

Abstract
Background
Treatment for pediatric posterior fossa group A (PFA) ependymoma with gain of chromosome 1q (1q+) has not improved over the past decade owing partially to lack of clinically relevant models. We described the first 2 1q+ PFA cell lines, which have significantly enhanced our understanding of PFA tumor biology and provided a tool to identify specific 1q+ PFA therapies. However, cell lines do not accurately replicate the tumor microenvironment. Our present goal is to establish patient-derived xenograft (PDX) mouse models.
Methods
Disaggregated tumors from 2 1q+ PFA patients were injected into the flanks of NSG mice. Flank tumors were then transplanted into the fourth ventricle or lateral ventricle of NSG mice. Characterization of intracranial tumors was performed using imaging, histology, and bioinformatics.
Results
MAF-811_XC and MAF-928_XC established intracranially within the fourth ventricle and retained histological, methylomic, and transcriptomic features of primary patient tumors. We tested the feasibility of treating PDX mice with fractionated radiation or chemotherapy. Mice tolerated radiation despite significant tumor burden, and follow-up imaging confirmed radiation can reduce tumor size. Treatment with fluorouracil reduced tumor size but did not appear to prolong survival.
Conclusions
MAF-811_XC and MAF-928_XC are novel, authentic, and reliable models for studying 1q+ PFA in vivo. Given the successful response to radiation, these models will be advantageous for testing clinically relevant combination therapies to develop future clinical trials for this high-risk subgroup of pediatric ependymoma.
Keywords: 1q+ PFA1 ependymoma, patient-derived orthotopic xenograft, preclinical model
Key Points.
Orthotopic patient-derived xenograft model of 1q+ PFA epenydmoma.
Robust model that can withstand fractionated radiation or daily drug treatments.
Importance of the Study.
Treatment for pediatric ependymoma has largely been unchanged for 20 years. Despite aggressive surgery and radiation, most patients will recur. Chemotherapy trials at relapse have failed, and current treatment at relapse does not benefit high-risk patients with PFA1 ependymoma. A subset of PFA1 patients, with chromosome gain of 1q, have particularly poor outcomes with early, often metastatic relapses. We were the first to develop 1q+ PFA1 cell lines, have gained considerable insight into the tumor biology, and have identified candidate inhibitors that may be effective. However, in vitro cell lines are not reliable preclinical models. Thus, there was an urgent need to develop in vivo modeling for 1q+ PFA1. We characterize 2 patient-derived orthotopic xenograft models harboring 1q+ and retaining PFA1 characteristics and show these models are efficacious to use for preclinical development of the next generation of combination therapy.
Ependymoma (EPN) is the third most common pediatric cancer of the brain and is still associated with poor outcomes and high morbidity. Nearly half of all patients with EPN will recur, and almost all those with relapses will eventually die.1 Standard of care at diagnosis is surgery and adjuvant radiation, benefiting a limited number of children, with less than 30% being progression free at 10 years.1 We now know there are 2 distinct molecular subgroups of posterior fossa (PF) ependymoma,2–4 PFA and PFB. PFA has been further subgrouped into 2 populations, PFA1 and PFA2.5 These two subgroups differ in tumor biology and have very different prognoses. PFA1 has a poor prognosis overall, and certain subtypes within PFA1 have a grim outlook, with few survivors at 10 years. One subtype of PFA1 has an extra-long arm of chromosome 1 (1q+), and several groups have associated 1q+ with very poor prognoses and early, often metastatic relapses.5,6
The poor outlook for 1q+ PFA with standard therapy suggests that novel therapy should be introduced for this group at diagnosis. Moreover, patients with 1q+ PFA relapse relatively quickly compared with other subtypes of PF EPN, so the effect of novel therapies can be assessed over a short interval.6 Until recently, the lack of preclinical models for PFA hindered understanding of tumor biology and thus the development of rational therapies. Our establishment of novel 1q+ PFA cell lines MAF-811 and MAF-928 has transformed our ability to study the molecular drivers of PFA. We have used them extensively to screen for potential therapeutic compounds, investigate tumor biology, and describe the specific immunophenotype of 1q+ PFA.7–9 These advancements have increased our collective knowledge, but in vitro studies still fail to reflect the complexity of treating intracranial tumors. A critical need for the development of an intracranial platform underscores the significance of this study as it describes a novel in vivo model of 1q+ PFA.
The goal of this study was to develop and characterize 2 patient-derived xenograft (PDX) orthotopic 1q+ PFA mouse models. These models are formed from disaggregated patient tumor samples collected at surgery and viably frozen. We have characterized these models by describing the methylome, transcriptome, and histology of both flank and intracranial tumors. We show that the xenograft model has RNA expression, methylation patterns, and immunohistochemistry features and had interleukin (IL)-6 secretion levels similar to the original patient tumors. These findings establish the preclinical significance of the model and suggest that it is fit for developing 1q+ PFA therapy. This novel animal model of pediatric 1q+ PFA will enhance targeted therapy development and biological understanding for this poor outcome subgroup.
Materials and Methods
Tumor Protocol and Study Approval
Both patient samples used to establish this model were obtained from surgery with consent (COMIRB 95–500). Freshly resected brain tumor was manually dissociated and viably frozen as previously described.7–9 MAF-811 was established from a patient with fourth recurrence of metastatic anaplastic EPN. MAF-928 was established from a patient with a local recurrence of anaplastic EPN. Both samples were positive for chromosome 1q gain (1q+) and displayed PFA1 genomic and methylomic profiles. These samples were also used to establish 2 long-term cell lines, which we have previously described.7 Original patient samples are denoted as MAF-811_5 and MAF-928_2.
