alter Pou5f1 mRNA expression (Figure S5B). Indeed, the absence of biotin dilution in aphidicolin-treated Hoxc6 and Pou5f1 chromatin suggests that the local dynamics of parental nucleosome segregation were frozen when S-phase progression was blocked.
Figure 4. Parental H3 nucleosome segregation is replication dependent.

(A) Experimental timeline whereby ESCs are synchronized at G1/S phase (0 hr) and released to continue the cell cycle or treated with 1 μg/ml aphidicolin to block S-phase progression. Cells were harvested at 12 hr and 24 hr. (B-E) Native biotin ChIP-qPCR analysis of the Hoxc6 peak primer set 22 (B), Pou5f1 peak primer set 9 (C), Pou5f1 peak primer set 22 (D), and Pou5f1 peak primer set 45 (E) in cells treated (green) or not treated (black) with aphidicolin following biotin enrichment during G1 synchronization and release into S-phase. All biotin enrichment levels are relative to input and normalized to Drosophila chromatin spike-in followed by subtraction of the minus-Dox (-Dox) control. Biotin-enriched aphidicolin chromatin was then normalized to biotin-H3 at 0 hr. Error bars represent standard error of three biological replicates. Statistical significance was determined using 2way ANOVA (***p<0.001 and **** p<0.0001).
Inheritance of repressed chromatin domains as a function of cellular differentiation
To maintain a pluripotent state, ESCs require that genes encoding lineage-specifying developmental regulators remain repressed, as their expression will promote differentiation to a specific lineage and consequently a change in cellular identity (Boyer et al., 2006; Margueron et al., 2009; Dowen et al., 2014). The GATA factors are such genes whose expression in ESCs leads to the differentiation of mesoderm and ectoderm-derived tissues (Lentjes et al., 2016). Since the retinoic acid (RA) signaling molecule can activate expression of the GATA factors (Zhang et al., 2015), we analyzed the segregation of parentally marked-nucleosomes upon RA treatment. As expected, RA addition to mESCs for 12 hr prior to G1-synchronization and release (Figure 5A), induces Gata2 and Gata6 gene expression (Figure 5B). Introducing gRNAs to target a 5-kb area upstream of the Gata2 and Gata6 genes (as in Figure 1D for the Gata6 locus) resulted in the recruitment of dCas9-DD-BirA and the subsequent biotinylation of parental H3.1- and H3.2-containing nucleosomes in active and repressed chromatin states (Figures 5C, E and S6A–B). As expected from our results with repressed chromatin domains (Figure 2), we found a discrete peak in the Gata2 and Gata6 loci of G1/S-blocked chromatin in cells not treated with RA (Figure 5C, E, and S6A, 0 hr, −RA). Similarly, the subsequent release of these G1/S-blocked mESCs showed the re-deposition of biotin parental nucleosomes within the vicinity of the initial Gata2 and Gata6 peak position by ChIP-qPCR [Figure 5C F, 12-24 hr (−RA)] and this phenomenon is highlighted by ChIP-seq on the Gata6 locus when cells have gone through one round of DNA replication [Figure S6B, +Dox 12 hr (−RA)].
Figure 5. Altering repressed to active domains changes local parental H3 recycling.

(A) Experimental timeline whereby ESCs are ± 1 μM RA for 12 hr prior to synchronizing at G1/S phase (0 hr) and released to continue the cell cycle. Cells were harvested at 12 hr and 24 hr. (B) Relative mRNA expression of Gata2 and Gata6 genes in ESCs treated or not treated with RA for 48 hr. Data was normalized to Actin expression and minus RA control. (C-F) Native mononucleosomal biotin ChIP-qPCR in mESCs ± RA, blocked in G1/S and then released following a 6 hr pulse of Dox and exogenous biotin, with BirA targeted to the Gata2 (C-D) and Gata6 (E-F) loci. Data shows the average of 3 biological replicates spanning a 35 kb area at a resolution of 500 bp. For (D and F), graph highlights the biotin enrichment taken from the highest peak of the corresponding assays: Gata2 primer set 54 (C) and Gata6 primer set 30 (E). Biotin enrichment levels are relative to input and normalized to Drosophila chromatin spike-in followed by subtraction of the minus-Dox (-Dox) control. Error bars represent standard error of three biological replicates. For panels (D and F), the dataset for 0 hr was set to 1 and the statistical significance determined using 2way ANOVA (***p<0.001).
Upon RA-induced transcription activation of Gata2 and Gata6 (Figure 5B), the levels of biotinylated parental H3.1 and H3.2 chromatin remained comparable to that of a repressed state, but the biotin enrichment across Gata2 or Gata6 was now broader [Figure 5C, E, and S6B, 0 hr (+RA)]. Of note, steady-state active chromatin (i.e. Ccna2, Pou5f1 and Nanog) exhibited a lower and much broader biotin signal (Figure 3) than observed when Gata2 and Gata6 loci were captured immediately upon RA-induced activation. Yet, similar to the non-local segregation of histones in steady-state active chromatin (Figure 3), Gata2- and Gata6-expressing cells showed a loss in biotinylated parental H3 chromatin at these loci by ChIP-qPCR [Figure 5C F, 12-24 hr (+RA)], and the biotin signal was decreased to background levels in ChIP-seq [Figure S6B, 12 hr (+RA)]. Quantitative assessment of the distribution of the original biotin peak observed in repressed chromatin showed that biotinylated parental nucleosomes were diluted from 1.00 to 0.34 for Gata2 (Figure 5D, 12 hr (−RA) and Table 1) and from 1.00 to 0.57 for Gata6 [Figure 5F, 12 hr (−RA) and Table 1]. In comparison, a significant drop in biotin enrichment was observed for active chromatin as biotin-labeled nucleosomes changed from 1.00 to 0.00 for Gata2 [Figure 5D, 12 hr (+RA) and Table 1] and from 1.00 to 0.08 for Gata6 [Figure 5F, 12 hr (+RA) and Table 1]. These results argue for the existence of distinct localized strategies for the re-deposition of parental nucleosomes in repressed versus active chromatin as changing the transcriptional state of Gata2 and Gata6 genes significantly alters the dynamics of parental H3 segregation [Figure 5C F and S6A–B, (±RA)].
Discussion
A key feature in the control of gene expression is embodied in the histones and their associated PTMs that modulate the structure, stability, and dynamics of the local chromatin state (Luger et al., 2012; Stillman, 2018). Although it is accepted that histone PTMs selectively impact a transcriptional program, it is unclear if histones and their PTMs are determinants to the heritable transmission of an ‘epigenetic’ state from one cell generation to the next. To address this question, we developed a system in which pre-existing H3.1- and H3.2-bearing nucleosomes are marked at a single gene and followed across DNA replication to assess their local segregation in active or repressed chromatin domains. We find a spatial conservation in the recycling of intact parental H3.1- and H3.2-nucleosomes within repressive chromatin domains, but this local re-deposition is absent in the case of nucleosomes associated with active chromatin (Figure 6 and Table 1). In the latter case, the chromatin is open (euchromatin), as evidenced by the broader area and decreased signals in our nucleosome labeling experiments of steady-state active genes (i.e. Ccna2, Pou5f1 and Nanog). In contrast to repressed chromatin (e.g. Hoxc6), this biotin-H3 enrichment is lost upon DNA replication (Figure 3, Table 1). When biotin enrichment levels of newly activated chromatin (i.e. Gata2 and Gata6 loci) are comparable to those of their respective, repressed state [Figure 5C F and S6A–B, 0 hr ±RA], a loss in biotin-H3 in the active chromatin is again observed, in contrast to their repressed states that retain the parental histone signals (Figure 5C F and S6A–B, 12 hr (±RA), and Table 1). Thus, re-deposition of parental H3.1- and H3.2-nucleosomes in active chromatin domains lacks heritable transmission. As the histone variant H3.3 and histone turnover are associated with a transcriptionally active state (Deal et al., 2010; Kraushaar et al., 2013), we predict that nucleosomes comprising H3.3 will also disperse upon DNA replication in a manner similar to those comprising active marks, as shown in this study. We propose that the histone PTMs associated with active chromatin function solely to facilitate transcription and can be readily restored subsequent to DNA replication upon re-engagement of the appropriate sequence-specific transcription factors to direct transcription (Ptashne and Gann, 2002; Reinberg and Vales, 2018). We conclude that it is the nucleosomes comprising a repressive chromatin state that segregate locally such that this signature is conveyed directly to the same domains on daughter DNA.
