

Abstract
Objective
To investigate the spectrum of antecedent infections in Chinese patients with Guillain‐Barré syndrome (GBS) and analyze the infections‐related clinical phenotypes locally.
Methods
A prospective case‐control study of 150 patients diagnosed with GBS and age‐ and sex‐matched neurological and healthy controls was performed to investigate recent infections of 14 pathogens serologically and collect the clinical data during a follow‐up of 12 months.
Results
In total, 53% of patients with GBS had a positive serology for recent infection, including Campylobacter jejuni (27%), influenza A (17%) and B (16%), hepatitis A virus (5%), dengue virus (3%), cytomegalovirus (3%), Epstein–Barr virus (3%), Mycoplasma pneumoniae (2%), herpes simplex virus (2%), varicella‐zoster virus (1%), and rubella virus (1%). Serology for infections of hepatitis E virus, Haemophilus influenzae, and Zika virus was negative. There was a higher frequency of C. jejuni, influenza A, influenza B, and hepatitis A virus infections in GBS patients than both the neurological and healthy controls. C. jejuni infection was more frequent in younger GBS patients and was associated with antibodies against GM1, GalNAc‐GD1a, and GM1:galactocerebroside complex. Influenza B infection was associated with a pure motor form of GBS.
Interpretation
C. jejuni, influenza A, influenza B, and hepatitis A virus serve as the most common cause of antecedent infections in GBS locally. Influenza B‐related GBS may represent a pure motor phenotype. Differences in the infectious spectrum worldwide may contribute to the geographical clinical heterogeneity of GBS.
Introduction
Guillain‐Barré syndrome (GBS) is an immune‐mediated polyradiculoneuropathy characterized by a rapidly progressive flaccid paresis. Recent evidence supports GBS as a spectrum disorder with regional variation and significant heterogeneity including clinical presentation, electrophysiology, and outcome.1, 2 Two thirds of the patients complained of antecedent infections before the onset of neurological signs.3 Some antecedent infections were associated with various clinical phenotypes in GBS. Typically, Campylobacter jejuni bearing the gangliosides‐like lipo‐oligosaccharides (LOS) accounts for the pathogenesis of axonal GBS, particularly acute motor axonal neuropathy.4 Cytomegalovirus (CMV) infection is associated with severe motor sensory deficits, demyelination, and antibodies to the ganglioside GM2.3 Mycoplasma pneumoniae infection is associated with anti‐galactocerebroside (GalC) antibodies and pediatric GBS.5 Global variation in infection burden may at least in part explain the regional differences in clinical presentation and subtype of GBS. In the 1990s, a study from Northern China reported axonal GBS as the major subtype in China associated with a high frequency of C. jejuni infection.6 More recent studies, however, showed that currently demyelinating GBS was the predominant subtype in both Northeastern and Southern China.7, 8 The rapid changes in the socioeconomic status of China may have influenced the exposure to infections and resulted in a shift of the predominant GBS subtype. Furthermore, many GBS patients developed liver dysfunction before treatment without obvious causes, that may be related to specific types of antecedent infections.9 The current study aimed to investigate the spectrum of GBS‐related antecedent infections in a Chinese local area and analyze the infection‐related clinical features.
Methods
Patients and blood samples
This study was performed in the Affiliated Hospital of Jining Medical University, a central hospital regionally in Southwest of Shandong Province, Northern China, where it has a population of 17.1 million with an urban–rural ratio of 1.19. Written informed consent was obtained from all participants, and study procedures were approved by the local Ethics Committee (reference 2013B017 and 2016B006). From October 2013 to June 2017, a total of 150 consecutive patients meeting the diagnostic criteria for GBS and its variants10, 11 from the Affiliated Hospital of Jining Medical University were included in this study, of whom 19 also participated in the International GBS Outcome Study.12 For each participant, the pretreatment serum was collected and kept at −80°C until use. The clinical data include: age, sex, upper respiratory tract infection or gastrointestinal infection within 4 weeks before developing neurological signs, motor and sensory deficits, cranial nerve involvement, ataxia, tendon reflex, pain, mechanical ventilation, nerve conduction study (NCS) within 2 weeks after onset,13 albuminocytological dissociation in cerebrospinal fluid (CSF), and GBS disability score (GBS‐DS)14 at nadir and 12 months. The disability score at 12 months was obtained from 146 (97%) of the patients by a telephone follow‐up or outpatient revisit. No follow‐up NCS was performed for the patients. Four patients were lost to follow‐up. To explore the relation between antecedent infection and liver function, data of liver function tests from patients before treatment were also collected. For the study of pretreatment liver dysfunction, 18 of the patients were excluded because of one or more of the reasons below: with a previous diagnosis of liver diseases, alcohol abuse or recent intake of liver‐toxic or liver enzyme‐inducing drugs, with definite factors resulting in muscle damage, and elevation of transaminases. The liver dysfunction was defined as either alanine aminotransferase or aspartate aminotransferase becoming 1.5 times higher than the upper limit of normal values.
