

SUMMARY
Sequences within 5′ UTRs dictate the site and efficiency of translation initiation. In this study, an unbiased screen designed to interrogate the 5′ UTR-mediated regulation of the growth-promoting gene MYC unexpectedly revealed the ribosomal pause relief factor eIF5A as a regulator of translation initiation codon selection. Depletion of eIF5A enhances upstream translation within 5′ UTRs across yeast and human transcriptomes, including on the MYC transcript, where this results in increased production of an N-terminally extended protein. Furthermore, ribosome profiling experiments established that the function of eIF5A as a suppressor of ribosomal pausing at sites of suboptimal peptide bond formation is conserved in human cells. We present evidence that proximal ribosomal pausing on a transcript triggers enhanced use of upstream suboptimal or non-canonical initiation codons. Thus, we propose that eIF5A functions not only to maintain efficient translation elongation in eukaryotic cells but also to maintain the fidelity of translation initiation.
In Brief
Translation initiation codon selection is a highly regulated process that can influence the sequence and activity of a translated protein. Here, Manjunath et al. demonstrate that loss of the translation elongation factor eIF5A triggers widespread translation initiation in 5′ UTRs, uncovering a role for this protein in start codon selection.
Graphical Abstract
INTRODUCTION
MYC encodes a transcription factor that, through the regulation of its target genes, promotes cell growth and malignant transformation. Overexpression or amplification of this gene and the consequent aberrant activation of the MYC transcriptional program are oncogenic events that occur frequently in diverse types of cancer (Gabay et al., 2014). Under normal physiologic conditions, MYC expression and activity are tightly regulated at all levels of gene expression, from the controlled induction of its transcription (Kelly et al., 1983) to the continuous degradation of MYC protein (Farrell and Sears, 2014; Sears, 2004). Accordingly, robust post-transcriptional regulation governs the stability and translation of the MYC mRNA, the cues for which are encoded within the well-conserved untranslated regions (UTRs) of the transcript (Dani et al., 1984; Fraser et al., 1996; Parkin et al., 1988; Yeilding et al., 1996).
In eukaryotes, initiation of translation is a highly regulated process that begins with recruitment of the pre-initiation complex (PIC), comprising the 40S small ribosomal subunit, initiator tRNA, and several eukaryotic initiation factors (eIFs), to the 5′ cap. The PIC then scans the transcript in a 5′ to 3′ direction for a codon in the optimal context for initiation (Jackson et al., 2010; Merrick and Pavitt, 2018). As the PIC traverses the 5′ UTR, this region of the transcript can potently affect the location and rate of translation initiation (Hinnebusch et al., 2016). For instance, many 5′ UTRs harbor non-canonical initiation codons that are engaged at lower frequencies or only under specific contexts, whose use can produce a new protein or have effects on translation of the downstream canonical open reading frame (ORF) (Kearse and Wilusz, 2017; Tang et al., 2017; Touriol et al., 2003). The MYC 5′ UTR, for example, contains a suboptimal CUG initiation codon upstream of the canonical AUG, which, when engaged, leads to the production of an N-terminally extended isoform of the protein (Hann et al., 1988). Such alternatively initiated proteins often have properties and functions that are distinct from their canonically initiated counterparts and add diversity to the repertoire of proteins within a cell (Tang et al., 2004). It has been suggested that the MYC N-terminal extension influences the preferred transcriptional targets of the protein (Benassayag et al., 2005; Hann et al., 1994; Sato et al., 2019). A similar configuration exists on other notable mRNAs, such as PTEN, in which the use of upstream non-canonical initiation codons produces proteins that adopt different functions, including regulating energy metabolism in mitochondria and influencing PI3K signaling as a secreted protein (Hopkins et al., 2013; Liang et al., 2017, 2014; Tzani et al., 2016).
The mechanisms that regulate alternative start codon selection are not fully understood. The classic model of translation initiation proposes that a scanning PIC will pause at an initiator codon in an appropriate sequence context, triggering a series of events that culminate in large subunit joining and formation of an elongation-competent ribosome (Hinnebusch, 2011). Stabilization of the PIC at a start codon can be influenced by several factors, including the initiator tRNA, initiation factors, and/or RNA elements within the 5′ UTR. Modulation of any of these determinants of translation initiation can alter start codon choice and either promote, or impede, the use of suboptimal start codons (Asano, 2014; Kearse and Wilusz, 2017; Tang et al., 2017). For example, an inhibitor of the translation initiation factor eIF5, known as eIF5-mimic protein 1 (5MP1; encoded by the BZW2 gene), is overexpressed in colorectal cancer and promotes the use of the MYC AUG initiation codon, an event that has been proposed to enhance tumorigenesis in this setting (Sato et al., 2019).
eIF5A (not to be confused with the unrelated protein eIF5 mentioned above) is a highly conserved translation factor whose molecular function has been studied extensively in yeast. Despite being initially classified as an initiation factor, it is now understood to function primarily during translation elongation as a ribosomal pause relief factor, facilitating peptide bond formation at sites at which the peptidyl transfer reaction occurs inefficiently (Dever and Ivanov, 2018; Gregio et al., 2009; Kemper et al., 1976; Pelechano and Alepuz, 2017; Saini et al., 2009; Schuller et al., 2017). eIF5A is the eukaryotic homolog of prokaryotic elongation factor P (EF-P) (Aoki et al., 1997), which also functions to suppress ribosomal pausing. For example, proline-proline (PP) peptide bonds are dependent on eIF5A (or EF-P in bacteria) for their efficient synthesis because this amino acid is a poor substrate for peptide bond formation due to its sterically constrained cyclic ring side chain (Doerfel et al., 2013; Gutierrez et al., 2013; Shin et al., 2017; Ude et al., 2013). A distinctive property of eIF5A is that it is the only known protein to contain the amino acid hypusine, a basic amino acid that is derived from the polyamine biosynthesis pathway and that is post-translationally incorporated into the protein by modification of a lysine residue (Cooper et al., 1983; Dever et al., 2014). Despite detailed characterization of the role of eIF5A in translation in yeast and EF-P in prokaryotes, the role of mammalian eIF5A in translation is not well understood, in part because of the technical difficulties of depleting this essential protein (depmap.org; Meyers et al., 2017; Park et al., 1998; Schnier et al., 1991).
In this study, we initially set out to genetically dissect mechanisms of post-transcriptional regulation of the MYC 5′ UTR, given the prominent role of this protein in tumorigenesis and its tight regulation under physiologic conditions. Through a fluorescent reporter-coupled genome-scale CRISPR/Cas9-mediated loss-of-function screen, we unexpectedly uncovered a role for eIF5A as a regulator of MYC translation initiation codon selection. Loss of eIF5A resulted in enhanced use of the upstream MYC CUG start codon. Through ribosome profiling experiments in cells depleted of eIF5A, we demonstrated that this increase in the use of upstream alternative start codons was not unique to MYC and occurred broadly throughout the human and yeast transcriptomes. Moreover, we established that the function of eIF5A as a ribosomal pause relief factor is conserved in human cells. Indeed, our findings suggest that ribosomal pausing immediately downstream of alternative initiation sites, an event promoted by depletion of eIF5A, triggers ribosome queuing and, consequently, increased use of upstream start codons on MYC and other transcripts. Thus, we propose that eIF5A is a translation elongation factor that also performs an essential function in maintaining appropriate start codon selection.
RESULTS
A Genome-Scale Loss-of-Function Screen Identifies eIF5A as a Regulator of MYC Translation Initiation Codon Selection
In order to study post-transcriptional regulation conferred by the MYC 5′ UTR, we generated reporter constructs in which a constitutive promoter drives expression of an EGFP-encoding transcript that either contained or lacked the MYC 5′ UTR (Figure 1A). These constructs were integrated into a defined locus (AAVS1/PPP1R12C) at single copy in the stably diploid human colon cancer cell line HCT116 to generate clonal MYC 5′ UTR and EGFP control reporter cell lines. Consistent with literature indicating that the MYC UTRs harbor repressive regulatory cues (Fraser et al., 1996; Parkin et al., 1988), cells expressing the 5′ UTR reporter construct were approximately 5 times dimmer than EGFP control reporter cells, as indicated by flow cytometry (Figure 1B). We therefore designed a genome-scale CRISPR/Cas9-mediated loss-of-function screen to identify the genes that mediate repression of the 5′ UTR reporter (Figure 1A).
Figure 1. A Genome-Scale Loss-of-Function Screen Identifies eIF5A as a Regulator of MYC Translation Initiation Codon Selection.

(A) Schematic of reporter constructs and CRISPR/Cas9 screening strategy.
(B) Flow cytometry analysis of EGFP fluorescence of reporter cell lines.
(C) Representative western blot analysis of HCT116 cells following lentiviral delivery of Cas9 and the indicated sgRNAs. In this and all subsequent western blots, α-TUB is included as a loading control. NT, non-targeting control sgRNA.
(D) Quantification of MYC1 protein relative to TUB (upper) or MYC1 relative to MYC2 (lower) from western blots after delivery of the indicated sgRNAs (n = 3 biological replicates, including blots shown in C).
(E) Schematic of the human MYC transcript.
(F) Western blot analysis of HCT116 cells following lentiviral delivery of Cas9 and the indicated sgRNAs. Hyp-eIF5A, hypusine-modified eIF5A.
(G) Quantification of MYC1 protein relative to TUB (upper) or MYC1 relative to MYC2 (lower) from western blots after delivery of the indicated sgRNAs (n = 3 biological replicates, including blots shown in F).
Error bars represent SD. *p < 0.05 and ***p < 0.001, calculated using two-tailed t test. See also Figures S1 and S2.
CRISPR/Cas9 screening was carried out in both MYC 5′ UTR and EGFP control reporter cell lines as previously described (Golden et al., 2017). Briefly, reporter cells were transduced with a lentiviral library at a low MOI to deliver Cas9 and single guide RNAs (sgRNAs) targeting more than 19,000 human genes, resulting in the generation of a pool of single gene knockouts. Fluorescence-activated cell sorting (FACS) was then used to isolate the brightest cells, thereby enriching for those with impaired repression of the MYC 5′ UTR. Finally, next-generation sequencing was used to detect sgRNAs that were enriched in the sorted versus the unsorted populations, leading to the identification of genes whose loss of function increased fluorescence in the MYC 5′ UTR reporter, but not the EGFP control reporter, cell line.
Analysis of the screening data revealed that chromatin modifiers (ASH2L, BRD4) and general transcription factors and coactivators (TAF1, TAF5, TAF7, CREBBP) that would be expected to regulate transcription of the reporter constructs were recovered as significant hits in both MYC 5′ UTR and EGFP control reporter screens (Figure S1A). On the basis of a stringent statistical cutoff, four genes were identified as 5′ UTR-specific hits: translation factors EIF5A/EIF5AL1 (enriched sgRNAs targeted both genes that encode nearly identical proteins) and EIF3L, the intron lariat debranching enzyme DBR1, and miR-889. Validation of screening results, performed by lentiviral delivery of Cas9 and individual sgRNAs, confirmed that EIF5A/EIF5AL1 and EIF3L loss of function increased fluorescence of the MYC 5′ UTR reporter cells compared with the EGFP control cell line (Figure S1B), while loss of DBR1 and miR-889 did not (data not shown). These data indicated that these translation factors were putative regulators of the MYC 5′ UTR and prompted us to investigate their effects on endogenous MYC.