Figure 6. Parental H3 nucleosomes segregate locally in repressed chromatin domains.

In repressed chromatin domains, a degree of spatial conservation in the re-deposition of intact pre-existing (H3.1-H4)2 or (H3.2-H4)2 tetrameric core-bearing histone PTMs (blue histones and solid arrows) is sufficient to transmit chromatin states to daughter cells if maintenance enzymes are available and can restore the transmitted modification(s) to neighboring newly synthesized histones (gray histones and dashed arrows) (Reinberg and Vales, 2018). Re-deposition of parental H3.1 or H3.2 nucleosomes is dispersed in active chromatin domains (see Discussion).
In a prokaryotic cell, gene regulation is not conveyed by structural changes to the DNA and its histone-like proteins (Ptashne and Gann, 2002; Dillon and Dorman, 2010). Instead, the genome is open and subject to the function of activators and repressors that control inducible operators to alter gene transcription (Ptashne and Gann, 1997; Ptashne and Gann, 2002). It is not until the advent of unicellular eukaryotes that histones and in particular, their amino-terminal tails afford a link to the direct regulation of gene transcription (Stillman, 2018). A key example is histone H4 and its acetylation at lysine 16 (H4K16ac), which facilitates open higher-order chromatin structures conductive to gene expression (Shogren-Knaak et al., 2006). However, similar to prokaryotes, unicellular eukaryotes require activators to initiate gene transcription. In multicellular organisms, master regulators establish transcriptional outputs through their sequence-specific DNA binding activity, which recruit coactivators and RNA polymerase II to promoter sequences (Patikoglou and Burley, 1997). RNA Polymerase II, in turn, recruits independent methyltransferases that methylate histone H3 at lysines 4 and 36. Methylated H3K4 and H3K36 are associated with active chromatin, functioning in combination with other proteins that recognize the methyl groups and further facilitate transcription (Sims et al., 2004). Thus, in eukaryotes, histones and their associated PTMs evolved to enable transcriptional changes, but sequence-specific transcription factors are required for transcription as in the case of prokaryotic cells and the histone modifications associated with active chromatin are a consequence of transcription activity.
Strikingly, it is through gene repression that histone PTMs show a heritable gene regulatory system in eukaryotic cells. For example, in S. cerevisiae, the repression of the silent mating type loci HMLα and HMRa requires the deacetylation of histone H4K16ac that is catalyzed by the silent information regulator (Sir) complex (Sir2-Sir3-Sir4), initially recruited through its interaction with the Sir1 adaptor (Johnson et al., 1990; Stillman, 2018). While Sir3 and Sir4 foster the expansion of this silencing, the histone deacetylase activity of Sir2 maintains the inheritance of the deacetylated version of H4K16 through multiple cell divisions in the absence of the initial signal from Sir1 (Johnson et al., 1990; Allshire and Madhani, 2018; Stillman, 2018).
In multicellular organisms, histone PTMs associated with repressed chromatin also mediate a heritable chromatin state. These heritable gene silencing histone PTMs include H3K9me3 catalyzed by Suv39h1/h2 and H3K27me2/me3 catalyzed by the Polycomb Repressive Complex 2 (PRC2). Both Suv39h1/h2 and PRC2 exhibit a bone fide “write and read” mechanism by which the polypeptide or complex, respectively, deposits their corresponding PTM and binds to the PTM resulting in an allosteric stimulation of the enzymatic activity (Margueron et al., 2009; Allshire and Madhani, 2018; Oksuz et al., 2018; Reinberg and Vales, 2018). Similar to Sir2 in yeast, both SUV39H1 and PRC2 can propagate and maintain a repressive chromatin state through multiple cell divisions in the absence of the initiating signal; an epigenetic phenomenon (Allshire and Madhani, 2018; Reinberg and Vales, 2018).
In addition to this “write and read” mechanism, the inheritance across DNA replication of a repressed chromatin state entails that the histone methyltransferase activity be in proximity to parental nucleosomes such that their associated PTMs are transmitted; otherwise, newly synthesized histones (naïve nucleosomes) that comprise half of replicative chromatin might attenuate a repressed state. Most importantly, the pre-existing histones and their PTMs must segregate to the same domains on daughter DNA strands. Therefore, our aim was to assess the segregation dynamics of parental H3.1- and H3.2-containing nucleosomes by establishing a system that irreversibly labels nucleosomes at a specific location and at a specific time. Our system allows for parental H3.1- and H3.2-nucleosomes to be biotinylated prior to DNA replication and their tagged density to be followed through chromatin duplication. These tagged densities could be detected through both ChIP-qPCR and ChIP-seq. However, as we are recruiting dCas9-DD-BirA to a 5 kb locus and biotinylating the nearby chromatin, we reason that the intended amount of candidate nucleosomes are not sufficient to quantitatively separate the signal from the whole-genome noise using the current sequencing technology. Nonetheless, it is through our technique that accurately scores for parental nucleosome position prior to DNA replication that the parental histone origin can be determined. ChIP-seq of histone PTMs and imaging systems in conjunction with synchronization experiments can detect the restoration dynamics of newly synthesized chromatin (Clement et al., 2018; Reveron-Gomez et al., 2018), however, such approaches cannot ascertain the locale of a parental nucleosome. Thus, our findings here can attribute the inheritance of a repressed state to both the local segregation of parental nucleosomes and to the “write and read” modules of the histone methyltransferase that deposit the repressive modification (Figure 6).
A feasible explanation for this local conservation of repressed chromatin domains versus active chromatin domains is that replication of repressed chromatin occurs late in S phase, in contrast to that of active chromatin, which is an early event. This timing difference is known to affect the rate of replication, such that euchromatin is replicated at a faster rate than heterochromatin (Rhind and Gilbert, 2013). The slower rate in the latter case, might facilitate parental histone segregation to the same domains. Whether these differences can account for the observed positional inheritance of repressive chromatin domains remains to be elucidated, but as replication timing and chromosomal domain structures are intertwined (Rhind and Gilbert, 2013), it is possible that active and repressed chromatin form self-interacting domains that set thresholds for the accessibility of factors promoting the appropriate chromatin organization during DNA replication. We speculate that distinct chaperones might operate during late S-phase, but are absent in early replicating chromatin. Our findings set the stage for investigating the veracity of such scenarios as they clearly demonstrate a fundamental feature of epigenetic inheritance: that nucleosomes associated with repressed chromatin segregate to the same chromatin domains whereas those associated with active chromatin exhibit a dispersed re-distribution.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Please direct inquiry to the Lead Contact, Danny Reinberg (danny.reinberg@nyumc.org). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The following were used in this study: Male KH2 mouse ESCs (Beard et al., 2006) and Female Human embryonic kidney 293T (293T) cells have been authenticated. KH2 ESCs were grown in DMEM supplemented with 15% FBS (for CRISPR/Cas9 targeting) or Tet-free FBS (Gemini 100-800, for biotinylation experiments), L-glutamine, penicillin/streptomycin, non-essential amino acids, 0.1 mM β-mercaptoethanol, LIF, and 2i inhibitors, which include 1 μM MEK1/2 inhibitor (PD0325901) and 3 μM GSK3 inhibitor (CHIR99021) on 0.1% gelatin coated plates. 293T cells were grown in DMEM supplemented with 10% FBS, L-glutamine and penicillin/streptomycin.
