

Abstract
Introduction:
Existing HIV therapy using oral antiretrovirals (ARVs) can result in pill fatigue and sub-optimal adherence. Microneedle array patches (MAPs) offer non-invasive, blood-free and painless drug delivery, and may improve patient adherence. The objective of this study was to develop a novel physiologically-based pharmacokinetic (PBPK) model to simulate the systemic pharmacokinetics of cabotegravir and rilpivirine MAPs using the intradermal route.
Methods:
The developed PBPK models were qualified against observed pharmacokinetic data after intramuscular (IM) and intradermal administration of long-acting nanoformulated rilpivirine to rats, and for IM administration of both drugs to healthy adults. Qualified models were then utilised to estimate suitable MAP characteristics (e.g. nanoformulation dose and release rates) and inform dosing strategies to maintain plasma concentrations above target trough concentrations for the designated dosing interval.
Results:
PBPK models simulated q4-weekly loading and maintenance doses of 360 mg and 180 mg for long-acting formulated cabotegravir between the release rates of 1×10−3 – 3×10−3 h−1 and 1×10−3 – 1.5×10−3 h−1 respectively, for a 70 kg adult. Estimated patch size was 60 cm2 for a 360 mg dose of cabotegravir. For q4-weekly dosing, rilpivirine required a 1080 mg loading dose and a 540 mg maintenance dose with release rates of 1.5×10−3 – 2.5×10−3 h−1 and 5×10−4 – 1×10−3 h−1, respectively. Weekly dosing was also evaluated to assess the potential application from a smaller patch size. The ability to self-administer via a patch that is only left in place for a short duration makes longer durations less important than for some other long-acting approaches. Weekly cabotegravir required 60 mg between release rates 7×10−3 – 9×10−3 h−1 and rilpivirine required 270 mg and 180 mg respectively between release rates of 7×10−3 – 9×10−3 h−1.
Discussion:
This model estimated optimal dose and release rates for cabotegravir and rilpivirine MAPs. Our approach provides a computational platform to support rational development of intradermal administration strategies to tackle problems associated with chronic oral ARV administration.
Keywords: PBPK, intradermal, microneedle, patch, antiretroviral, long-acting
Graphical Abstract
Introduction
Antiretrovirals (ARVs) have improved the average lifespan of infected individuals and also found clinical application in HIV pre-exposure prophylaxis (PrEP), preventing transmission to high risk individuals. The majority of existing ARVs are available only as oral formulations. They require daily administration and chronic dosing, often resulting in sub-optimal adherence and pill fatigue (1, 2). Alternative dosing strategies such as long-acting injectables (LAIs) have been utilised for the administration of antipsychotics (e.g. paliperidone palmitate) and contraceptives (e.g. medroxyprogesterone), effectively addressing adherence issues associated with chronic oral administration (3). The recent development of cabotegravir and rilpivirine LAI intramuscular (IM) nanoformulations has stimulated interest in this strategy (4).
The existing injectable ARV formulations face challenges that may hinder development, acceptability, and widespread implementation although they have proven very successful for other indications. Administration of IM injections typically requires a skilled healthcare worker. For LAI rilpivirine, two large volume injections are necessary and the formulation requires a cold-chain, thus hindering clinical implementation (5). Importantly, cold chain is required because of stability of an excipient rather than the drug itself and thus this may not be a widespread problem for the approach. Nonetheless, the need for cold chain increases cost and, in low income countries, may reduce or even prevent access to treatments. In addition, some studies noted that 20% of people administered with IM injection reportedly suffer from needle phobia. This may reduce the number of patients accepting injectables, as seen for vaccines and routine dental procedures (6). It should be noted that all existing HIV patient attitude surveys for LAI approaches have shown a high level of enthusiasm for the approach, but complimentary approaches warrant further investigation (7–9).
Microneedle array patches (MAPs) can be used for minimally-invasive intradermal delivery of micro- or nanoformulated drug into the skin (10). The intradermal route of administration has multiple advantages compared to oral intake, avoiding gastrointestinal degradation and first-pass metabolism, resulting in reduced total dose and may improve patient adherence (11). MAPs disrupt the stratum corneum, the major skin barrier to drug delivery, and can deliver drugs painlessly and without drawing blood into the upper skin layers (12), thus avoiding local pain, bruising, discomfort or bleeding (13). Drugs deposited in particulate form by MAPs in viable skin layers can be absorbed systemically by the rich dermal microcirculation upon release into the interstitial fluid (13). Hence, MAPs represent a promising strategy for chronic administration as long as the doses necessary for adequate pharmacokinetic exposure can be achieved, and may be compatible with nanoformulation strategies being investigated for long-acting drug delivery (14, 15).
Various types of MAPs exist for intradermal drug delivery (e.g. hollow, dissolvable and solid) and these have been extensively studied in the delivery of various drugs, including large molecules and vaccines. Hollow stainless steel microneedles (AdminPen® template) have been used to deliver liquid formulations (16). MAPs can be used for the delivery of biomacromolecules (17), transcutaneous immunization (18) and hormones (19).
Polymeric MAPs are suitable for LA systemic delivery of small molecules. Hydrogel-forming MAPs control drug release through in-skin hydrogel swelling (20). Micro- or nanoformulations loaded into dissolving MAPs deliver the formulations to the viable skin layers upon dissolution of the needles. Subsequent controlled drug release from the polymeric micro- or nanoparticles can then achieve LA release for sustained time intervals (21, 22). Unlike hydrogel-forming MAPs, drug release is therefore possible for weeks or even months after removal of the patch, thus potentially improving patient compliance.
Preclinical and clinical evaluation of novel formulations and modes of delivery is hindered by numerous challenges. Computational simulations can support design of successful administration strategies and rational optimisation. Physiologically-based pharmacokinetic (PBPK) modelling is based on the mathematical description of anatomical, physiological and molecular processes describing pharmacokinetics through the integration of drug and patient specific data (23). PBPK modelling has been increasingly used in new chemical entity (NCE) applications and in investigation of clinical scenarios, suggesting reliability in pharmacokinetic predictions. PBPK models have been used to inform drug-drug interactions, CYP induction/ inhibition, pharmacogenetics and therapy optimisation in special patient populations (24).
The aim of this paper was to develop a novel intradermal PBPK model for ARV administration using MAPs. The developed model was used to identify minimum dose and a range of release rates for the LA administration of cabotegravir and rilpivirine nanoformulations to maintain plasma concentrations above established antiretroviral targets throughout dosing intervals of one to four weeks.
Methods
A whole-body PBPK model was used to assess the intradermal release from MAPs in healthy adults. An intradermal compartment was appended to an earlier PBPK model using Simbiology® v.4.3.1., a product of MATLAB® v.8.2 (MathWorks, Natick, MA, USA 2013) (25) Drug distribution was described using blood flow-limited, first-order kinetics (26). Instant and uniform drug distribution across each tissue and organ was assumed. Ethical approval was not required for this study as the data was computer generated.