Despite increasing EGFP fluorescence in a MYC 5′ UTR-specific manner, sgRNAs targeting EIF3L did not alter steady-state MYC protein levels in HCT116 cells (Figure S1C). It is possible that an effect on endogenous MYC was masked by the multiple redundant mechanisms that regulate MYC abundance, and therefore examination of the EIF3L-MYC interaction in other cellular contexts is warranted. Nevertheless, here we chose to focus on the top hit from the screen, EIF5A/EIF5AL1. Notably, significantly enriched sgRNAs in the screen targeted both EIF5A and its processed pseudogene EIF5AL1, which encodes a nearly identical protein. Importantly, EIF5AL1 was not expressed in HCT116 cells (Figure S1D), and sgRNAs that selectively target EIF5A were sufficient to deplete the protein, while those selectively targeting EIF5AL1 had no effect on protein levels (Figure S1E). Thus, EIF5A is the relevant eIF5A-encoding gene in this cell line.
Although eIF5A is an essential protein in eukaryotic cells (depmap.org; Meyers et al., 2017; Park et al., 1998; Schnier et al., 1991), lentiviral delivery of CRISPR components produced cell populations with nearly complete loss of detectable eIF5A protein (Figure 1C). This resulted in a surprising effect on endogenous MYC protein in multiple cell lines, specifically, enhanced production of a larger protein isoform, termed MYC1, initiated from a non-canonical CUG initiation codon in the MYC 5′ UTR (Figures 1C–1E and S1F). We confirmed that the more slowly migrating MYC isoform is indeed the N-terminally extended MYC1 protein by introducing a sgRNA that targeted the MYC1 initiation codon (Figures S2A and S2B). As expected, this resulted in a loss of the larger MYC protein isoform in the presence or absence of EIF5A-targeting small interfering RNAs (siRNAs) (Figure S2C).
Hypusine, or hydroxyputrescine-lysine, is synthesized by the polyamine biosynthesis pathway and post-translationally incorporated into eIF5A, replacing lysine 50 in the protein (Park and Wolff, 2018). Knockout of deoxyhypusine hydrolase (DOHH), which catalyzes the terminal step in this pathway, phenocopied the loss of eIF5A and resulted in enhanced production of MYC1 (Figures 1F, 1G, and S1F). Depletion of DOHH significantly reduced hypusine-modified but not total eIF5A levels (Figure 1F), indicating that the hypusine-modified form of eIF5A functions to regulate use of the non-canonical MYC initiation codon. Importantly, MYC mRNA levels and polysome association (Figures S2D–S2F), as well as MYC1 and MYC2 protein stability (Figure S2G), were not significantly altered upon genetic ablation of EIF5A or DOHH. Overall, these data reveal that hypusine-modified eIF5A functions as a regulator of non-canonical translation initiation in the MYC 5′ UTR.
Given that the predominant effect of loss of eIF5A on endogenous MYC was altered translation initiation codon selection, it was surprising that eIF5A depletion resulted in an increase in fluorescence of the MYC 5′ UTR EGFP reporter. We speculate that the discrepant behavior of the reporter and endogenous MYC may arise from a difference in reliance upon eIF5A for efficient translation of the EGFP and MYC ORFs. As shown below, eIF5A is required for efficient translation of PP and proline-glycine (PG) dipeptides. The MYC coding sequence has six such ribosomal pause sites, while EGFP has none. Thus, it is expected that efficient translation of MYC, but not EGFP, would be highly dependent upon eIF5A activity, offering a mechanistic explanation for why an increase in expression of the reporter does not correspond to an increase in endogenous MYC protein levels. Nevertheless, for the purposes of this study, rather than further investigating the behavior of the reporter system, we chose to focus our subsequent efforts on dissecting the role of eIF5A in regulating endogenous MYC and other transcripts. Ultimately, as shown below, this allowed us to generate new reporters that more accurately recapitulated the behavior of endogenous MYC.
eIF5A Relieves Ribosomal Pausing at PP and PG Dipeptides during Translation Elongation in Human Cells
In order to determine whether eIF5A regulates translation initiation more broadly across the transcriptome, we performed ribosome profiling under eIF5A loss-of-function conditions. Ribosome profiling experiments provide a snapshot of the position of ribosomes on mRNAs across the transcriptome, enabling the visualization of elongating ribosomes, as well as ribosomes that are stalled on transcripts (Brar and Weissman, 2015; Ingolia, 2016). Pools of EIF5A and DOHH knockout HCT116 cells were generated using two independent sgRNAs per gene. Upon mapping of ribosome footprints to the human transcriptome, a clear periodicity of reads was observed (Figure S3), consistent with the movement of ribosomes along a transcript one codon at a time and indicating the generation of a high-quality ribosome profiling dataset.
In bacteria and yeast, eIF5A homologs have been demonstrated to relieve ribosomal pausing at inefficiently translated peptide sequences, the most prominent of these being PP dipeptides (Pelechano and Alepuz, 2017; Schuller et al., 2017; Woolstenhulme et al., 2015). Thus, we first examined our ribosome profiling dataset to assess whether this function of eIF5A is conserved in human cells. Indeed, we observed that transcripts containing two or more PP dipeptide sequences exhibited increased ribosome occupancy upon loss of eIF5A or DOHH, consistent with an increase in stalled ribosomes on these mRNAs (Figures 2A and 2B). In order to directly visualize whether ribosome occupancy is increased at PP sites, we generated meta-codon plots in which transcriptome-wide ribosome occupancy on PP codons and their surrounding sequences within mRNAs were visualized. A clear increase in ribosome occupancy at PP dipeptides in EIF5A and DOHH knockout cells was observed (Figure 2C), demonstrating that hypusine-modified eIF5A functions to relieve ribosomal pausing at these sequences in human cells.
Figure 2. eIF5A Relieves Ribosomal Pausing at PP and PG Dipeptides during Translation Elongation in Human Cells.

(A and B) Cumulative distribution function (CDF) plots of coding sequence (CDS) ribosome occupancy on transcripts that have either ≤1 or >1 PP dipeptide in their ORF in EIF5A−/− (A) or DOHH−/−
(B) cells relative to cells expressing non-target (NT) sgRNA. CDS occupancy is defined as ribosome occupancy in CDS (RPKM) normalized to transcript expression level derived from RNA-seq (FPKM). Number of genes in the analysis (that fulfilled minimum coverage criteria; see STAR Methods): in (A), 2,467 (≤1 PP) and 2,309 (> 1 PP); in (B), 2,597 (≤1PP) and 2,482 (>1 PP). p values for this and all subsequent CDF plots were calculated using one-sided Kolmogorov-Smirnov tests.
(C) Meta-codon plot of ribosome occupancy in a 100 nt window surrounding all PP dipeptides (28,571 total) in EIF5A−/−, DOHH−/−, and control sgNT cells. Mean ribosome occupancy of two biological replicates is shown.
(D and E) Motif analysis of amino acid enrichment within the E, P, and A sites of paused ribosomes in EIF5A−/− (D) and DOHH−/− (E) cells, compared with sgNT control cells.
(F) Top tripeptide pause motifs identified in EIF5A−/− cells (having pause scores > 3 and ranked by ratio of pause score in EIF5A−/− versus sgNT). PP/PG-containing tripeptides are indicated with red text. Ribosome pause sites identified in both EIF5A−/− and DOHH−/− cells are shaded gray.
(G) Meta-codon plot of ribosome occupancy in a 100 nt window surrounding the top 20 tripeptide pause motifs in EIF5A−/− cells compared with sgNT (32,856 total). Mean ribosome occupancy of two biological replicates is shown.
See also Figure S3.
Recent evidence from yeast demonstrates that several additional peptide motifs, beyond polyproline sequences, may also require eIF5A for efficient peptide bond formation (Pelechano and Alepuz, 2017; Schuller et al., 2017). A de novo motif analysis was therefore performed to identify tripeptide sequences that exhibit increased ribosome occupancy in EIF5A or DOHH knockout cells (Figures 2D–2F). The tripeptide sequences at which ribosomes are most likely to stall in the absence of eIF5A or DOHH show an overrepresentation of PP and PG residues, consistent with previously reported ribosomal stalling sequences in yeast and bacteria (Doerfel et al., 2013; Peil et al., 2013; Pelechano and Alepuz, 2017; Schuller et al., 2017; Woolstenhulme et al., 2015). Additional amino acids were also highly represented, with proline-aspartic acid (PD) dipeptides and several tryptophan-containing motifs among the most enriched (Figure 2F). Visualizing ribosome occupancy at the most highly enriched tripeptide stall motifs identified in the human dataset and their surrounding sequences through a meta-codon plot confirmed that pausing is enhanced at these sites in EIF5A or DOHH knockout cells (Figure 2G). Collectively, these data demonstrate that the role of eIF5A in relieving ribosomal pausing during translation elongation is conserved from bacteria and yeast to humans.
Loss of Function of eIF5A Increases Upstream Translation Initiation
Loss of hypusine-modified eIF5A resulted in enhanced upstream translation initiation on the MYC mRNA at a non-canonical start codon. To investigate whether this occurs more broadly throughout the transcriptome, we examined upstream translation within the 5′ UTRs of transcripts in both yeast and human cells depleted of eIF5A using previously published ribosome profiling data (Schuller et al., 2017) and the data generated in this study. Analysis of data from eIF5A-depleted yeast revealed a dramatic increase in upstream translation of transcripts that fulfilled minimal coverage cutoffs for reliably detectable 5′ UTR translation (Figure 3A). These genes included classically studied genes known to harbor upstream ORFs (uORFs) such as GCN4, CPA1, and HAP4 (Zhang and Dietrich, 2005; Figures 3B and S4A; Table S1).
Figure 3. Loss of Function of eIF5A Increases Upstream Translation Initiation.

(A) CDF plot of 5′ UTR translation in EIF5A knockdown (KD) and wild-type (WT) yeast. 5′ UTR translation defined as normalized ribosome occupancy (RPKM) in 5′ UTR/CDS (1,338 genes included on the basis of minimal coverage criteria). Data for this and all subsequent yeast analyses are from Schuller et al. (2017).
(B) Ribosome occupancy on the yeast GCN4 transcript in EIF5A KD and WT yeast. Region of upstream translation is marked with an asterisk. Previously reported uORFs are denoted by gray boxes (Zhang and Dietrich, 2005).
(C) CDF plot of 5′ UTR translation in EIF5A−/− and control sgNT HCT116 cells. 5′ UTR translation is defined as ribosome occupancy (RPKM) in 5′ UTR normalized to transcript expression level derived from RNA sequencing (RNA-seq) (FPKM; 3,447 genes included on the basis of minimal coverage criteria).