METHOD DETAIL
Cell Line Generation
To generate dCas9-DD-BirA expressing stable cell lines, 2 μg of pINTA-dCas9-DD-BirA plasmid was transfected with Lipofectamine 2000 (Thermo Fisher Scientific) into KH2 ESCs. After 2 weeks of 200 μg/ml of Zeocin selection, single colonies were selected. dCas9-DD-BirA inducibility was tested and cells were provided with FUCCI reporter (Sladitschek and Neveu, 2015) for testing in synchronization experiments. The desired stable clone was expanded for CRISPR/Cas9 targeting of Flag-BAP knockins to H3.1 and H3.2 copies in the Hist1h3 cluster (see below).
Cell Synchronization and in vivo biotinylation
For G1 synchronization experiments, mESCs were plated at 50-60% confluency and pulsed for 18 hr with 7-8 μM thymidine, followed by a PBS wash and a 7-8 hr release into 2i Tet-free media. The cells were then given a second thymidine treatment at a 5-6 μM concentration for 12-13 hr. The G1-block was confirmed by staining DNA with propidium iodide. For the biotinylation experiments, a 6 hr pulse of 2 μg/ml of doxycycline and 1 μg/ml of exogenous biotin was given during the latter half of the second thymidine treatment. For proteasome inhibition experiments, 1, 5, or 10 μM of MG-132 was added during this 6 hr pulse of doxycycline. Next, mESCs were either harvested at the G1-block (starting time point, 0 hr) or washed with PBS and released to 2i Tet-free media for the indicated time points. In S phase-block experiments, 1 μg/ml of aphidicolin was added to the 2i Tet-free media following release from the G1-block and for the transcription inhibition experiments, 1 μM of Triptolide was added following release into S-phase. Lastly, for Retinoic Acid (RA) differentiation, mESCs were given 1 μM RA 12 hr prior to the double thymidine synchronization and kept in RA-media throughout G1-synchronization and the experimental time course.
Cloning
Cloning of the pINTA-N3-dCas9-DD-BirA was performed in several steps, as follows. The cDNA for dCas9 was initially cloned from EF1-dCas9-KRAS (pHAGE EF1α dCas9-KRAB was a gift from Rene Maehr and Scot Wolfe, Addgene plasmid #50919) (Kearns et al., 2014) into pINTO-C-HF (pcDNA4/TO from Invitrogen with a C-terminus HA-Flag tag) using EcoRl and Notl. Then, BirA cDNA with a stop codon was cloned from pCDNA3.1 BirA-GFP (a gift from Vasily Ogryzko) (Kulyyassov et al., 2011) into pInto-dCas9 using Not1 and XhoI. For insertion of the FKBP(DD), the sequence was amplified from pBMN FKBP(DD)-YFP (a gift from Thomas Wandless, Addgene plamid #31763) (Banaszynski et al., 2006) using primers designed to leave Not1 overhangs. Digestion and ligation of the Not1 site in pINTO-dCas9-(Not1)-BirA and ligation of the NotI flanked FKBP(DD) fragment gave in-frame dCas9-DD-BirA. Lastly, the dCas9-DD-BirA cDNA was moved into the pINTA-N3 vector, based on the Tet-On 3G system (Clontech) through Gibson cloning to derive pINTA-dCas9-DD-BirA for expression in KH2 ESCs. See Table S1 for cloning primers.
CRISPR-mediated genome editing
GFP was replaced for BFP in the SpCas9-2A-GFP plasmid [pSpCas9(BB)-2A-GFP (PX458 was a gift from Feng Zhang, Addgene plasmid # 48138) (Ran et al., 2013)] by Gibson cloning and made SpCas9-2A-BFP (see Table S1 for cloning primers). Then, an optimal gRNA target sequence closest to the genomic target site was chosen using the http://crispr.mit.edu design tool. The individual gRNAs (Table S2) were cloned into the SpCas9-2A-BFP plasmid via BbsI digestion and insertion (Ran et al., 2013). For knock-in of Flag-BAP, ~750 bp gBlock design were ordered from IDT or Genscript, that included: 1) a homology arm corresponding to the ~350-450 bp of the H3 promoter area, 2) the Flag-BAP sequence placed after the start codon (Flag-BAP sequence: GACTACAAAGACGATGACGACAAGGGCCTGACAAGAATCCTGGAAGCTCAGAAGATCGTGAGAGGAGGCCTCGAG), and 3) a homology arm corresponding to ~200-300 bp of the H3 coding sequence. The PAM sequences of the gBlocks were mutated for correct Cas9 digestion of genomic DNA within cells (see Table S2 for primers corresponding to gBlock amplification). ESCs were then transfected with 0.5 μg of the Cas9-gRNA-BFP plasmid and 0.5 μg of amplified gBlock in Lipofectamine 2000 containing 2i media. After transfections, the medium to high BFP populations were FACS sorted and seeded at 20,000 cells per 15 cm plate. 7-10 days later, single ESC clones were selected and plated onto individual wells of a 96-well plate for genotyping. Genomic DNA was harvested via QuickExtract (Epicentre) DNA extraction, and genotyping PCRs were performed using primers surrounding the target site (Table S2), in addition to screening against Cas9 insertions with the genotyping primers FWD: ACCCAGAGAAAGTTCGACAATC and REV: GTGGTGGTAGTTGTTGATCTCG. The PCR products of the Flag-BAP positive clones were purified and sequenced to verify the presence and correct sequence of a Flag-BAP-H3 insertion.
For gRNA recruitment of dCas9-DD-BirA to 5 kb of a candidate locus in ESCs containing Flag-BAP-H3 chromatin, individual gRNAs were inserted into the pKLV-U6gRNA-EF(BbsI)-PGKpuro2ABFP (a gift from Hiroshi Ochiai, Addgene plasmid #62348) (Ochiai et al., 2015) (Table S3). Lentiviruses for each gRNA were generated in 293T cells using the packaging plasmids pMD2.G and psPAX2. Briefly, 300 K 293T cells were seeded per well in a 6-well plate the day prior to transfections. For transfections, 1 μg of pAX/MD2g/gRNA vector (0.375 μg/0.125 μg/0.5 μg) was added to Lipofectamine 2000 media for overnight incubation. Fresh media was then added and 48 hr viral supernatants were collected and frozen at −80 °C. For KH2 ESCs transductions, 5000 cells were seeded in 100 μl of 2i media for a 96-well plate. The following morning, tandem viral transductions were done with Polybrene containing 10 ul of each viral gRNA supernatant corresponding to ~1-16 gRNAs (total volume 160 ul) and spinoculated 2000 rpm for 90 min at 32 °C. After a 6 hr incubation in a temperature (37 °C)- and CO2-controlled chamber, the transductions were repeated with 10 μl of viral supernatants from ~17-35 gRNAs (total volume of 180 μl). Fresh 2i media was added and cells were allowed to recover for 3-5 days before FACS sorting a BFP positive population. This method allowed for a 90-95% efficiency of ES cell transduction, with ~35 gRNAs targeting a 5 kb area of a candidate locus.
