

Abstract
Post-translational modifications of histone proteins regulate all biological processes requiring access to DNA. Monoubiquitination of histone H2B is a mark of actively transcribed genes in all eukaryotes that also plays a role in DNA replication and repair. Solution and structural studies of the mechanism by which histone ubiquitination modulates these processes depend on the ability to generate homogeneous preparations of nucleosomes containing ubiquitin conjugated to a specific lysine residue. We describe here methods for generating milligram quantities of histone H2B with ubiquitin (Ub) conjugated to Lys 120 via either a non-hydrolyzable, dichloroacetone linkage or a cleavable isopeptide bond. H2B-Ub with an isopeptide linkage is generated by a combination of intein-fusion protein derivatization and native chemical ligation, yielding a fully native ubiquitinated lysine that can be cleaved by Ub isopeptidases. We also describe how to reconstitute nucleosomes containing ubiquitinated H2B.
Keywords: histone modifications, intein, ubiquitin, chromatin
1. Introduction
Eukaryotic genomes are packaged into a nucleoprotein complex knows as chromatin, which organizes the genome within the confines of the nucleus and regulates access to DNA. The fundamental organizational unit of chromatin is the nucleosome, which comprises ~146 base pairs of duplex DNA wrapped around a protein core containing two copies each of histones H2A, H2B, H3 and H4 (Andrews & Luger, 2011). Histone proteins are decorated with post-translational modifications such as acetylation, methylation, and ubiquitination, forming a language of genomic metadata known as the histone code (Jenuwein & Allis, 2001). These histone marks are deposited by “writer” enzymes, “read” by other chromatin-binding proteins that bind to the histone mark and removed by “eraser” enzymes. The impracticality of purifying homogeneously modified histones from endogenous sources presents a barrier to biochemical and structural studies of individual histone modifications. Fortunately, semi-synthetic protein chemistry methodologies have emerged to efficiently construct uniformly modified histone proteins.
Small histone side-chain modifications such as acetylation, methylation, and phosphorylation can, in some cases, be mimicked by side-chain substitutions, incorporated using amber codon suppression (Chin et al., 2003) or introduced by assembly of peptides generated by solid-phase synthesis (Bondalapati, Jbara, & Brik, 2016). Site-specific incorporation of ubiquitin (Ub), however, represents a greater challenge, as it involves covalent attachment of the C-terminal carboxylate of the 76-amino acid Ub protein to a lysine residue via an amide (isopeptide) linkage (Holt & Muir, 2015; Jbara, Sun, Kamnesky, & Brik, 2018; Kumar, Spasser, Ohayon, Erlich, & Brik, 2011; Long, Furgason, & Yao, 2014). Histone H2B ubiquitinated at lysine 120 (in humans and X. laevis; lysine 123 in yeast) is a hallmark of actively transcribed genes and is required for histone H3K4 and H3K79 trimethylation (Weake & Workman, 2008) – two other marks of active transcription. H2B-Ub also plays a role in regulating nucleosome assembly during DNA replication to stimulate homologous recombination (HR) and replication fork progression, promoting DNA damage tolerance (Hung, Wong, Ulrich, & Kao, 2017). H2A monoubiquitinated at lysine 119, by contrast, is a mark of transcriptionally silent heterochromatin (Wang et al., 2004). Studies of the mechanism by which ubiquitinated nucleosomes interact with a variety of enzymes and other cellular proteins, as well as the effect of monoubiquitination on chromatin compaction, require methods for generating ubiquitinated nucleosomes at a scale and homogeneity that makes it possible to carry out in vitro biochemistry and structural studies. While this article focuses on methods to generate nucleosomes containing ubiquitinated histone H2B, these approaches also can be used to generate nucleosomes with H2A-Ub (Jbara, Laps, et al., 2018).
A number of methods have been developed for generating homogeneously ubiquitinated histone H2B containing a fully native ubiquitinated lysine, cleavable analogues of ubiquitinated lysine or a non-hydrolyzable mimic (Jbara, Sun, et al., 2018). Cleavable H2B-Ub is essential for studies of the relevant deubiquitinating enzymes, while non-cleavable linkages can be used to study binding of nucleosomes containing H2B-Ub to deubiquitinating enzymes or other proteins that interact with ubiquitinated nucleosomes. Muir and colleagues (Chatterjee, McGinty, Pellois, & Muir, 2007) first reported a semi-synthetic method for generating H2B-Ub from intein-derivatized protein fragments Ub(1-75) and H2B(1-116), plus a synthetic peptide that encodes the rest of the H2B C-terminus and contains a photocleavable protective group at the ubiquitination site (K120). This reagent, which contains a fully native ubiquitinated lysine, was used in biochemical assays to produce direct evidence that Dot1L methylation of H3K79 is stimulated by the presence of H2B-Ub (McGinty, Kim, Chatterjee, Roeder, & Muir, 2008).
This semisynthetic approach has been further developed to make it possible to generate milligram quantities of H2B-Ub while eliminating the need for photochemistry (Kumar et al., 2011). Multiple groups have recently developed new linkage chemistries for site-specific ubiquitination, including non-cleavable and activity-based linkages used to study DUB activity on PCNA (Gong et al., 2018; Yang et al., 2016) and hydrazide chemistry that produces modified lysine analogs that mimic methyllysine, acetyllysine, and ubiquitinated lysine (Bhat et al., 2018). Another non-cleavable mimic of the isopeptide linkage can be generated by substituting both the C-terminal Ub glycine and H2B-K120 with cysteine and then cross-linking the cysteines with 1,3-dichloroacetone (DCA) (Long, Furgason, et al., 2014; Wiener, Zhang, Wang, & Wolberger, 2012; Yin, Krantz, Russell, Deshpande, & Wilkinson, 2000). An advantage of the DCA linkage is that it is chemically stable in the presence of reducing agents, which is important for studies of proteins that require reducing agent for stability. Cross-linking Ub to lysine residues using DCA was first introduced by Wilkinson as a non-hydrolyzable isopeptide mimic that can be used to affinity purify DUBs (Yin et al., 2000) and has been used in several studies (Borodovsky et al., 2001; Morgan & Wolberger, 2018; Wiener et al., 2012; Wilson et al., 2016; Zukowski, Al-Afaleq, Duncan, Yao, & Johnson, 2018).
2. Preparation of dichloroacetone-linked H2B-Ub
Nucleosomes containing H2B-Ub that is linked by a non-hydrolyzable DCA bond were used to determine the structure of the heterotetrameric SAGA deubiquitinating module bound to a nucleosome containing H2B-Ub (Morgan et al., 2016). A similar strategy has been used to create various diubiquitin linkages (Yin et al., 2000), H2AK119-Ub (Long, Furgason, et al., 2014), and H2AK15-Ub (Wilson et al., 2016). In the SAGA DUB module structure (Morgan et al., 2016), substitution of the active site cysteine of the Ubp8 subunit with alanine caused the DUB module to form a stable complex with H2B-UbDCA nucleosomes that could be purified by size-exclusion chromatography, whereas the wild type enzyme did not form a stable complex. A subsequent study (Morrow et al., 2018) revealed that the cysteine to alanine substitution removes a steric block in the active site, thus allowing the free carboxylate in the Ub C-terminus to form hydrogen bonds with neighboring backbone residues. Several other cysteine protease deubiquitinating enzymes bearing a cysteine to alanine substitution also bind tightly to substrate (Morrow et al., 2018), suggesting that this active site mutation could be exploited to drive tight binding of other DUBs to ubiquitinated nucleosomes containing the non-hydrolyzable DCA linkage.