Controls
After the inclusion of each patient with GBS, a sex‐ and age‐matched inpatient with other neurological diseases (OND) and sex‐ and age‐matched healthy donors in the same period were, respectively, selected from the local biological sample bank of the hospital established from July 2012. Among the 150 OND controls were included patients with a cerebral infarction (n = 23), cerebral hemorrhage (n = 27), peripheral vertigo (n = 28), Bell's palsy (n = 39), migraine (n = 15), myasthenia gravis (n = 4), meningitis (n = 3), Parkinson disease (n = 6), and epilepsy (n = 5). The 150 healthy controls were recruited from the same hospital and in all heathy controls, organic diseases were ruled out by the routine medical examination.
Antecedent infectious spectrum detection
A total of 14 infectious agents were selected according to previous report3 and unpublished data from Department of Infectious Diseases, Chinese Centre for Disease Control, including C. jejuni, M. pneumoniae, Haemophilus influenzae, influenza A and B virus, herpes simplex virus, varicella‐zoster virus, dengue virus, rubella virus, CMV, Epstein–Barr virus (EBV), hepatitis A virus, hepatitis E virus, and Zika virus. The details of ELISA kits for detecting the infectious agents are shown in Table S1. The serum samples were tested according to the manufactory instructions. To detect IgM antibodies, the serum was pretreated to remove the IgG antibodies and prevent false positivity. C. jejuni, influenza A, and influenza B virus infections were defined as the presence of IgA and/or IgM antibodies. H. influenzae infection was defined as the presence of IgG antibodies. Infections of M. pneumoniae, hepatitis A virus, herpes simplex virus, varicella‐zoster virus, EBV, dengue virus, rubella virus, CMV, hepatitis E virus, and Zika virus were defined as the presence of IgM antibodies. The serology for specific infections in the controls was detected only if more than five GBS patients were positive for this infection as previously described.3
Anti‐glycolipid antibody assay
The serum samples of patients with GBS were tested in duplicate for IgM and IgG antibodies against GM1, GM2, GM3, GM1b, GD1a, N‐acetylgalactosaminyl GD1a (GalNAc‐GD1a), GD1b, GT1a, GQ1b, GalC, sulfatide, GM1:GalC, GM1:sulfatide, GalC:cholesterol, and GalC:sulfatide complexes as previously described.15 In general, a vinyl ELISA plate (Corning, ME, USA) was coated with the glycolipid or glycolipid complex (5 pmol per well; for glycolipid complex, 2.5 pmol each). The serum diluted in phosphate buffered saline (PBS) (0.1 M, pH 7.4) containing 0.5% casein (1:500) was added into the plates for incubation overnight at 4°C. After three times of washing with 0.1 M PBS containing 0.05% Tween 20, the plates were added with horseradish peroxidase‐conjugated goat anti‐human IgG (gamma chain) (Thermo Scientific, Rockford, IL, USA) (1:3000 in PBS containing 0.5% casein) or horseradish peroxidase‐conjugated goat anti‐human IgM (heavy chain) (Thermo Scientific) (1:10000) and incubated at 37°C for 1 h. After washing the plates with the same washing buffer, the binding of IgG or IgM antibodies was visualized by O‐phenylenediamine (Sigma, MO, USA) developing solution in the darkness for 15 min and then the reaction was stopped by 2N hydrochloride. The absorbance at 492 nm/630 nm (as reference) was measured using a ChroMate® Microplate Reader (Awareness Technology, Palm City, USA). Each sample was tested with a blank and negative control for quality control. With reference to a blank control, the optical density value over 0.1 was considered positive. A positive serology for anti‐glycolipid antibodies was defined as the presence of either IgG or IgM or both.
Data availability
Our data will be shared by request from any qualified investigator for scientific purposes.
Statistical analysis
Normally distributed continuous data were presented as means and standard deviations. The categorical variables were shown as n (%). The difference in the frequency of infections in patients with GBS and the controls was compared by the Chi‐square test or Fisher's exact test. For patients with and without infections, the difference in demographic and clinical features, anti‐glycolipid antibodies, and clinical subtypes were compared by Chi‐square test or Fisher's exact test. The analysis was performed with the SPSS 20.0 analysis software (IBM, Armonk, NY). A two‐sided P < 0.05 was considered to be significant.