(D and E) Ribosome occupancy on the human MYC (D) and PLOD3 (E) transcripts in EIF5A−/−, DOHH−/−, and control sgNT cells. Region of upstream translation is marked with an asterisk.
(F) PLOD3 reporter constructs used for luciferase assays, consisting of the PLOD3 5′ UTR and beginning of the PLOD3 ORF fused to a P2A-luciferase sequence (left). Because the PLOD3 5′ UTR has multiple potential upstream initiation codons in all three reading frames, the P2A-luciferase sequence was fused in all three reading frames, with all upstream stop codons removed. Following transfection of reporters into EIF5A−/− or control (sgNT-infected) HCT116 cells, upstream translation was calculated by first normalizing firefly luciferase activity to co-transfected renilla luciferase and then normalizing to activity produced by a construct with the canonical PLOD3 AUG-initiation codon. Data are represented as mean ± SD (n = 6 biological replicates). **p < 0.001 and ***p < 0.00001, calculated using two-tailed Welch’s t test.
We next examined ribosome profiling data from HCT116 cells to assess whether similar effects were detectable in human cells. A global increase in upstream translation in eIF5A- or DOHH-depleted cells was not observed (Figures 3C and S4B). Instead, 5′ UTR translation was enhanced in a subset of genes that included MYC and other transcripts such as PLOD3 and CCDC94 (Figures 3D, 3E, and S4C). In total, 295 of 3,447 genes that fulfilled minimal coverage requirements for reliable detection showed at least a 2-fold increase in 5′ UTR translation in EIF5A knockout cells (Table S2).
An examination of the genes whose upstream translation is increased in eIF5A-depleted cells revealed an overrepresentation of genes associated with the response to cellular stress in both human and yeast ribosome profiling datasets (Figure S4D). Activation of the eukaryotic integrated stress response (ISR) pathway, which coordinates the cellular response to diverse stress stimuli and whose defining feature is phosphorylation of eIF2α on serine 51, is known to broadly induce upstream translation (Andreev et al., 2015; Starck et al., 2016). We therefore assessed whether loss of hypusine-modified eIF5A activated the ISR, which could account for the observed increase in 5′ UTR translation. Accumulation of phosphorylated eIF2α, however, was not observed in EIF5A−/− or DOHH−/−cells (Figure S4E), demonstrating that regulation of upstream translation by eIF5A is independent of the ISR pathway. Because transcripts whose translation is activated by the ISR are enriched for upstream initiation codons and uORFs, this class of genes may be particularly reliant upon eIF5A activity to suppress 5′ UTR translation under non-stress conditions.
Loss of eIF5A Promotes Upstream Translation Initiation in the 5′ UTR of PLOD3
As an initial validation of the ribosome profiling data, we designed reporter constructs to assess non-canonical translation initiation in the 5′ UTR of PLOD3, a transcript that exhibited upstream translation in EIF5A−/− and DOHH−/− cells (Figure 3E; Table S2). Because the PLOD3 5′ UTR has multiple potential upstream initiation codons (AUG or single nucleotide variants thereof) in all three reading frames, we designed three separate reporters in which a P2A-luciferase sequence was fused to the 5′ UTR and the proximal segment of the PLOD3 ORF in all three reading frames (with all potential stop codons removed), with the canonical AUG initiation codon mutated or frameshifted. Luciferase activity was normalized to translation of a reporter that initiated translation at the canonical PLOD3 AUG start codon. Consistent with the ribosome profiling analysis, increased upstream translation initiation in all three reading frames, but particularly in frame 3, was observed in EIF5A−/− cells (Figure 3F).
Ribosomal Pausing Promotes Increased Use of the MYC1 Initiation Codon
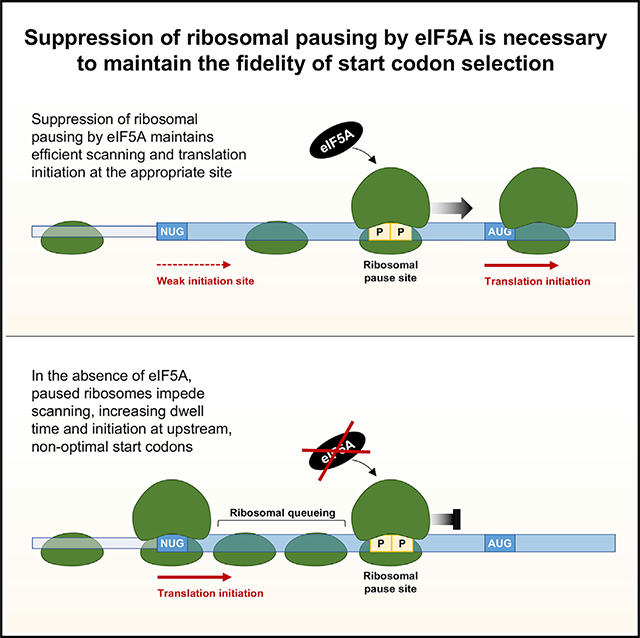
Given that the principal effect of eIF5A depletion is increased ribosomal pausing at difficult to translate peptide motifs, we hypothesized that increased 5′ UTR translation under these conditions could be a consequence of inefficient translation elongation. Multiple observations supported this concept. First, it was recently reported that ribosomal pausing in the 5′ UTR of the mouse Azin1 mRNA promoted non-canonical translation initiation within a short distance upstream of the pause site (Ivanov et al., 2018). The authors proposed that this enhanced alternative initiation could be attributed to the queuing of scanning ribosomes upstream of a paused ribosome, resulting in the enhanced positioning of ribosomes at a non-canonical initiation codon. This model is further supported by the demonstration that ribosome stalling induced by a stable RNA secondary structure downstream of a weak translation initiation site enhanced upstream initiation (Kozak, 1990; Liang et al., 2017). Similarly, protein synthesis inhibitors that impair ribosome elongation enhance the use of non-canonical start codons (Kearse et al., 2019). Interestingly, we noticed that a strong ribosomal pause motif (PPA) is located between the MYC1 and MYC2 initiation sites on the MYC transcript (Figure 4A). Thus, we postulated that ribosome stalling in the MYC leader sequence may promote enhanced use of the MYC1 translation initiation site upon eIF5A loss of function.
Figure 4. Ribosomal Pausing Promotes Increased Use of the MYC1 Initiation Codon.

(A) Model of ribosome pause-induced upstream initiation on the MYC transcript. Enhanced pausing at the PPA tripeptide motif within the MYC leader in EIF5A−/− cells may lead to queuing of scanning ribosomes upstream, thereby promoting increased use of the MYC1 initiation codon.
(B) MYC reporter constructs used for luciferase assays, consisting of a 5′ segment of the MYC transcript fused to a P2A-luciferase sequence (left). The frameshift (F/S) reporter was constructed by deleting a single nucleotide six codons upstream of the PPA pause motif, thus altering all encoded amino acids downstream until the P2A sequence. Following transfection of reporters into EIF5A−/− or control (sgNT-infected) HCT116 cells, CUG-initiated translation was calculated by first normalizing firefly luciferase activity to co-transfected renilla luciferase and then normalizing to activity produced by an otherwise identical AUG-initiated construct. Data are represented as mean ± SD (n = 6 biological replicates). **p < 0.001 and ***p < 0.00001, calculated using two-tailed Welch’s t test.
See also Figure S5.
To test this model, luciferase constructs that report MYC1 translation were generated (Figure 4B). These constructs consisted of a fragment of the MYC transcript beginning upstream of the MYC1 initiation codon and extending into the body of the MYC ORF followed by a P2A-luciferase sequence. To specifically measure MYC1-initiated translation, the canonical MYC2 AUG initiation codon was mutated to UUU. Control constructs in which the MYC1 CUG initiation codon was mutated to AUG were also generated. Normalization of the luciferase signal produced by CUG-initiated constructs to the signal produced by AUG-initiated derivatives ensured that only the efficiency of non-canonical initiation was measured, and any effects on translation elongation or other downstream events were discounted. Consistent with the behavior of endogenous MYC, we observed increased CUG-initiated translation in EIF5A−/− cells (Figure 4B). This effect was driven by a specific increase in activity of CUG-initiated reporters and was not a normalization artifact due to altered activity of AUG-initiated reporters (Figure S5A). Mutating the PPA tripeptide motif in the MYC leader sequence to PAA partially reduced the enhanced CUG-initiated translation observed in EIF5A−/− conditions. Moreover, a frameshift mutation that eliminated the PPA motif and altered the surrounding amino acids strongly impaired CUG-initiated translation. These data support the model that ribosomal pausing in the proximal segment of the MYC transcript in eIF5A-deficient cells promotes upstream translation initiation at the non-canonical MYC1 initiation codon. The behavior of the frameshifted reporter suggests that pausing in this region of the transcript is mediated not only by the PPA tripeptide motif but also by other N-terminal amino acids encoded in this sequence. This is in accordance with previous reports showing that amino acids upstream and downstream of pause sites, as well as amino acids within the nascent peptide chain in the exit channel of the ribosome, can influence the extent of ribosomal pausing (Buskirk and Green, 2017; Peil et al., 2013).
Proximal Ribosomal Pausing Triggers Widespread Upstream Translation Initiation
We next examined ribosome profiling data to determine whether proximal ribosomal pausing influences start codon selection more broadly throughout the transcriptome. Transcripts were segregated into two groups on the basis of their predicted behavior according to our pausing-associated upstream initiation model (Figure 5A). Group 1 comprised genes that were predicted to show increased 5′ UTR translation in EIF5A−/− cells, specifically genes whose transcripts contained a canonical (AUG) or non-canonical (any single nucleotide variant of AUG) initiation site upstream of the annotated start site and a downstream in-frame ribosomal pause site within 67 codons. This window was chosen because of evidence indicating that this was the maximal distance that allowed effective ribosome queuing-mediated effects on alternative translation initiation (Ivanov et al., 2018). All other genes that did not satisfy these criteria were placed into group 2. 5′ UTR translation in these transcripts under conditions of eIF5A or DOHH loss of function was then assessed.
Figure 5. Proximal Ribosomal Pausing Triggers Widespread Upstream Translation Initiation.

(A) Genes were classified into those that contained an upstream AUG or single-nucleotide variant thereof within 67 codons of an in-frame pause site (group 1) and all others (group 2).
(B and C) CDF plots of 5′ UTR translation in EIF5A−/− cells compared with sgNT control, with pause sites defined as PP dipeptides (B) or the top 20 tripeptide pause motifs identified in EIF5A−/− cells compared to sgNT control (C). 5′ UTR translation was defined as ribosome occupancy (RPKM) in 5′ UTR normalized to transcript expression level derived from RNA-seq (FPKM). Genes included on the basis of minimal coverage requirements: 2,877 in group 1 and 570 in group 2 for (B) and 2,860 in group 1 and 587 in group 2 for (C).