We describe here the protocol that was used to generate DCA-linked H2B-Ub that was then incorporated into nucleosomes and used for structural studies (Morgan et al., 2016). Proteins containing cysteine substitutions in H2B, H2BK120C, and Ub, UbG76C, are used to generate the final H2B-Ub. In order to facilitate later purification steps, an N-terminal hexahistidine tag followed by a Tobacco Etch Virus (TEV) protease cleavage site was added to Ub-G76C. Since DCA will cross-link any two proteins bearing cysteines (Yin et al., 2000), a mix of hetero- and homodimers will result from any cross-linking reaction. When using DCA to crosslink Ub and histone H2B (each containing a single cysteine), one should expect the H2B-Ub species to be formed at a 20% yield. The Ub and histone will also form crosslinked homodimers that must be removed by subsequent purification steps (see Figure 1), resulting in a final yield of ~10% of the starting material. Since both the H2B and Ub mutant can be easily expressed and purified, we recommend working at a large scale in order to overcome the inherent inefficiency of the reaction. We suggest using ~100 mg of H2BK120C and ~80 mg of His6TEV-UbG76C of starting material (both quantities are easily obtained from a single preparation of each protein).
Figure 1:
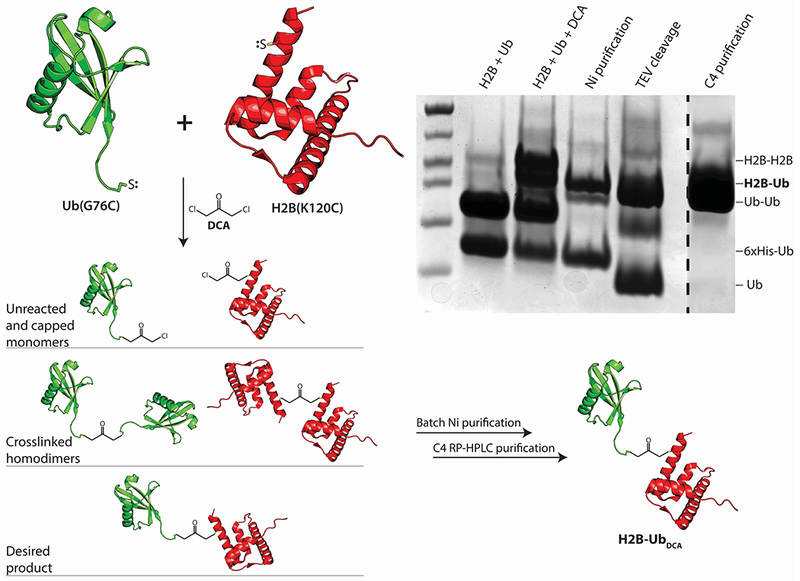
Overview schematic describing the crosslinking of His6TEV-UbG76C (green) and H2BK120C (red) using 1,3-dichloroacetone (DCA). Each protein must be separately purified, then crosslinked under conditions favoring cysteine deprotonation. The crosslinking reaction produces a mixture of H2B-UbDCA, crosslinked H2BK120C and His6TEV-UbG76C dimers, and unreacted monomers that must be purified by the protocol described in Section 2.
In the case of H2B-UbDCA, His6TEV-UbG76C and H2BK120C are separately purified and then crosslinked with 1,3-dichloroacetone at a pH close to the pKa of cysteine (pH 8.1). For H2BK120C, the histone is purified under denaturing conditions using the same purification protocol for recombinantly expressed wild-type histones, as described previously (Luger, Rechsteiner, & Richmond, 1999). In brief, H2BK120C is expressed in bacterial inclusion bodies, which are washed, then unfolded in Guanidinium-HCl and exchanged into a urea buffer by size exclusion chromatography. H2BK120C is further purified using cation exchange chromatography under denaturing conditions, followed by exhaustive dialysis to remove the urea, lyophilization, and storage. Readers should refer to the protocol described in Luger, Rechsteiner, & Richmond, 1999 for details. The plasmid encoding H2BK120C based on the Xenopus laevis sequence is available from the Addgene repository (plasmid #75332). The next section describes the purification of His6TEV-UbG76C, followed by a protocol to purify H2B-UbDCA from the unwanted products that are the crosslinked Ub dimers, crosslinked histone dimers, and unreacted monomers.
2.1. Expression and purification of His-tagged UbG76C
Hexahistidine-tagged Ub containing a cysteine substitution at G76 (His6TEV-UbG76C) enables site-specific crosslinking of Ub to residue 120 on H2B, with the cleavable tag providing a useful purification step after crosslinking. The plasmid encoding His6TEV-UbG76C can be obtained from the Addgene repository (plasmid #75299). This construct features a tobacco etch virus (TEV) protease cleavage site (ENLYFQGS) with an N-terminal extension for optimal cleavage (DYDIPTT). Cleavage with TEV protease produces a 2 amino acid scar (GS) at the N-terminus of UbG76C. See Figure 1 for an overview of the protocol below, and the expected outcomes from each step of the reaction and purification.
2.1.1. Equipment
Temperature-controlled shaker and flasks for cell growth
Centrifuge and bottles
Gravity flow column body (at least 50 mL)
Microfluidizer, sonicator, or French press
HisPur Ni-NTA resin (Thermo Fisher), gravity flow column body, and stopcock or HisTrap HP column (GE Healthcare) if using an AKTA (GE Life Sciences) for affinity purification step
0.22 µm sterile filters
Dialysis tubing (3-8 kD molecular weight cutoff (MWCO))
Centrifugal concentrators (3-10 kD MWCO)
HiTrap SP-XL column (5 mL), AKTA FPLC (GE Life Sciences), and fraction collector
HPLC and semi-preparative C4 RP-HPLC column (such as Higgins Analytical, PROTO C4 5 μm 250x10mm)
2.1.2. Buffers and reagents
BL21(DE3) Rosetta2-pLysS transformation-competent E. coli cells
Ni-NTA Lysis Buffer: 50 mM Tris-HCl pH 7.8, 500 mM NaCl, 20 mM Imidazole, 1 mM phenylmethane sulfonyl fluoride (PMSF), and 5 mM 2-mercaptoethanol (BME)
Ni-NTA Elution Buffer: 50 mM Tris-HCl pH 7.8, 500 mM NaCl, 150 mM Imidazole, 1 mM PMSF, and 5 mM BME
SP Column Buffer A: 50 mM ammonium acetate (pH 4.5), 1 mM DTT
SP Column Buffer B: 50 mM ammonium acetate (pH 4.5), 1 M NaCl, 1 mM DTT
2.1.3. Procedure
BL21(DE3)Rosetta2-pLysS E. coli cells are transformed with the His6TEV-UbG76C plasmid using standard methods and colonies are grown on agar plates containing ampicillin and chloramphenicol (Amp/Chlor).
~20 colonies are picked and grown in a 100 mL starter culture for approximately 2 hours to an OD600 of 0.4, then distributed evenly between 12 x 1 L flasks each containing 1 L of sterile LB supplemented with a final concentration of 100 µg/mL Amp and 25 µg/mL Chlor. Each flask should be grown to OD600 of ~0.8 and then induced with a final concentration of 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) and incubated with shaking at 16°C overnight.
Cells expressing His6TEV-UbG76C are collected by centrifugation at 5000 x g for 30 minutes, followed by resuspension in Ni-NTA lysis buffer. If not immediately lysing the cells, store at −80°C.
Lyse cells using sonication, French press, or microfluidizer according to established protocols. (See Note 1). Immediately centrifuge the lysate at 27000 x g for 30 minutes and collect the soluble fraction, which contains His6TEV-UbG76C. Filter the lysate with a 0.22 µm cutoff filter.
Equilibrate 10 mL HisPur Ni-NTA resin (Thermo Fisher) with Ni-NTA lysis buffer and incubate with soluble lysate at 4°C for 1 hour to bind the protein to the resin.
Wash the resin with 5 column volumes (CV) of lysis buffer to remove residual proteins. (See Note 2)
Elute bound proteins from the resin with lysis buffer supplemented with 150 mM imidazole with 5 x 1 CV elution fractions.
Dialyze the eluant containing hexahistidine-tagged Ub (G76C) into 4 L of 50 mM ammonium acetate pH 4.5 and 1 mM DTT at 4°C overnight (See Note 3).
Filter the resultant dialysate and load onto a HiTrap SP-HP column (GE Healthcare), followed by elution with a 0-1M NaCl gradient in 50 mM ammonium acetate pH 4.5 and 1 mM DTT.