Microneedle patch
In this exploratory study, we used a single dissolving MAP design as a starting point, based on some of our previous work (27). The thickness of stratum corneum, viable epidermis and dermis were in the range of 12-20 μm (28), 50-80 μm (29) and 300-4000 μm (30) respectively. An 11 × 11 array of microneedles 600 μm long with an interspacing of 300 μm on a 0.49 cm2 baseplate were considered as the base of a single microneedle array (27). A mean penetration depth of the microneedles after skin application was assumed to be 430 ± 6.0 μm, with a pore width of 224 ± 5.0 μm made during penetration for each microneedle (27). A maximum of 32.7 mg formulation (including drug and excipients) was assumed per MAP (made up of 16.33 individual microneedle arrays), with an area of 8 cm2. An average of 3.55 ml/min/100g tissue (2.8-4.3 ml/min/100g tissue) (31) and 8 × 10−6 cm3/s/cm3 of tissue (31) in the adult forearm were considered as the respective blood and lymphatic flow rates to the intradermal compartment.
Intradermal PBPK model
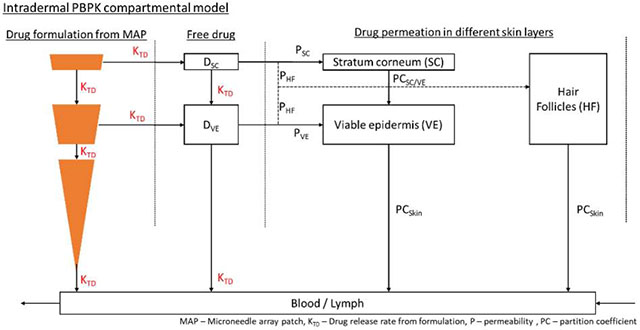
The intradermal compartment was adapted and modified from Gajewska et al. (32), as shown in Figure 1. The intradermal compartment has been divided into four sub-compartments, namely stratum corneum, viable epidermis, hair follicles and dermis. The inserted microneedle was divided into three different compartments having a relative proportion of the total drug amount depending on insertion length and pore width of each skin layer as shown in Figure 2.
Figure 1.
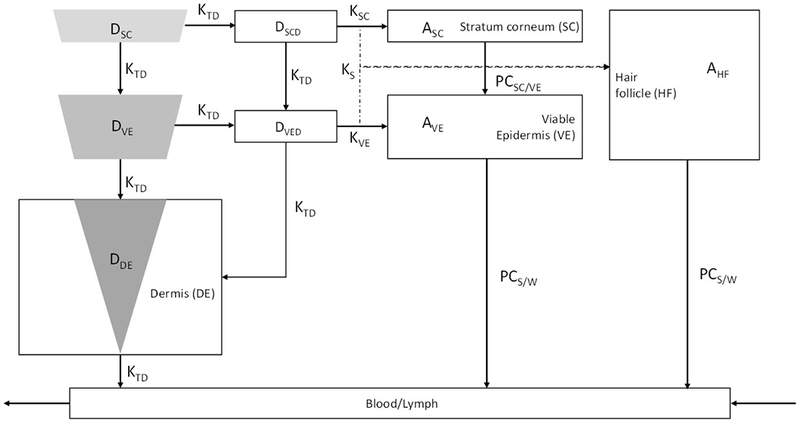
Drug release pathway from the microneedles in the intradermal compartment reaching the blood and lymphatic circulation. AHF, ASC, AVE – drug amount penetrating the hair follicles, stratum corneum and viable epidermis respectively; DDE, DSC, DVE – microneedle drug depot in the dermis, stratum corneum and viable epidermis respectively; DSCD, DVED – amount of released drug from the nanoparticles in the stratum corneum and viable epidermis respectively; Ks, KSC, KVE – rate of drug permeation from microneedle to skin, microneedle to stratum corneum and microneedle to viable epidermis respectively; KTD – drug release rate from the encapsulated nanoparticles; PCSC/VE, PCS/W – drug partition coefficient between stratum corneum & viable epidermis and skin & water respectively.
Figure 2.
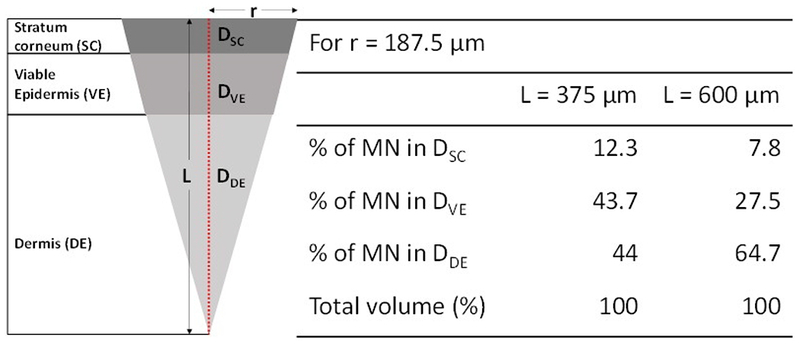
Representative drug deposition subsequent to microneedle insertion in different layers of the skin at time t = 0 h for varying insertion depths. L - microneedle insertion depth, r – radius of the created pore; DSC, DVE, DDE – amount of drug present in stratum corneum, viable epidermis and dermis respectively.
The following assumptions were made in the skin compartment: 1) unidirectional drug flow from the top layer (stratum corneum) to blood circulation; 2) hair follicles cover 0.1 % of the total skin (as previously described, it was assumed that hair follicles have access to one-third of the drug traversing laterally, with one-fourth of the fine skin’s blood-flow reaching the hair follicles (32)); 3) diffusion and partition coefficients across different skin layers and nanoparticle release rate were kept constant throughout the kinetic process; 4) only the free drug released from the nanoparticles diffuse through the layers of skin to reach blood circulation. Drug flow through the intradermal compartment is shown in Figure 2. The equations used in the intradermal compartment are as follows:
Drug present at the microneedle depot in the stratum corneum:
Where DSC – total drug present at the microneedle depot in the stratum corneum, KTD – drug release rate from the encapsulated formulation, DSCD – total free drug present at the microneedle depot in the stratum corneum at time t, KSC – permeability rate constant to the stratum corneum
Drug present at the microneedle depot in the viable epidermis:
Where DVE is the total drug present at the microneedle depot in the viable epidermis at time t, DVED – total free drug present at the microneedle depot in the viable epidermis at time t, PCS/W – partition coefficient between skin and water, KVE – permeability rate constant to the viable epidermis, KS – permeability rate constant to the skin/hair follicles.
Drug present at the microneedle depot in the dermis:
Where DDE is the total drug amount present at the microneedle depot in the dermis at time t.