(D) CDF plot of 5′ UTR translation in EIF5A KD yeast compared with WT. Pause sites defined as PP dipeptides. 5′ UTR translation was defined as normalized ribosome occupancy (RPKM) in 5′ UTR/CDS. One hundred forty-nine genes were included in group 1 and 1,189 genes were included in group 2 on the basis of minimal coverage criteria.
See also Figure S5.
We first defined group 1 transcripts as those containing a PP dipeptide, the minimal, classically studied eIF5A-associated pause motif, downstream of putative 5′ UTR initiation sites. Notably, of the 295 genes that showed at least a 2-fold increase in upstream translation in EIF5A knockout cells described above, 259 fulfilled these criteria and were classified as group 1. Consistent with the behavior of these 259 genes, group 1 transcripts exhibited an overall increase in 5′ UTR translation upon eIF5A and DOHH loss of function (Figures 5B and S5B). We also detected an increase in upstream translation in EIF5A−/− cells when we reclassified group 1 to comprise genes that contain an upstream initiation site followed by an in-frame pause site defined as any of the top 20 pause motifs identified in our de novo analysis (Figures 2F and 5C). A similar trend was also documented in DOHH−/− cells, although the effect did not reach statistical significance under these conditions (Figure S5C). Last, when this model was applied to yeast ribosome profiling data, similar effects were observed (Figures 5D and S5D). Collectively, these data support the model that hypusine-modified eIF5A is required to suppress widespread proximal ribosomal pausing in order to maintain efficient scanning and accurate start codon selection, in a manner that is conserved in yeast and human cells.
DISCUSSION
Accumulating evidence from multiple tumor types has established that altered translational control plays an important role in cancer pathogenesis (Truitt and Ruggero, 2016). This principle is exemplified by the growth-promoting mTOR pathway, which is activated in many malignancies and promotes translation of specific mRNAs through multiple mechanisms (Hsieh et al., 2012; Mossmann et al., 2018; Nandagopal and Roux, 2015; Roux and Topisirovic, 2018). For example, translation of MYC and other mRNAs whose 5′ UTRs contain complex secondary structures is stimulated by the mTOR pathway in response to growth factors, nutrients, and oncogenic signaling. Activity of the mTORC1 complex, through phosphorylation of its protein targets S6K and eIF4E-BPs, promotes the assembly of the cap-binding complex (comprising eIF4E, eIF4G, and eIF4A) and thus facilitates translation of difficult to translate messages, including MYC.
Given the importance of regulation of MYC expression at multiple post-transcriptional levels, including translational control, we performed an unbiased screen to identify new regulators of the MYC 5′ UTR. These experiments led to the discovery of an unexpected role for eIF5A as a regulator of start codon selection. In the case of the MYC transcript, eIF5A depletion triggered the enhanced production of the N-terminally extended MYC1 protein initiated at an upstream CUG start codon. Similar effects, specifically enhanced translation initiation at suboptimal or non-canonical initiation codons in 5′ UTRs, occurred broadly throughout the human and yeast transcriptomes under conditions of eIF5A loss of function. Ribosome profiling experiments demonstrated that the function of eIF5A as a suppressor of ribosomal pausing at difficult to translate peptide motifs is conserved in human cells and provided strong evidence that ribosomal pausing promotes the use of upstream initiation codons in eIF5A-deficient cells. These findings extend our understanding of the functions of this deeply conserved translation factor and uncover a role for eIF5A in maintaining the fidelity of translation initiation.
Reporter-coupled CRISPR/Cas9 screening represents a powerful approach for dissecting mechanisms of post-transcriptional gene regulation. This work was initiated with a genome-wide screen to interrogate regulators of the MYC 5′ UTR. Two important translation factors emerged from the screen: eIF3L and eIF5A. Our finding that eIF3L is a putative repressor of the MYC 5′ UTR is in line with reports suggesting that in addition to a general role for the large multi-subunit eIF3 complex in translation initiation, specialized eIF3 complexes may direct the translational repression of a subset of transcripts, such as BTG1 (Lee et al., 2015). Although EIF3L knockout did not increase steady-state MYC protein levels in HCT116 cells, possibly because of multiple redundant levels of MYC regulation in this cell line, further examination of this putative regulatory interaction in other contexts remains an interesting direction for future work.
Loss of eIF5A, the top hit from our screen, resulted in enhanced EGFP fluorescence produced by the MYC 5′ UTR reporter but, unexpectedly, altered endogenous MYC translation initiation codon selection rather than increasing total MYC protein levels. As discussed above, these apparently discordant results may have arisen in part from differences in the requirement for eIF5A to facilitate translation elongation through the EGFP and MYC ORFs. The MYC coding sequence has six strong PP or PG ribosomal pause sites, while EGFP has none, suggesting that efficient translation through the MYC ORF may be highly dependent on eIF5A. Rather than further investigating the behavior of the reporter, however, we chose to focus on dissecting the role of this protein in regulating endogenous MYC and other transcripts. Ultimately, our transcriptome-wide analyses of eIF5A function not only allowed us to elucidate a role for eIF5A in translation initiation but also enabled the generation of new reporters that, unlike the EGFP reporter, accurately recapitulated the behavior of endogenous MYC (Figure 4).
The established function of eIF5A, derived from an extensive body of work studying the yeast and bacterial orthologs, is to relieve ribosomal pausing at sites of inefficient peptide bond formation (Pelechano and Alepuz, 2017; Schuller et al., 2017; Woolstenhulme et al., 2015). Here we globally analyzed translation in human cells lacking this protein, demonstrating that this molecular function is conserved and supporting a role for eIF5A as a universal pause relief factor. We observed that hypusine-modified eIF5A alleviates ribosomal pausing primarily at PP and PG dipeptides in human cells. This generally agrees with previously identified pause signatures and is consistent with literature implicating these amino acids as suboptimal substrates for peptide bond formation (Doerfel et al., 2013; Pavlov et al., 2009; Peil et al., 2013; Pelechano and Alepuz, 2017; Schuller et al., 2017; Shin et al., 2017). This functional conservation of eIF5A is a direct reflection of the remarkable degree to which the homologs of these proteins are conserved in sequence and structure across kingdoms (Dever et al., 2014; Magdolen et al., 1994). Notably, our data are also consistent with prior literature indicating that the hypusine modification is critical for eIF5A function (Cano et al., 2008; Schnier et al., 1991).
Suppression of ribosomal pausing by eIF5A is necessary to maintain efficient translation elongation. Interestingly, it has been suggested that depletion of EF-P, the bacterial eIF5A ortholog, not only impairs elongation but also inhibits translation initiation on certain transcripts, specifically those harboring start codons located upstream of strong ribosomal pause sites (Woolstenhulme et al., 2015). Ribosome queuing upstream of these pause sites was proposed to sterically block access of initiating ribosomes to start codons on these mRNAs. Our analysis of human and yeast eIF5A also supports a role for this protein in regulating translation initiation in an analogous manner. Unlike the data from bacterial systems, however, our findings suggest that ribosome queuing upstream of pause sites in eIF5A-depleted eukaryotic cells increases the use of suboptimal or non-canonical start codons in 5′ UTRs. This model is supported by the demonstration that sites of inefficient peptide bond formation, thermostable RNA structures, or pharmacologic agents that impede ribosomal elongation can similarly enhance translation initiation from AUG codons in suboptimal Kozak contexts or non-AUG codons in both viral (Ventoso et al., 2006) and endogenous transcripts (Ivanov et al., 2018; Kearse et al.,2019; Kozak, 1990; Liang et al., 2017). Taken together, the existing data from eukaryotic systems suggest that in the absence of eIF5A, stalled ribosomes in 5′ UTRs or proximal coding sequences lead to ribosome queuing, which impedes the efficient scanning of PICs before they reach downstream, optimal start codons (as illustrated by the MYC transcript in Figure 4A). The subsequent increase in dwell time of these complexes in the 5′ UTR may increase the probability of translation initiation at upstream, non-optimal initiation codons.
Whether there is a physiologic role for enhanced upstream translation initiation triggered by proximal ribosomal pausing is an interesting question for future study. Analysis of yeast ribosome profiling data revealed a widespread increase in 5′ UTR translation across the transcriptome upon depletion of eIF5A, an effect that occurred on a more restricted set of human transcripts. Nevertheless, in both yeast and human cells, transcripts encoding mediators of the cellular stress response, which have been documented to harbor suboptimal or non-canonical translation initiation sites within their 5′ UTRs, exhibited a dependence on eIF5A for maintenance of appropriate translation initiation. Taken together with the fact that widespread and early (within 50–65 codons of initiation) ribosomal pausing has been documented under specific stress conditions (Liu et al., 2013; Shalgi et al., 2013), our data suggest that the mechanism of proximal pause-induced upstream initiation may be pervasively used to enhance translation of mRNAs encoding stress response factors.
Multiple lines of evidence also suggest the intriguing possibility that enhanced upstream translation initiation, caused by activation of the ISR or impaired eIF5A activity, may contribute to cancer pathogenesis. In a mouse model of squamous cell carcinoma, for example, elevated eIF2α phosphorylation, indicative of activation of the ISR, was associated with the use of an eIF2A-driven translational program that facilitated preferential translation of oncogenic mRNAs initiated from upstream start codons (Sendoel et al., 2017). eIF5A loss of function may trigger a similar effect in several human cancers. EIF5A is located within the frequently deleted human 17p locus that also contains the tumor suppressor TP53. Approximately 50% of all human cancers exhibit inactivation of TP53, and a majority of these malignancies contain at least one large 17p deletion, thus concurrently ablating EIF5A. The functional relevance of TP53-EIF5A co-deletion was substantiated by the demonstration that loss of function of EIF5A, or other members of the polyamine-hypusine pathway, accelerated MYC-driven lymphomagenesis in mice (Liu et al., 2016; Scuoppo et al., 2012). Whether these effects are driven by enhanced production and oncogenic activity of MYC1 in this model, or the more widespread induction of ribosomal pausing and upstream translation, remains to be established. Additionally, it is important to note that overexpression of EIF5A has been observed in some tumor types (Tunca et al., 2013), raising the possibility that more efficient suppression of ribosomal pausing, and potentially reduced production of MYC1, may be favored in some cancer contexts.
We have demonstrated that ribosomal pausing and increased upstream translation on MYC and other transcripts can be induced by depletion of total or hypusine-modified eIF5A. Thus, it also is likely that upstream signals that modulate cellular polyamine levels, and consequently levels of hypusine-modified eIF5A, can similarly influence alternative initiation. Polyamines—putrescine, spermidine, and spermine—are essential cations that, in addition to serving as obligate precursors for the production of hypusine, have myriad cellular functions that support the demands of cellular growth, including chromatin maintenance, scavenging of free radicals, and binding to ribosomal components to promote translation (Casero and Marton, 2007; Dever and Ivanov, 2018). Accordingly, oncogenic signaling can induce polyamine production, and MYC itself promotes transcription of several enzymes in the polyamine-biosynthesis pathway (Bachmann and Geerts, 2018). Although modulation of cellular polyamine levels would be expected to have pleiotropic effects on cell growth and homeostasis, our findings reveal that start codon selection may be broadly affected under these conditions as well. Thus, the effect of polyamine availability on translational reprogramming through modulation of eIF5A activity is an interesting area for further investigation.