Establishment of Subcutaneous Tumors
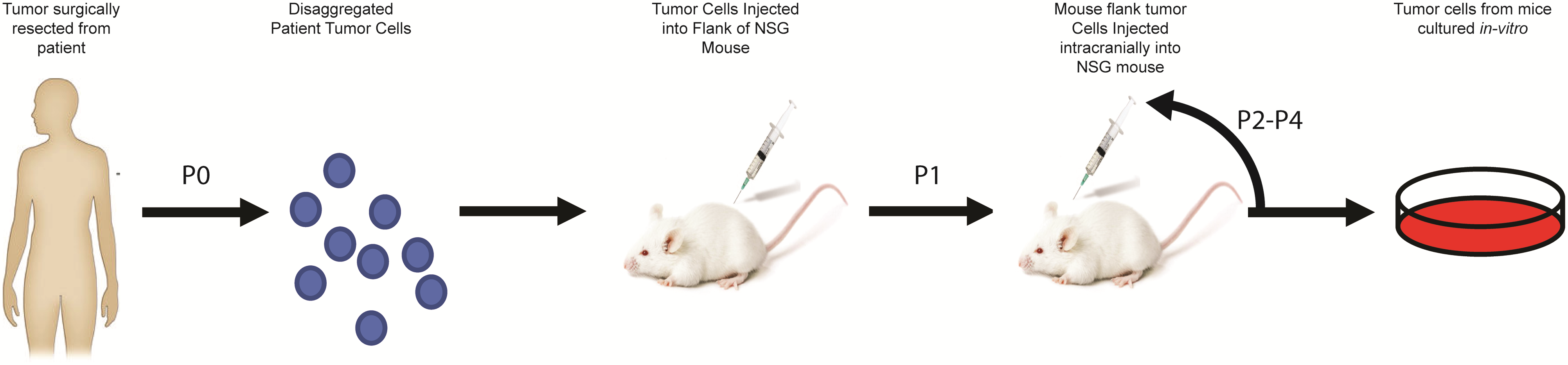
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC 00192). Viably frozen disaggregated patient tumor cells were thawed and resuspended to 250K cells/100 μL in serum-free OptiMEM minimal essential medium. NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice, aged 6 weeks, were obtained through Jackson Laboratories and housed in the University of Colorado (UC) Denver vivarium. Mice were subcutaneously injected with 100 μL of cell suspension and monitored for weight and tumor burden. When tumors reached an area of 2 cm2, mice were euthanized. Flank tumor was removed aseptically and preserved in formalin for immunohistochemistry, snap-frozen for biochemical analysis, and disaggregated and viably frozen for serial transplant. Tumors from subcutaneous transplanted mice are denoted as MAF-811_XF and MAF-928_XF and are designated as passage 1 (P1).
Establishment of Orthotopic Xenograft Tumors
P1 flank tumors were resected from euthanized donor mice and manually dissociated under sterile conditions (Supplementary Figure 1). Cells were resuspended to 250K/3 μL in serum-free media. Intracranial injections were performed as previously described10 using 0.000 mm lateral and 6.00 mm posterior to bregma; 4.300 mm ventral to the skull surface for the fourth ventricle. For the lateral ventricle, we used 1.500 mm lateral and 2.000 mm posterior to bregma; 3.000 mm ventral to the skull surface. Mice were weighed weekly and monitored for neurological distress. Animals were euthanized when weight loss reached 15% or any neurologic impairment (ataxia, head tilt, seizures) was observed (IACUC 00192). Orthotopic mice are denoted as MAF-811_XC and MAF-928_XC and metastatic mice as MAF-811_XV and MAF-928_XV. Intracranial tumors engrafted from P1 flank tumors are designated as P2. We have now successfully serial-transplanted intracranial tumors up to passage 4, the highest passage number used to minimize genetic drift.
Magnetic Resonance Imaging
Mice underwent MRI scans when showing signs of tumor burden (weight loss or neurological distress). Rapid acquisition with relaxation enhancement T2-weighted MR images were acquired using Bruker 4.7 Tesla PharmaScan and Bruker 9.4 Tesla BioSpec animal scanners (UC Animal Imaging Shared Resource). Mice were anesthetized with isoflurane and maintained on isoflurane/O2 (1.5% vol/vol) throughout image acquisition. Twenty-four contiguous anatomic slices (in both transaxial and sagittal planes with fat suppression) were collected (MR parameters: field of view = 3 × 3 cm; repetition time = 4000 ms; echo time = 80 ms; slice thickness = 0.7 mm; 256 × 256 matrix size; 4 averages). All images were acquired and analyzed using Bruker ParaVision v4.0 and NEO. The quantification of tumor burden was achieved by hands-free drawing of the region of interest. The total of region-of-interest areas on all affected slices was multiplied by the slice thickness to achieve 3D assessment of tumor burden.