Results
Antecedent infectious spectrum in patients with GBS
The demographic and clinical features of the patients with GBS are shown in Table 1. There was no difference in age or sex between the patients with GBS and the controls. The mean age of patients with GBS was 51.0; interquartile range (IQR) was 41 to 64 and the male to female ratio was 1.2 (81/69). For patients with OND and the healthy controls, the mean age was 51.9 (IQR, 42–64) and 51.3 (IQR, 42–64), respectively; the sex ratio was both the same 1.2 (81/69). Of the 150 patients with GBS, 53% (80/150) had positive serology for either C. jejuni (n = 40, 27%), influenza A (n = 26, 17%), influenza B (n = 24, 16%), hepatitis A virus (n = 7, 5%), dengue virus (n = 4, 3%), CMV (n = 4, 3%), EBV (n = 4, 3%), M. pneumoniae (n = 3, 2%), herpes simplex virus (n = 3, 2%), varicella‐zoster virus (n = 2, 1%), and rubella virus (n = 1, 1%). There were significant higher frequencies of C. jejuni, influenza A, influenza B, and hepatitis A virus infections in patients with GBS than in the controls (Table 2). Twenty‐six (17%) patients had more than one infection (Fig. 1). Eighty percent of patients (120/150) were from rural areas. There was no difference in the frequency of antecedent infections in patients with GBS between urban and rural areas (Table S2). None of the patients had positive serology for hepatitis E virus, H. influenza, and Zika virus.
Table 1.
Demographic and clinical characteristics of 150 patients with Guillain‐Barré syndrome.
| Characteristic | n (%) |
|---|---|
| Age, mean (standard deviation) | 51.0 (16.1) |
| Male/female ratio | 1.2 (81/69) |
| Antecedent infection within 4 weeks | |
| Upper respiratory tract infection | 36 (24) |
| Gastrointestinal infection | 19 (13) |
| Motor deficits | |
| Upper and lower limb weakness | 111 (74) |
| Upper limb weakness only | 5 (3) |
| Lower limb weakness only | 11 (7) |
| None | 23 (15) |
| Sensory deficits | 62 (41) |
| Cranial nerve involvement | |
| Oculomotor weakness | 23 (15) |
| Facial weakness | 30 (20) |
| Bulbar weakness | 29 (19) |
| None | 68 (45) |
| Ataxia | 6 (4) |
| Tendon reflex at the nadir | |
| Hyporeflexia or areflexia | 128 (85) |
| Normal | 22 (15) |
| Pain | 13 (9) |
| Disability score at the nadir | |
| 1 | 35 (23) |
| 2 | 31 (21) |
| 3 | 20 (13) |
| 4 | 45 (30) |
| 5 | 17 (11) |
| 6 | 2 (1) |
| Albuminocytological dissociation in CSF | 95/123 (77) |
| Single nerve conduction study | |
| Primary demyelinating | 41/120 (34) |
| Primary axonal | 35/120 (29) |
| Unclassified | 27/120 (23) |
| Normal | 17/120 (14) |
| Disability score at 12 months | |
| 0–1 | 113/146 (77) |
| 2 | 13/146 (9) |
| 3 | 8/146 (6) |
| 4 | 5/146 (3) |
| 6 | 7/146 (5) |
| Pretreatment liver dysfunction | 13 (17/132) |
CSF, cerebrospinal fluid.
Table 2.
Frequency of antecedent infections in patients with Guillain‐Barré syndrome and the controls.
| GBS (n = 150) | OND controls (n = 150) | OR (95% CI) | P value |
HC (n = 150) |
OR (95% CI) | P value | |
|---|---|---|---|---|---|---|---|
| No infection | 70 (47) | 128 (85) | Reference | 132 (88) | Reference | ||
| Campylobacter jejuni | 40 (27) | 10 (7) | 7.3 (3.5, 15.5) | <0.001 | 13 (9) | 5.8 (2.9, 11. 6) | <0.001 |
| Influenza A | 26 (17) | 8 (5) | 5.9 (2.6, 13.8) | <0.001 | 10 (7) | 4.9 (2.2, 10.7) | <0.001 |
| Influenza B | 24 (16) | 7 (5) | 6.3 (2.6, 15.3) | <0.001 | 9 (6) | 5.0 (2.2, 11.4) | <0.001 |
| Hepatitis A virus | 7 (5) | 3 (2) | 4.3 (1.1, 17.0) | 0.027 | 0 (0) | ‐ | ‐ |
| Dengue virus | 4 (3) | ||||||
| Cytomegalovirus | 4 (3) | ||||||
| Epstein–Barr virus | 4 (3) | ||||||
| Mycoplasma pneumoniae | 3 (2) | ||||||
| Herpes simplex virus | 3 (2) | ||||||
| Varicella‐zoster virus | 2 (1) | ||||||
| Rubella virus | 1 (1) |
The data are shown as n (%).
OR, odds ratio; CI, confidence interval; GBS, Guillain‐Barré syndrome; OND, other neurological disease; HC, healthy controls. No infection of the hepatitis E virus, Haemophilus influenzae, and Zika virus was detected.
Figure 1.