In conclusion, we have demonstrated that suppression of ribosomal pausing by eIF5A is a highly conserved function that is required not only to maintain efficient translation within ORFs but also to suppress the use of upstream initiation codons. These findings highlight the broad influence of ribosomal pausing on multiple steps of translation. Further study of the stimuli that cause an elongating ribosome to stall, including eIF5A-associated pause cues, as well as other triggers of pausing such as tRNA abundance and RNA secondary structure, will enable the dissection of subsequent effects on the transcriptome and proteome and the physiological contexts in which these mechanisms are used for gene regulation.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joshua Mendell (joshua.mendell@utsouthwestern.edu). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines used in this study were obtained from ATCC and confirmed to be free of mycoplasma contamination. HCT116 cells (human) were cultured in McCoy’s 5A medium supplemented with 10% fetal bovine serum (FBS) and 1X antibiotic/antimycotic (ThermoFisher). HEK293T cells (human) were cultured in DMEM supplemented with 10% FBS and 1X antibiotic/antimycotic. HCT116 is a male cell line and HEK293T is most likely female due to the presence of multiple X chromosomes and no detectable Y chromosome.
METHOD DETAILS
Construction of MYC-EGFP reporter constructs
The sequences of all oligonucleotides used in this study are provided in Table S3. The following plasmids were obtained: AAVS1 hPGK-PuroR-pA donor (Addgene plasmid #22072), pMSCV-Hygro (Clontech), and pEGFP-N1 (Clontech). First, PGK-Puro was excised from AAVS1 hPGK-PuroR-pA donor by HindIII digestion and replaced with PGK-Hygro (amplified from pMSCV-Hygro with primer pair HM284/HM285) using the In Fusion HD Cloning Kit (Takara) to generate the AAVS1-Donor-PGK-Hygro plasmid. During this cloning, FseI and MluI sites were introduced 5′ to the PGK promoter, and SalI, EcoRV and HindIII sites were introduced 3′ of the hygromycinB resistance gene. Next, a synthetic polyA signal (Seibler et al., 2007) was synthesized, and inserted downstream of the hygromycinB resistance gene using the SalI and EcoRV sites. Third, the CMV-EGFP-SV40polyA cassette was amplified from pEGFP-N1 (using primer pair HM280/HM281) and cloned into the AAVS1-Donor-PGK-Hygro-pA plasmid using FseI and MluI sites, to generate the AAVS1-Donor-Hygro-EGFP plasmid. Lastly, the EGFP ORF was amplified (using the primer pair HM320/HM321) and cloned back into this vector (AAVS-Donor-Hygro-EGFP) using KpnI and XbaI sites to generate the final EGFP reporter donor vector (pAAVS-EGFP-DONOR). This was done to simplify in-frame cloning upstream of the EGFP ORF in this vector. The MYC 5′ UTR was amplified from human genomic DNA (using primers HM322/HM324) and cloned into pAAVS-EGFP-DONOR digested with KpnI using the In Fusion HD Cloning kit to generate the MYC 5′ UTR reporter donor plasmid (pAAVS-5′ UTR-DONOR). The complete sequence of the MYC 5′ UTR used in the reporter construct is provided in Table S3.
Generation of clonal fluorescent reporter cell lines
A published TALEN pair targeting the human AAVS1/PPP1R12C locus (Sanjana et al., 2012) was used, in combination with the either pAAVS1-EGFP-DONOR or pAAVS1–5′ UTR-DONOR, to generate MYC 5′ UTR reporter and EGFP reporter knock-in HCT116 cell lines. The three plasmids were transfected into HCT116 cells using FugeneHD transfection reagent (Promega), using the following molar ratio: Left TALEN:Right TALEN:Donor vector for homology-directed repair = 1:1:8. Two days post-transfection, the cells were split into 0.5 mg/mL hygromycinB (Thermofisher Scientific). The cells were selected for at least 7 days, before plating into 96-well plates at single cell density. Colonies arising from single cells were picked and expanded, and genomic DNA was extracted using the DNeasy Blood and Tissue Kit (QIAGEN). Clonal cell lines were then genotyped by PCR to detect the AAVS1 wild-type and the EGFP knock-in alleles, using the primer pairs HM356/HM357 and HM356/HM359 respectively. Heterozygous knock-in clones were used for all experiments.
Genome-wide CRISPR/Cas9 screening
Fluorescent-reporter coupled CRISPR/Cas9-mediated loss-of-function screening was carried out as previously described in detail (Golden et al., 2017). A brief overview is provided here.
The human GeCKO v2 libraries A and B (Addgene pooled libraries ##1000000048, #1000000049) were amplified in bacteria and purified, and then used to prepare lentivirus in HEK293T cells. Genome-wide screening was performed with libraries A and B in the MYC 5′ UTR and EGFP reporter cell lines, with two replicates for each cell line. Reporter cells were transduced with lentivirus at a multiplicity of infection between 0.2 – 0.4. Two days later, the cells were seeded into 1 μg/mL puromycin to select for cells that had been successfully transduced. After passaging for 13 days in puromycin, cells were subjected to FACS on a MoFlo cell sorter (Beckman Coulter). The brightest 0.5% of cells were collected by FACS, pelleted, and frozen at −80°C.
sgRNA coverage was maintained at 500X during infection and at all subsequent stages of screening. 120 ×106 cells were infected per library and at least 100 ×106 cells were maintained per library during selection. 60–80 ×106 cells were sorted per replicate per library, and 1.5 ×105 – 2.5 ×105 cells were collected post sorting. 40 ×106 unsorted cells were also pelleted and frozen at −80°C. Genomic DNA was extracted from the sorted and unsorted cell pellets using either a gDNA isolation method that has been previously described (Golden et al., 2017) for the sorted cells or the DNeasy Blood and Tissue Kit (QIAGEN) for the unsorted cells. Two sequential rounds of PCR were performed using the isolated genomic DNA as template to generate PCR amplicon libraries. Library DNA was then purified using Agencourt AMPure XP beads (Beckman Coulter Life Sciences). DNA concentration of the libraries was quantified by using the Qubit dsDNA BR Assay Kit (Thermofisher Scientific), as well as by qPCR using the KAPA Library Quantification Kit for Illumina platforms (Kapa Biosystems). Amplicon library size and integrity were also assessed via the Bioanalyzer High Sensitivity DNA Analysis Kit (Agilent). Following these quality checks, library amplicons were sequenced on an Illumina NextSeq500 with 75 bp single-end reads. Approximately 20 ×106 reads were obtained per sample.
Knockout of genes via lentiviral delivery of CRISPR/Cas9 components
Generation of lentivirus
sgRNAs targeting genes of interest, as well as non-target (NT) sgRNAs (Table S3), were cloned into the lentiCRISPRv2 (LCv2) vector (Addgene plasmid #52961) according to the LCv2 cloning protocol (Shalem et al., 2014). LCv2-sgRNA lentivirus was generated in HEK293T cells as follows: Cells were seeded at a density of 600,000 cells per well of a 6-well dish. The next day, cells were transfected with 0.5 μg LCv2-sgRNA plasmid, 0.3 μg psPAX2 (Addgene #12260), and 0.2 μg pMD2.G (Addgene #12259) lentiviral packaging plasmids using FugeneHD transfection reagent (Promega). Media was changed 24 hours post-transfection, and viral collection was performed on the two subsequent days. Media containing virus was filtered through a 0.45 μm SFCA sterile filter and either utilized immediately for transduction or frozen at −80°C in single use aliquots.
Infection of cells and generation of knockout pools
Cells to be infected were seeded in an equal mix of fresh media and viral supernatant with polybrene (EMD Millipore) at a final concentration of 8 μg/ml. Cells were infected at densities such that they achieved confluency 48 hours post-transduction. Media was changed the day after infection and cells were split into media containing puromycin at a concentration of 1 μg/ml 48 hours after infection.
For all experiments using EIF5A−/− and DOHH−/− pools, cells transduced with the appropriate LCv2-sgRNA virus were harvested for analysis after 6 days of selection in puromycin. Control pools used in these experiments were cells transduced with LCv2-NT sgRNA virus, selected in puromycin for 6 days and harvested in the same manner as EIF5A−/− and DOHH−/− knockout pools.
Genome editing of the MYC1 initiation codon
Generation of MYC1 knockout pools and siRNA transfection
HCT116 cells were infected with lentiCRISPRv2 virus selectively targeting MYC1 (sgRNA sequence listed in Table S3) and selected in puromycin as described above. On the third day of selection, cells infected with MYC1 sgRNA and NT sgRNA were reverse transfected with siRNA (either targeting EIF5A, or NT controls listed in Table S3, Dharmacon) at a concentration of 20 nM, using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s instructions. Cells were harvested for gDNA isolation and western blot analysis at 72 hours post-transfection of siRNA (corresponding to 6 days of puromycin selection).
Genotyping of MYC1-sgRNA targeting site by next-generation sequencing
Cells infected with lentiCRISPR-MYC1 sgRNA or lentiCRISPR-NT sgRNA and then transfected with NT siRNA were harvested for genomic DNA isolation using the DNeasy Blood and Tissue Kit (QIAGEN). Amplicon libraries (of the sgRNA target site) were generated by two sequential rounds of PCR using Phusion High-Fidelity DNA Polymerase (NEB). PCR1 was performed using gene specific primers HM685/HM686, and PCR2 was performed using Illumina TruSeq CD Indexes D508 (Fwd)/D706–709 (Rev). Amplicons were sequenced using a MiSeq instrument with at least 100,000 reads per sample.