Bioluminescence Development
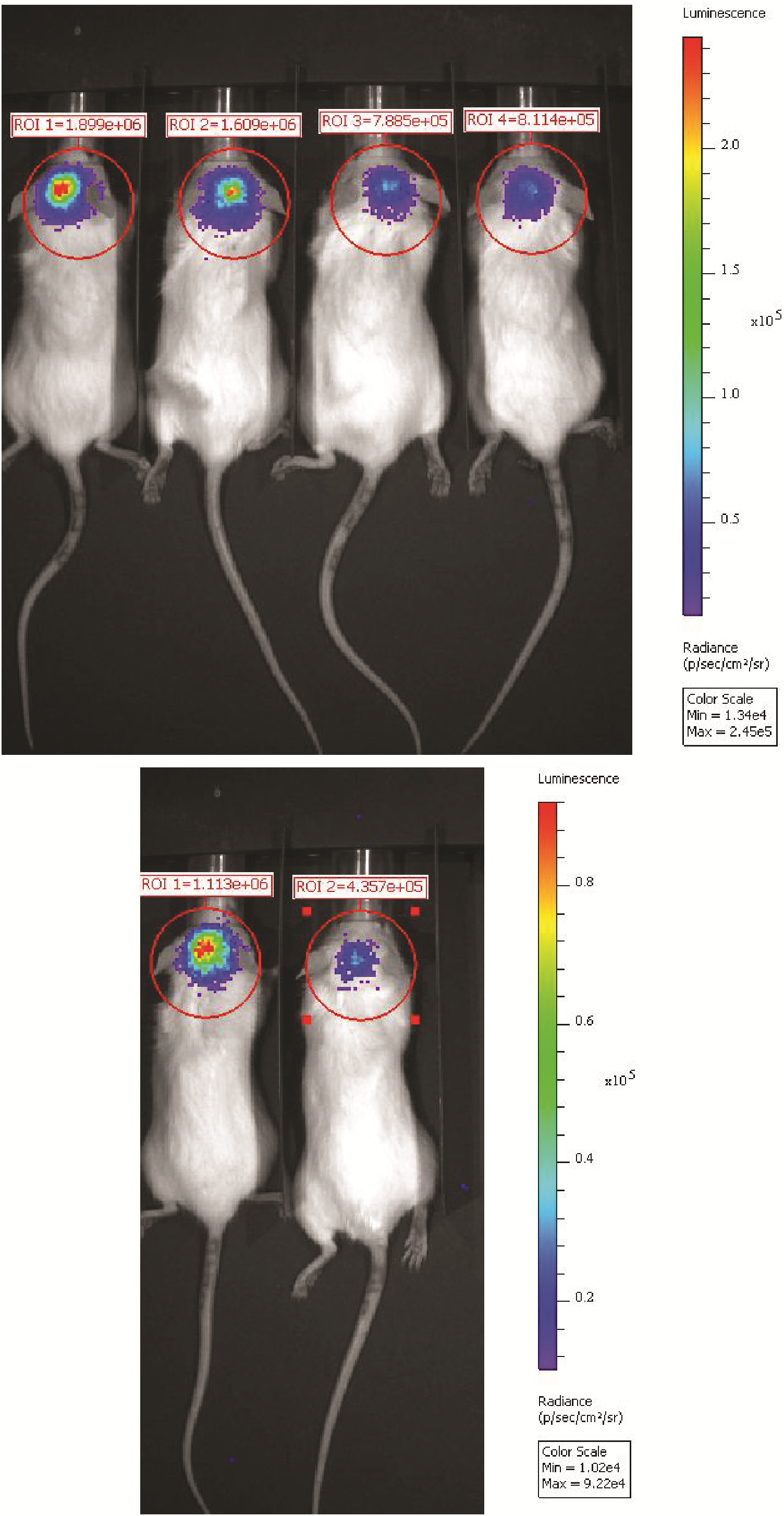
P1 MAF-928_XF tumor cells resuspended at 300K cells/200 μL serum-free media on an ultra-low attachment plate. EFS-Luc2-T2A-tGF lentivirus was added at 25 μL virus/250K cells and 1 μg/mL polybrene. Cells were incubated overnight, washed in serum-free media, and centrifuged. Medium was aspirated and cells resuspended in serum-free media. Injected subcutaneously into NSG mice were 250K cells. When tumors reached 2 cm2, mice were euthanized and tumors extracted as above. Flank tumors were mechanically dissociated and 250K serially transplanted into the fourth ventricle of NSG mice (P3). Mice were imaged on PerkinElmer Spectrum IVIS/Quantum microCT following intraperitoneal (i.p.) injection of luciferin at 10 μL/g body weight.
Histology and Immunohistochemistry
Following euthanasia, brains were removed and fixed in 10% buffered formalin phosphate. Tumor tissue was processed in paraffin, sectioned horizontally at 5 μm, and mounted on slides. After deparaffinization, sections were stained for hematoxylin and eosin or processed for immunohistochemical staining by the UC Denver Histology Core. Immunohistochemistry was performed on an automated Ventana Benchmark Ultra stainer using an Optiview detection kit (Ventana). Primary antibodies used included glial fibrillary acidic protein (GFAP; 1:2500 dilution; Dako), epithelial membrane antigen (EMA; 1:1 dilution; Ventana), H3K27 trimethylation (H3K27me3; 1:200 dilution; Cell Signaling, antigen retrieval 10 min at 110°C; Borg), and CXorf67 (1:200 dilution; Sigma, antigen retrieval citrate, 10 min at 110°C). Sections were counterstained with hematoxylin.
DNA Methylation Analysis
DNA methylation and analysis were performed as previously described.9 Copy number analysis data were generated from unnormalized signal intensities using ChAMP module with default control samples. Resulting IDAT files were uploaded to MolecularNeuropathology.org (https://www.molecularneuropathology.org/mnp), which provided subgrouping into PFA1. Further subgrouping was performed by our collaborators.5
Transcriptomic Analysis
RNA was isolated from snap frozen tumor specimens as previously described.2,8–10 Extracted mRNA quality was assessed for library preparation using DNA Analysis ScreenTape (Aligent Technologies). Libraries for cDNA were generated using the Illumina TrueSEq Stranded mRNA Sample Prep Kit. Libraries were sequenced using Illumina HiSeq4000 platform with paired-end reads (2 × 151). Forty million reads/sample were collected. Resulting sequences were filtered and trimmed, removing low-quality bases (Phred score <15), and analyzed using a custom computational pipeline11–14 consisting of open-source gSNAP, Cufflinks. R was used for differential gene expression alignment and discovery. Reads were mapped to the human genome (GRCh38) by gSNAP. Gene expression (fragments/kilobase/million) was derived by Cufflinks. DAVID (http://david.abcc.ncifcrf.gov) was used to measure GOTerm gene lists’ enrichment, providing gene ontology mappings directly annotated by the source database.
IL-6 Detection in Cerebral Spinal Fluid
Cerebral spinal fluid (CSF) from patients with PFA1 or PFA2 EPN was collected at surgery (COMIRB 95–500). CSF was spun down to remove any cells or debris and frozen at −80°C. For MAF-928_XC, MRI was used to evaluate hydrocephalus in the mouse due to tumor obstructing the ventricles. Following euthanasia, the skull was carefully removed, exposing the brain. A 28-gauge insulin needle was carefully inserted just to the right of midline, approximately one third the distance from the bregma to frontal lobe and approximately 3 mm deep. Obtained from the PDX was 10–20 μL of CSF fluid. Fluid was spun down to remove tissue and frozen at −80°C. CSF fluid was loaded onto a human IL-6 enzyme-linked immunosorbent assay (R&D Biosystems) specific to human cytokine. Values were normalized per manufacturer protocol.