Immunofluorescence
For immunofluorescence of Cas9, we followed the previously described protocol (Jeppesen, 2000 #97) to isolate native chromatin from synchronized cells. Briefly, mESCs were synchronized with a double Thymidine block (as described above) and cells harvested at desired time points. Using a hypotonic KCl solution for cell swelling, the harvested pellet was resuspended and cytospun on glass slides. A KCM treatment follows thereafter, allowing for the removal of cytoplasmic components and preservation of native nuclei. The slides were then incubated with Cas9 antibody (Millipore MAC133-clone 7A9) and a goat anti-mouse Alexa Fluororphore 488 secondary antibody. To image the nuclei, Nikon Eclipse Ti2 was used. For quantitation of the Cas9 focus, ImageJ was used to obtain the mean fluorescence signal of an area of interest minus the mean fluorescence of background readings and the significance determined by Student’s t-test with *** p<0.001 and **** denotes p<0.0001.
ChIP-qPCR and ChIP-seq
For native MNase chromatin preparation, nuclei were harvested from ESCs by hypotonic lysis TMSD buffer (40 mM Tris, 5 mM MgCl2, 0.25 M Sucrose with protease inhibitors), and pelleted for 10 min at 3600 rpm at 4 °C. Nuclei were resuspended in NIB-250 (15 mM Tris-HCl, pH 7.5, 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 1 mM CaCl2, 250 mM Sucrose with protease inhibitors) containing 0.3% NP-40. A chromosome pellet was isolated by centrifuging the solubilized nuclei for 5 min at 600 rcf at 4 °C. The chromosome pellet was washed with NIB-250 buffer, then resuspended with MNase digestion buffer (10 mM Hepes, 50 mM NaCl, 5 mM MgCl2, 5 mM CaCl2 with protease inhibitors) and treated with MNase (Sigma) until a DNA fragment size of 150-300 bp (1-2 nucleosomes) was attained. MNase digestion was stopped with 15 μl of EGTA (0.5 M) per 1 ml pellet and then spun at 600 rcf for 5 min at 4 °C. The supernatant was placed in a fresh tube and the chromatin pellet was further processed by adding the same volume of BC500 (40 mM Tris, 5 mM MgCl2, 500 mM NaCl and 5% glycerol, and protease inhibitors) and EGTA (0.5 M) as in the MNase buffer plus EGTA step. The soluble chromosomes in BC500 and EGTA were then incubated for 30 min while rotating at 4 °C, allowing for further enrichment of desired chromatin. Samples were pelleted at 600 rcf and the BC500 supernatant was pooled with an equal volume of MNase-treated supernatant to acquire the starting chromatin material for chromatin immunoprecipitations (IPs).
For native Flag, H3K27me3, or biotin chromatin IPs, dynabeads protein G (Invitrogen) were pre-blocked with PBS and 0.3% BSA for 1 hr, washed 5x with TE, and used to pre-clear the prepared MNase-treated chromatin for 1 hr at 4 °C. For IP set up, 100 μg of pre-cleared chromatin was used with 10 μg of biotin antibody (Bethyl A150-109A), 4 μg of H3K27me3 (Cell Signaling C36B11), or FLAG-M2 agarose beads (Sigma) and IP was performed overnight with slow rotation at 4 °C. For quantitative analysis of ChIPs, 0.5 μg of Drosophila S2 chromatin was spiked-in to 100 μg of sample (1:200 concentration). The following morning, pre-blocked dynabeads protein G were added to the IPs and incubated for 2-3 hr at 5 μl slurry/1 pg antibody with 0.2 μl of Drosophila-specific H2Av antibody for spike-in control in each sample. IPs were then washed three times with 1 ml BC300 buffer (40 mM Tris, 5 mM MgCl2, 300 mM NaCl and 5% glycerol with protease inhibitors), once with 1 ml BC100 buffer (40 mM Tris, 5 mM MgCl2, 100 NaCl and 5% glycerol with protease inhibitors), and a quick wash with 1 ml TE + 50 mM NaCl. Except for Flag-ChIPs, beads were then resuspended in 125 μl of TE, 3 μl of 10% SDS (TES) and incubated at 65 °C for 1 hr. For Flag-ChIPs, the bound complexes were eluted with 0.25 μg FLAG peptide (Sigma) in 100 μl BC100 buffer overnight with slow rotation at 4 °C. The Flag-ChIPs and TES-treated chromatin were then removed from their respective beads and the input was thawed and resuspended in 125 μl TES. Input and samples were then digested for 2-4 hr with 8 μg of proteinase K at 55 °C while shaking. All samples were PCR column purified, eluted in 50 μl, and diluted 1:4 with water for further qPCR studies. For qPCR quantification, 5 μl SYBR Green I Master mix (Roche), ROX reference dye, 1 μl 5 μM primer pair, 4 μl-diluted DNA were mixed for PCR amplification, and detected by a Stratagene Mx3005p instrument. The data was then quantified and described in the corresponding figure legends. For Drosophila S2 chromatin, primers were FWD: TGGCTAGACTTTTGCGTCCT and REV: TACCAAAAGCCGTCCAAATC and other qPCR primers are listed in Table S4.
For Library preparation of native Flag, H3K27me3, or biotin ChIP-Seq, DNA was eluted in 30 μl elution buffer and libraries were prepared according to the manufacturer’s instructions (Illumina). Briefly, IP’ed DNA (~1-30 ng) was end-repaired using End-It Repair Kit (Epicenter), tailed with deoxyadenine using Klenow exo- (New England Biolabs), and ligated to custom adapters with T4 Rapid DNA Ligase (Enzymatics). Fragments of 200-400 bp were size-selected using Agencourt AMPure XP beads, and subjected to PCR amplification using Q5 DNA polymerase (New England Biolabs). Libraries were quantified by qPCR using primers annealing to the adapter sequence and sequenced on an Illumina HiSeq. Barcodes were utilized for multiplexing.
Fixed chromatin IP experiments were described previously (Gao et al., 2012). Briefly, cells were fixed with 1% Formaldehyde. Nuclei were isolated using buffers in the following order: LB1 (50 mM HEPES, pH 7.5 at 4 °C, 140 mM NaCl, 1 mM EDTA, 10% Glycerol, 0.5% NP40, 0.25% Triton X; 10 min at 4 °C), LB2 (10 mM Tris, pH 8 at 4 °C, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA; 10 min at RT), and LB3 (10 mM Tris, pH 7.5 at 4 °C, 1 mM EDTA, 0.5 mM EGTA, and 0.5% N-Lauroylsarcosine sodium salt). Chromatin was fragmented to an average size of 250 bp using a Diagenode Bioruptor. For fixed chromatin, Cas9 antibody from Millipore (MAC133-clone 7A9) was used and for enrichment of Cas9 at the Gata6, Hoxc6, and Gadph loci, primers sets were as follows, Gata6 FWD: CATGTCTGGAGCGGTTTTGT and REV: CTTCGTGGCGAGTCCTTTTT, Hoxc6 FWD: CTCCAATGCTGAAGTGCAAA and REV: GGCATGTTCAACTGCTGAGA, and Gapdh FWD: GTGGAGAGTTGGGACGTGAG and REV: CTGATCTCACCCTGTGTCCA.
RNA Purification and Quantitative PCR
Total RNA extractions were performed using Roche High pure RNA isolation kit. Superscript III Reverse Transcription Reagents (Invitrogen) and random hexamers were used to prepare cDNAs. For qPCR quantification, 5 μl SYBR Green I Master mix (Roche), ROX reference dye, 1 μl IDT PrimeTime Primer set for corresponding assays, 1 μl cDNA were mixed for PCR amplification, and detected by a Stratagene Mx3005p instrument. For quantitative analysis, Pou5f1, Gata2, and Gata6 expression was normalized to Actb expression and for Ccna2, the absolute value was compared to the untreated Triptolide control. For RNA-seq of triptolide experiments, polyA+ RNA was isolated using Dynabeads Oligo(dT)25 (Invitrogen) and Superscript III Reverse Transcription Reagents (Invitrogen) was used to prepare cDNAs. Libraries were then constructed from cDNAs and prepared according to the manufacturer’s instructions (Illumina) and as described in the ChIP-seq Library prepartion section above.