Drug permeation from the microneedle to the adjacent skin layers (33) is given by:
Where M is the amount of drug permeating across the skin, A is the surface area of drug in contact with the skin, Pe is the effective permeability and Cb is the drug concentration at the depot site.
Drug traversing through the stratum corneum:
Where ASC is the amount present in the stratum corneum at time t, PCSC/VE – partition coefficient between stratum corneum and viable epidermis.
Drug traversing through the viable epidermis:
Where AVE is the amount present in the viable epidermis at time t, DVED – total free drug present at the microneedle depot in the viable epidermis at time t, PCS/W – partition coefficient between skin and water.
Drug traversing through the hair follicles:
Where AHF is the amount present in the hair follicles at time t
Lateral diffusion from the microneedle depot was based on the drug permeability rate and the diffusion across the skin was dependent on the drug partition coefficient between the layers. Permeability rate constants – KS, KSC, KVE and drug partition coefficients - PCSC/VE, PCS/W were derived using quantitative structure-property relationships (QSPR) informed by a previous publication (32) (included in the supplementary document). A summary of the various QSPR equations used for parameter computation is provided in the supplementary section. An average value was considered from the numerous equations.
Human physiological parameters
Key characteristics of both male and female (50:50) adults such as age ranging between 18 and 60 years, weight and body mass index (BMI) were defined initially, and other key characteristics such as body surface area (BSA) and height were computed using allometric equations. Organ weights, volumes, and blood flow rates were computed using anthropometric equations from the literature (34, 35). The drug distribution across tissues and organs were described using first-order differential equations (36, 37). Physicochemical and drug specific properties are presented in Table 1.
Table 1.
Physicochemical properties, in vitro and population pharmacokinetic data of anti-HIV drugs
| Rilpivirine (rat) | Rilpivirine (human) | Cabotegravir (human) | |
|---|---|---|---|
| log Po:w | 4.32 (53) | 4.32 (53) | 2.2 (23) |
| Protein binding | 99.7% (53) | 99.7% (53) | 99.3% (23) |
| pKa | 3.26 (53) | 3.26 (53) | 4.14 (23) |
| Blood-to-plasma ratio | 0.67 (53) | 0.67 (53) | 0.441 (23) |
| †4.5 (1A1) / 2.2 (1A9) | |||
| Plasma clearance | 1.3 L/kg/h | †2.04 (53) | (23) |
| IM release rate (h−1) | 2.6 × 10−2 | *9 × 10−4 | 4.54 × 10−4 (23) |
| Intradermal release rate (h−1) | ‡2 × 10−4 | - | - |
| Dose | 5, 20 mg/kg IM, 120 mg TD | - | - |
Values are presented as mean (reference). log Po:w – Partition coefficient between octanol and water; pKa – logarithmic value of the dissociation constant;
Release rate followed a linear increase with respect to time shown in the following equation: (0.005/1344)*time+0.0002, time in hours.
Values represent intrinsic clearance in μl/min/pmol, rilpivirine is metabolised by CYP3A4, and cabotegravir by UGT1A1 and UGT1A9.
Release rate observed for an old formulation of rilpivirine derived using the PBPK model (38).
Model qualification
The model was initially qualified in vivo for IM nanoformulated rilpivirine in rats followed by human data as shown in supplementary figure 1. The drug specific parameters of rilpivirine utilised in the PBPK model were qualified against available IM pharmacokinetic data followed by the qualification of the intradermal compartmental model using available in vivo data in rats from an earlier pharmacokinetic study (38). A single IM injection with two different doses - 5 mg/kg and 20 mg/kg and new in vivo experiments performed in rats with a 120 mg intradermal microneedle patch administered for 8 weeks were used for model qualification. For the intradermal patch, only 57.45% of the total available drug was assumed to be effectively delivered over the dosing interval, i.e., the total amount of drug present in the microneedles excluding the drug present in the baseplates, as determined in previous in vivo experiments (27). PBPK models were considered as qualified if the mean simulated Cmax (maximum plasma concentration), Ctrough (trough plasma concentration) and AUC (area under the curve) were within 50% of the mean reported in vivo data. Subsequently, the intradermal compartment was appended to a previously qualified rilpivirine PBPK model in humans (23) for pharmacokinetic predictions.
PBPK analysis
PBPK predictions were performed initially for a daily oral dosing for four weeks (10 mg and 25 mg once daily for cabotegravir and rilpivirine, respectively) to reach steady-state concentrations, followed by a q4-weekly loading dose with a single microneedle patch and then 11 consecutive q4-weekly maintenance patches, for a total of 336 consecutive days of drug exposure. Pharmacokinetics were predicted for various combinations of dose (180 mg, 360 mg, 540 mg, 720 mg, 900 mg and 1080 mg) and release rates (5×10−4, 1×10−3, 1.5×10−3, 2×10−3, 2.5×10−3 and 3×10−3 h−1) for both cabotegravir and rilpivirine. Considering smaller patch sizes, q-weekly MAP doses were also modelled using loading doses – 30mg, 60 mg , 90 mg, 180 mg, 270 mg and 360 mg -- and maintenance doses – 30mg, 60mg, 90 mg, 180 mg and 270 mg -- at various release rates (7×10−3, 8×10−3, 9×10−3, 10×10−3, 11×10−3 and 12×10−3 h−1) after achieving steady state concentrations from oral administration. The optimal dose and release rate were identified by implementing trial and error approach by assessing various combinations of doses and release rates. Minimum doses and a range of suitable release rates of nanoformulations were estimated such that plasma concentrations remained above minimally effective concentrations throughout the dosing interval – 1.2 mg/L for a 10 mg daily oral Ctrough for cabotegravir (39), and 70 ng/ml for a 25 mg daily oral Ctrough for rilpivirine (39).
The effect of varying penetration depth was assessed at a constant pore width of 224 ± 5.0 μm and varying pore radius. The effect of pore size on the release rate pharmacokinetics of rilpivirine was also assessed at a constant microneedle length of 430 ± 6.0 μm and varying pore width at a constant dose of 720 mg and release rate of 1.5×10−3 h−1.
Results
PBPK qualification
Comparisons of observed and simulated pharmacokinetic parameters at the end of eight weeks are shown in Table 2. The percentage difference of the simulated Cmax and AUC against in vivo data is less than 50% for model qualification. Simulated pharmacokinetics from the designed intradermal PBPK model also satisfied the qualification limit against experimental data. Qualification of IM cabotegravir and rilpivirine human PBPK models is presented in the supplementary section.
Table 2.