Fractionated Radiation of MAF-928_XC
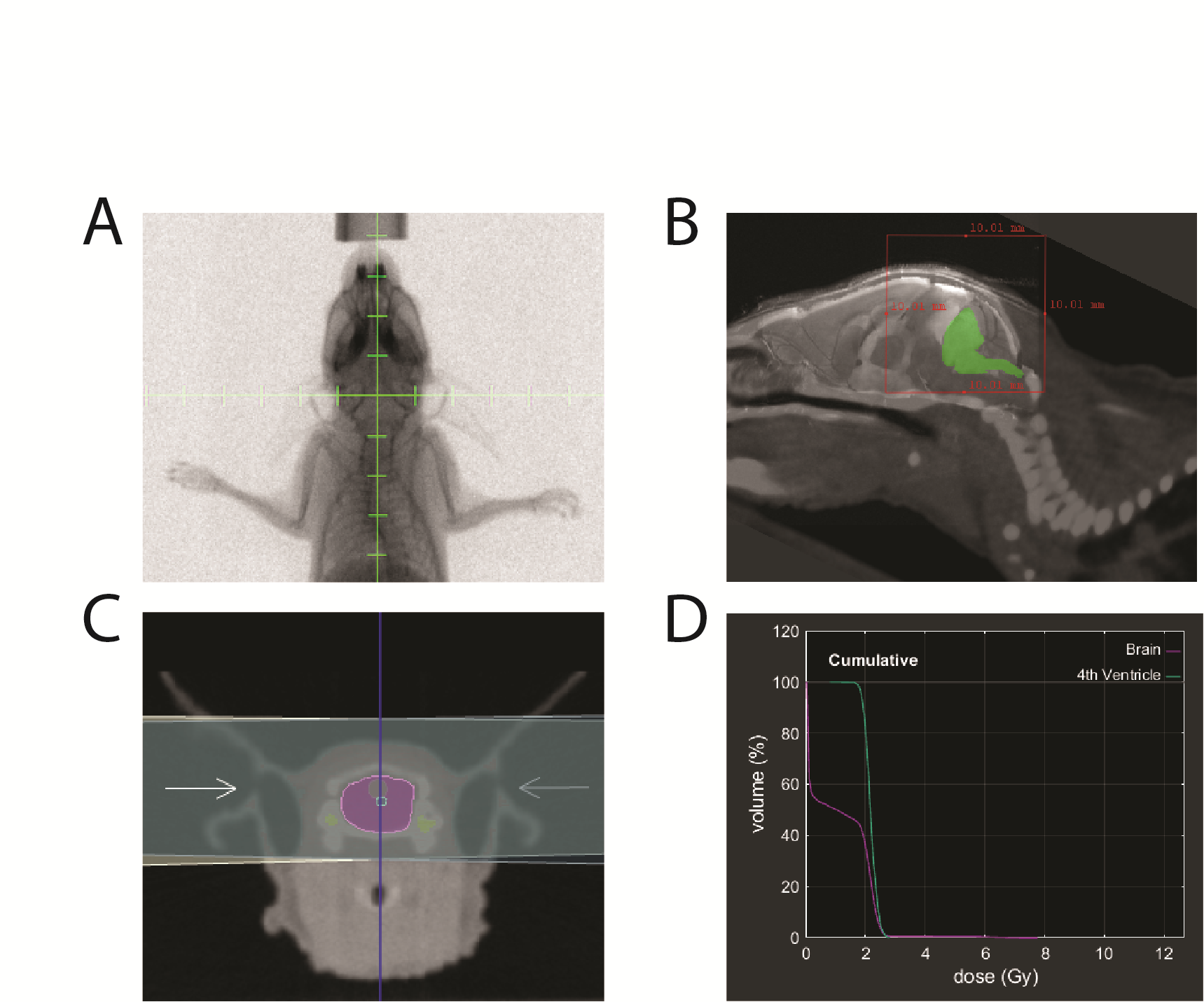
P3 MAF-928_XC cells were injected into the fourth ventricle of 6 NSG mice. MRI confirmed tumor burden in 5 of 6 mice. Irradiation was performed under IACUC protocol 00784. Using the X-RAD SmART image-guided irradiator (Precision X-Ray) at 225 kVp with a 0.3 mm copper filter, mice received 10 or 20 Gy in 5 daily fractions at a dose rate of 5.9 Gy/min. Under isoflurane anesthesia, each mouse was positioned in the prone orientation and aligned to the isocenter in 2 orthogonal planes by fluoroscopy (Supplementary Figure 2A, B). Half of the dose was delivered from each side in opposing, lateral beams (Supplementary Figure 2C). Monte Carlo simulation in SmART-ATP (SmART Scientific Solutions) shows 95% of the target volume (fourth ventricle) receiving the prescribed dose (Supplementary Figure 2D). Body weight and general appearance were monitored weekly. Mice were euthanized when weight loss exceeded 15% or neurological symptoms developed (IACUC 00784).
Chemotherapy Trial Modeling Using MAF-928_XC
P4 MAF-928_XC cells were injected into the fourth ventricle of 8 NSG mice. Due to a temporary shutdown of the animal imaging facility, imaging at the start of therapy was not possible. Mice were divided into 2 groups at random based on mouse identification number. Pharmaceutical grade fluorouracil (5FU) was prepared in dimethyl sulfoxide at a concentration of 50 mg/mL and stored in small volume aliquots at −80°C. Stock 5FU was diluted in sterile phosphate buffered saline (PBS) to 5 mg/mL on each treatment day. Sterile PBS was used as a vehicle control. Mice received 30 mg/kg 5FU or equivalent PBS volume once weekly via i.p. injections for a total of 7 doses. Body weight and general appearance were monitored weekly. Mice were euthanized when weight loss exceeded 15% or neurological symptoms developed (IACUC 00784).