Western blot analysis
Cell lysates were prepared by harvesting in ice cold 1X RIPA buffer. Proteins were visualized and signals were quantified using an infrared fluorescent antibody detection system (LI-COR). Blocking was performed in 10% BSA for phospho-antibodies or 10% non-fat milk for all other antibodies. Primary antibodies were utilized at a concentration of 1:1000, diluted in 5% BSA in TBST (for phospho-antibodies) or 5% non-fat milk in TBST (for all other antibodies), and are listed in the Key Resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti c-Myc (MYC) | Cell Signaling | Cat# 5605; RRID:AB_1903938 |
| Mouse monoclonal anti α-Tubulin | Millipore Sigma | Cat# T6199; RRID:AB_477583 |
| Rabbit monoclonal anti eIF5A | Abcam | Cat# ab32407; RRID:AB_2230904 |
| Rabbit polyclonal anti DOHH | Millipore Sigma | Cat# HPA041953; RRIDABJ0795386 |
| Rabbit polyclonal anti Hypusine | Millipore Sigma | Cat# ABS1064; RRID:AB_2631138 |
| Rabbit polyclonal anti phospho-eIF2α | Cell Signaling | Cat# 9721; RRID:AB_330951 |
| Rabbit polyclonal anti eIF2α | Cell Signaling | Cat# 9722; RRID:AB_2230924 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| FuGENE® HD Transfection Reagent | Promega Corporation | Cat# E2311 |
| Hygromycin B | Thermofisher Scientific | Cat# 10687010 |
| Puromycin (Puromycin Dihydrochloride) | Thermofisher Scientific | Cat# A11138–03 |
| Polybrene Infection/Transfection Reagent | Millipore Sigma | Cat# TR-1003-G |
| TRIzol | Thermofisher Scientific | Cat# 15596026 |
| Critical Commercial Assays | ||
| In-Fusion® HD Cloning Kit | Takara (Clontech) | Cat# 639649 |
| DNeasy Blood & Tissue Kit | QIAGEN | Cat# 69504 |
| Agencourt AMPure XP | Beckman Coulter Life Sciences | Cat# A63882 |
| Qubit dsDNA BR Assay Kit | Thermofisher Scientific | Cat# Q32853 |
| Bioanalyzer High Sensitivity DNA Analysis | Agilent | Cat# 5067–4626 |
| KAPA Library Quantification Kit for Illumina platforms (KK4835) | Kapa Biosystems (Roche) | Cat# 07960204001 |
| Direct-zol RNA Miniprep Kit | Zymo Research | Cat# R2051 |
| SuperScript III First-Strand Synthesis SuperMix | SuperScript III First-Strand Synthesis SuperMix | Cat# 18080400 |
| SYBR Green PCR Master Mix | Thermofisher Scientific | Cat# 4309155 |
| Lipofectamine RNAiMAX Transfection Reagent | Thermofisher Scientific | Cat# 13778075 |
| Phusion® High-Fidelity DNA Polymerase | New England Biolabs | Cat# M0530S |
| QuikChange Lightning Site-Directed Mutagenesis Kit | Agilent | Cat# 210518 |
| NEBuilder HiFi DNA Assembly Master Mix | New England Biolabs | Cat# E2621S |
| Dual-Luciferase Reporter Assay System | Promega Corporation | Cat# E1910 |
| Deposited Data | ||
| CRISPR screening data | This paper | GEO: GSE132010 |
| Raw and processed ribosome profiling data | This paper | GEO: GSE132010 |
| RNA-seq data (complementary to ribosome profiling) | This paper | GEO: GSE132010 |
| Experimental Models: Cell Lines | ||
| HCT116 | ATCC | CCL-247 |
| HEK293T | ATCC | CRL-3216 |
| HCT116 MYC 5’ UTR reporter | This paper | N/A |
| HCT116 EGFP reporter | This paper | N/A |
| Oligonucleotides | ||
| Sequences of oligonucleotides used in this study provided in Table S3 | This paper | N/A |
| Recombinant DNA | ||
| AAVS1 hPGK-PuroR-pA donor | Addgene (Rudolf Jaenisch) | Plasmid #22072 (Hockemeyer et al., 2009) |
| pMSCV-Hygro | Clontech | Cat# 634401 |
| pEGFP-N1 | Clontech | Cat# 6085–1 |
| pAAVS-EGFP-DONOR | This paper | N/A |
| pAAVS-5’ UTR-DONOR | This paper | N/A |
| Human CRISPR Knockout Pooled Library (GeCKO v2) | Addgene (Feng Zhang) | Pooled Library #1000000048, #1000000049 (Sanjana et al., 2014) |
| lentiCRISPR v2 | Addgene (Feng Zhang) | Plasmid #52961 (Sanjana et al., 2014) |
| psPAX2 | Addgene (Didier Trono) | Plasmid #12260 |
| pMD2.G | Addgene (Didier Trono) | Plasmid #12259 |
| pGL3-Control | Promega Corporation | Cat# E1741 |
| pGL3-Control_NP2 | This paper | N/A |
| pRL-SV40 | Promega Corporation | Cat# E2231 |
| pcDNA3.1(+) | Invitrogen | Cat# V790–20 |
| Software and Algorithms | ||
| Bowtie2 | Langmead and Salzberg, 2012 | N/A |
| MAGeCK | Li etal., 2014 | N/A |
| HISAT2 | Kim et al., 2015 | N/A |
RNA isolation and qRT-PCR
Cells at subconfluent densities were harvested in Trizol (Thermo Fisher Scientific). RNA was isolated using the Direct-zol RNA miniprep kit (Zymoresearch). The on-column DNase digestion step was performed according to manufacturer’s instructions. SuperScript III First-strand Synthesis SuperMix (Thermo Fisher Scientific) was used to synthesize cDNA from RNA, using 1 μg of RNA per reaction. All qPCR assays were performed in technical triplicate, using the SybrGreen 2X PCR Master Mix (Applied Biosystems). Primers used for qRT-PCR are listed in Table S3.
Ribosome profiling
HCT116 cells were transduced with lentiCRISPRv2 virus targeting EIF5A or DOHH, or expressing NT sgRNA, and selected for 6 days in puromycin as described above to generate knockout pools. Cells were harvested and ribosome profiling was performed exactly as described (McGlincy and Ingolia, 2017), with the following modifications:
Ribosome footprints were size selected by gel electrophoresis as described previously (Ingolia et al., 2012), and fragments between 24–35nt were excised for library preparation.
Samples were not pooled after linker ligation, and the subsequent steps were performed individually for each sample.
In order to effectively separate 3′ linker-ligated RNA fragments from unligated linker, we utilized gel electrophoresis, followed by gel extraction as previously described (Ingolia et al., 2012), instead of an enzymatic depletion.
Arsenite treatment
800,000 HCT116 cells were seeded in each well of a 6-well dish. The following day, the cells were treated with 250 μM sodium arsenite for up to 3 hours. Cells were then harvested and analyzed by western blotting as described above.
Luciferase reporter assays
Construction of luciferase reporters
The firefly luciferase ORF in pGL3-Control (Promega) was first modified to remove N-terminal/proximal pause sites. A proline-glycine dipeptide (aa 37–38) and a proline-proline dipeptide (aa 173–174) were mutated to proline-alanine using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent) and the primer pairs HM673/HM674 and HM677/HM678, respectively, to generate pGL3-Control_NP2. MYC 5′ UTR reporter fragments were then cloned into the HindIII and NcoI sites in pGL3-Control_NP2 using the NEBuilder HiFi DNA Assembly Master Mix (NEB). The MYC 5′ UTR reporter fragments used in the assembly were designed to contain a fragment of the MYC transcript beginning 15 nt upstream of the MYC1 CUG initiation codon and extending 201 nt downstream of the MYC2 AUG initiation codon into the main MYC ORF (as depicted in Figure 4B), followed by a P2A sequence. The MYC2 initiation codon was mutated from AUG to a non-initiating UUU in all constructs. The frameshifted reporter was generated by introducing a single nucleotide deletion 6 aa upstream of the PPA pause motif. Appropriate homology arms to facilitate HiFi assembly were added and fragments were obtained as gBlocks synthesized by IDT (sequences provided in Table S3).
PLOD3 5′ UTR luciferase reporter constructs were designed and constructed in a manner similar to the MYC 5′ UTR luciferase reporters. Fragments of the PLOD3 transcript containing the entire PLOD3 5′ UTR and a small segment of the ORF were synthesized as gene fragments and cloned into a plasmid vector by Genewiz (sequences provided in Table S3). Synthesized fragments were then amplified using primers that added a left homology arm, and a P2A element and a homology arm on the right to facilitate cloning into pGL3-Control_NP2 using the NEBuilder HiFi DNA Assembly Master Mix (NEB) at HindIII and NcoI sites, as described above. Three PLOD3 constructs that each reported translation in a different reading frame were generated by synthesizing segments of the PLOD3 transcript that differed in length by one nucleotide (the first 498, 499, or 500 nucleotides of the transcript) and fusing them to the P2A-luciferase cassette. In each reporter, all stop codons contained within the PLOD3 transcript segments that were in the same reading frame as the luciferase ORF were mutated to alanine codons to ensure productive read out of translation. Further, the AUG start codon (contained in the Frame 1 construct) was mutated to the non-initiating UUU. Lastly, a luciferase reporter containing the unaltered AUG start codon (and similar to the Frame 1 reporter in all other respects) was generated to use as a control to monitor canonical AUG translation. Primers used for amplifying the PLOD3 fragments were as follows (sequences provided in Table S3): Frame 1 – HM705/HM706; Frame 2 – HM705/HM707; Frame 3 – HM705/HM710; ATG-Control – HM705/HM706.)
Dual luciferase reporter assays
HCT116 cells were infected with lentiCRISPRv2 virus containing EIF5A-targeting or NT sgRNAs as described above. On the fourth day of selection in puromycin, cells were plated in 24-well dishes at densities such that they would be 50% confluent in 24 hours. The following day, cells were transfected with luciferase reporters using FugeneHD transfection reagent (Promega). 300 ng of DNA was transfected/well, comprising 2 ng of pRL-SV40 (Promega) as a transfection control, 198 ng of empty pcDNA3.1(+) (Invitrogen), and 100 ng of a pGL3–5′ UTR reporter. Cells were harvested 24 hours later and luciferase activity was measured with the Dual-Luciferase Reporter Assay System (Promega).
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis of genome-scale CRISPR/Cas9 screening data
The GeCKO v2 library sgRNA reference files were obtained from Addgene and identical sgRNAs targeting more than one gene were removed. Bowtie 2 was used to map reads obtained from the demultiplexed FASTQ files to the modified reference library, allowing no mismatches, and normalized read counts were calculated as described previously (Shalem et al., 2014). MAGeCK was used to identify statistically significant hits from the screening data (Li et al., 2014). We detected contaminating reads in the library corresponding to sgRNAs targeting NFE2L2 (arising from cloning of individual sgRNAs into the LCv2 vector) and this gene was therefore removed from our analysis.
Bioinformatic analysis of ribosome profiling data
Read preparation and mapping
For analysis of reads from the human ribosome profiling dataset, reads from two separate sequencing runs for each sample were first combined. Next, the adaptor, inline barcode, and the random-mer incorporated into the library amplicon were trimmed for each read. Then, reads from noncoding RNAs were removed by mapping to known rRNA (Quast et al., 2013), tRNA (Chan and Lowe, 2016), and snRNA (Kuksa et al., 2019) sequences using HISAT2 (Kim et al., 2015). Reads that did not map to these databases were subsequently mapped to the human reference genome (GRCh38) (Frankish et al., 2019) using HISAT2. Reads with lengths between 24 – 31 nt were used for all analyses. Annotation of transcripts was based on GENCODE v27 (Frankish et al., 2019) and for each gene with multiple isoforms, the longest transcript was used.
Reads from yeast ribosome profiling data were processed as follows: First, the adaptor was trimmed for each read. Reads with quality score less than 20 at any position or that mapped to known noncoding RNAs were removed. The remaining reads were then mapped to the yeast reference genome (Engel et al., 2014) using HISAT2. Only reads between 25 – 34 nt were used for subsequent analyses. Annotation of transcripts was based on a published dataset (Nagalakshmi et al., 2008).