Validation of the PBPK model for IM (38) and intradermal rilpivirine formulations in rats in vivo
| Observed | Simulated (n =100) | % difference simulated vs. clinical | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Route of administration and dose | Cmax | AUC0-last | Ctrough | Cmax | AUC0-last | Ctrough | Cmax | AUC0-last | Ctrough |
| Intramuscular (5 mg/kg, single injection) | 71 | 3840 | - | 55.9 ± 6.43 | 5.67 ± 1.25 | - | −21.3 | 47.6 | - |
| Intramuscular (20 mg/kg, single injection) | 158 | 15300 | - | 222 ± 25.5 | 22.4 ± 4.64 | - | 40.5 | 46.3 | - |
| Intradermal † (120 mg, microneedle patch) | 416 | - | 26.5 | 481 ± 42.9 | 286 ± 28.1 | 38.7 ± 4.45 | 24.5 | - | 46.0 |
Values are represented as arithmetic mean ± standard deviation wherever applicable; AUC0-last – area under the concentration-time curve, Cmax – maximum plasma concentration, Ctrough – trough plasma concentration; Cmax and Ctrough are expressed as ng/ml and AUC is expressed as μg × h/ml;
PBPK model is assumed to be qualified if % difference is less than 100.
Only 57.45 % of the total administered drug was assumed to be delivered using microneedles (27).
Intradermal dose and release rates
The Ctrough of cabotegravir and rilpivirine at various q4-weekly doses and release rates (Figure 3) and weekly intradermal loading and maintenance dose and release rates (Figure 4) are shown. Our calculations indicate that a minimum q4-weekly loading dose of 360 mg with a release rate between 1×10−3 – 3×10−3 h−1, and a maintenance dose of 180 mg with a release rate of 1×10−3 −1.5×10−3 h−1 would be required for an intradermal cabotegravir MAP to maintain plasma concentrations above a target Ctrough of 1.2 μg/ml.
Figure 3.
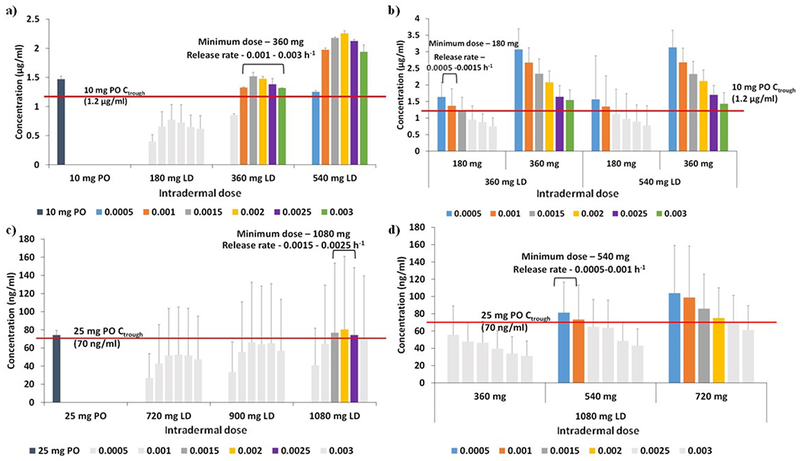
Ctrough values of cabotegravir (a, b) and rilpivirine (c, d) – 4-weekly loading (a, c) and maintenance (b, d) doses for various release rates. The red lines represent the target concentrations considered for this dose optimisation study. LD – loading dose, PO – once daily. Error bars represent the standard deviation.
Figure 4.

Ctrough values of cabotegravir (a, b) and rilpivirine (c, d) – weekly loading (a, c) and maintenance (b, d) doses for various release rates. The red lines represent the target concentrations considered for this dose optimisation study. LD – loading dose. Error bars represent the standard deviation.
For rilpivirine, a q4-weekly regimen requires a 1080 mg loading dose with release rates of 1.5×10−3 – 2.5×10−3 h−1, and a maintenance dose of 540 mg release rates of 5×10−4 – 1×10−3 h−1 to maintain a target Ctrough of 70 ng/ml.
Cabotegravir q-weekly MAP administration requires a minimum loading and maintenance dose of 60 mg between the release rates of 7×10−3 and 12×10−3 h−1. But if the loading dose is increased to 90 mg, a maintenance dose of 30 mg would be enough to have plasma concentrations over the target Ctrough. For rilpivirine a loading dose of 270 mg and a maintenance dose of 180 mg, with a nanoformulation release rate ranging from 7×10−3 – 9×10−3 h−1. This is significantly higher than the optimal release rate observed for q4-weekly formulations. However, if the loading dose is increased to 360 mg, the required maintenance dose falls to 90 mg to sustain the required Ctrough.
Effect of needle length, pore radius and release rate
The effect on pharmacokinetics of rilpivirine for varying penetration depths and needle pore sizes at a constant dose and release rate are shown in Figure 5a & b. The pharmacokinetic parameters Cmax and AUC increased as the penetration depth increased. However, there was no significant difference in the simulated Ctrough. Neither a significant difference nor a trend was observed in the pharmacokinetic parameters as the pore radius increased (Figure 5b).
Figure 5.
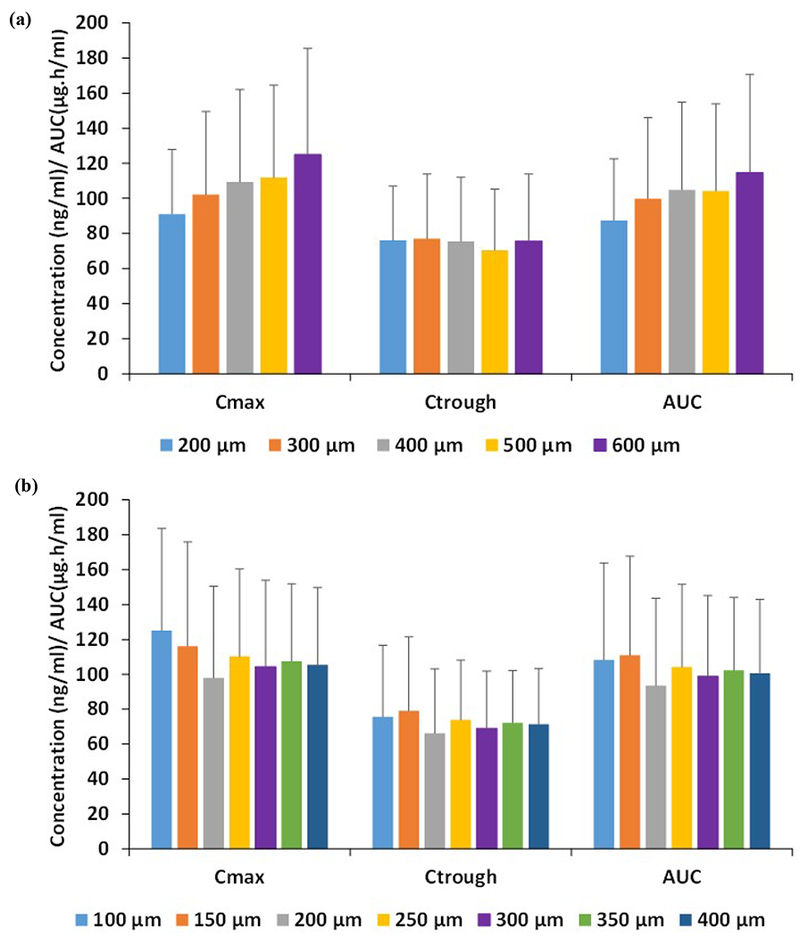
Pharmacokinetic parameters AUC, Cmax and Ctrough of rilpivirine for various (a) penetration depths and (b) microneedle pore radius. Error bars represent the standard deviation.