Statistical Analysis
For all tests, statistical significance was defined as P < 0.05 using Student’s t-test R bioinformatics, GraphPad Prism v7, and Microsoft Excel software.
Results
Development of Orthotopic Xenograft Tumors
To best model high-risk human pediatric EPN, 1q+ PFA1 tumor samples were utilized. We performed flank injections of viably frozen dissociated tumor samples into NSG, and all mice presented with noticeable flank tumors 4–8 months post injection (Supplementary Figure 3). Serial flank-to-flank transplantation of the PDX cells significantly shortened time to tumor formation to an average of 4–6 weeks with 100% transplant success. We harvested cells from the flank tumors (P1) for immediate re-injections into either the fourth or lateral ventricle of NSG mice and viably froze aliquots for future characterization and xenograft studies. For MAF-811_XC, we transplanted 3–6 mice and achieved 80% transplant success in 6–8 months. MAF-928_XC tumor cells transplant much quicker, with 80% of the 6–10 mice developing tumors within 4 months. The maximum volume and cell number that can be injected is 330K cells in 4 μL; higher cell numbers and volumes caused rapid hydrocephalus, and mice had to be euthanized within 24 hours of surgery. Serial passaging MAF-928 intracranially has decreased time to tumor formation to 2 months. However, serial transplanting MAF-811 intracranial-to-intracranial has not evidently enhanced the rate of tumor formation.
Imaging of Xenograft Tumor Burden in Mouse Brains
Following intracranial transplant of P1 MAF-811 and MAF-928, tumors formed within the fourth ventricle, confirmed by mouse T2-MRI brain scans, mimicking MRI appearance seen in human EPN (Fig. 1A). The fourth ventricle is expanded, the brainstem is pushed forward, and the cerebellum deflected backward (Fig. 1B). Tumors extend through the foramen of Magendi, also as seen in humans. Gain of chr 1q (1q+) PFA1 often metastasizes to the lateral ventricles (Fig. 1C). Injection of both MAF-811 and MAF-928 tumor cells into the lateral ventricle resulted in metastatic spread to the ventricles (Fig. 1D). While MRI brain scans are critical for determining tumor location and metastases throughout the CNS, development of high-throughput imaging is critical for large-scale preclinical studies. We therefore established a bioluminescent tagged P3 MAF-928_XC. After 6 weeks, we were able to detect strong luciferase signal in the brains of all 6 mice injected (Supplementary Figure 4). We are in the process of developing MAF-811_XC into a bioluminescent model.
Fig. 1.

Radiographic imaging of orthotopic and metastatic PDX. (A) Sagittal plane, T2 MRI of MAF-811 original patient tumor. (B) Sagittal plane, T2 scans of MAF-811_XC, MAF-928_XC in NSG mice. (C) Coronal, T2 of MAF-811 patient with metastatic relapse in lateral ventricle. (D) Axial, T2 scans of MAF-811_XV, MAF-928_XV with tumor growth in lateral ventricle. Tumors outlined in yellow.
Histopathologic Fidelity Observed Between Mouse and Human Tumor
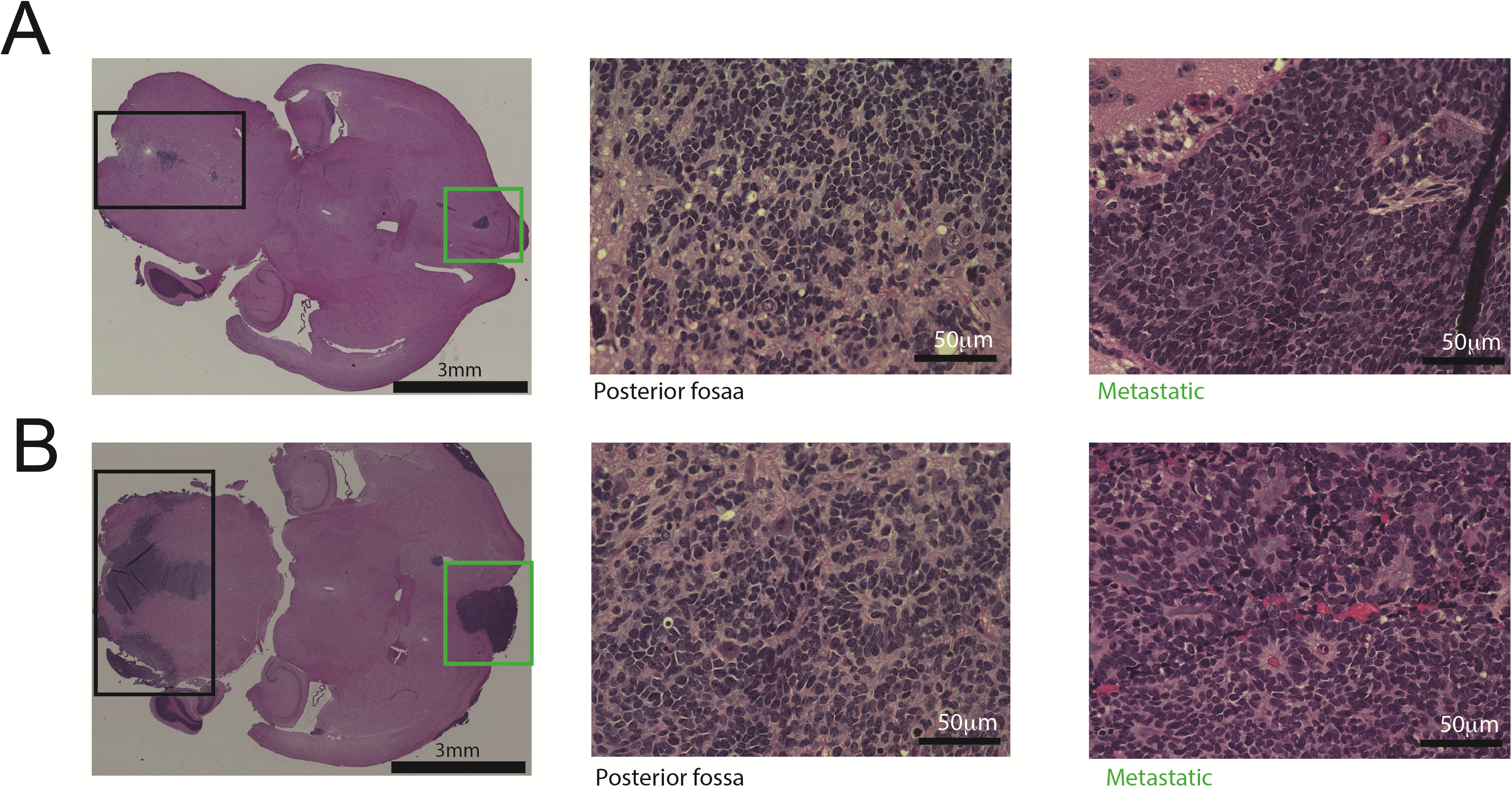