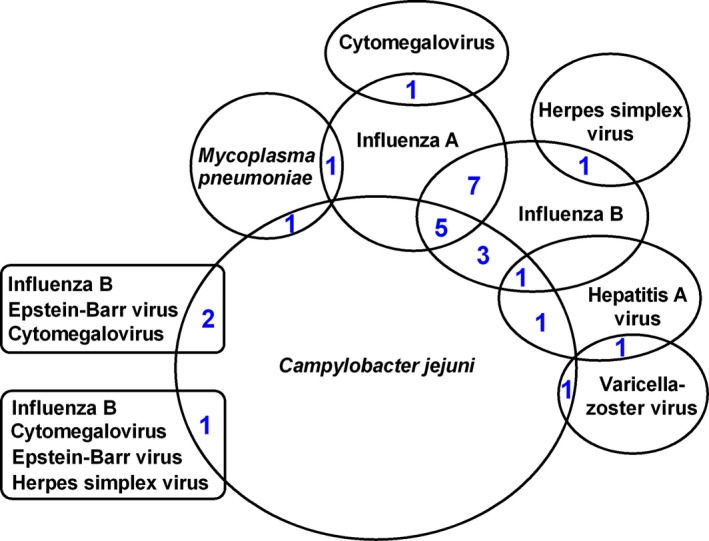
The number of patients with more than one infection.
Infection‐related clinical features
As shown in Table 3, the comparison was made among the GBS patients with infection of C. jejuni, influenza A, and influenza B as well as patients with no infections. There were significantly younger age and higher frequency of antecedent diarrhea complaints in patients with C. jejuni infection than the controls. All seven patients with preceding influenza B infection had a pure motor GBS without sensory deficits, which differed from the patients with infection of C. jejuni, influenza A, and patients without infection (P = 0.037). In addition, these patients with influenza B infection had no ataxia (0/7), no pain (0/7), no demyelinating subtypes (0/5) and high frequency of mechanical ventilation (2/7, 29%), and pretreatment liver dysfunction (2/7, 29%). There was no mechanical ventilation (0/11) in GBS patients following influenza A infection. There was no pretreatment liver dysfunction (0/25) in GBS patients following C. jejuni infection. With regard to patients with C. jejuni, influenza A and B virus as well as without infection, there was no difference in frequency of patients on cranial nerve involvement, ataxia, tendon reflex at nadir, pain, mechanical ventilation, electrophysiological classification, and pretreatment liver dysfunction.
Table 3.
Antecedent infections and clinical characteristics of patients with Guillain‐Barré syndrome.
| Characteristic | No infection (n = 70) | Campylobacter jejuni (n = 25) | Influenza A (n = 11) | Influenza B (n = 7) | P value* |
|---|---|---|---|---|---|
| Age, mean (standard deviation) | 51.8 (15.3) | 44.6 (15.3) | 60.6 (10.3) | 54.3 (19.3) | 0.03 |
| Antecedent infection within 4 weeks | |||||
| Upper respiratory tract infection | 16 (23) | 7 (28) | 3 (27) | 2 (28) | 0.032 |
| Gastrointestinal infection | 6 (9) | 8 (32) | 0 (0) | 0 (0) | |
| None | 48 (68) | 10 (40) | 8 (73) | 5 (72) | |
| Motor deficits | 61 (87) | 21 (84) | 9 (82) | 6 (86) | 0.969 |
| Sensory deficits | 35 (50) | 8 (32) | 6 (55) | 0 (0) | 0.037 |
| Cranial nerve involvement | 41 (59) | 15 (60) | 6 (55) | 6 (86) | 0.766 |
| Ataxia | 4 (6) | 1 (4) | 1 (9) | 0 (0) | 0.846 |
| Hyporeflexia or areflexia at nadir | 62 (89) | 24 (96) | 9 (82) | 5 (71) | 0.276 |
| Pain | 3 (4) | 3 (12) | 2 (18) | 0 (0) | 0.227 |
| Mechanical ventilation | 9 (13) | 3 (12) | 0 (0) | 2 (29) | 0.353 |
| Disability score at nadir | |||||
| ˂4 | 35 (50) | 13 (52) | 8 (73) | 3 (43) | 0.524 |
| ≥4 | 35 (50) | 12 (48) | 3 (27) | 4 (57) | |
| Albuminocytological dissociation in CSF | 41/58 (71) | 18/20 (90) | 9/9 (100) | 5/5 (100) | 0.053 |
| Single nerve conduction study | |||||
| Primary demyelinating | 21/57 (37) | 5/17 (29) | 3/9 (33) | 0/5 (0) | 0.661 |
| Primary axonal | 14/57 (25) | 7/17 (41) | 2/9 (22) | 2/5 (40) | |
| Unclassified | 13/57 (23) | 2/17 (12) | 3/9 (33) | 1/5 (20) | |
| Normal | 9/57 (16) | 3/17 (18) | 1/9 (11) | 2/5 (40) | |
| Disability score at 12 months | |||||
| ˂2 | 52/68 (76) | 18 (72) | 9/10 (90) | 5 (71) | 0.708 |
| ≥2 | 16/68 (24) | 7 (28) | 1/10 (10) | 2 (29) | |
| Pretreatment liver dysfunction | 12/63 (19) | 0/25 (0) | 2/11 (18) | 2/7 (29) | 0.109 |
CSF, cerebrospinal fluid. If not specified, the data are shown as n (%).