Simulated intradermal administration of rilpivirine predicted a rise in Cmax as the release rate increased. However, Ctrough increased until a release rate of 1.5χ10−3 h−1 and faster release rates were characterised by decreasing Ctrough (Figure 6a). At a constant release rate, as the dose increased, rilpivirine pharmacokinetics increased accordingly (6b).
Figure 6.
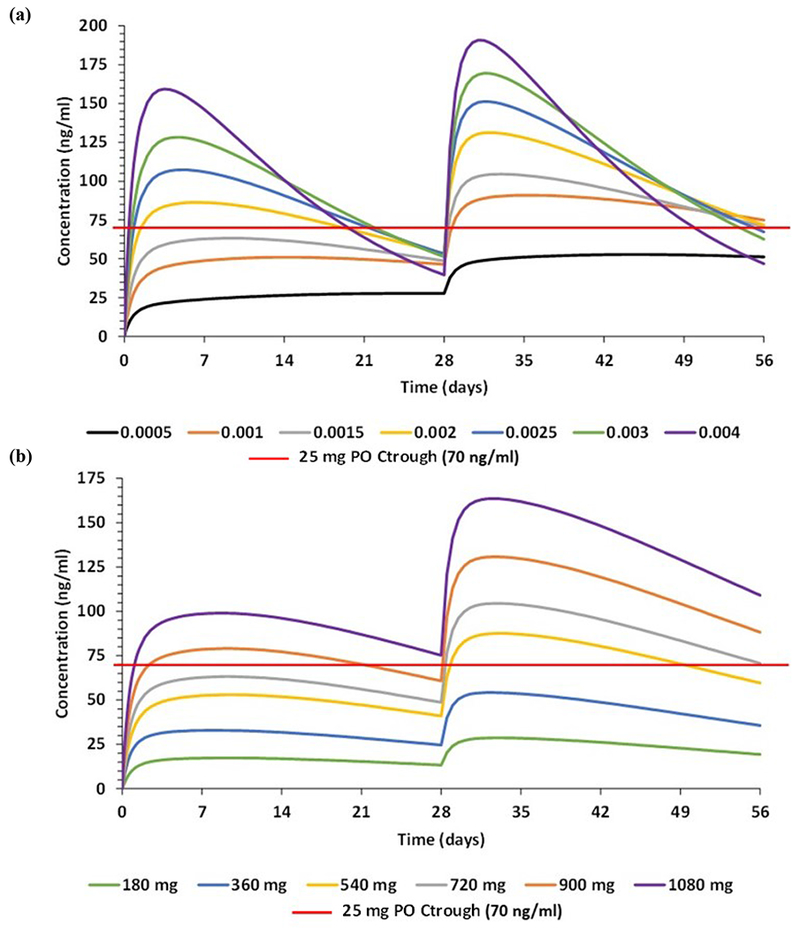
Effect on plasma concentration of rilpivirine at (a) various release rates in h−1 at a constant dose of 720 mg and (b) varying doses at a constant release rate of 1.5×10−3 h−1.
Discussion
Non-invasive intradermal MAPs may represent effective novel drug delivery vehicles for chronic administration of ARVs. In this study, a compartmental PBPK model was designed and integrated with a previously published whole-body PBPK model (25). This was first qualified with observed data in rats and humans followed by informing nanoformulation characteristics in humans for cabotegravir and rilpivirine MAPs.
Model qualification against observed data showed satisfactory results with differences less than 50% in line with convention for this approach. Subsequently, the models were utilised to simulate administration of rilpivirine and cabotegravir LA formulations using MAPs. A range of release rates were evaluated to identify the optimal dosing values that would sustain plasma concentrations over established targets throughout the dosing interval at the lowest possible doses.
The simulations of cabotegravir MAPs indicate an initial minimum monthly dose of 360 mg and a maintenance dose of 180 mg. Consequently, a MAP with high drug loading will be required. In the past, several examples of MAPs containing drug loading between 50 and 90% have been reported (40, 41). Additionally, in order to maximize drug loading in the needle tips, differently shaped MAPs may be used. Patches containing a higher needle density (19×19) while keeping the needle geometry (600 μm height and 300 μm base diameter) have been previously used (42–45). These MAPs showed similar insertion depths as the 11×11 MAPs (approximately 450 μm using manual insertion) (45) but have three times more needles in the same MAP area. Therefore, with drug loading of 50% to 90%, the density of common polymeric materials used to prepare MAPs (1.2 g/cm3) (46), and the volume of the needles tips, all the drug present in the needles can be successfully delivered (40, 41). The estimated patch size for the loading dose of cabotegravir will range between 70 and 90 cm2 (based on a loading of 32.7 mg of formulation per 8 cm2). On the other hand, the maintenance dose patch will have a size ranging from 35 to 45 cm2. Although the required patch sizes seem large, smaller patches can be achieved with higher drug loading and since these are dissolvable patches, they can be removed within a short span of time (<24 h) subsequent to their application.
Following a similar rationale, rilpivirine could be formulated in q-weekly patches (weekly loading and maintenance doses of 270 mg and 180 mg, respectively). Our model suggests that the doses required for q4-weekly patches of RPV will require unrealistically large patches between 130 – 260 cm2. If a 19×19 RPV MAP can be prepared, the patch sizes will range between 25 and 44 cm2 for the q-weekly RPV loading dose and between 15 and 30 cm2 for the maintenance dose. High variability in dose predictions were observed due to the high variation in the abundance of cytochrome P450 enzyme across the population that affects the systemic clearance of rilpivirine.
The presented data indicate that a weekly combination CAB and RPV may be achievable for a self-administered therapy. These patch sizes seem large, but it is important to note that conventional transdermal patches with sizes up to 140 cm2 can be found in the market for post-herpetic neuralgia (47). Moreover, it has been reported that manual application of larger MAPs has been carried out successfully in human volunteers (11). However, the patient must be properly instructed as to how these patches should be applied (11). It should also be noted that the required patch size could be reduced by using more potent ARV formulations, or higher drug loading.
The parametrisation of drug partition coefficients and permeability rates between skin tissues were based on previous models, and when multiple parameters were available, an average value was selected (correlations shown in the supplementary document). However, standardised experimental methodology for the characterisation of these parameters and in vitro - in vivo extrapolation would support a more reliable parametrisation with application across different computational models.