Dialyze peak fractions in 4L of 10 mM Tris-HCl pH 8 and 0.5 mM tris(2-carboxyethyl)phosphine (TCEP) overnight, recover the sample, and concentrate (2 mM is a useful working concentration).
2.2. DCA Linkage reaction
2.2.1. Buffers and reagents
His6TEV-UbG76C purified as in section 2.1
X. laevis H2BK120C purified as described (Luger et al., 1999)
Dimethyl formamide (DMF)
2 mM 1,3-dichloroacetone (DCA) in DMF (see Note 4)
2x DxR buffer: 100 mM Borate pH 8.1, 2 mM TCEP
Degassed water
2.2.2. Procedure
To generate H2B-Ub with a non-hydrolyzable dichloroacetone (DCA) cross-link between cysteine residues of His6TEV-UbG76C and H2BK120C, it is important to recognize that redox states determine the effectiveness of the crosslinking reaction. Solutions should therefore be exhaustively degassed so that cysteines are fully reduced. Alternative approaches, such as preparing the materials in an argon environment, are also effective; however, we find that degassing solutions with a stir bar under vacuum is sufficient.
Separately degas water and 2x DxR buffer (overnight)
Mix H2B-K120C and His6TEV-UbG76C at a final concentration of 100 µM each, with half of the final volume composed of 2x DxR buffer. Use the degassed water to dilute the mixture to 1x DxR buffer (50 mM Borate pH 8.1, 1 mM TCEP). and incubate in a water bath at 50°C for 1 hour to reduce cysteines. Cool on ice for 1 hour.
Add 2 mM DCA dissolved in dimethyl formamide (DMF) to a final concentration of 100 μM and incubate for 1 hour on ice. After addition of the DCA, ensure homogeneity by gently inverting the tube 5 times.
The reaction is quenched with 50 mM β-mercaptoethanol, frozen, and lyophilized until further purification. The resultant product contains a mixture of the desired H2B-UbDCA, crosslinked H2B-K120C and His6TEV-UbG76C dimers, and unreacted histone and ubiquitin.
2.3. Purification of H2B-UbDCA
2.3.1. Buffers and reagents
9 M urea (deionized, see Note 5)
Ni-NTA-6MU: 6 M deionized Urea, 50 mM Tris-HCl pH 7.8, 500 mM NaCl, 10 mM β-mercaptoethanol, 20 mM imidazole, and 0.1 mM PMSF
TEV protease (purchased or purified from Addgene plasmid #8831 following the protocol in Kapust et al (Kapust et al., 2001))
TEV cleavage buffer: 50 mM Tris-HCl pH 8.0, 0.5 mM EDTA, and 1 mM DTT
C4 solvent A: 0.1% trifluoroacetic acid (TFA) in water
C4 solvent B: 0.1% TFA in 90% acetonitrile (ACN)
7 M guanidine-HCl
2.3.2. Procedure
Take the lyophilized reaction mixture, which contains H2B-UbDCA, Ub dimers, H2B dimers, and unreacted proteins, and resuspend the mixture in Ni-NTA-6MU buffer.
Apply the sample to a Ni-NTA column equilibrated in the same buffer, retaining all products that contain the His-tagged Ub while allowing H2B monomers and cross-linked H2B dimers to flow through.
Elute bound proteins with Ni-NTA-6MU supplemented with 150 mM imidazole.
Dialyze the resultant mixture of species containing Ub into TEV cleavage buffer overnight.
Remove the N-terminal hexahistidine tag from Ub by adding TEV protease to a final concentration of 0.1 mg/mL and incubating the mixture overnight at 4˚C (See Note 6).
Dialyze the resulting products into Ni-NTA-6MU buffer for 3 hours at 4˚C
Add 1 mL Ni-NTA resin per 20 mg of protein in the dialyzed product.
Load the resin in a gravity column and collect the flow-through. Uncleaved ubiquitinated species and TEV remain bound to the resin, while H2B-UbDCA, Ub dimers (UbG76C2), and unreacted Ub flow-through (See Note 7).
Dialyze the flow-through into 4x4L of 5 mM β-mercaptoethanol and 0.1 PMSF, freeze and lyophilize.
Resuspend lyophilized mixture of Ub, UbG76C2, and H2B-UbDCA in 7 M Guanidine-HCL and purify using a semi-preparative RP-HPLC column (Higgins Analytical, PROTO C4 5 µm 250x10mm or similar) with the following acetonitrile gradient in 0.1% TFA (run at 4 mL/min), yielding purified H2B-UbDCA. Figure 2 shows a typical chromatogram produced from the gradient described in Table 1 (see Note 8).
Following freezing and lyophilization, the product can be reconstituted into nucleosomes using standard methods (Luger et al., 1999).
Figure 2:
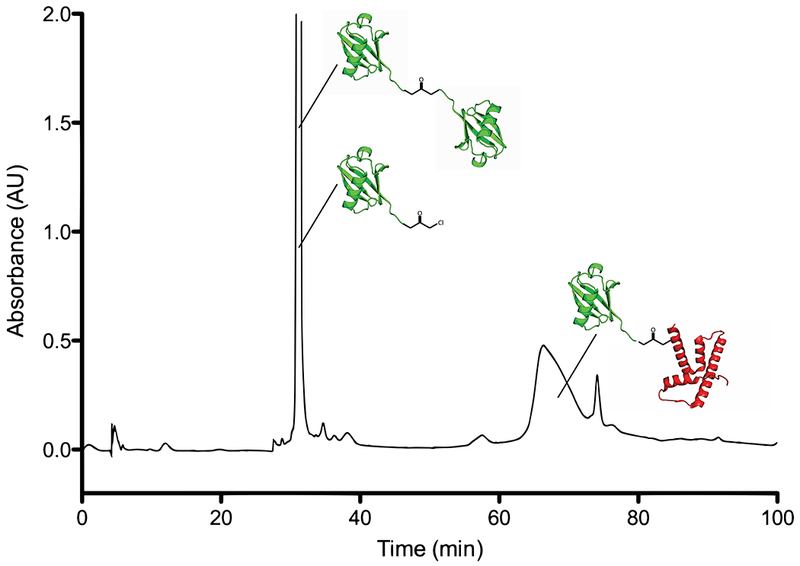
Chromatogram (230 nm) from HPLC purification of H2B-UbDCA by semipreparative C4 RP-HPLC. Crosslinked complexes comprised solely of ubiquitin elute in a single sharp peak, with H2B-UbDCA eluting in a broad peak late in the gradient. Fractions can be lyophilized and run on a gel to confirm separation.
Table 1:
Gradient used to purify H2B-UbDCA using semi-preparative C4 Higgins PROTO column. Time is in minutes.
| Time | %A | %B |
|---|---|---|
| 0 | 100 | 0 |
| 20 | 100 | 0 |
| 25 | 57 | 43 |
| 45 | 57 | 43 |
| 95 | 49 | 51 |
| 96 | 0 | 100 |
| 106 | 0 | 100 |
| 107 | 100 | 0 |
| 117 | 100 | 0 |
3. Preparation of native H2B-Ub
Semisynthesis of H2B-Ub with a native isopeptide linkage involves sequential ligation of three peptide fragments: 1) An N-terminal H2B(1-114) fragment produced by MESNa derivatization from a fusion protein with a GyrA intein fused to a chitin-binding domain (CBD). The H2B fragment is cleaved from the intein and derivatized at its C-terminus using sodium 2-mercaptoethanesulfonate (MESNa); 2) a synthetic peptide encoding the C-terminal fragment of H2B bearing a thiazolidine-δ-mercaptolysine (δK) residue at the site of the isopeptide bond; and 3) full-length Ub (1-76) derivatized at its C-terminus with MES using a previously described chemoenzymatic approach that takes advantage of the ability of E1 Ub ligase to modify the C-terminus of Ub (El Oualid et al., 2010). Below, we first describe the expression and purification of each of the three peptide components, followed by a protocol to assemble H2B-Ub (Nune et al., 2018). While we used X. laevis histone sequences in this example, this protocol may be adapted for homologs from other organisms.