Periodicity analysis (Figure S3)
For each mapped read in the human dataset, the codon occupying the P-site of the ribosome was defined as nucleotides 13–15 from the 5′ end of the mapped read. This offset length was calculated by identifying the modal distance between the 5′ end of reads mapped to annotated translation start sites and the corresponding AUG start codons. Ribosome occupancy at each position was calculated by determining the number of reads whose 5′ ends mapped 12 nt upstream, and thus represented P-site occupancy of the ribosome in all analyses. The numbers of reads at each position (corresponding to P-site ribosome occupancy at that position) were normalized to the total number of mapped reads per sample (in millions) to yield reads per million mapped reads (RPM). Normalized read counts (RPM) at each position were aggregated across all transcripts to generate periodicity plots.
Meta-codon plots (Figures 2C and 2G)
Proline-proline (PP) motifs from all coding sequence (CDS) regions were considered, and those with another PP motif located within 50 nt upstream were discarded. RPM values were aggregated at each position within a 100 nt region centered on PP motifs and the values at the first nucleotide of each codon (corresponding to in-frame reads) were plotted.
A similar analysis was performed to generate a meta-codon plot centered on the top 20 tripeptide pause motifs in EIF5A−/− cells compared to sgNT. Identification of those tripeptides is described below.
Pause motif analysis (Figures 2D–2F)
Genes whose CDS fulfilled a minimum coverage requirement of at least 128 reads in each replicate of each sample were used in this analysis of human ribosome profiling data. The first 100 nt and the last 100 nt of the CDS were excluded from consideration in pause motif calculations. The ratio of average ribosome occupancy at each codon triplet (tripeptide motif) to average ribosome occupancy of the CDS was calculated and defined as the pause score. The average pause score for each tripeptide motif across all considered CDSs was calculated. Tripeptides that occurred > 50 times in the considered CDSs, and whose average pause score in EIF5A−/− samples was > 3 in both replicates (listed in Figure 2F) were used to construct a position weighted matrix, where tripeptides were weighted by the ratio of their average pause score in EIF5A−/− to sgNT. An identical analysis was performed to identify a tripeptide pause motif signature in DOHH−/− cells.
CDF plots (Figures 2A, 2B, 3A, 3C, S4B, 5B–5D, and S5B–S5D)
For reads obtained from yeast ribosome profiling data, the 15th nucleotide from the 3′ end was defined as the first nucleotide of the P-site codon (offset calculated from reads mapping to annotated start codons, as described above). For both human and yeast datasets, reads were assigned to the 5′ UTR or CDS based on the location of the calculated P-site. Reads whose P-site mapped 3 nt upstream or downstream of the start codon were not utilized for these analyses. Reads per kilobase per million mapped reads, or RPKM, was used to represent ribosome occupancy of a 5′ UTR or CDS.
For CDF plots generated from human ribosome profiling data comparing EIF5A−/− to sgNT, genes that were covered by at least 128 reads in the RNA-seq experiment in both sgNT or in both EIF5A−/− samples, and that additionally fulfilled the following coverage criteria were used in the analysis: For plots calculating CDS ribosome occupancy, genes covered by at least 128 reads in the CDS in both sgNT samples or in both EIF5A−/− samples were included. For CDF plots calculating 5′ UTR translation, genes whose 5′ UTRs were covered by at least 16 reads in both sgNT samples or in both EIF5A−/− samples were included. Similar coverage requirements were applied when comparing DOHH−/− to sgNT samples. For CDF plots comparing EIF5A knockdown (KD) to WT yeast, genes whose CDS was covered by at least 64 reads in both WT or in both EIF5A KD samples, and genes whose 5′ UTRs were covered by at least 6 reads in both WT or both EIF5A KD samples were included.
DATA AND CODE AVAILABILITY
All high-throughput sequencing data generated in this study (CRISPR screening, RNA-seq, and ribosome profiling) have been deposited in GEO under accession GEO: GSE132010.
Supplementary Material
Highlights.
A CRISPR screen reveals that eIF5A regulates start codon selection on MYC mRNA
Ribosome profiling demonstrates that eIF5A suppresses ribosomal pausing in human cells
Loss of eIF5A causes widespread translation initiation in 5′ UTRs in human and yeast
Ribosomal pausing in eIF5A-deficient cells promotes upstream translation initiation
ACKNOWLEDGMENTS
We thank Rudolph Jaenisch, Didier Trono, and Feng Zhang and for plasmids; Vanessa Schmid, Rachel Bruce, and Caitlin Eaton at the McDermott Center Next Generation Sequencing Core for assistance with high-throughput sequencing; Angie Mobley and the University of Texas (UT) Southwestern (UTSW) Flow Cytometry Core facility for FACS; Frank Gillett for experimental assistance; and Stephen Johnson for software implementation. We also thank Michael Buszczak, Kathryn O’Donnell, and members of the Mendell laboratory for helpful comments on the manuscript. This work was supported by grants from CPRIT (RP160249 to J.T.M. and RP150596 for the UTSW Bioinformatics Core Facility), the NIH (R35CA197311, P30CA142543, and P50CA196516 to J.T.M.), and the Welch Foundation (I-1961-20180324 to J.T.M.). J.T.M. is an investigator of the Howard Hughes Medical Institute.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPORTING CITATIONS
The following references appear in the Supplemental Information: Carmona-Saez et al. (2007); Huang et al. (2009); Nogales-Cadenas et al. (2009); Tabas-Madrid et al. (2012).
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.129.
REFERENCES
- Andreev DE, O’Connor PBF, Fahey C, Kenny EM, Terenin IM, Dmitriev SE, Cormican P, Morris DW, Shatsky IN, and Baranov PV (2015). Translation of 5′ leaders is pervasive in genes resistant to eIF2 repression. eLife 4, e03971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki H, Dekany K, Adams SL, and Ganoza MC (1997). The gene encoding the elongation factor P protein is essential for viability and is required for protein synthesis. J. Biol. Chem 272, 32254–32259. [DOI] [PubMed] [Google Scholar]
- Asano K (2014). Why is start codon selection so precise in eukaryotes? Translation (Austin) 2, e28387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann AS, and Geerts D (2018). Polyamine synthesis as a target of MYC oncogenes. J. Biol. Chem 293, 18757–18769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benassayag C, Montero L, Colombié N, Gallant P, Cribbs D, and Morello D (2005). Human c-Myc isoforms differentially regulate cell growth and apoptosis in Drosophila melanogaster. Mol. Cell. Biol 25, 9897–9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar GA, and Weissman JS (2015). Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat. Rev. Mol. Cell Biol 16, 651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buskirk AR, and Green R (2017). Ribosome pausing, arrest and rescue in bacteria and eukaryotes. Philos. Trans. R. Soc. Lond. B Biol. Sci 372, 20160183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano VSP, Jeon GA, Johansson HE, Henderson CA, Park JH, Valentini SR, Hershey JWB, and Park MH (2008). Mutational analyses of human eIF5A-1—identification of amino acid residues critical for eIF5A activity and hypusine modification. FEBS J. 275, 44–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona-Saez P, Chagoyen M, Tirado F, Carazo JM, and Pascual-Montano A (2007). GENECODIS: a web-based tool for finding significant concurrent annotations in gene lists. Genome Biol. 8, R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casero RA Jr., and Marton LJ (2007). Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov 6, 373–390. [DOI] [PubMed] [Google Scholar]
- Chan PP, and Lowe TM (2016). GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 44 (D1), D184–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper HL, Park MH, Folk JE, Safer B, and Braverman R (1983). Identification of the hypusine-containing protein hy+ as translation initiation factor eIF-4D. Proc. Natl. Acad. Sci. U S A 80, 1854–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani C, Blanchard JM, Piechaczyk M, El Sabouty S, Marty L, and Jeanteur P (1984). Extreme instability of myc mRNA in normal and transformed human cells. Proc. Natl. Acad. Sci. U S A 81, 7046–7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever TE, and Ivanov IP (2018). Roles of polyamines in translation. J. Biol. Chem 293, 18719–18729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever TE, Gutierrez E, and Shin BS (2014). The hypusine-containing translation factor eIF5A. Crit. Rev. Biochem. Mol. Biol 49, 413–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerfel LK, Wohlgemuth I, Kothe C, Peske F, Urlaub H, and Rodnina MV (2013). EF-P is essential for rapid synthesis of proteins containing consecutive proline residues. Science 339, 85–88. [DOI] [PubMed] [Google Scholar]
- Engel SR, Dietrich FS, Fisk DG, Binkley G, Balakrishnan R, Costanzo MC, Dwight SS, Hitz BC, Karra K, Nash RS, et al. (2014). The reference genome sequence of Saccharomyces cerevisiae: then and now. G3 (Bethesda) 4, 389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell AS, and Sears RC (2014). MYC degradation. Cold Spring Harb. Perspect. Med 4, a014365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47 (D1), D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser SD, Wilkes-Johnston J, and Browder LW (1996). Effects of c-myc first exons and 5′ synthetic hairpins on RNA translation in oocytes and early embryos of Xenopus laevis. Oncogene 12, 1223–1230. [PubMed] [Google Scholar]
- Gabay M, Li Y, and Felsher DW (2014). MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med 4, a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden RJ, Chen B, Li T, Braun J, Manjunath H, Chen X, Wu J, Schmid V, Chang TC, Kopp F, et al. (2017). An Argonaute phosphorylation cycle promotes microRNA-mediated silencing. Nature 542, 197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregio APB, Cano VPS, Avaca JS, Valentini SR, and Zanelli CF (2009). eIF5A has a function in the elongation step of translation in yeast. Biochem. Biophys. Res. Commun 380, 785–790. [DOI] [PubMed] [Google Scholar]
- Gutierrez E, Shin BS, Woolstenhulme CJ, Kim JR, Saini P, Buskirk AR, and Dever TE (2013). eIF5A promotes translation of polyproline motifs. Mol. Cell 51, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hann SR, King MW, Bentley DL, Anderson CW, and Eisenman RN (1988). A non-AUG translational initiation in c-myc exon 1 generates an N-terminally distinct protein whose synthesis is disrupted in Burkitt’s lymphomas. Cell 52, 185–195. [DOI] [PubMed] [Google Scholar]