Several percutaneous in silico models have been published to describe drug absorption kinetics for topical or systemic delivery. Some relevant examples include a percutaneous model for different topical formulations of diclofenac (48), as well as quantitative structure-permeation relationship (QSPR) models, porous models, transient models such as compartment models, and slow partition kinetic models, to describe transdermal drug delivery (49). Additionally, multiple pharmacokinetic dermal models for the simulation of drug permeation through the skin for topical applications have been developed (32). However, no intradermal models for the simulation of LA formulation administration using MAPs has yet been reported.
The described intradermal PBPK model provides a valuable estimate of formulation requirements for MAP delivery but is characterised by several limitations. The dose and release rates were simulated in optimal conditions and do not include any loss of drug during application. This includes the assumption of 100% bioavailability during dose predictions and lower fraction can increase the dose required thus increasing the size of the microneedle patch. A canonical shaped needle has been simulated in this study; changes in the shape and size of the microneedle such as pyramidal or tetrahedral, or an increase in microneedle density could be used to tune the release rate and diffusion following application. Ethnicity and sex are additional patient factors that can influence pharmacokinetics following intradermal administration. However, their effect on release kinetics has not been evaluated in clinical studies to date. Lag time after the application of MAPs has not been considered in this model, and a delay could be observed in the Cmax and tmax as a consequence. The human dermis is highly vascularized, and vasodilation or vasoconstriction could lead to altered drug release from the depot, affecting plasma pharmacokinetics (50, 51). Highly lipophilic drugs and particles less than 100 nm tend to enter the lymphatic circulation rather than blood, which may impact pharmacokinetics (52). Drug transport proteins play a key role in defining the pharmacokinetic profile of many drugs, but limited data relating to this site of delivery restricted their inclusion in the PBPK model. Complexity in the design of nanoformulations with the estimated release rates from current technology used in this model could also prove to be a limiting factor.
The qualified intradermal PBPK model has been cross-checked only against rat data for one drug, as this was the only relevant in vivo data in the published literature. Qualification against observed human data and diverse formulations will further improve confidence in the predictive value of the model. Long-term stability and tolerability of drug formulation at the site of administration in human skin must be investigated thoroughly to prevent any unwanted side effects. Transdermal drug administration is attractive for neonatal and paediatric applications, but this model only investigated the kinetics of drug delivery in average sized adults.
Conclusion
PBPK models have been successfully qualified for oral and IM rilpivirine and cabotegravir. A novel intradermal model has been qualified against observed data in rats and nanoformulation design has been informed for MAP administration in humans. Optimal doses between ranges of release rates suitable for intradermal delivery, and preferred dosing intervals were simulated such that plasma concentrations remained above target trough concentrations throughout dosing intervals. Based on our simulations, q4-weekly cabotegravir and q-weekly cabotegravir and rilpivirine MAPs are feasible with patch sizes less than 60 cm2, and q-monthly rilpivirine would be possible with a denser MAP. This model could provide a useful platform to inform the design of novel formulations for chronic transdermal drug administration.
Supplementary Material
Acknowledgements
This work was presented at the 25th Conference on Retroviruses and Opportunistic Infections, Boston, MA, USA, 2018 (Poster 485)
Funding
This study was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R24 AI 118397).
Transparency declarations
Dr. Rajoli, Dr. Curley, Mr. Chiong, Dr. Larrañeta and Prof. Donnelly do not have any conflict of interests; Prof. Flexner reports grants from Gilead Sciences, personal fees from ViiV Healthcare, personal fees from Janssen Pharmaceuticals, personal fees from Merck Laboratories, personal fees from Mylan Pharmaceuticals, personal fees from Cipla Pharmaceuticals, outside the submitted work; In addition, Prof. Flexner has a patent Semi-Solid Prodrug SDNs pending. Prof. Owen reports grants from Merck, personal fees from Merck, grants from ViiV Healthcare, personal fees from ViiV Healthcare, grants from Janssen, non-financial support from Janssen, grants from AstraZeneca, outside the submitted work; In addition, Dr. Owen has a patent drug delivery issued, and a patent drug delivery pending. Dr. Siccardi reports grants from ViiV, grants from Janssen, outside the submitted work;
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhang H, Jin R, Yao C, Zhang T, Wang M, Xia W, et al. Combination of long-acting HIV fusion inhibitor albuvirtide and LPV/r showed potent efficacy in HIV-1 patients. AIDS Research and Therapy. 2016;13(1):8. doi: 10.1186/s12981-016-0091-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Claborn KR, Meier E, Miller MB, Leffingwell TR. A Systematic Review of Treatment Fatigue among HIV-infected Patients Prescribed Antiretroviral Therapy. Psychology, health & medicine. 2015;20(3):255–65. doi: 10.1080/13548506.2014.945601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Landovitz RJ, Kofron R, McCauley M. The promise and pitfalls of long-acting injectable agents for HIV prevention. Current Opinion in HIV and AIDS. 2016;11(1):122–8. doi: 10.1097/coh.0000000000000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trezza C, Ford SL, Spreen W, Pan R, Piscitelli S. Formulation and pharmacology of long-acting cabotegravir. Curr Opin HIV AIDS. 2015;10(4):239–45. doi: 10.1097/coh.0000000000000168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barnhart M Long-Acting HIV Treatment and Prevention: Closer to the Threshold. Global Health: Science and Practice. 2017;5(2):182–7. doi: 10.9745/ghsp-d-17-00206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taddio A, Ipp M, Thivakaran S, Jamal A, Parikh C, Smart S, et al. Survey of the prevalence of immunization non-compliance due to needle fears in children and adults. Vaccine. 2012;30(32):4807–12. doi: 10.1016/j.vaccine.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 7.Parsons JT, Rendina HJ, Whitfield THF, Grov C. Familiarity with and Preferences for Oral and Long-Acting Injectable HIV Pre-exposure Prophylaxis (PrEP) in a National Sample of Gay and Bisexual Men in the U.S. AIDS and Behavior. 2016;20(7):1390–9. doi: 10.1007/s10461-016-1370-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Williams J, Sayles HR, Meza JL, Sayre P, Sandkovsky U, Gendelman HE, et al. Long-acting parenteral nanoformulated antiretroviral therapy: interest and attitudes of HIV-infected patients. Nanomedicine. 2013;8(11):1807–13. doi: 10.2217/nnm.12.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meyers K, Rodriguez K, Moeller RW, Gratch I, Markowitz M, Halkitis PN. High Interest in a Long-Acting Injectable Formulation of Pre-Exposure Prophylaxis for HIV in Young Men Who Have Sex with Men in NYC: A P18 Cohort Substudy. PLOS ONE. 2014;9(12):e114700. doi: 10.1371/journal.pone.0114700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donnelly RF, Larrañeta E. Microarray patches: potentially useful delivery systems for long-acting nanosuspensions. Drug Discovery Today. 2018;23(5):1026–33. doi: 10.1016/j.drudis.2017.10.013. [DOI] [PubMed] [Google Scholar]