The P value showed the difference among the four groups.
Antecedent infections and the clinical variants
The patients were classified into GBS (n = 125, 83%), Miller Fisher syndrome (MFS) (n = 4, 3%), acute ophthalmoparesis (n = 4, 3%), GBS/MFS overlap (n = 2, 1%), bifacial weakness with paraesthesias (n = 2, 1%), acute pharyngeal weakness (n = 2, 1%), and pure sensory subtype (n = 11, 7%). As shown in Figure 2, there was no association between these variants of GBS and the presence of specific types of preceding infection.
Figure 2.
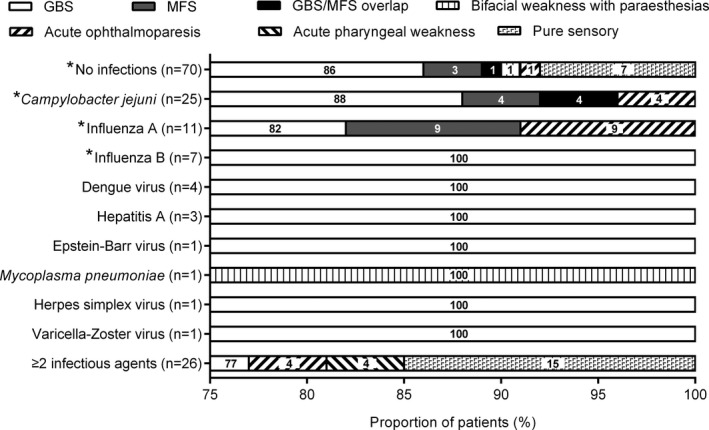
Antecedent infections and the clinical variants of Guillain‐Barré syndrome. As shown, Guillain‐Barré syndrome (GBS) constitutes the major subtypes in all of the patients with and without infections except that one patient with Mycoplasma pneumoniae had the bifacial weakness with paraesthesias. *There was no difference in frequency of GBS among the patients with and without infections (partition of Chi‐square test, P = 0.607). MFS, Miller Fisher syndrome.
Antecedent infections and anti‐glycolipid antibodies
As shown in Table S3, the frequency of antibodies against glycolipids and glycolipids complex in 150 of patients with GBS was anti‐GM1 (39%), anti‐GM1b (1%), anti‐GD1a (11%), anti‐GD1b (19%), anti‐GalNAc‐GD1a (11%), anti‐GQ1b (13%), anti‐GM1:GalC complex (23%), anti‐GM1:sulfatide complex (20%), and anti‐GalC:sulfatide complex (1%), respectively. There was significantly higher frequency of antibodies against GM1, GalNAc‐GD1a, and GM1:GalC complex in patients following C. jejuni infection than patients without infections (P = 0.002, P < 0.001, and P = 0.019, respectively). The anti‐GalC:sulfatide complex antibodies were IgG subtype and were detected in one patient with influenza A and M. pneumoniae infections. For patients with anti‐GM1 IgG antibodies, 40% (23/58) had cross‐reaction with GM1:GalC complex and 60% solely bound to GM1. For patients with anti‐GM1:GalC complex IgG antibodies, 26% (9/34) showed complex‐independent, 24% (8/34) showed complex‐enhancement, 18% (6/34) showed complex‐attenuated, and 32% (11/34) showed complex‐dependent. For patients with anti‐GM1:sulfatide complex IgG antibodies, 43% (13/30) showed complex‐independent, 13% (4/30) showed complex‐enhancement, 17% (5/30) showed complex‐attenuated, and 27% (8/30) showed complex‐dependent.
Discussion
We hereby demonstrated that C. jejuni, influenza A, influenza B, and hepatitis A virus currently served as the most common cause of antecedent infections in GBS in the Southwest of Shandong Province, Northern China. The proportion of patients following infections of CMV, EBV, and M. pneumoniae only accounted for less than 5% of the total patients, respectively. Infections of dengue virus, herpes simplex virus, varicella‐zoster virus, and rubella virus were also detected but only in a minority of patients and not higher than in controls. The proportion of patients with C. jejuni infection in our region (27%) was similar to those patients reported in Dutch (32%)3 and the UK(26%)16 but lower than those reported in Bangladesh (57%)17 and a previous study from Northern China (66%).6 From 2000 to 2018, the average life expectancy locally increased from 73.9 to 78.1 (unpublished data). The changes in the proportion of GBS patients with C. jejuni infection in China may reflect the improved healthy conditions of this country and be related to the rapid development of society and the economy.18 The C. jejuni infection has a strong relation with axonal GBS.6, 17 Reduced proportion of patients with C. jejuni infection may contribute to the predominant GBS subtype in China changed from axonal GBS in the 1990s to demyelinating GBS in 2010s. Notably, seven patients with C. jejuni infection complained of upper respiratory tract symptoms. The infection of C. jejuni may breakdown the host immune balance and increase the host susceptibility to the infectious diseases.19 It is possible that C. jejuni infection may cause other infections beyond our study, which accounts for the upper respiratory tract symptoms in the patients.