- Hann SR, Dixit M, Sears RC, and Sealy L (1994). The alternatively initiated c-Myc proteins differentially regulate transcription through a noncanonical DNA-binding site. Genes Dev. 8, 2441–2452. [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG (2011). Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev 75, 434–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG, Ivanov IP, and Sonenberg N (2016). Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science 352, 1413–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, Katibah GE, Amora R, Boydston EA, Zeitler B, et al. (2009). Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol 27, 851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins BD, Fine B, Steinbach N, Dendy M, Rapp Z, Shaw J, Pappas K, Yu JS, Hodakoski C, Mense S, et al. (2013). A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 341, 399–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, et al. (2012). The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Ingolia NT (2016). Ribosome footprint profiling of translation throughout the genome. Cell 165, 22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, and Weissman JS (2012). The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc 7, 1534–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov IP, Shin BS, Loughran G, Tzani I, Young-Baird SK, Cao C, Atkins JF, and Dever TE (2018). Polyamine control of translation elongation regulates start site selection on antizyme inhibitor mRNA via ribosome queuing. Mol. Cell 70, 254–264.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CU, and Pestova TV (2010). The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol 11, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse MG, and Wilusz JE (2017). Non-AUG translation: a new start for protein synthesis in eukaryotes. Genes Dev. 31, 1717–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse MG, Goldman DH, Choi J, Nwaezeapu C, Liang D, Green KM, Goldstrohm AC, Todd PK, Green R, and Wilusz JE (2019). Ribosome queuing enables non-AUG translation to be resistant to multiple protein synthesis inhibitors. Genes Dev. 33, 871–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly K, Cochran BH, Stiles CD, and Leder P (1983). Cell-specific regulation of the c-myc gene by lymphocyte mitogens and platelet-derived growth factor. Cell 35, 603–610. [DOI] [PubMed] [Google Scholar]
- Kemper WM, Berry KW, and Merrick WC (1976). Purification and properties of rabbit reticulocyte protein synthesis initiation factors M2Balpha and M2Bbeta. J. Biol. Chem 251, 5551–5557. [PubMed] [Google Scholar]
- Kim D, Langmead B,and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M (1990). Downstream secondary structure facilitates recognition of initiator codons by eukaryotic ribosomes. Proc. Natl. Acad. Sci. U S A 87, 8301–8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuksa PP, Amlie-Wolf A, Katanić Ž, Valladares O, Wang LS, and Leung YY (2019). DASHR 2.0: integrated database of human small non-coding RNA genes and mature products. Bioinformatics 35, 1033–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AS, Kranzusch PJ, and Cate JH (2015). eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature 522, 111–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, He S, Yang J, Jia X, Wang P, Chen X, Zhang Z, Zou X, McNutt MA, Shen WH, and Yin Y (2014). PTENa, a PTEN isoform translated through alternative initiation, regulates mitochondrial function and energy metabolism. Cell Metab. 19, 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Chen X, Yin Q, Ruan D, Zhao X, Zhang C, McNutt MA, and Yin Y (2017). PTENβ is an alternatively translated isoform of PTEN that regulates rDNA transcription. Nat. Commun 8, 14771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Han Y, and Qian SB (2013). Cotranslational response to proteotoxic stress by elongation pausing of ribosomes. Mol. Cell 49, 453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Chen C, Xu Z, Scuoppo C, Rillahan CD, Gao J, Spitzer B, Bosbach B, Kastenhuber ER, Baslan T, et al. (2016). Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature 531, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magdolen V, Klier H, Wöhl T, Klink F, Hirt H, Hauber J, and Lottspeich F (1994). The function of the hypusine-containing proteins of yeast and other eukaryotes is well conserved. Mol. Gen. Genet 244, 646–652. [DOI] [PubMed] [Google Scholar]
- McGlincy NJ, and Ingolia NT (2017). Transcriptome-wide measurement of translation by ribosome profiling. Methods 126, 112–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick WC, and Pavitt GD (2018). Protein synthesis initiation in eukaryotic cells. Cold Spring Harb. Perspect. Biol 10, a033092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossmann D, Park S, and Hall MN (2018). mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 18, 744–757. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, and Snyder M (2008). The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320, 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandagopal N, and Roux PP (2015). Regulation of global and specific mRNA translation by the mTOR signaling pathway. Translation (Austin) 3, e983402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales-Cadenas R, Carmona-Saez P, Vazquez M, Vicente C, Yang X, Tirado F, Carazo JM, and Pascual-Montano A (2009). GeneCodis: interpreting gene lists through enrichment analysis and integration of diverse biological information. Nucleic Acids Res. 37, W317–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MH, and Wolff EC (2018). Hypusine, a polyamine-derived amino acid critical for eukaryotic translation. J. Biol. Chem 293, 18710–18718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MH, Joe YA, and Kang KR (1998). Deoxyhypusine synthase activity is essential for cell viability in the yeast Saccharomyces cerevisiae. J. Biol. Chem 273, 1677–1683. [DOI] [PubMed] [Google Scholar]
- Parkin N, Darveau A, Nicholson R, and Sonenberg N (1988). cis-acting translational effects of the 5′ noncoding region of c-myc mRNA. Mol. Cell. Biol 8, 2875–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov MY, Watts RE, Tan Z, Cornish VW, Ehrenberg M, and Forster AC (2009). Slow peptide bond formation by proline and other N-alkylamino acids in translation. Proc. Natl. Acad. Sci. U S A 106, 50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peil L, Starosta AL, Lassak J, Atkinson GC, Virumäe K, Spitzer M, Tenson T, Jung K, Remme J, and Wilson DN (2013). Distinct XPPX sequence motifs induce ribosome stalling, which is rescued by the translation elongation factor EF-P. Proc. Natl. Acad. Sci. U S A 110, 15265–15270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, and Alepuz P (2017). eIF5A facilitates translation termination globally and promotes the elongation of many non polyproline-specific tripeptide sequences. Nucleic Acids Res. 45, 7326–7338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, and Glöckner FO (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, and Topisirovic I (2018). Signaling pathways involved in the regulation of mRNA translation. Mol. Cell. Biol 38, e00070–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini P, Eyler DE, Green R, and Dever TE (2009). Hypusine-containing protein eIF5A promotes translation elongation. Nature 459, 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Cong L, Zhou Y, Cunniff MM, Feng G, and Zhang F (2012). A transcription activator-like effector toolbox for genome engineering. Nat. Protoc 7, 171–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Masuda T, Hu Q, Tobo T, Gillaspie S, Niida A, Thornton M, Kuroda Y, Eguchi H, Nakagawa T, et al. (2019). Novel oncogene 5MP1 reprograms c-Myc translation initiation to drive malignant phenotypes in colorectal cancer. EBioMedicine 44, 387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnier J, Schwelberger HG, Smit-McBride Z, Kang HA, and Hershey JW (1991). Translation initiation factor 5A and its hypusine modification are essential for cell viability in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol 11, 3105–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller AP,Wu CC, Dever TE, Buskirk AR, and Green R (2017). eIF5A functions globally in translation elongation and termination. Mol. Cell 66, 194–205.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scuoppo C, Miething C, Lindqvist L, Reyes J, Ruse C, Appelmann I, Yoon S, Krasnitz A, Teruya-Feldstein J, Pappin D, et al. (2012). A tumour suppressor network relying on the polyamine-hypusine axis. Nature 487, 244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears RC (2004). The life cycle of C-myc: from synthesis to degradation. Cell Cycle 3, 1133–1137. [PubMed] [Google Scholar]
- Seibler J, Kleinridders A, Küter-Luks B, Niehaves S, Brüning JC, and Schwenk F (2007). Reversible gene knockdown in mice using a tight, inducible shRNA expression system. Nucleic Acids Res. 35, e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sendoel A, Dunn JG, Rodriguez EH, Naik S, Gomez NC, Hurwitz B, Levorse J, Dill BD, Schramek D, Molina H, et al. (2017). Translation from unconventional 5′ start sites drives tumour initiation. Nature 541, 494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, and Zhang F (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalgi R, Hurt JA, Krykbaeva I, Taipale M, Lindquist S, and Burge CB (2013). Widespread regulation of translation by elongation pausing in heat shock. Mol. Cell 49, 439–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin BS, Katoh T, Gutierrez E, Kim JR, Suga H, and Dever TE (2017). Amino acid substrates impose polyamine, eIF5A, or hypusine requirement for peptide synthesis. Nucleic Acids Res. 45, 8392–8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starck SR, Tsai JC, Chen K, Shodiya M, Wang L, Yahiro K, Martins-Green M, Shastri N, and Walter P (2016). Translation from the 5′ untranslated region shapes the integrated stress response. Science 351, aad3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas-Madrid D, Nogales-Cadenas R, and Pascual-Montano A (2012). GeneCodis3: a non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res. 40, W478–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang HL, Yeh LS, Chen NK, Ripmaster T, Schimmel P, and Wang CC (2004).Translation of a yeast mitochondrial tRNA synthetase initiated at redundant non-AUG codons. J. Biol. Chem 279, 49656–49663. [DOI] [PubMed] [Google Scholar]
- Tang L, Morris J, Wan J, Moore C, Fujita Y, Gillaspie S, Aube E, Nanda J, Marques M, Jangal M, et al. (2017). Competition between translation initiation factor eIF5 and its mimic protein 5MP determines non-AUG initiation rate genome-wide. Nucleic Acids Res. 45, 11941–11953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touriol C, Bornes S, Bonnal S, Audigier S, Prats H, Prats AC, and Vagner S (2003). Generation of protein isoform diversity by alternative initiation of translation at non-AUG codons. Biol. Cell 95, 169–178. [DOI] [PubMed] [Google Scholar]
- Truitt ML, and Ruggero D (2016). New frontiers in translational control of the cancer genome. Nat. Rev. Cancer 16, 288–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunca B, Tezcan G, Cecener G, Egeli U, Zorluoglu A, Yilmazlar T, Ak S, Yerci O, Ozturk E, Umut G, and Evrensel T (2013). Overexpression of CK20, MAP3K8 and EIF5A correlates with poor prognosis in early-onset colorectal cancer patients. J. Cancer Res. Clin. Oncol 139, 691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzani I, Ivanov IP, Andreev DE, Dmitriev RI, Dean KA, Baranov PV, Atkins JF, and Loughran G (2016). Systematic analysis of the PTEN 5′ leader identifies a major AUU initiated proteoform. Open Biol. 6, 150203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ude S, Lassak J, Starosta AL, Kraxenberger T, Wilson DN, and Jung K (2013). Translation elongation factor EF-P alleviates ribosome stalling at polyproline stretches. Science 339, 82–85. [DOI] [PubMed] [Google Scholar]
- Ventoso I, Sanz MA, Molina S, Berlanga JJ, Carrasco L, and Esteban M (2006).Translational resistance of late alphavirus mRNA to eIF2α phosphorylation: a strategy to overcome the antiviral effect of protein kinase PKR. Genes Dev. 20, 87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolstenhulme CJ, Guydosh NR, Green R, and Buskirk AR (2015). High-precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep. 11, 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeilding NM, Rehman MT, and Lee WM (1996). Identification of sequences in c-myc mRNA that regulate its steady-state levels. Mol. Cell. Biol 16, 3511–3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, and Dietrich FS (2005). Identification and characterization of upstream open reading frames (uORF) in the 5′ untranslated regions (UTR) of genes in Saccharomyces cerevisiae. Curr. Genet 48, 77–87. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All high-throughput sequencing data generated in this study (CRISPR screening, RNA-seq, and ribosome profiling) have been deposited in GEO under accession GEO: GSE132010.