- 11.Ripolin A, Quinn J, Larrañeta E, Vicente-Perez EM, Barry J, Donnelly RF. Successful application of large microneedle patches by human volunteers. International Journal of Pharmaceutics. 2017;521(1–2):92–101. doi: 10.1016/j.ijpharm.2017.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donnelly RF, Singh TRR, Garland MJ, Migalska K, Majithiya R, McCrudden CM, et al. Hydrogel-Forming Microneedle Arrays for Enhanced Transdermal Drug Delivery. Advanced Functional Materials. 2012;22(23):4879–90. doi: 10.1002/adfm.201200864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ita K Transdermal Delivery of Drugs with Microneedles—Potential and Challenges. Pharmaceutics. 2015;7(3):90–105. doi: 10.3390/pharmaceutics7030090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bakshi RP, Tatham LM, Savage AC, Tripathi AK, Mlambo G, Ippolito MM, et al. Long-acting injectable atovaquone nanomedicines for malaria prophylaxis. Nature Communications. 2018;9(1):315. doi: 10.1038/s41467-017-02603-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tatham LM, Savage AC, Dwyer A, Siccardi M, Scott T, Vourvahis M, et al. Towards a Maraviroc Long-Acting Injectable Nanoformulation. European Journal of Pharmaceutics and Biopharmaceutics. 2018. doi: 10.1016/j.ejpb.2018.04.009. [DOI] [PubMed] [Google Scholar]
- 16.Aoyagi S, Izumi H, Isono Y, Fukuda M, Ogawa H. Laser fabrication of high aspect ratio thin holes on biodegradable polymer and its application to a microneedle. Sensors and Actuators A: Physical. 2007;139(1):293–302. doi: 10.1016/j.sna.2006.11.022. [DOI] [Google Scholar]
- 17.Ling M-H, Chen M-C. Dissolving polymer microneedle patches for rapid and efficient transdermal delivery of insulin to diabetic rats. Acta Biomaterialia. 2013;9(11):8952–61. doi: 10.1016/j.actbio.2013.06.029. [DOI] [PubMed] [Google Scholar]
- 18.Ding Z, Verbaan FJ, Bivas-Benita M, Bungener L, Huckriede A, van den Berg DJ, et al. Microneedle arrays for the transcutaneous immunization of diphtheria and influenza in BALB/c mice. Journal of Controlled Release. 2009;136(1):71–8. doi: 10.1016/j.jconrel.2009.01.025. [DOI] [PubMed] [Google Scholar]
- 19.Prausnitz MR, Langer R. Transdermal drug delivery. Nature Biotechnology. 2008;26:1261–8. doi: 10.1038/nbt.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim M, Jung B, Park J-H. Hydrogel swelling as a trigger to release biodegradable polymer microneedles in skin. Biomaterials. 2012;33(2):668–78. doi: 10.1016/j.biomaterials.2011.09.074. [DOI] [PubMed] [Google Scholar]
- 21.Yang P-Y, Zou H, Chao E, Sherwood L, Nunez V, Keeney M, et al. Engineering a long-acting, potent GLP-1 analog for microstructure-based transdermal delivery. Proceedings of the National Academy of Sciences. 2016;113(15):4140–5. doi: 10.1073/pnas.1601653113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen W, Tian R, Xu C, Yung BC, Wang G, Liu Y, et al. Microneedle-array patches loaded with dual mineralized protein/peptide particles for type 2 diabetes therapy. Nature Communications. 2017;8(1):1777. doi: 10.1038/s41467-017-01764-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rajoli RKR, Back DJ, Rannard S, Meyers CF, Flexner C, Owen A, et al. In Silico Dose Prediction for Long-Acting Rilpivirine and Cabotegravir Administration to Children and Adolescents. Clin Pharmacokinet. 2018;57(2):255–66. doi: 10.1007/s40262-017-0557-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshida K, Budha N, Jin JY. Impact of Physiologically Based Pharmacokinetic Models on Regulatory Reviews and Product Labels: Frequent Utilization in the Field of Oncology. Clinical Pharmacology & Therapeutics. 2017;101(5):597–602. doi: 10.1002/cpt.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajoli RKR, Back DJ, Rannard S, Freel Meyers CL, Flexner C, Owen A, et al. Physiologically Based Pharmacokinetic Modelling to Inform Development of Intramuscular Long-Acting Nanoformulations for HIV. Clin Pharmacokinet. 2015;54(6):639–50. doi: 10.1007/s40262-014-0227-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nestorov I Whole Body Pharmacokinetic Models. Clin Pharmacokinet. 2003;42(10):883–908. doi: 10.2165/00003088-200342100-00002. [DOI] [PubMed] [Google Scholar]
- 27.Garland MJ, Migalska K, Tuan-Mahmood T-M, Raghu Raj Singh T, Majithija R, Caffarel-Salvador E, et al. Influence of skin model on in vitro performance of drug-loaded soluble microneedle arrays. International Journal of Pharmaceutics. 2012;434(1):80–9. doi: 10.1016/j.ijpharm.2012.05.069. [DOI] [PubMed] [Google Scholar]
- 28.Böhling A, Bielfeldt S, Himmelmann A, Keskin M, Wilhelm KP. Comparison of the stratum corneum thickness measured in vivo with confocal Raman spectroscopy and confocal reflectance microscopy. Skin Research and Technology. 2014;20(1):50–7. doi: 10.1111/srt.12082. [DOI] [PubMed] [Google Scholar]
- 29.Sandby-Moller J, Poulsen T, Wulf HC. Epidermal thickness at different body sites: Relationship to age, gender, pigmentation, blood content, skin type and smoking habits. Acta Dermato-Venereologica. 2003;83(6):410–3. doi: 10.1080/00015550310015419. [DOI] [PubMed] [Google Scholar]
- 30.Bergman Ronald A. AK A, Heidger Paul M. Jr. Atlas of Microscopic Anatomy - A Functional Approach. . 2nd edition ed: W B Saunders Co; 1999. [Google Scholar]
- 31.Ibrahim R, Nitsche JM, Kasting GB. Dermal Clearance Model for Epidermal Bioavailability Calculations. Journal of Pharmaceutical Sciences. 2012;101(6):2094–108. doi: 10.1002/jps.23106. [DOI] [PubMed] [Google Scholar]
- 32.Gajewska M, Worth A, Urani C, Briesen H, Schramm KW. Application of physiologically-based toxicokinetic modelling in oral-to-dermal extrapolation of threshold doses of cosmetic ingredients. Toxicology Letters. 2014:227(3):189–202. doi: 10.1016/j.toxlet.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 33.Sinko PJ, Leesman GD, Amidon GL. Predicting Fraction Dose Absorbed in Humans Using a Macroscopic Mass Balance Approach. Pharmaceutical Research. 1991;8(8):979–88. doi: 10.1023/a:1015892621261. [DOI] [PubMed] [Google Scholar]
- 34.Bosgra S, Eijkeren Jv, Bos P, Zeilmaker M, Slob W. An improved model to predict physiologically based model parameters and their inter-individual variability from anthropometry. Crit Rev Toxicol. 2012;42(9):751–67. doi: 10.3109/10408444.2012.709225. [DOI] [PubMed] [Google Scholar]
- 35.Williams LR. Reference values for total blood volume and cardiac output in humans. 1994. Available from: http://web.ornl.gov/info/reports/1994/3445606042010.pdf. Accessed: 18/01/2018.