QUANTIFICATION AND STATISTICAL ANALYSIS
ChIP-seq and RNA-seq data analysis
ChIP-seq were analyzed as previously reported (Stafford et al., 2018) Briefly, the quality of sequencing was assessed with FastQC v0.11.4 (www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads having less than 80% of quality scores above 25 were removed with NGSQC- Toolkit v2.3.3 (Patel et al. 2012), using the command IlluQC.pl -se $file_path N A -l $percent -s $threshold -p $nb_cpu -o $out_path. Sequence reads were then aligned to the mouse mm10 reference genome from Illumina igenomes UCSC collection using Bowtie v1.0.0 (Langmead et al., 2009) allowing 3 mismatches and keeping uniquely aligned reads (bowtie -q -v 3 -p $nb_cpu -m 1 -k 1 -best—sam—seed 1 $bowtie_index_path “$file_path”). Sam ouputs were converted to Bam with Samtools v1.0.6 (Li et al. 2009) (samtools view -S -b $file_path -o $out_path/$file_name.bam) and sorted with Picard tools v1.88 (http://broadinstitute.github.io/picard:Java-jarSortSamjarSO=coordinateI=$input_bamO=$output_bam). Data were further processed with Pasha v0.99.21 (Fenouil et al. 2016) with the following parameters: WIGvs = TRUE, incrArtefactThrEvery = 7000000, elongationSize = NA. Fixed steps wiggle files were converted to bigwigs with the script wigToBigWig available on the UCSC Genome Browser website (http://hgdownload.soe.ucsc.edu/admin/exe/). Spike-in normalization was achieved using Drosophila Melanogaster DNA aligned to Illumina igenomes dm3 (Orlando et al., 2014). Endogenous and exogenous scaling factors were computed from the bam files (1/(number_mapped_reads/1000000)). Endogenous scaling factors were applied to the data before input subtraction (without scaling). The RPM normalization was then reversed before scaling with exogenous factors. The scripts used to perform spike-in scaling have been integrated in a bioconductor package ChIPseqSpike (https://bioconductor.org/packages/3.7/bioc/html/ChIPSeqSpike.html). We used the default settings on UCSC genome browser for visualizing the ChIP-seq tracks. Recruitment and biotinylation significance at the Gata6 locus was determined by a Mann-Whitney test, p<0.0001 comparing normalized tag densities divided into 500 bp bins ± Dox conditions. For RNA-seq analysis, to calculate TPM values, Salmon (Patro et al, 2017) Galaxy version 0.11.2 (Afgan et al, 2018) was used with the reference transcriptome vM20 (GRCm38.p6) from Gencode (Frankish et al, 2019). The tool was run on a paired dataset collection with the “validate mapping” option. All other parameters were left to default. Genes having expression of 0 in all conditions and outliers are removed. For triptolide experiments, significance was determined by a Mann-Whitney-Wilcoxon test (p<10-19).
ChIP-qPCR and statistical significance
Native biotin enrichment levels were normalized to 5% input followed by Drosophila chromatin spike-in levels. For time course experiments, the minus-Dox (-Dox) control was subtracted from the data and error bars represent standard deviation of three biological replicates. GraphPad Prism 7.0 was used for statistical analysis (2way ANOVA or student t-test). A p value ≤ 0.05 was considered statistically significant with * p<0.05, ** p<0.01, *** p<0.001 and **** denotes p<0.0001.
DATA AND CODE AVAILABILITY
The ChIP-seq and RNA-seq data has been deposited in the Gene Expression Omnibus (GEO) under GSE120056 and GSE136245. Source data for gel images can be found in Medeley Data: https://data.mendeley.com/datasets/kkvz8zd767/draft?a=b267dbed-a5ca-40c7-9d95-aacb97fa9c90
Supplementary Material
Figure S1. Endogenous tagging of H3.1 and H3.2 histones with Flag - Biotin Acceptor Peptide (BAP) in KH2 mESCs containing dCas9-DD-BirA. Related to Figure 1. (A) Overview and genotype of KH2 mESC lines containing 13 copies of Flag-BAP knock-ins to H3.1 and H3.2 loci. (B) Western blot analysis for acid extracted histone H3 from cell lines with numerous Flag-BAP-H3 copies. The upper band corresponds to the endogenous number of Flag-BAP-H3 copies in the cell and the red asterisk denotes the cell line used for further studies. (C) Flag IPs and flow-through (FT) of KH2 cells containing Flag-BAP tags and controls. Analysis aacb97fa9c90 highlights the pull-down of H3K4me3 and H3K27me3 chromatin. (D) Flow cytometry analysis showing DNA content of mESCs following double thymidine G1 synchronization. (E) Western blot analysis for Cas9 (top panel) and Tubulin from RIPA extracts of mESCs containing dCas9-DD-BirA treated or not treated with 2 μg/ml doxycycline (Dox) and 1 μM, 5 μM, or 10 μM of MG-132. Dox was added to the cultures for 6 hr (lanes 2-5 and 7) or 12 hr for lane 6. Lane 7 represents transient overexpression (ox) of dCas9-DD-BirA in the 293TREX system. 25 μg of total RIPA extracts was loaded, except for lane 7 having 2.5 μg loaded.
Table S3. Repressed and Active 5kb target gRNAs primers, Related to STAR Methods. List of primer sets for the construction of gRNAs cloned into the pKLV-U6gRNA-EF(BbsI)-PGKpuro2ABFP plasmid for active and repressed candidate loci.
Table S4. Repressed and Active loci qPCR primers, Related to STAR Methods. List of primer sets for ChIP-qPCR analysis.
Figure S2. Inducibility of dCas9-DD-BirA during cell division in mESCs. Related to Figure 1. (A-B) Cas9 ChIP-qPCR analysis of G1/S-blocked mESCs that were released following a 6 hr pulse with minimal doxycycline (Dox) and exogenous biotin in cells, targeting dCas9-DD-BirA to the Gata6 (A) and Hoxc6 (B) loci, compared to Gapdh controls. Data are relative to input, normalized to Drosophila chromatin spike-in, and compared to -Dox control, which was set to 1. Error bars represent standard error of three biological replicates. (C) ChIP-seq analysis of G1/S-blocked cells at the HoxC cluster following dCas9-DD-BirA recruitment. Native Flag (top panel) and biotin (bottom two panels). (D) Native ChIP-qPCR analysis of biotin-H3 in G1/S-blocked mESCs showing biotin enrichment at the Hoxc6 locus compared to Ebf1, Meis2, Ccna2, Gapdh and IgG controls. (E) ChIP-seq analysis of cells at the Ebf1 locus following dCas9-DD-BirA recruitment. Native Flag (top panel) and biotin (bottom two panels). (F) Native ChIP-qPCR analysis of biotin-H3 in G1/S-blocked mESCs, validating biotin enrichment at the Ebf1 locus compared to Hoxc6, Meis2, Ccna2, Gapdh and IgG controls. For ChIP-seq tracks, the data were normalized using Drosophila chromatin spike-in and the tracks show a scaling factor processed with the bioconductor package ChIPSeqSpike (see Methods). For ChIP-qPCR, the data were normalized to 5% input and error bars represent standard error of three biological replicates and Student’s t-test is used to determine statistical significance with * p<0.05, ** p<0.01.