Necropsies were performed on P2 orthotopic PDXs to isolate brains from mice. The brains were enlarged and distorted and had distinct focal tumors. The xenografts maintained histopathologic and immunophenotypic characteristics of the original patient tumors (Fig. 2F, L). Xenografts in both MAF-811_XC (Fig. 2A–E) and MAF-928_XC (Fig. 2G–K) showed the classic ependymoma histology with a homogeneous population of cells with round monomorphic nuclei with a stippled (salt and pepper) chromatin pattern. The tumor cells formed characteristic true ependymal rosettes with a halo of tumor cells surrounding an empty lumen (Fig. 2B, C, I; arrows) as well as pseudorosettes with tumor cells arranging around blood vessels (Fig. 2B, C, I; arrows). The metastatic lesion in MAF-811_XC also retained classic ependymoma histology (Fig. 2E). The growth characteristics were also typical of ependymoma with focal leptomeningeal spread (Fig. 2H, arrows). As in the original tumors (designated anaplastic EPN, World Health Organization grade III), proliferation rate was high in the xenografts, and mitosis was readily seen. Immunohistochemical profile was typical of ependymoma with diffuse positivity of GFAP (Fig. 2D, J) and dot-like positivity of EMA stain (Fig. 2K).
Fig. 2.

Histologic characterization of orthotopic PDX tumors. (A–G) Representative images of MAF-811. (A) Axial section of mouse brainstem showing P2 MAF-811 xenograft tumor in fourth ventricle (black square, injection site) and temporal horn of lateral ventricle (green square, metastatic lesion). (B–C) MAF-811_XC xenograft showing classic features of ependymoma: small round blue cells with stippled chromatin pattern, forming true ependymal rosettes (arrows) and pseudorosettes (arrowheads). (D) Immunohistochemical staining against GFAP showing diffuse positivity in tumor cells. (E) MAF-811 xenograft metastases to lateral ventricle. (F) Original patient tumor from MAF-811_5 showing xenograft had high degree of morphologic similarity with original. (G–K) Representative images of MAF-928_XC. G. Whole mount of mouse cerebellum showing ependymoma (square). (H) Leptomeningeal spread of the tumor (arrows). (I) Higher magnification view of xenograft showed homogeneous tumor with tumor cells arranged around blood vessels (perivascular pseudorosettes). (J–K) Tumor cells were positive for GFAP and exhibited dot-like (arrows) positivity on EMA immunohistochemical staining. (L) Original patient tumor from MAF-928_2. Scale bars indicated on images.
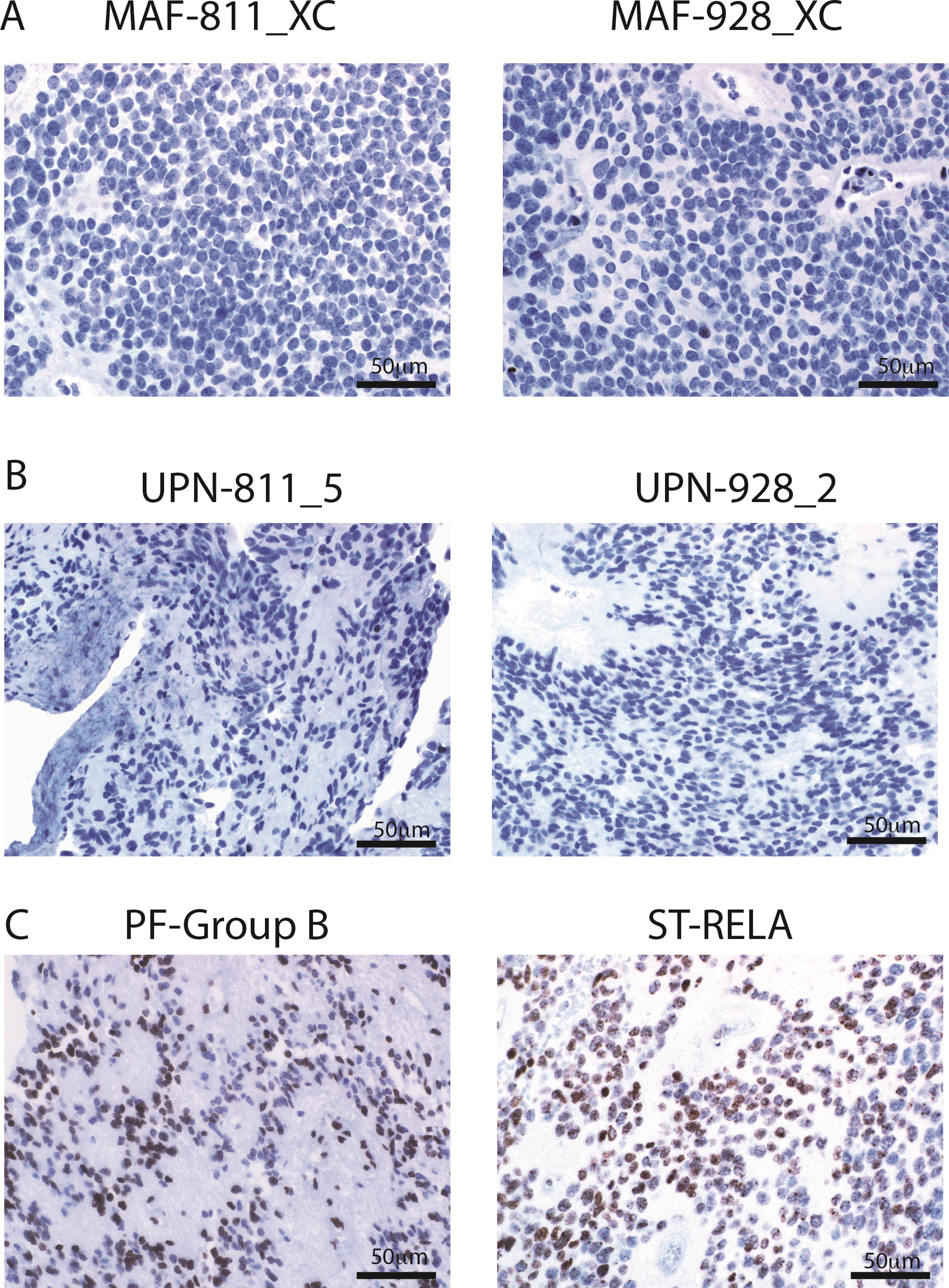
PFA can be distinguished from PFB and supratentorial EPN by lack of H3K27 trimethylation staining,5,15 a feature that has been proposed to select PFA tumors for an upcoming clinical trial. As predicted, both the xenograft tumors and the original patient tumor stained negative for H3K27 trimethylation (Supplementary Figure 5A, B). For positive controls, we used a PFB EPN and a supratentorial EPN with the fusion of v-rel avian reticuloendotheliosis viral on-cogene homolog A (RELA), both of which had widespread staining of H3K27 trimethylation (Supplementary Figure 5C). We also observed a lack of H3K27 trimethylation in the metastases (Supplementary Figure 6). The lack of H3K27 trimethylation of both human and xenograft suggests these tumors are PFA.