Patients with GBS in our cohort study displayed infection‐related clinical features. Being similar to the Dutch study,3 patients with C. jejuni infection had more often complaints of diarrhea than other patients. Although a cohort study from Bangladesh showed no association between the age of patients with GBS and C. jejuni infection,17 our study showed that C. jejuni infection was more frequent in younger patients with GBS, which may reflect a special dietary construct increasing the risk of C. jejuni infection in the youth population. Similarly, a recent international study in GBS showed that patients with the axonal subtype are younger than other patients.2 However, in our study, there was no difference in frequency of electrophysiological subtypes between patients with C. jejuni and other infections. Notably, GBS patients following influenza B infections displayed pure motor deficits and high risk of needing mechanical ventilation (2/7, 29%) while GBS patients following influenza A infections had no need for mechanical ventilation (0/11) and relatively low frequency of patients with GBS‐DS ≥ 4 at nadir. Our study supported influenza B‐related GBS as a severe phenotype of pure motor deficits while influenza A‐related GBS mainly presents as a mild clinical phenotype. Our results were similar to a previous study that none of influenza A‐related GBS (0/8) needed mechanical ventilation while half of the GBS patients following influenza B infection (2/4) needed mechanical ventilation.20 The exposure to different types of preceding infection in combination with host factors among different regional or ethnic groups may result in the geographical clinical heterogeneity in GBS worldwide.
Previously, the relation between C. jejuni infection and antibodies against gangliosides, including GM1, GM1b, GD1a, GalNAc‐GD1a, and GQ1b, has been well established in GBS.21, 22 In our region, there was a strong correlation between C. jejuni infection and anti‐GM1, anti‐GalNAc‐GD1a, and anti‐GM1:GalC complex antibodies. The gene polymorphism of cst‐II, encoding a sialyltransferase in C. jejuni, leads to the biosynthesis of different gangliosides‐mimicking LOS.22, 23 C. jejuni strains with cst‐II (Asn51) regularly express the GQ1b‐like LOS while the strains with cst‐II (Thr51) express more GM1‐like and GD1a‐like LOS.22 Typically, IgG antibodies against GQ1b were associated with MFS while the IgG antibodies against GM1 and GD1a were associated with axonal GBS.24, 25 High incidence of MFS was previously reported in East Asia, especially Japan (up to 26%);2, 26 however, there was much lower frequency of MFS in both our current study (3%) and a large multicenter study from Southern China (12%).8 Our results supported C. jejuni with cst‐II (Thr51), bearing GM1‐like LOS, as one of the causative pathogens in our region, which deserves a further study using C. jejuni samples from the patients. Moreover, none of the patients in our cohort were detected with H. influenzae infection, which was frequently seen in patients with MFS.27 The regional infectious spectrum may partly explain why there was a low frequency of MFS in China.
Liver injury in patients following influenza A and B infections has been described in both the cohort study and case report.28, 29 Some GBS‐related pathogens including EBV, varicella‐zoster virus, hepatitis A virus, and hepatitis E virus were able to trigger the autoimmune response against liver.30, 31, 32 Although pretreatment liver dysfunction has been reported for a long time,9 it remains unknown that the pretreatment liver dysfunction in GBS was caused by antecedent infections or GBS itself. In this study, 13% (17/132) of the patients were detected with pretreatment liver dysfunction at entry. Although the difference in frequency of pretreatment liver dysfunction among patients with and without infections was not significant, GBS patients following influenza B virus infection displayed more susceptibility to pretreatment liver dysfunction. Our study supported antecedent infection as one cause of pretreatment liver dysfunction in GBS patients. However, 19% of the patients without infection also had pretreatment liver dysfunction. It cannot be excluded that some other pathogens beyond our study were associated with liver dysfunction in patients with GBS. None of the GBS patients with C. jejuni infection had pretreatment liver dysfunction, which needs further confirmation by other cohort studies.
In conclusion, this is the first study to report the antecedent infectious spectrum in patients with GBS in China. We firstly reported influenza B‐related GBS as a pure motor phenotype. C. jejuni serves as the predominant cause of antecedent infections in patients with GBS in our region, but the frequency is much lower than 30 years ago. The regional infectious spectrum contributed to the clinical heterogeneity of GBS. Our results deserve further confirmation by other larger cohort studies.
Author Contributions
Y. Wang developed the concept and design of the article. Y. Hao, W. Wang and B. Qiao and D. Liu performed the experiment. W. Wang and X. Feng collected the clinical data. M. Chen and Y. Wang analyzed all of the data. Y. Hao and Y. Wang wrote the first draft, B.C. Jacobs and Y. Wang performed the critical revision and all the authors critically evaluated the manuscript.