Figure S3. Candidate repressed chromatin domains in mESCs. Related to Figure 2. (A) Cell-cycle analysis corresponding to G1/S-blocked and released mESCs at time 0 hr -1 cell, 12 hr -2 cell, 24 hr - 4 cells, and 48 hr - 16 cells. (B-D) Native Flag (top panel) and H3K27me3 ChIP-seq analysis of G1/S-blocked cells at the Hoxc cluster (B), Ebf1 (C), and Meis2 (D) candidate loci. The data were normalized with Drosophila chromatin spike-in and the tracks show a scaling factor processed with the bioconductor package ChIPSeqSpike (see STAR methods).
Figure S4. Biotin-H3 segregation in Ccna2 active chromatin is dynamic during S-phase progression. Related to Figure 3. (A) Cell cycle analysis corresponding to G1/S-blocked and released mESCs at time 0 hr - 1 cell, 6 hr -1 cell (late S-phase), and 12 hr - 2 cells. (B) Native biotin ChIP-qPCR from G1/S-blocked mESCs that were released following a 6 hr pulse of minimal Dox and exogenous biotin, with BirA targeted to the Ccna2 locus. Cells were released into S-phase with or without 1 μM triptolide for the following times: 0 hr -1 cell, 6 hr - Late S phase, and 12 hr - 2 cells. Data is the average of two biological replicates spanning a 25 kb area at a resolution of 500 bp. All biotin enrichment levels are relative to input and normalized to Drosophila chromatin spike-in followed by subtraction of the -Dox control. Error bars represent standard error of two biological replicates. (C) Box plots showing global expression analysis in cells that were released into S-phase ±1 μM triptolide (Trp) at 3 hr or 6 hr. Mann-Whitney-Wilcoxon test was performed and *** p<10-19. (D) Ccna2 and Actb mRNA expression following treatment with 1 μM triptolide for cells released into S-phase at 0 hr, 3 hr, and 6 hr. Data is absolute quantification in which untreated expression was set to 1.0 and error bars represent standard deviation of three biological replicates with statistical significance determined using 2way ANOVA (**** p<0.0001). (E) Western blot analysis for Ccna2 or H3 from RIPA extracted mESCs lysates in which cells were treated with or without 1 μM Trp when released into S-phase for 6 hr.
Figure S5. Aphidicolin treatment sustains mESCs in S-phase and does not interfere with gene expression. Related to Figure 4. (A) Cell-cycle analysis corresponding to G1/S-blocked and released ESCs treated with 1 μg/ml of aphidicolin for 12 hr or 24 hr. (B) Pou5f1 mRNA expression from mESCs treated with or without 1 μg/ml aphidicolin for 12 hr or 24 hr. Data was normalized to Actb and compared to 0 hr with error bars representing standard deviation of three biological replicates.
Figure S6. Differing H3 nucleosome segregation of Gata6 in a repressed and active chromatin state. Related to Figure 5. (A and B) Native ChIP-seq for biotin and IgG in mESCs under −RA (A) or +RA (B) conditions. Targeting BirA to the Gata6 locus, cells were blocked in G1/S and then released following a 6 hr pulse of Dox and exogenous biotin. The following times correspond to cells at G1/S (0 hr -1 cell) or cells released into S phase (12 hr - 2 cells). Recruitment significance for (A) and (B) was determined by a Mann-Whitney test (p<0.0001). The data were normalized with Drosophila chromatin spike-in and the tracks show a scaling factor processed with the bioconductor package ChIPSeqSpike (see STAR methods).
Table S1. Recombinant DNA cloning primers, Related to STAR Methods. List of primer sets for (1) the construction of dCa9-DD-BirA into the pINTA plasmid and (2) subcloning of BFP into pX458-SpCas9-2A-GFP, replacing GFP.
Table S2. CRISPR-Cas9 targeting of Hist1h3 cluster, Related to STAR Methods. Primer sets for (1) construction of gRNAs cloned into pX458-SpCas9-2A-BFP (2) gBlock primers for the knockin of Flag-BAP and (3) genotyping of CRISPR knockin clones.
Acknowledgments:
We thank Drs. S. Krishnan, E. Campos, S. Tu, and A. Loyola for thoughtful discussions. We thank Dr. L. Vales for discussion and revision of the manuscript, D. Hernandez for excellent technical assistance, and Dr. Laura Taylor for technical assistance in imaging of immunofluorescence experiments. We also are grateful to Brian J. Abraham and Ann Boija for ChIP-seq discussions. We are also appreciative for the expertise offered at the following facilities at New York University Langone Health: the Genome Technology Center for help with sequencing and the Cytometry and Cell Sorting core facility for cell sorting/flow cytometry technologies.
D.R. dedicates this study to the memory of his mentor, Jerry Hurwitz, a legendary biochemist.
Funding: This work in the D.R. Lab was supported by the Howard Hughes Medical Institute and the National Cancer Institute (NCI NIH 9R01CA199652-13A1). T.M.E was supported by NCI NIH 3R01CA199652-14S1. The NYU Langone’s Cytometry and Cell Sorting Laboratory is supported in part by grant P30CA016087 from the National Institutes of Health/National Cancer Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: Danny Reinberg is a cofounder of Constellation Biotechnology and Fulcrum Biotechnology
References
- Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Cech M, Chilton J, Clements D, Coraor N, Gruning BA, et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res 46, W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabert C, Bukowski-Wills JC, Lee SB, Kustatscher G, Nakamura K, de Lima Alves F, Menard P, Mejlvang J, Rappsilber J, and Groth A (2014). Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nature cell biology 16, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabert C, and Groth A (2012). Chromatin replication and epigenome maintenance. Nat Rev Mol Cell Biol 13, 153–167. [DOI] [PubMed] [Google Scholar]
- Allshire RC, and Madhani HD (2018). Ten principles of heterochromatin formation and function. Nat Rev Mol Cell Biol 19, 229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida M, Pintacuda G, Masui O, Koseki Y, Gdula M, Cerase A, Brown D, Mould A, Innocent C, Nakayama M, et al. (2017). PCGF3/5-PRC1 initiates Polycomb recruitment in X chromosome inactivation. Science 356, 1081–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziato AT (2013). Assembling chromatin: the long and winding road. In Biochim Biophys Acta, pp. 196–210. [DOI] [PubMed] [Google Scholar]
- Banaszynski LA, Chen LC, Maynard-Smith LA, Ooi AG, and Wandless TJ (2006). A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 126, 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard C, Hochedlinger K, Plath K, Wutz A, and Jaenisch R (2006). Efficient method to generate single-copy transgenic mice by site-specific integration in embryonic stem cells. Genesis 44, 23–28. [DOI] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. (2006). Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441, 349–353. [DOI] [PubMed] [Google Scholar]
- Campos EI, and Reinberg D (2009). Histones: annotating chromatin. Annual review of genetics 43, 559–599. [DOI] [PubMed] [Google Scholar]
- Campos EI, Stafford JM, and Reinberg D (2014). Epigenetic inheritance: histone bookmarks across generations. Trends in cell biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, et al. (2013). Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155, 1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement C, Orsi GA, Gatto A, Boyarchuk E, Forest A, Hajj B, Mine-Hattab J, Garnier M, Gurard-Levin ZA, Quivy JP, et al. (2018). High-resolution visualization of H3 variants during replication reveals their controlled recycling. Nat Commun 9, 3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper S, Grijzenhout A, Underwood E, Ancelin K, Zhang T, Nesterova TB, Anil-Kirmizitas B, Bassett A, Kooistra SM, Agger K, et al. (2016). Jarid2 binds monoubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat Commun 7, 13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusick ME, Herman TM, DePamphilis ML, and Wassarman PM (1981). Structure of chromatin at deoxyribonucleic acid replication forks: prenucleosomal deoxyribonucleic acid is rapidly excised from replicating simian virus 40 chromosomes by micrococcal nuclease. Biochemistry 20, 6648–6658. [DOI] [PubMed] [Google Scholar]
- Deal RB, Henikoff JG, and Henikoff S (2010). Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science 328, 1161–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon SC, and Dorman CJ (2010). Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat Rev Microbiol 8, 185–195. [DOI] [PubMed] [Google Scholar]