Molecular Subgrouping of Xenograft Tumors
DNA was isolated from PDX samples and processed for 850K methylation analysis. Following normalization as previously described,9 methylation files were uploaded into the DNA methylation CNS tumor classifier (MolecularNeuropathology.org)16 platform for subgroup classification. Both the original patient tumor and the corresponding xenograft for MAF-811 were classified as PFA (Fig. 3A, B). We further analyzed the methylation profiles according to Patjler et al5 and found that both the original MAF-811 patient tumor and MAF-811_XC tumors are predicted to be PFA1c subgroup with maximum prediction scores of 0.999853 and 0.9999, respectively (Supplementary Figure 7, Supplementary Table 1). Copy number variation (CNV) profile showed gain of chromosome 1q and loss of chromosome 6 in both (Fig. 3C and D). MAF-811_XC also harbored loss of chromosome 22 (Fig. 3D). Original patient tumor for MAF-928_2 was classified as PFA1 (Fig. 3E) and could be further subgrouped into PFA1d with a maximum prediction score of 0.937721 (Supplementary Figure 7, Supplementary Table 1). MAF-928_XC was unmatchable with any tumor type using the online classifier (Fig. 3F) but showed a weak association (prediction score 0.390954) with PFA1d (Supplementary Figure 7, Supplementary Table 1). Comparing the CNV profiles of the xenograft tumor and the original MAF-928 tumor, the xenografts retain the chromosome 1q+ and loss of chromosome 6 (Fig. 3G, H).
Fig. 3.

Methylation subgroup classification of PDX tumors. (A–B) Human patient tumor UPN811_5 and mouse MAF-811_XC; subgrouping with methylation class PFA using German Brain Tumor Classifier. (C–D) CNV profile of UPN811_5, MAF-811_XC. (E–F) Human patient tumor UPN928_2 and mouse MAF-928_XC; subgrouping with methylation class PFA using DNA methylation CNS tumor classifier (MolecularNeuropathology.org).16 (G–H) CNV profile of UPN928_2, MAF-928_XC.
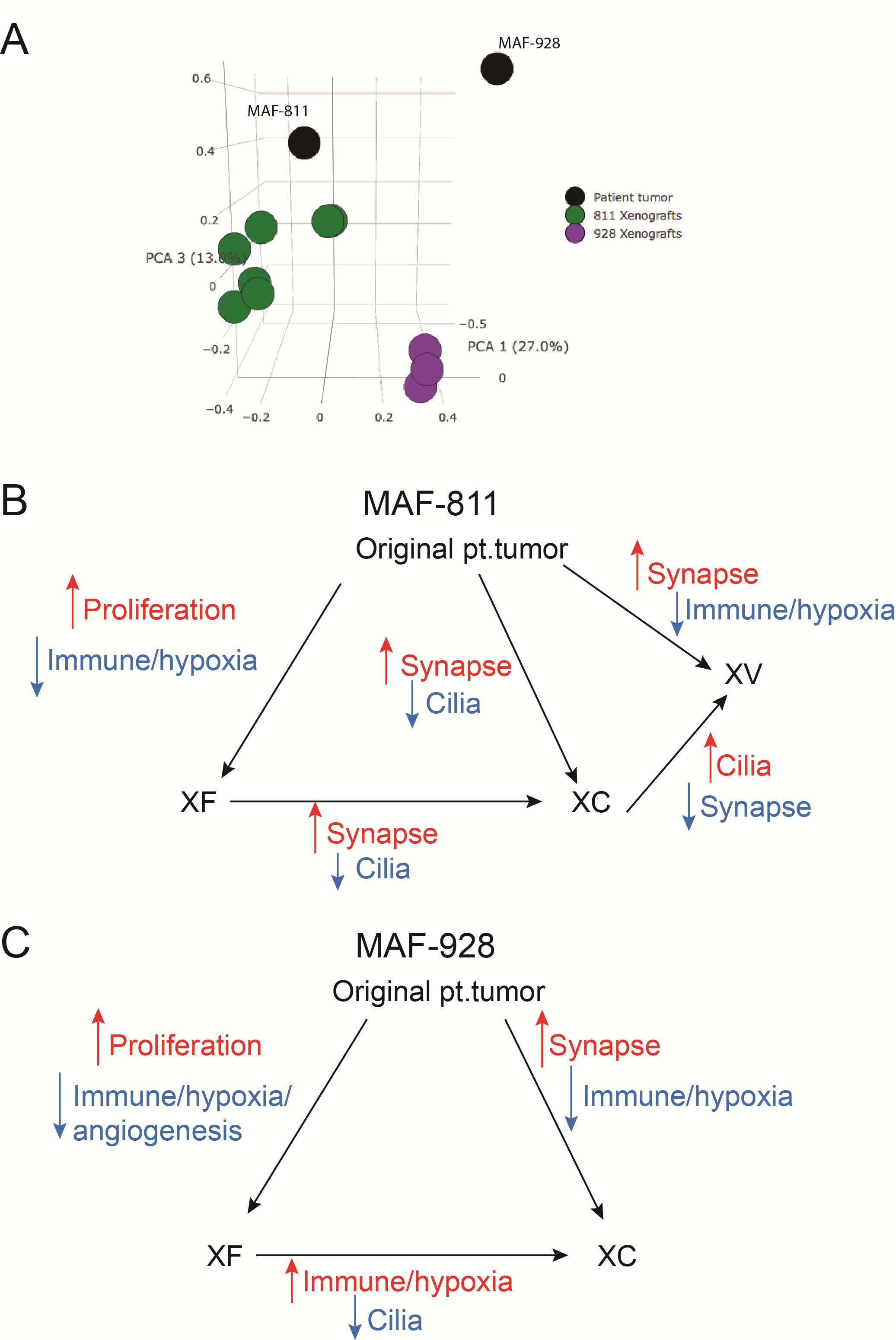
To establish PDXs, we passaged the patient tumor cells subcutaneously before transplanting into the brains of mice. Methylation profiles varied little among flank, intracranial, and original patient samples; however, it is known that passaging through the flank can alter the gene expression profiling of the tumor cells. We performed principal components analysis of the original patient tumor, flank xenografts, posterior fossa xenografts, and metastases and found there are changes in gene expression (Supplementary Figure 8A). We used GOTerm Enrichment analysis (DAVID) to show changes in gene expression programs between the xenograft tumors and original patient tumors. In both PDX models, flank tumors showed increase in proliferation-associated gene expression and decreased hypoxia/angiogenesis compared with original patient samples (Supplementary Figure 8B, C; Supplementary Table 2). The posterior fossa xenografts showed increased synapse-associated gene expression compared with original patient tumor. Metastatic tumor showed an increase in cilia-associated genes and decrease in synapse-associated genes compared with posterior fossa xenografts (Supplementary Fig. 8B, C; Supplementary Table 2). The increase in cilia-associated genes in the metastatic tumor is associated with a more differentiated phenotype and can be visualized by histology, which shows more rosettes in the metastatic tumor compared with the posterior fossa site (Fig. 2E, Supplementary Figure 9). Immune-related gene signatures were decreased in all xenograft tumors compared with original patient tumors, which is not surprising as the xenografts are generated in immune-incompetent mice.