- 36.Peters S Evaluation of a Generic Physiologically Based Pharmacokinetic Model for Lineshape Analysis Clin Pharmacokinet. 2008;47(4):261–75. doi: 10.2165/00003088-200847040-00004. [DOI] [PubMed] [Google Scholar]
- 37.Poulin P, Theil FP. Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution. J Phann Sci. 2002;91(1):129–56. 10.1002/jps.10005. [DOI] [PubMed] [Google Scholar]
- 38.van ’t Klooster G, Hoeben E, Borghys H, Looszova A, Bouche M-P, van Velsen F, et al. Pharmacokinetics and Disposition of Rilpivirine (TMC278) Nanosuspension as a Long-Acting Injectable Antiretroviral Formulation. Antimicrobial Agents Chemotherapy. 2010;54(5):2042–50. 10.1128/aac.01529-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Margolis DA, Gonzalez-Garcia J, Stellbrink H-J, Eron JJ, Yazdanpanah Y, Podzamczer D, et al. Long-acting intramuscular cabotegravir and rilpivirine in adults with HIV-1 infection (LATTE-2): 96-week results of a randomised, open-label, phase 2b, non-inferiority trial. Lancet. 2017;390(10101):1499–510. doi: 10.1016/S0140-6736(17)31917-7. [DOI] [PubMed] [Google Scholar]
- 40.McCrudden MTC, Alkilani AZ, McCrudden CM, McAlister E, McCarthy HO, Woolfson AD, et al. Design and physicochemical characterisation of novel dissolving polymeric microneedle arrays for transdermal delivery of high dose, low molecular weight drugs. Journal of Controlled Release. 2014;180:71–80. doi: 10.1016/j.jconrel.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.González-Vázquez P, Larrañeta E, McCrudden MTC, Jarrahian C, Rein-Weston A, Quintanar-Solares M, et al. Transdermal delivery of gentamicin using dissolving microneedle arrays for potential treatment of neonatal sepsis. Journal of Controlled Release. 2017;265:30–40. doi: 10.1016/j.jconrel.2017.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larrañeta E, Lutton REM, Brady AJ, M. Vicente-Pérez E, David WA, Singh TRR, et al. Microwave-Assisted Preparation of Hydrogel-Forming Microneedle Arrays for Transdermal Drug Delivery Applications. Macromolecular Materials and Engineering. 2015;300(6):586–95. doi: 10.1002/mame.201500016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kennedy J, Larrañeta E, McCrudden MTC, McCrudden CM, Brady AJ, Fallows SJ, et al. In vivo studies investigating biodistribution of nanoparticle-encapsulated rhodamine B delivered via dissolving microneedles. Journal of Controlled Release. 2017;265:57–65. doi: 10.1016/j.jconrel.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vicente-Perez EM, Larrañeta E, McCrudden MTC, Kissenpfennig A, Hegarty S, McCarthy HO, et al. Repeat application of microneedles does not alter skin appearance or barrier function and causes no measurable disturbance of serum biomarkers of infection, inflammation or immunity in mice in vivo. European Journal of Pharmaceutics and Biopharmaceutics. 2017;117:400–7. doi: 10.1016/j.ejpb.2017.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Donnelly RF, McCrudden MTC, Zaid Alkilani A, Larrañeta E, McAlister E, Courtenay AJ, et al. Hydrogel-Forming Microneedles Prepared from “Super Swelling” Polymers Combined with Lyophilised Wafers for Transdermal Drug Delivery. PLOS ONE. 2014;9(10):e111547. doi: 10.1371/journal.pone.0111547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parekh G, Pattekari P, Joshi C, Shutava T, DeCoster M, Levchenko T, et al. Layer-by-layer nanoencapsulation of camptothecin with improved activity. International Journal of Pharmaceutics. 2014;465(1):218–27. doi: 10.1016/j.ijpharm.2014.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prausnitz MR, Mitragotri S, Langer R. Current status and future potential of transdermal drug delivery. Nature Reviews Drug Discovery. 2004;3:115–24. doi: 10.1038/nrd1304. [DOI] [PubMed] [Google Scholar]
- 48.Polak S, Ghobadi C, Mishra H, Ahamadi M, Patel N, Jamei M, et al. Prediction of concentration & time profile and its inter-individual variability following the dermal drug absorption. Journal of Pharmaceutical Sciences. 2012;101(7):2584–95. doi: 10.1002/jps.23155. [DOI] [PubMed] [Google Scholar]
- 49.Mitragotri S, Anissimov YG, Bunge AL, Frasch HF, Guy RH, Hadgraft J, et al. Mathematical models of skin permeability: An overview. International Journal of Pharmaceutics. 2011;418(1):115–29. doi: 10.1016/j.ijpharm.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 50.Sugibayashi K, Yanagimoto G, Hayashi T, Seki T, Juni K, Morimoto Y. Analysis of skin disposition of flurbiprofen after topical application in hairless rats. Journal of Controlled Release. 1999;62(1):193–200. doi: 10.1016/S0168-3659(99)00038-3. [DOI] [PubMed] [Google Scholar]
- 51.Ita KB. Transdermal drug delivery: progress and challenges. Journal of Drug Delivery Science and Technology. 2014;24(3):245–50. doi: 10.1016/S1773-2247(14)50041-X. [DOI] [Google Scholar]
- 52.Tegenge M, Mitkus R. A physiologically-based pharmacokinetic (PBPK) model of squalene-containing adjuvant in human vaccines. J Pharmacokinet Pharmacodyn. 2013;40(5):545–56. doi: 10.1007/s10928-013-9328-y. [DOI] [PubMed] [Google Scholar]
- 53.Clinical Pharmacology and Biopharmaceutics Review(s). Application number: 202022Orig1s000. Center for Drug Evaluation and Research, 2011. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202022Orig1s000ClinPharmR.pdf. Accessed: 17/07/2018. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.