- Dowen JM, Fan ZP, Hnisz D, Ren G, Abraham BJ, Zhang LN, Weintraub AS, Schujiers J, Lee TI, Zhao Kv et al. (2014). Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 159, 374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenouil R, Descostes N, Spinelli L, Koch F, Maqbool MA, Benoukraf T, Cauchy P, Innocenti C, Ferrier P, and Andrau JC (2016). Pasha: a versatile R package for piling chromatin HTS data. Bioinformatics 32, 2528–2530. [DOI] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 47, D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Zhang J, Bonasio R, Strino F, Sawai A, Parisi F, Kluger Y, and Reinberg D (2012). PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol Cell 45, 344–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SI, and Moazed D (2003). Heterochromatin and epigenetic control of gene expression. Science 301, 798–802. [DOI] [PubMed] [Google Scholar]
- Herman TM, DePamphilis ML, and Wassarman PM (1981). Structure of chromatin at deoxyribonucleic acid replication forks: location of the first nucleosomes on newly synthesized simian virus 40 deoxyribonucleic acid. Biochemistry 20, 621–630. [DOI] [PubMed] [Google Scholar]
- Jeppesen P (2000). Immunofluorescence in cytogenetic analysis: method and applications. Genet Mol Biol 23, 1107–1114. [Google Scholar]
- Johnson LM, Kayne PS, Kahn ES, and Grunstein M (1990). Genetic evidence for an interaction between SIR3 and histone H4 in the repression of the silent mating loci in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 87, 6286–6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns NA, Genga RM, Enuameh MS, Garber M, Wolfe SA, and Maehr R (2014). Cas9 effector-mediated regulation of transcription and differentiation in human pluripotent stem cells. Development 141, 219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraushaar DC, Jin W, Maunakea A, Abraham B, Ha M, and Zhao K (2013). Genome-wide incorporation dynamics reveal distinct categories of turnover for the histone variant H3.3. Genome Biol 14, R121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulyyassov A, Shoaib M, Pichugin A, Kannouche P, Ramanculov E, Lipinski M, and Ogryzko V (2011). PUB-MS: a mass spectrometry-based method to monitor protein-protein proximity in vivo. Journal of proteome research 10, 4416–4427. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentjes MH, Niessen HE, Akiyama Y, de Bruine AP, Melotte V, and van Engeland M (2016). The emerging role of GATA transcription factors in development and disease. Expert Rev Mol Med 18, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Dechassa ML, and Tremethick DJ (2012). New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat Rev Mol Cell Biol 13, 436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ 3rd, Voigt P, Martin SR, Taylor WR, De Marco V, et al. (2009). Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461, 762–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, and Reinberg D (2011). The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, and Surani MA (2011). The transcriptional and signalling networks of pluripotency. Nature cell biology 13, 490–496. [DOI] [PubMed] [Google Scholar]
- Ochiai H, Sugawara T, and Yamamoto T (2015). Simultaneous live imaging of the transcription and nuclear position of specific genes. Nucleic Acids Res 43, e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksuz O, Narendra V, Lee CH, Descostes N, LeRoy G, Raviram R, Blumenberg L, Karch K, Rocha PP, Garcia BA, et al. (2018). Capturing the onset of PRC2-mediated repressive domain formation. Mol Cell 70, 1149–1162 e1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, and Guenther MG (2014). Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep 9, 1163–1170. [DOI] [PubMed] [Google Scholar]
- Patel RK, and Jain M (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7, e30619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patikoglou G, and Burley SK (1997). Eukaryotic transcription factor-DNA complexes. Annu Rev Biophys Biomol Struct 26, 289–325. [DOI] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pengelly AR, Copur O, Jackle H, Herzig A, and Muller J (2013). A histone mutant reproduces the phenotype caused by loss of histone-modifying factor Polycomb. Science 339, 698–699. [DOI] [PubMed] [Google Scholar]
- Petryk N, Dalby M, Wenger A, Stromme CB, Strandsby A, Andersson R, and Groth A (2018). MCM2 promotes symmetric inheritance of modified histones during DNA replication. Science 361, 1389–1392. [DOI] [PubMed] [Google Scholar]
- Ptashne M, and Gann A (1997). Transcriptional activation by recruitment. Nature 386, 569–577. [DOI] [PubMed] [Google Scholar]
- Ptashne M, and Gann A (2002). Genes & signals (Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; ). [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nature protocols 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinberg D, and Vales LD (2018). Chromatin domains rich in inheritance. Science 361, 33–34. [DOI] [PubMed] [Google Scholar]
- Reveron-Gomez N, Gonzalez-Aguilera C, Stewart-Morgan KR, Petryk N, Flury V, Graziano S, Johansen JV, Jakobsen JS, Alabert C, and Groth A (2018). Accurate recycling of parental histones reproduces the histone modification landscape during DNA replication. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N, and Gilbert DM (2013). DNA replication timing. Cold Spring Harb Perspect Biol 5, a010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoaib M, Kulyyassov A, Robin C, Winczura K, Tarlykov P, Despas E, Kannouche P, Ramanculov E, Lipinski M, and Ogryzko V (2013). PUB-NChIP--”in vivo biotinylation” approach to study chromatin in proximity to a protein of interest. Genome research 23, 331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, and Peterson CL (2006). Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847. [DOI] [PubMed] [Google Scholar]
- Simon JA, and Kingston RE (2013). Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol Cell 49, 808–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ 3rd, Belotserkovskaya R, and Reinberg D (2004). Elongation by RNA polymerase II: the short and long of it. Genes Dev 18, 2437–2468. [DOI] [PubMed] [Google Scholar]
- Sladitschek HL, and Neveu PA (2015). MXS-Chaining: A highly efficient cloning platform for imaging and flow cytometry approaches in mammalian systems. PLoS One 10, e0124958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford JM, Lee CH, Voigt P, Descostes N, Saldana-Meyer R, Yu JR, Leroy G, Oksuz O, Chapman JR, Suarez F, et al. (2018). Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci Adv 4, eaau5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stillman B (2018). Histone Modifications: Insights into Their Influence on Gene Expression. Cell 175, 6–9. [DOI] [PubMed] [Google Scholar]
- Tagami H, Ray-Gallet D, Almouzni G, and Nakatani Y (2004). Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 116, 51–61. [DOI] [PubMed] [Google Scholar]
- Takahashi K, and Yamanaka S (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- Xu M, Long C, Chen X, Huang C, Chen S, and Zhu B (2010). Partitioning of histone H3-H4 tetramers during DNA replication-dependent chromatin assembly. Science 328, 94–98. [DOI] [PubMed] [Google Scholar]
- Yu C, Gan H, Serra-Cardona A, Zhang L, Gan S, Sharma S, Johansson E, Chabes A, Xu RM, and Zhang Z (2018). A mechanism for preventing asymmetric histone segregation onto replicating DNA strands. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zee BM, Britton LM, Wolle D, Haberman DM, and Garcia BA (2012). Origins and formation of histone methylation across the human cell cycle. Molecular and cellular biology 32, 2503–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Gao Y, Yu M, Wu H, Ai Z, Wu Y, Liu H, Du J, Guo Z, and Zhang Y (2015). Retinoic acid induces embryonic stem cell differentiation by altering both encoding RNA and microRNA expression. PLoS One 10, e0132566. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Endogenous tagging of H3.1 and H3.2 histones with Flag - Biotin Acceptor Peptide (BAP) in KH2 mESCs containing dCas9-DD-BirA. Related to Figure 1. (A) Overview and genotype of KH2 mESC lines containing 13 copies of Flag-BAP knock-ins to H3.1 and H3.2 loci. (B) Western blot analysis for acid extracted histone H3 from cell lines with numerous Flag-BAP-H3 copies. The upper band corresponds to the endogenous number of Flag-BAP-H3 copies in the cell and the red asterisk denotes the cell line used for further studies. (C) Flag IPs and flow-through (FT) of KH2 cells containing Flag-BAP tags and controls. Analysis aacb97fa9c90 highlights the pull-down of H3K4me3 and H3K27me3 chromatin. (D) Flow cytometry analysis showing DNA content of mESCs following double thymidine G1 synchronization. (E) Western blot analysis for Cas9 (top panel) and Tubulin from RIPA extracts of mESCs containing dCas9-DD-BirA treated or not treated with 2 μg/ml doxycycline (Dox) and 1 μM, 5 μM, or 10 μM of MG-132. Dox was added to the cultures for 6 hr (lanes 2-5 and 7) or 12 hr for lane 6. Lane 7 represents transient overexpression (ox) of dCas9-DD-BirA in the 293TREX system. 25 μg of total RIPA extracts was loaded, except for lane 7 having 2.5 μg loaded.