3.1. MESNa-mediated cleavage of H2B(1-114)-GyrA-CBD intein to produce H2B-MES
The C-terminus of H2B(1-114) must be functionalized with MES, which is necessary for downstream ligation chemistry. In brief, the H2B(1-114)-GyrA intein-CBD fusion is bound to a chitin resin, followed by cleavage with MESNa, which traps the H2B peptide as it interconverts between the side-chain and peptide backbone of the N-terminal cysteine (N-S shift) of the GyrA intein. This batch purification releases the H2B(1-114)-MES (H2B-MES) fragment into solution, leaving the GyrA-CBD protein fragment bound to the chitin resin. While the histone fragment is soluble when fused with GyrA-CBD, it is less soluble once cleaved and in the form of H2B-MES. The protein is therefore purified further by SP-HP ion exchange chromatography under denaturing conditions, followed by C4 RP-HPLC purification. Below, we describe the purification of H2B-MES.
3.1.1. Equipment
Temperature-controlled water bath
Temperature-controlled shaker and flasks for cell growth
Sealable, 300-500 mL gravity column
Nutating or rotating platform
HPLC and semi-preparative C4 RP-HPLC column (such as Higgins Analytical, PROTO C4 5 um 10 x 250 mm)
3.1.2. Buffers and reagents
LB growth media
Chitin resin (NEB)
Intein Cell Resuspension Buffer: 50 mM Tris-HCl pH 7.5, 200 mM NaCl, 1 mM EDTA, 0.25 mM TCEP, and 0.1 mM PMSF
Intein Wash Buffer 1: 50 mM Tris-HCl pH 7.2, 200 mM NaCl, 1 mM EDTA, 0.25 mM TCEP, and 0.1 mM PMSF
Intein Wash Buffer 2: 50 mM Tris-HCl pH 7.4, 200 mM NaCl, 1 mM EDTA, 0.25 mM TCEP, and 0.1 mM PMSF
Intein Cleavage Buffer: 50 mM Tris-HCl pH 7.4, 200 mM NaCl, 250 mM MESNa, 1 mM EDTA, 0.25 mM TCEP, and 0.1 mM PMSF
SAU-0 Buffer: 20 mM Sodium Acetate pH 5.2, 7 M Urea, 1 mM EDTA, 0.25 mM TCEP, and 0.1 mM PMSF
SAU-1000 Buffer: 20 mM Sodium Acetate pH 5.2, 7 M Urea, 1 M NaCl, 1 mM EDTA, 0.25 mM TCEP, and 0.1 mM PMSF
C4 solvent A: 0.1% TFA in water
C4 solvent B: 0.1% TFA in 90% acetonitrile (ACN)
7M guanidine-HCl
3.1.3. Procedure
Transform cells with the H2B(1-114)-GyrA-intein-CBD plasmid. Grow the cells and express the protein as described in sections 2.2.1–2.
Induce the cells with 0.5 mM IPTG at 16°C and shake overnight. Recover the cells by centrifugation at 5000 x g for 30 minutes. Decant the growth medium and resuspend the cell paste with 20 mL of intein cell resuspension buffer per liter of growth medium.
Lyse cells according to established protocol for available equipment (See section 2.1.3) and clarify the lysate by centrifugation at 27000 x g for 30 minutes and collect the soluble fraction. Unlike typical bacterial expression of recombinant histones, which are expressed in inclusion bodies, the chimeric intein protein will partition to the soluble fraction.
Filter the lysate with a 0.22 µm filter and reserve a sample for future SDS-PAGE analysis.
Equilibrate 50-100 mL of chitin resin with 5 CV intein cell resuspension buffer and load into a gravity flow column (See Note 9).
Load lysate containing H2B(1-114)-GyrA-intein-CBD onto the chitin resin and incubate at room temperature for 1 hour with gentle agitation. Collect the flow-through after incubation and reserve a sample to run on a gel (See Note 10).
Wash the resin with 6 CV of Intein Wash Buffer 1. With the final CV of the wash, resuspend the resin and take a sample of the loaded resin, then drain the wash buffer and collect a sample for the gel.
Wash the resin with 20 CV of Wash Buffer 2. As with the previous step, use the final CV of the wash to collect resin and wash samples.
Add 1 CV of Intein Cleavage Buffer to the resin and incubate for 16 hours at room temperature.
Drain the cleavage buffer from the column, which contains the first fraction of MES-derivatized H2B-MES. Wash residual H2B-MES from the resin with 2 CV of cleavage buffer, then add 1 CV of cleavage buffer to the resin and continue the cleavage reaction for another 16 hours at room temperature. Keep each fraction at 4°C until cleavage is complete.
Due to the propensity of H2B-MES to aggregate and non-specifically adhere to the chitin resin, the final wash is done under denaturing conditions with 6 M Urea to disrupt the aggregates and enable their recovery. Thus, the beads should be resuspended with 1 CV SAU-1000 buffer, incubated for 1 hour, then eluted. Save a sample of the resin and eluate as in previous steps to monitor cleavage over the course of the purification.
Dialyze the combined fractions containing H2B-MES (other than the SAU-1000 eluate, which should be dialyzed in a separate vessel in the same 4 L of SAU-0) against 4 L of SAU-0 overnight. Make sure to keep the SAU-1000-eluted fraction separate from the other elutions, as the SAU-1000 tends to elute more impurities.
The following day, load the dialyzed protein onto an SP-HP column equilibrated with SAU-0 buffer and elute with a gradient of 0-60% SAU-1000 over 15 CV. Check fractions by SDS-PAGE and pool fractions that contain suitably pure H2B-MES.
Dialyze the pooled fractions against 4 x 4 L H2O, 0.1 mM PMSF, and 0.25 mM PMSF (minimum 3 hours of dialysis before changing), freeze the dialyzed protein with liquid nitrogen, and lyophilize the product.
Resuspend lyophilized H2B-MES in a small volume of 7M Guanidine-HCL and purify as described in section 2.3.2 using the same C4 purification gradient.
3.2. Peptide synthesis of Cys(116-122)H2B fragment
Ub-MES, H2B-MES, and a synthetic H2B C-terminal peptide fragment can be combined to form H2B-Ub using sequential chemical ligation (Holt & Muir, 2015; Maity, Jbara, & Brik, 2016). The following section describes a protocol designed to produce H2B-Ub with a chemically native isopeptide bond involving native chemical ligation (NCL) (Bondalapati et al., 2016; Dawson, Muir, Clark-Lewis, & Kent, 1994) coupled with palladium-mediated Cys decaging (Jbara, Laps, et al., 2018; Jbara, Maity, Seenaiah, & Brik, 2016; Maity et al., 2016) and desulfurization (Wan & Danishefsky, 2007) (Figure 3). First a peptide is synthesized encoding the C-terminal portion of histone H2B and containing a protected mercaptolysine at the ubiquitination site (K120). The peptide is ligated to H2B-MES, followed by deprotection and ligation of the Ub and mercaptolysine residue.