Conflict of Interest
The authors report no disclosures relevant to the manuscript.
Supporting information
Table S1. Details of ELISA kits of antecedent infections assay.
Table S2. Urban and rural distribution of antecedent infections in patients with Guillain‐Barré syndrome.
Table S3. Antecedent infections and antibodies to glycolipids and glycolipid complex in patients with Guillain‐Barré syndrome.
Acknowledgments
We thank Dr. Xinke Chen (Clinic Laboratory, Affiliated Hospital of Jining Medical University, Jining, Shandong Province, China) for technical assistance.
Funding Information
This work was supported by the National Natural Science Foundation of China (81771298, 81771360, and 81301072), Shandong Province Natural Science Fund Project (ZR2017LH034), Shandong Medical and Healthy Science Technology Development Plan (2016WS0184), and the Technology boosting new and old kinetic energy conversion projects of Jining City (2017SMNS002).
Funding Statement
This work was funded by National Natural Science Foundation of China grants 81771298, 81771360, and 81301072; Technology boosting new and old kinetic energy conversion projects of Jining City grant 2017SMNS002; Shandong Province Natural Science Fund Project grant ZR2017LH034; Shandong Medical and Healthy Science Technology Development Plan grant 2016WS0184.
References
- 1. Willison HJ, Jacobs BC, van Doorn PA. Guillain‐Barré syndrome. Lancet 2016;388:717–727. [DOI] [PubMed] [Google Scholar]
- 2. Doets AY, Verboon C, van den Berg B, et al. Regional variation of Guillain‐Barré syndrome. Brain 2018;141:2866–2877. [DOI] [PubMed] [Google Scholar]
- 3. Jacobs BC, Rothbarth PH, van der Meché FG, et al. The spectrum of antecedent infections in Guillain‐Barré syndrome: a case‐control study. Neurology 1998;51:1110–1115. [DOI] [PubMed] [Google Scholar]
- 4. Yuki N, Taki T, Inagaki F, et al. A bacterium lipopolysaccharide that elicits Guillain‐Barré syndrome has a GM1 ganglioside‐like structure. J Exp Med 1993;178:1771–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meyer Sauteur PM, Huizinga R, Tio‐Gillen AP, et al. Mycoplasma pneumoniae triggering the Guillain‐Barré syndrome: a case‐control study. Ann Neurol 2016;80:566–580. [DOI] [PubMed] [Google Scholar]
- 6. Ho TW, Mishu B, Li CY, et al. Guillain‐Barré syndrome in northern China. Relationship to Campylobacter jejuni infection and anti‐glycolipid antibodies. Brain 1995;118(Pt 3):597–605. [DOI] [PubMed] [Google Scholar]
- 7. Ye Y, Wang K, Deng F, Xing Y. Electrophysiological subtypes and prognosis of Guillain‐Barré syndrome in northeastern China. Muscle Nerve 2013;47:68–71. [DOI] [PubMed] [Google Scholar]
- 8. Liu S, Xiao Z, Lou M, et al. Guillain‐Barré syndrome in southern China: retrospective analysis of hospitalised patients from 14 provinces in the area south of the Huaihe River. J Neurol Neurosurg Psychiatry 2018;89:618–626. [DOI] [PubMed] [Google Scholar]
- 9. Oomes PG, van der Meché FG, Kleyweg RP. Dutch Guillain‐Barré Study Group. Liver function disturbances in Guillain‐Barré syndrome: a prospective longitudinal study in 100 patients. Neurology 1996;46:96–100. [DOI] [PubMed] [Google Scholar]
- 10. Asbury AK, Cornblath DR. Assessment of current diagnostic criteria for Guillain‐Barré syndrome. Ann Neurol 1990;27(Suppl):S21–24. [DOI] [PubMed] [Google Scholar]
- 11. Wakerley BR, Uncini A, Yuki N. Guillain‐Barré and miller fisher syndromes–new diagnostic classification. Nat Rev Neurol 2014;10:537–544. [DOI] [PubMed] [Google Scholar]
- 12. Jacobs BC, van den Berg B, Verboon C, et al. International Guillain‐Barré Syndrome Outcome Study: protocol of a prospective observational cohort study on clinical and biological predictors of disease course and outcome in Guillain‐Barre syndrome. J Peripher Nerv Syst 2017;22:68–76. [DOI] [PubMed] [Google Scholar]
- 13. Hadden RD, Cornblath DR, Hughes RAC, et al. Plasma Exchange/Sandoglobulin Guillain‐Barré Syndrome Trial Group. Electrophysiological classification of Guillain‐Barré syndrome: clinical associations and outcome. Ann Neurol 1998;44:780–788. [DOI] [PubMed] [Google Scholar]
- 14. Fokke C, van den Berg B, Drenthen J, et al. Diagnosis of Guillain‐Barré syndrome and validation of Brighton criteria. Brain 2014;137:33–43. [DOI] [PubMed] [Google Scholar]