Table S3. Repressed and Active 5kb target gRNAs primers, Related to STAR Methods. List of primer sets for the construction of gRNAs cloned into the pKLV-U6gRNA-EF(BbsI)-PGKpuro2ABFP plasmid for active and repressed candidate loci.
Table S4. Repressed and Active loci qPCR primers, Related to STAR Methods. List of primer sets for ChIP-qPCR analysis.
Figure S2. Inducibility of dCas9-DD-BirA during cell division in mESCs. Related to Figure 1. (A-B) Cas9 ChIP-qPCR analysis of G1/S-blocked mESCs that were released following a 6 hr pulse with minimal doxycycline (Dox) and exogenous biotin in cells, targeting dCas9-DD-BirA to the Gata6 (A) and Hoxc6 (B) loci, compared to Gapdh controls. Data are relative to input, normalized to Drosophila chromatin spike-in, and compared to -Dox control, which was set to 1. Error bars represent standard error of three biological replicates. (C) ChIP-seq analysis of G1/S-blocked cells at the HoxC cluster following dCas9-DD-BirA recruitment. Native Flag (top panel) and biotin (bottom two panels). (D) Native ChIP-qPCR analysis of biotin-H3 in G1/S-blocked mESCs showing biotin enrichment at the Hoxc6 locus compared to Ebf1, Meis2, Ccna2, Gapdh and IgG controls. (E) ChIP-seq analysis of cells at the Ebf1 locus following dCas9-DD-BirA recruitment. Native Flag (top panel) and biotin (bottom two panels). (F) Native ChIP-qPCR analysis of biotin-H3 in G1/S-blocked mESCs, validating biotin enrichment at the Ebf1 locus compared to Hoxc6, Meis2, Ccna2, Gapdh and IgG controls. For ChIP-seq tracks, the data were normalized using Drosophila chromatin spike-in and the tracks show a scaling factor processed with the bioconductor package ChIPSeqSpike (see Methods). For ChIP-qPCR, the data were normalized to 5% input and error bars represent standard error of three biological replicates and Student’s t-test is used to determine statistical significance with * p<0.05, ** p<0.01.
Figure S3. Candidate repressed chromatin domains in mESCs. Related to Figure 2. (A) Cell-cycle analysis corresponding to G1/S-blocked and released mESCs at time 0 hr -1 cell, 12 hr -2 cell, 24 hr - 4 cells, and 48 hr - 16 cells. (B-D) Native Flag (top panel) and H3K27me3 ChIP-seq analysis of G1/S-blocked cells at the Hoxc cluster (B), Ebf1 (C), and Meis2 (D) candidate loci. The data were normalized with Drosophila chromatin spike-in and the tracks show a scaling factor processed with the bioconductor package ChIPSeqSpike (see STAR methods).
Figure S4. Biotin-H3 segregation in Ccna2 active chromatin is dynamic during S-phase progression. Related to Figure 3. (A) Cell cycle analysis corresponding to G1/S-blocked and released mESCs at time 0 hr - 1 cell, 6 hr -1 cell (late S-phase), and 12 hr - 2 cells. (B) Native biotin ChIP-qPCR from G1/S-blocked mESCs that were released following a 6 hr pulse of minimal Dox and exogenous biotin, with BirA targeted to the Ccna2 locus. Cells were released into S-phase with or without 1 μM triptolide for the following times: 0 hr -1 cell, 6 hr - Late S phase, and 12 hr - 2 cells. Data is the average of two biological replicates spanning a 25 kb area at a resolution of 500 bp. All biotin enrichment levels are relative to input and normalized to Drosophila chromatin spike-in followed by subtraction of the -Dox control. Error bars represent standard error of two biological replicates. (C) Box plots showing global expression analysis in cells that were released into S-phase ±1 μM triptolide (Trp) at 3 hr or 6 hr. Mann-Whitney-Wilcoxon test was performed and *** p<10-19. (D) Ccna2 and Actb mRNA expression following treatment with 1 μM triptolide for cells released into S-phase at 0 hr, 3 hr, and 6 hr. Data is absolute quantification in which untreated expression was set to 1.0 and error bars represent standard deviation of three biological replicates with statistical significance determined using 2way ANOVA (**** p<0.0001). (E) Western blot analysis for Ccna2 or H3 from RIPA extracted mESCs lysates in which cells were treated with or without 1 μM Trp when released into S-phase for 6 hr.
Figure S5. Aphidicolin treatment sustains mESCs in S-phase and does not interfere with gene expression. Related to Figure 4. (A) Cell-cycle analysis corresponding to G1/S-blocked and released ESCs treated with 1 μg/ml of aphidicolin for 12 hr or 24 hr. (B) Pou5f1 mRNA expression from mESCs treated with or without 1 μg/ml aphidicolin for 12 hr or 24 hr. Data was normalized to Actb and compared to 0 hr with error bars representing standard deviation of three biological replicates.
Figure S6. Differing H3 nucleosome segregation of Gata6 in a repressed and active chromatin state. Related to Figure 5. (A and B) Native ChIP-seq for biotin and IgG in mESCs under −RA (A) or +RA (B) conditions. Targeting BirA to the Gata6 locus, cells were blocked in G1/S and then released following a 6 hr pulse of Dox and exogenous biotin. The following times correspond to cells at G1/S (0 hr -1 cell) or cells released into S phase (12 hr - 2 cells). Recruitment significance for (A) and (B) was determined by a Mann-Whitney test (p<0.0001). The data were normalized with Drosophila chromatin spike-in and the tracks show a scaling factor processed with the bioconductor package ChIPSeqSpike (see STAR methods).
Table S1. Recombinant DNA cloning primers, Related to STAR Methods. List of primer sets for (1) the construction of dCa9-DD-BirA into the pINTA plasmid and (2) subcloning of BFP into pX458-SpCas9-2A-GFP, replacing GFP.
Table S2. CRISPR-Cas9 targeting of Hist1h3 cluster, Related to STAR Methods. Primer sets for (1) construction of gRNAs cloned into pX458-SpCas9-2A-BFP (2) gBlock primers for the knockin of Flag-BAP and (3) genotyping of CRISPR knockin clones.
Data Availability Statement
The ChIP-seq and RNA-seq data has been deposited in the Gene Expression Omnibus (GEO) under GSE120056 and GSE136245. Source data for gel images can be found in Medeley Data: https://data.mendeley.com/datasets/kkvz8zd767/draft?a=b267dbed-a5ca-40c7-9d95-aacb97fa9c90