Figure 3:
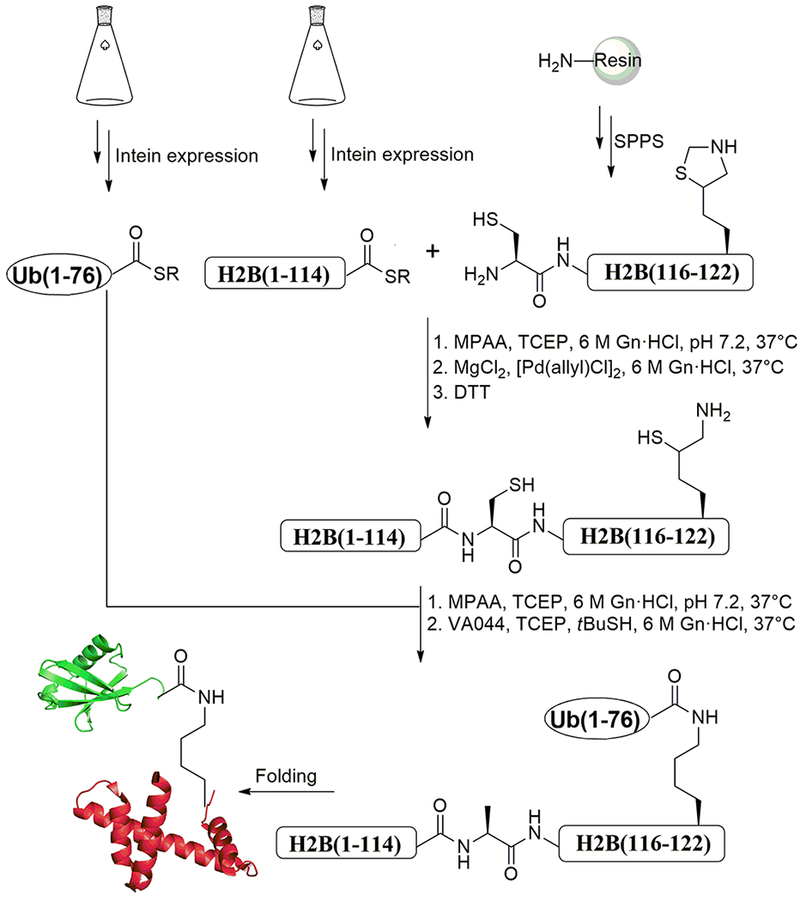
Schematic representation of semisynthesis of H2BK120Ub. Synthetic fragment Cys-H2B(116-122) is ligated with the H2B-MES fragment under native chemical ligation (NCL) conditions. “SR” represents a generic thiol, which in the case of this protocol is MESNa. Upon completion of the ligation reaction, the thiazolidine protection on Lys120 of H2B is deprotected, followed by NCL reaction with Ub(1-76)-MES and desulfurization reaction to provide native H2BK120Ub. SPPS, solid-phase peptide synthesis.
3.2.1. Chemical Synthesis of Cys(116-122)H2B peptide fragment:
The C-terminal H2B peptide is synthesized with a thiazolidine protected δ-mercaptolysine amino acid in place of the lysine to be modified with Ub, the protected form of which enables the chemoselective ligation reaction of the two H2B fragments. Following this step, thiazolidine is decaged to enable ligation of the Ub thioester to H2B, which yields an isopeptide (amide) linkage following the desulfurization reaction. The synthetic peptide is made by fluorenylmethyloxycarbonyl solid-phase peptide synthesis (Fmoc-SPPS), which can then be ligated with the complementary H2B part and Ub thioester fragments prepared via intein methods to create H2BK120Ub that is sufficiently pure for nucleosome reconstitution and structural studies (Jbara, Laps, et al., 2018; Jbara et al., 2016; Morgan et al., 2016). The following protocol describes the preparation of the synthetic fragment at milligram scale.
3.2.1.1. Equipment
Teflon-fritted polypropylene solid-phase extraction (SPE) tubes with piston
Platform shaker
Vortex mixer
HPLC system
Preparative C18 column suitable for peptide purification (pore size: 300 Å; particle size: 10 μm)
Lyophilizer
Syringe filters
Refrigerated tabletop centrifuge
Automated peptide synthesizer or custom peptide synthesis service
Manual peptide-synthesis vessels
3.2.1.2. Materials
N,N-Dimethylformamide (DMF)
Dichloromethane (DCM)
Piperidine
Trifluoroacetic acid (TFA)
N-ethyldiisopropylamine (DIEA)
Acetonitrile (ACN)
Rink amide Knorr resin, low substitution, 1% (wt/wt) divinylbenzene (DVB), 0.4-0.43 mmol/g
2-(6-Chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminiumhexa-fluorophosphate (HCTU)
O-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU)
Methanol (MeOH)
Triisopropylsilane (TIPS)
Diethyl ether, anhydrous (Et2O)
Nitrogen gas cylinder, regulator, and tubing
Fmoc-Ala-OH (A)
Boc-Cys(Trt)-OH (C)
Fmoc-Ser(tBu)-OH (S)
Fmoc-Thr(tBu)-OH (T)
Fmoc-Tyr(tBu)-OH (Y)
Fmoc-Val-OH (V)
Fmoc-Lys(Boc)-OH (K)
Fmoc-thiazolidine-δ-mercaptolysine (δK)
3.2.1.3. Procedure
For a 0.1 mmol scale of peptide synthesis, weigh 250 mg of Knorr Rink amide resin (0.4 mmol g −1 loading) into a 20 mL syringe equipped with a Teflon filter.
Wash the resin by adding 20 mL of DMF with gentle mixing. Perform this step three times.
Swell the resin by adding 20 mL of 50% DCM/50% DMF for 30 min at room temperature.
Drain the solvent and wash the resin three times with 20 mL DMF.
Drain the DMF from the fritted syringe and add 10 mL of 20% (vol/vol) piperidine in DMF, gently mix, leave for 3 min and then drain the syringe.
Add 10 mL of 20% (vol/vol) piperidine with gentle mixing. Incubate for 5 min and then drain the syringe.
Add 10 mL of 20% (vol/vol) piperidine in DMF, gently mix, incubate for 3 min and then drain the syringe.
Drain the fritted syringe, wash the resin as described in Step 2 and keep the resin in DMF.
Place 0.1 mmol Fmoc-Rink amide resin prepared in Steps 1-8 into the manual peptide-synthesis vessel.
Run the sequence on the peptide synthesizer, CVT(δK)YTSAK, using a single-coupling method for all the amino acids. Weigh 4 equiv. of each Fmoc-protected amino acid with respect to the initial loading of the resin in 10 mL peptide synthesizer tubes and dissolve with 5 mL DMF. (4 equiv. of HCTU and 8 equiv. DIEA is needed for each coupling, see Note 11).
Couple the Fmoc-thiazolidine-δ-mercaptolysine-OH using 2.5 equiv. of the amino acid, 2.5 equiv. of HATU and 5 equiv. DIEA at 2.5 mL DMF.
Transfer the resin from the peptide-synthesis vessels to a syringe equipped with a Teflon filter.
Wash the resin three times each with 20 mL DMF, 20 mL MeOH and 20 mL DCM sequentially and let the resin dry.
Cleave the peptide from the resin with a mixture of 15 mL of TFA, 0.35 mL of H2O and 0.35 mL TIPS for 2 h. The cleavage mixture should be freshly prepared before using.
Slowly filter the cleavage mixture through a syringe filter and collect the solution dropwise into two 50-mL Falcon tubes containing ~30 mL of ice-chilled diethyl ether each.
Centrifuge the mixture at 3,350 x g for 10 min at −10 °C to precipitate the peptide.
Separate the supernatant and the precipitate. Dry the precipitate with nitrogen gas flow for one minute then dissolve the dried peptide in 10 mL of 50% (vol/vol) ACN in water containing 0.1% (vol/vol) TFA. Note: the precipitant could be directly dissolved and lyophilized.
Freeze the peptide solution using liquid nitrogen and lyophilize the crude peptide for 48 hrs. Stored at 4°C, the lyophilized product is stable for several months.
Purify the peptide via preparative HPLC using a preparative C18 column (Phenomenex Jupiter 21.2 x 250 mm; pore size: 300 Å; particle size: 10 μm) developed with a gradient of 0-50% (vol/vol) B over 60 min. A: deionized H2O containing 0.1% (vol/vol) TFA; B: ACN containing 0.1% (vol/vol) TFA. Flow rate: 15 mL min1. Pure fractions are identified via mass spectrometry (MALDI or ESI-MS)
Lyophilize the purified peptide. The expected yield is 40%-50%.
3.3. Ub-MES derivatization
3.3.1. Materials
Human E1 Ub-activating enzyme (purchased or expressed from Addgene plasmid #34965 and purified as described in (Berndsen & Wolberger, 2011)).
2x derivatization buffer: 100 mM sodium phosphate buffer pH 8.0, 200 mM MESNa, 20 mM ATP, 20 mM MgCl2
SEC buffer: 50 mM ammonium acetate pH 5.2, 0.5 mM TCEP
Purified Ub (Pickart & Raasi, 2005)
Centrifugal concentrators
FPLC and size exclusion column (Superdex 75 16/60, GE Life Science or similar)
Lyophilizer
3.3.2. Procedure
Mix 2x derivatization buffer, Ub, E1 enzyme and water such that the final concentrations are 100 µM Ub, and 250 nM E1 concentration and 1x derivatization buffer.