- 15. Shahrizaila N, Kokubun N, Sawai S, et al. Antibodies to single glycolipids and glycolipid complexes in Guillain‐Barré syndrome subtypes. Neurology 2014;83:118–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rees JH, Soudain SE, Gregson NA, Hughes RAC. Campylobacter jejuni infection and Guillain‐Barré syndrome. N Engl J Med 1995;333:1374–1379. [DOI] [PubMed] [Google Scholar]
- 17. Islam Z, Jacobs BC, van Belkum A, et al. Axonal variant of Guillain‐Barré syndrome associated with Campylobacter infection in Bangladesh. Neurology 2010;74:581–587. [DOI] [PubMed] [Google Scholar]
- 18. Altekruse SF, Stern NJ, Fields PI, Swerdlow DL. Campylobacter jejuni–an emerging foodborne pathogen. Emerg Infect Dis 1999;5:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kuijf ML, Samsom JN, van Rijs W, et al. TLR4‐mediated sensing of Campylobacter jejuni by dendritic cells is determined by sialylation. J Immunol 2010;185:748–755. [DOI] [PubMed] [Google Scholar]
- 20. Sivadon‐Tardy V, Orlikowski D, Porcher R, et al. Guillain‐Barré syndrome and influenza virus infection. Clin Infect Dis 2009;48:48–56. [DOI] [PubMed] [Google Scholar]
- 21. Yuki N, Ho TW, Tagawa Y, et al. Autoantibodies to GM1b and GalNAc‐GD1a: relationship to Campylobacter jejuni infection and acute motor axonal neuropathy in China. J Neurol Sci 1999;164:134–138. [DOI] [PubMed] [Google Scholar]
- 22. Koga M, Takahashi M, Masuda M, et al. Campylobacter gene polymorphism as a determinant of clinical features of Guillain‐Barré syndrome. Neurology 2005;65:1376–1381. [DOI] [PubMed] [Google Scholar]
- 23. Gilbert M, Karwaski MF, Bernatchez S, et al. The genetic bases for the variation in the lipo‐oligosaccharide of the mucosal pathogen, Campylobacter jejuni. Biosynthesis of sialylated ganglioside mimics in the core oligosaccharide. J Biol Chem 2002;277:327–337. [DOI] [PubMed] [Google Scholar]
- 24. Chiba A, Kusunoki S, Shimizu T, Kanazawa I. Serum IgG antibody to ganglioside GQ1b is a possible marker of Miller Fisher syndrome. Ann Neurol 1992;31:677–679. [DOI] [PubMed] [Google Scholar]
- 25. Sekiguchi Y, Uncini A, Yuki N, et al. Antiganglioside antibodies are associated with axonal Guillain‐Barré syndrome: a Japanese‐Italian collaborative study. J Neurol Neurosurg Psychiatry 2012;83:23–28. [DOI] [PubMed] [Google Scholar]
- 26. Mitsui Y, Kusunoki S, Arimura K, et al. A multicentre prospective study of Guillain‐Barré syndrome in Japan: a focus on the incidence of subtypes. J Neurol Neurosurg Psychiatry 2015;86:110–114. [DOI] [PubMed] [Google Scholar]
- 27. Koga M, Gilbert M, Li J, et al. Antecedent infections in Fisher syndrome: a common pathogenesis of molecular mimicry. Neurology 2005;64:1605–1611. [DOI] [PubMed] [Google Scholar]
- 28. Jayashree K, Rao S, Kamath N. Influenza B virus triggering macrophage activation syndrome in an infant. Indian J Crit Care Med 2017;21:802–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Papic N, Pangercic A, Vargovic M, et al. Liver involvement during influenza infection: perspective on the 2009 influenza pandemic. Influenza Other Respir Viruses 2012;6:e2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vento S, Guella L, Mirandola F, et al. Epstein‐Barr virus as a trigger for autoimmune hepatitis in susceptible individuals. Lancet 1995;346:608–609. [DOI] [PubMed] [Google Scholar]
- 31. Al‐Hamoudi WK. Severe autoimmune hepatitis triggered by varicella zoster infection. World J Gastroenterol 2009;15:1004–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huppertz HI, Treichel U, Gassel AM, et al. zum Büschenfelde KH. Autoimmune hepatitis following hepatitis A virus infection. J Hepatol 1995;23:204–208. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Details of ELISA kits of antecedent infections assay.
Table S2. Urban and rural distribution of antecedent infections in patients with Guillain‐Barré syndrome.
Table S3. Antecedent infections and antibodies to glycolipids and glycolipid complex in patients with Guillain‐Barré syndrome.
Data Availability Statement
Our data will be shared by request from any qualified investigator for scientific purposes.