Incubate 6 hours at 37°C.
Concentrate the sample to a volume suitable for injection onto size exclusion column
Equilibrate size exclusion column with SEC buffer.
Purify the Ub-MES by size exclusion. The derivatized Ub comprises the major peak, eluting before a peak of small molecules that elute at the end of the column volume. RP-HPLC may also be used to purify the derivatized Ub.
Dialyze peak fractions against 4 L of 0.5% TFA in water, freeze the sample, and lyophilize overnight. Store lyophilized Ub-MES at −20°C until further use (see Note 12).
3.4. Semi-synthesis of native H2BK120Ub
The synthetic peptide, Cys-H2B(116-122), prepared via Fmoc-SPPS as described in section 3.2 is ligated with the H2B-MES fragment under NCL conditions (6 M Gn·HCl, 200 mM Na2HPO4 buffer pH~7, containing MPAA and TCEP additives). Once the ligation reaction is complete, the thiazolidine protecting group on Lys120 of H2B is removed by addition of MgCl2 and [Pd(AllylCl)]2 followed by DTT addition. The H2B(1-122) intermediate is then ligated to Ub(1-76)-MES. Finally, the desulfurization reaction is performed by adding TCEP, VA-044 and t-butyl thiol to yield native H2BK120Ub (Figure 3).
3.4.1. Equipment
HPLC system
Analytical C4 HPLC column
Semi-preparative C4 HPLC column suitable for peptide purification
1.5 mL low adhesion centrifuge tubes (USA Scientific item # 1415-2600)
Dialysis cassettes
Lyophilizer
Storage dewar
3.4.2. Buffers and reagents
Trifluoroacetic acid (TFA)
Acetonitrile (ACN)
Allylpalladium chloride dimer, 98% (vol/vol) ([Pd(allyl)Cl]2)
Tris(2-carboxyethyl)phosphine hydrochloride (TCEP)
2,2-Azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA044)
tert-Butylthiol (tBuSH)
4-mercaptophenylacetic acid (MPAA)
Sodium hydroxide (5N NaOH)
Hydrochloric acid, 35% (vol/vol) (HCl)
Dithiothreitol (DTT)
Guanidine hydrochloride (Gn·HCl)
Sodium phosphate, dibasic (Na2HPO4)
3.4.3. Procedure
Weigh 30 mg of H2B-MES (2.3 x 10 −3 mmol) in a 1.5 mL low-adhesion centrifuge tube. Add 750 μl (H2B-MES concentration, 3 mM) of 6 M Gn·HCl, 200 mM Na2HPO4 buffer containing 20 equiv. of MPAA (7.8 mg) and 10 equiv. of TCEP (6.6 mg) at pH ~7 and dissolve the mixture by gentle shaking.
Add this mixture to a 1.5 mL low-adhesion centrifuge tube containing 4 mg of the fragment Cys-H2B(116-122) (3.3 x 10 −3 mmol).
Shake the tube to dissolve the solid and incubate at 37 °C.
Monitor the progress of the ligation by adding 2 μl of the reaction mixture to 20 µl of a 50% (vol/vol) ACN/H2O mixture, containing 0.1% (vol/vol) TFA. Inject this mixture into an analytical HPLC C4 column. Collect the targeted peaks and check them by ESI-MS; the reaction typically goes to completion within 1-2 h.
Add 31 mg of MgCl2 (100 equiv.) dissolved in 100 μl of 6 M Gn·HCl,200 mM Na2HPO4 buffer, to the reaction mixture, shake the tube and incubate it at 37°C for 5 min.
Dissolve 18 mg (15 equiv.) of [Pd(AllylCl)]2 in 100 μl of 6 M Gn·HCl, 200 mM Na2HPO4 buffer and shake for 5 min. Add this solution to the reaction mixture, shake the tube vigorously and incubate at 37 °C.
Monitor the progress of the reaction as described in Step 4; the reaction is typically complete in 1 h. Samples of the reaction should be taken at 20-minute intervals and analyzed with ESI-MS to confirm completion of the reaction.
After the reaction is complete, add 38 mg (75 equiv.) of DTT to quench the reaction and precipitate the palladium. Allow the mixture to react for 5-10 min to maximize the precipitation of the palladium.
Centrifuge the reaction mixture at 10,050 x g for 1 min at room temperature and collect the supernatant. Wash the precipitate twice with 6 M Gn·HCl, 200 mM Na2HPO4 buffer at pH~7, centrifuge at 10,050 x g for 1 min at room temperature and combine the supernatant solutions.
Purify the final protein by preparative HPLC according to the conditions described in step 19 (below) and lyophilize. The expected yield is 45%, or 14 mg of H2B(1-122) at this scale.
Weigh 12 mg H2B(1-122) (8.5 x 10−4 mmol), in a 1.5-mL low-adhesion centrifuge tube. Add 400 μl (H2B concentration, 2 mM) of 6 M Gn·HCl, 200 mM Na2HPO4 buffer containing 50 equiv. of MPAA (7 mg) and 25 equiv. of TCEP (6 mg) at pH ~7 and dissolve the mixture by gentle shaking.
Add this mixture to a 1.5-mL low-adhesion centrifuge tube containing 20 mg of fragment Ub(1-76)-MES (1.7 x 10−3 mmol).
Shake the tube to dissolve the solid and incubate at 37 °C.
Monitor the progress of the ligation by adding 2 μl of the reaction mixture to a 50% (vol/vol) ACN/H2O mixture containing 0.1% (vol/vol) TFA. Inject this mixture into an analytical HPLC C4 column. Collect the targeted peaks and check them by ESI-MS; the reaction is typically going to completion within 1-2 h.
After completion of the ligation, dialyze the reaction mixture against 500 mL of 6 M Gn·HCl, 200 mM Na2HPO4 buffer at pH 7 for 12 h.
Perform the desulfurization reaction by first dissolving 50 equivalents of TCEP (0.25 M) and 28 mg of VA044 (50 equivalents per sulfur atom) in 100 μL of 6 M Gn·HCl, 200 mM Na2HPO4 buffer, then add the dissolved reagents to the 400 μl previously dialyzed ligation reaction mixture (giving a total volume of 500 μl). Finally, add 50 μl of tBuSH (10% of the total volume) giving a final reaction volume of 550 μl.
Shake the tube to dissolve the solid and incubate at 42 °C.
Monitor the progress of the desulfurization by adding 2 μl of the reaction mixture to 20 μl of a 50% (vol/vol) ACN/H2O mixture containing 0.1% (vol/vol) TFA. Inject this mixture into an analytical HPLC C4 column. Collect the targeted peaks and check them by ESI-MS; the reaction is typically complete within 2-3 h.
Purify the final protein by semi-preparative HPLC, Phenomenex, Jupiter C4 column (10 x 250 mm; pore size: 300 Å; particle size: 10 μm) Flow rate: 4 mL min1. Pure fractions can be identified with ESI-MS or MALDI (see Note 13). The HPLC gradient described in Table 3 produces a chromatogram such as that shown in Figure 4.
Freeze the peptide solution using liquid nitrogen and lyophilize it for 48 hrs to yield 4 mg of H2BK120Ub in a 30% yield.
Table 3:
Gradient used to purify H2BK120Ub using preparative C4 Phenomenex Jupiter column
| Time | %A | %B |
|---|---|---|
| 0 | 100 | 0 |
| 5 | 100 | 0 |
| 15 | 75 | 25 |
| 20 | 75 | 25 |
| 55 | 35 | 65 |
| 60 | 25 | 75 |
| 65 | 5 | 95 |
| 75 | 5 | 95 |
| 80 | 100 | 0 |
Figure 4:

Representative HPLC chromatogram (230 nm) from purification of H2BUb by semipreparative C4 RP-HPLC. Ligated product (H2BUb) elutes as the major product, Ub thioester (Ub-MPAA) and hydrolyzed Ub (Ub-OH) elute earlier in the gradient. (#) - MPAA thiol used in ligation reaction.
Summary and Conclusion
The choice of whether to generate chemically native H2B-Ub or the non-hydrolyzable H2B-UbDCA mimic, H2B-UbDCA , is driven by the requirements of the planned experiments. Since generating H2B-UbDCA is more straightforward and less technically demanding than the procedure for semisynthesis of H2B-Ub with a native linkage, this approach should be used if possible. H2B-UbDCA incorporated into nucleosomes can be used for structural and binding studies (Cooper et al., 2016; Long, Thelen, et al., 2014; Morgan et al., 2016; Wilson et al., 2016; Zukowski et al., 2018) where a cleavable linkage is not needed, or even undesirable. An important caveat is that the non-native character of the DCA linkage, with its additional bond length and carboxylate (Figure 1), may make this material unsuitable for applications in which a fully native isopeptide bond is needed. In structural studies of the SAGA DUB module bound to H2B-UbDCA nucleosomes (Morgan et al., 2016), the active site cysteine of Ubp8, the USP-class DUB subunit, was substituted with alanine out of concern that the Ub C-terminal carboxylate would clash with other atoms in the active site. SAGA DUB module containing the Ubp8 C146A substitution indeed binds more tightly than wild type DUB module to nucleosomes, as judged by the ability of the mutant DUB module to form a stable complex with H2B-UbDCA nucleosomes (Morgan et al., 2016). Although the DCA linkage could not be resolved in the electron density maps (Morgan et al., 2016), a subsequent study suggests that the active site C to A substitution may have driven tighter binding by enabling the Ub C-terminal carboxylate to form additional contacts within the active site (Morrow et al., 2018). Similar tight binding would therefore be expected for other USP class DUBs with an active site alanine substitution.
The approaches described here can be used to generate other ubiquitinated histone substrates. As first described by Yin, Wilkinson and colleagues (Yin et al., 2000), the DCA linkage can be used to mimic any ubiquitinated lysine, with the important caveat that the protein contain no cysteines other than the one substituted for the target lysine. Comparable approaches to our procedure for producing H2B-UbDCA have been applied to chemically ubiquitinate histone H2A, which is ubiquitinated at K119 in Polycomb repressed genomic loci (Long, Furgason, et al., 2014) and at K13 and K15 at sites of DNA damage (Wilson et al., 2016). A semisynthetic approach similar to that described here can also be used to generate H2A-K119Ub (Whitcomb et al., 2012), but is not appropriate for other Ub modifications that are not located near the histone C-terminus. The other available approaches for installing cleavable mimics of the ubiquitinated lysine may be appropriate in these cases (Bhat et al., 2018; Debelouchina, Gerecht, & Muir, 2017; Yang et al., 2016).
Table 2:
Gradient used to purify Cys-H2B(116-122) using preparative C18 Phenomenex Jupiter column
| Time | %A | %B |
|---|---|---|
| 0 | 100 | 0 |
| 5 | 100 | 0 |
| 8 | 95 | 5 |
| 48 | 55 | 45 |
| 60 | 50 | 50 |
| 65 | 45 | 55 |
| 80 | 10 | 90 |
| 85 | 95 | 5 |
Acknowledgements
Supported by National Institute of General Medical Sciences grant GM095822 (C.W.) and the U.S.-Israel Binational Science Foundation (C.W. and A.B.).
4. Notes
Progress of lysis can be monitored by observing progressive reduction in OD600 values throughout lysis. When OD600 values cease to change, lysis is complete.
Progress of wash step can be monitored by checking OD230 values throughout the wash. When this value ceases to change, the wash is complete.
It is normal to observe substantial precipitation during this step. The precipitate primarily contains contaminants.
2 mM DCA in DMF can be made by weighing ~50 mg of DCA in a tube. Calculate the volume of DMF to add and dissolve to make a 1 M stock. Dilute 2 µL of the 1M stock in 1 mL DMF to make a 2 mM working stock. DCA solutions should always be prepared shortly before use.
Urea is in slow equilibrium with isocyanate, and isocyanate can spontaneously react with lysine side chains. After dissolving 9 M urea, one can add Mixed Bed Resin (Sigma) to the urea (stir for 1 hr and remove) to prevent this modification of lysines. Urea solutions can be used for up to 24 hours after deionizing.
TEV protease can be added to the mixture before dialysis, i.e. in the presence of urea. As the urea concentration is reduced, TEV refolds, regaining its activity.
Cleavage should be 100%, but a second passage over the Ni resin is needed to remove TEV protease, which would otherwise coelute with H2B-UbDCA from the C4 RP-HPLC. Additionally, TEV protease migrates on SDS-PAGE gels similarly to hexahistidine-tagged H2B-UbDCA, and this can be misleading when judging TEV cleavage completion.
Binding capacity of NEB chitin resin is approximately 2 mg/mL resin, and 50-100 mL resin is an advisable volume of resin to use for a 12 L growth of the H2B(1-114)-GyrA-CBD construct. Whatever the chosen volume of chitin resin is, this volume is considered 1 CV.
If it is later determined that the fusion protein is in this fraction, the resin is likely saturated, and the flow-through can be reapplied to fresh chitin resin and processed in the same manner as the original lysate.
The gradient described is designed to be useful for a wide range of protein injection quantities. Under these conditions, injections of 10 mg of total protein is recommended and sequential runs may be needed to purify all material. Different C4 columns may produce varying results.
Activation of the amino acid with HCTU should not exceed 5 min in order to prevent the isomerization of the activated amino acids.
Ub-MES is stable at pH < 6.5 – higher pH results in hydrolysis of the C-terminal derivatization and reductions in yield for subsequent steps. Lyophilized Ub-MES can be stored in the freezer, but preferably the material is used quickly; after a month of storage at −20°C, >50% of the derivatization is lost.
Samples of each fraction can also be lyophilized and run on a gel, however this does not confirm desulfurization chemistry.
References cited
- Andrews AJ, & Luger K (2011). Nucleosome structure(s) and stability: variations on a theme. Annu Rev Biophys, 40, 99–117. doi: 10.1146/annurev-biophys-042910-155329 [DOI] [PubMed] [Google Scholar]
- Berndsen CE, & Wolberger C (2011). A spectrophotometric assay for conjugation of ubiquitin and ubiquitin-like proteins. Anal Biochem, 418(1), 102–110. doi: 10.1016/j.ab.2011.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat S, Hwang Y, Gibson MD, Morgan MT, Taverna SD, Zhao Y, Cole PA (2018). Hydrazide Mimics for Protein Lysine Acylation To Assess Nucleosome Dynamics and Deubiquitinase Action. J Am Chem Soc, 140(30), 9478–9485. doi: 10.1021/jacs.8b03572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondalapati S, Jbara M, & Brik A (2016). Expanding the chemical toolbox for the synthesis of large and uniquely modified proteins. Nat Chem, 8(5), 407–418. doi: 10.1038/nchem.2476 [DOI] [PubMed] [Google Scholar]
- Borodovsky A, Kessler BM, Casagrande R, Overkleeft HS, Wilkinson KD, & Ploegh HL (2001). A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. EMBO J, 20(18), 5187–5196. doi: 10.1093/emboj/20.18.5187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee C, McGinty RK, Pellois JP, & Muir TW (2007). Auxiliary-mediated site-specific peptide ubiquitylation. Angew Chem Int Ed Engl, 46(16), 2814–2818. doi: 10.1002/anie.200605155 [DOI] [PubMed] [Google Scholar]
- Chin JW, Cropp TA, Anderson JC, Mukherji M, Zhang Z, & Schultz PG (2003). An expanded eukaryotic genetic code. Science, 301(5635), 964–967. doi: 10.1126/science.1084772 [DOI] [PubMed] [Google Scholar]
- Cooper S, Grijzenhout A, Underwood E, Ancelin K, Zhang T, Nesterova TB, Brockdorff N (2016). Jarid2 binds mono-ubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat Commun, 7, 13661. doi: 10.1038/ncomms13661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson PE, Muir TW, Clark-Lewis I, & Kent SB (1994). Synthesis of proteins by native chemical ligation. Science, 266(5186), 776–779. [DOI] [PubMed] [Google Scholar]
- Debelouchina GT, Gerecht K, & Muir TW (2017). Ubiquitin utilizes an acidic surface patch to alter chromatin structure. Nat Chem Biol, 13(1), 105–110. doi: 10.1038/nchembio.2235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Oualid F, Merkx R, Ekkebus R, Hameed DS, Smit JJ, de Jong A, … Ovaa H (2010). Chemical synthesis of ubiquitin, ubiquitin-based probes, and diubiquitin. Angew Chem Int Ed Engl, 49(52), 10149–10153. doi: 10.1002/anie.201005995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong P, Davidson GA, Gui WJ, Yang K, Bozza WP, & Zhuang ZH (2018). Activity-based ubiquitin-protein probes reveal target protein specificity of deubiquitinating enzymes. Chemical Science, 9(40), 7859–7865. doi: 10.1039/c8sc01573b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt M, & Muir T (2015). Application of the protein semisynthesis strategy to the generation of modified chromatin. Annu Rev Biochem, 84, 265–290. doi: 10.1146/annurev-biochem-060614-034429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung SH, Wong RP, Ulrich HD, & Kao CF (2017). Monoubiquitylation of histone H2B contributes to the bypass of DNA damage during and after DNA replication. Proc Natl Acad Sci U S A, 114(11), E2205–E2214. doi: 10.1073/pnas.1612633114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jbara M, Laps S, Morgan M, Kamnesky G, Mann G, Wolberger C, & Brik A (2018). Palladium prompted on-demand cysteine chemistry for the synthesis of challenging and uniquely modified proteins. Nat Commun, 9(1), 3154. doi: 10.1038/s41467-018-05628-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jbara M, Maity SK, Seenaiah M, & Brik A (2016). Palladium Mediated Rapid Deprotection of N-Terminal Cysteine under Native Chemical Ligation Conditions for the Efficient Preparation of Synthetically Challenging Proteins. J Am Chem Soc, 138(15), 5069–5075. doi: 10.1021/jacs.5b13580 [DOI] [PubMed] [Google Scholar]
- Jbara M, Sun H, Kamnesky G, & Brik A (2018). Chemical chromatin ubiquitylation. Curr Opin Chem Biol, 45, 18–26. doi: 10.1016/j.cbpa.2018.02.001 [DOI] [PubMed] [Google Scholar]
- Jenuwein T, & Allis CD (2001). Translating the histone code. Science, 293(5532), 1074–1080. doi: 10.1126/science.1063127 [DOI] [PubMed] [Google Scholar]
- Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, & Waugh DS (2001). Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng, 14(12), 993–1000. [DOI] [PubMed] [Google Scholar]
- Kumar KS, Spasser L, Ohayon S, Erlich LA, & Brik A (2011). Expeditious chemical synthesis of ubiquitinated peptides employing orthogonal protection and native chemical ligation. Bioconjug Chem, 22(2), 137–143. doi: 10.1021/bc1004735 [DOI] [PubMed] [Google Scholar]
- Long L, Furgason M, & Yao T (2014). Generation of nonhydrolyzable ubiquitin-histone mimics. Methods, 70(2–3), 134–138. doi: 10.1016/j.ymeth.2014.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long L, Thelen JP, Furgason M, Haj-Yahya M, Brik A, Cheng D, Yao T (2014). The U4/U6 recycling factor SART3 has histone chaperone activity and associates with USP15 to regulate H2B deubiquitination. J Biol Chem, 289(13), 8916–8930. doi: 10.1074/jbc.M114.551754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Rechsteiner TJ, & Richmond TJ (1999). Preparation of nucleosome core particle from recombinant histones. Methods Enzymol, 304, 3–19. [DOI] [PubMed] [Google Scholar]
- Maity SK, Jbara M, & Brik A (2016). Chemical and semisynthesis of modified histones. J Pept Sci, 22(5), 252–259. doi: 10.1002/psc.2848 [DOI] [PubMed] [Google Scholar]
- McGinty RK, Kim J, Chatterjee C, Roeder RG, & Muir TW (2008). Chemically ubiquitylated histone H2B stimulates hDot1L-mediated intranucleosomal methylation. Nature, 453(7196), 812–816. doi: 10.1038/nature06906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MT, Haj-Yahya M, Ringel AE, Bandi P, Brik A, & Wolberger C (2016). Structural basis for histone H2B deubiquitination by the SAGA DUB module. Science, 351(6274), 725–728. doi: 10.1126/science.aac5681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MT, & Wolberger C (2018). Competition Assay for Measuring Deubiquitinating Enzyme Substrate Affinity. Methods Mol Biol, 1844, 59–70. doi: 10.1007/978-1-4939-8706-1_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow ME, Morgan MT, Clerici M, Growkova K, Yan M, Komander D, Wolberger C (2018). Active site alanine mutations convert deubiquitinases into high-affinity ubiquitin-binding proteins. EMBO Rep. doi: 10.15252/embr.201745680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nune M, Morgan MT, Connell Z, McCullough L, Jbara M, Sun H, Wolberger C (2018). FACT and Ubp10 collaborate to modulate H2B deubiquitination and nucleosome dynamics. 397653. doi: 10.1101/397653 %J bioRxiv [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM, & Raasi S (2005). Controlled synthesis of polyubiquitin chains. Methods Enzymol, 399, 21–36. doi: 10.1016/S0076-6879(05)99002-2 [DOI] [PubMed] [Google Scholar]
- Wan Q, & Danishefsky SJ (2007). Free-radical-based, specific desulfurization of cysteine: a powerful advance in the synthesis of polypeptides and glycopolypeptides. Angew Chem Int Ed Engl, 46(48), 9248–9252. doi: 10.1002/anie.200704195 [DOI] [PubMed] [Google Scholar]
- Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, & Zhang Y (2004). Role of histone H2A ubiquitination in Polycomb silencing. Nature, 431(7010), 873–878. doi: 10.1038/nature02985 [DOI] [PubMed] [Google Scholar]
- Weake VM, & Workman JL (2008). Histone ubiquitination: triggering gene activity. Mol Cell, 29(6), 653–663. doi: 10.1016/j.molcel.2008.02.014 [DOI] [PubMed] [Google Scholar]
- Whitcomb SJ, Fierz B, McGinty RK, Holt M, Ito T, Muir TW, & Allis CD (2012). Histone monoubiquitylation position determines specificity and direction of enzymatic cross-talk with histone methyltransferases Dot1L and PRC2. J Biol Chem, 287(28), 23718–23725. doi: 10.1074/jbc.M112.361824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener R, Zhang X, Wang T, & Wolberger C (2012). The mechanism of OTUB1-mediated inhibition of ubiquitination. Nature, 483(7391), 618–622. doi: 10.1038/nature10911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MD, Benlekbir S, Fradet-Turcotte A, Sherker A, Julien JP, McEwan A, Durocher D (2016). The structural basis of modified nucleosome recognition by 53BP1. Nature, 536(7614), 100–103. doi: 10.1038/nature18951 [DOI] [PubMed] [Google Scholar]
- Yang K, Li G, Gong P, Gui W, Yuan L, & Zhuang Z (2016). Chemical Protein Ubiquitylation with Preservation of the Native Cysteine Residues. Chembiochem, 17(11), 995–998. doi: 10.1002/cbic.201600042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L, Krantz B, Russell NS, Deshpande S, & Wilkinson KD (2000). Nonhydrolyzable diubiquitin analogues are inhibitors of ubiquitin conjugation and deconjugation. Biochemistry, 39(32), 10001–10010. [DOI] [PubMed] [Google Scholar]
- Zukowski A, Al-Afaleq NO, Duncan ED, Yao T, & Johnson AM (2018). Recruitment and allosteric stimulation of a histone-deubiquitinating enzyme during heterochromatin assembly. J Biol Chem, 293(7), 2498–2509. doi: 10.1074/jbc.RA117.000498 [DOI] [PMC free article] [PubMed] [Google Scholar]