Xenografts Retain PFA1 Molecular Characteristics
We have extensively studied the tumor biology of PFA1 and recently identified a PFA1-specific signaling cascade that drives tumor biology and downstream immunobiology.8,9 Consistent with human PFA1 tumors, MAF-811_XC and MAF-928_XC retain silenced gene expression of leucine zipper protein downregulated in cancer 1 (LDOC1) (Fig. 4A). LDOC1, as we have reported, is a negative regulator of nuclear factor-kappaB (NF-κB) activation. Silenced LDOC1 in PFA1 results in constitutively active NF-κB activation and chronic IL-6 secretion. We were able to measure human IL-6 in CSF taken from a mouse harboring MAF-928_XC tumor (Fig. 4B). The mouse CSF had greater concentration of IL-6 compared with CSF from PFA2 patients and close to the average IL-6 in PFA1 patient CSF (Fig. 4B). CXorf67 overexpression has been implicated as a potential oncogenic driver of PFA.5 Both xenografts have high gene expression values for CXorf67 similar to both PFA subgroups (Fig. 4C). Consistent with the transcript levels, protein levels of CXorf67 were also high in both xenografts (Fig. 4D). These data further support the PDX model as a strong representation of PFA1.
Fig. 4.

Molecular characterization of 1q+ PFA1 PDX. (A) LDOC1 transcript from RNAseq profiles. Values are in fragments per kilobase of transcript per million mapped reads (FPKM). (B) Human IL-6 concentrations (pg/mL) in CSF. (C) CXorf67 transcript from RNAseq analysis. Values are in FPKM. *P < 0.05. Error bars present standard error of the mean. (D) Immunohistochemical staining of CXorf67 protein in PDX lines. Brown denotes cells stained positive for CXorf67; slides were counterstained with hematoxylin. Scale bars indicated on images.
Preclinical Modeling
Radiation is standard of care for posterior fossa EPN patients over 12 months of age. In order to more accurately model pediatric treatment in which radiation is given in fractions and localized to the tumor region, we used image-guided fractionated irradiation on the X-RADmART (Supplementary Figure 2). P2 MAF-928_XC were imaged to confirm tumor burden. Mice were assigned to 2 groups based on a radiation dose of either 10 Gy or 20 Gy. The mice receiving 10 Gy radiation were given 2 Gy fractions daily for 5 days. Similarly, the 20 Gy radiation group was given 4 Gy fractions daily for 5 days. Mice were re-imaged after 2 and 4 weeks following radiation. Both radiation groups had significant reduction in tumor burden by MRI, with 10 Gy achieving partial tumor elimination (Fig. 5A middle panel, 46% decrease) and 20 Gy almost complete tumor elimination (Fig. 5B middle panel, approximately 95% decrease). All irradiated mice recovered to normal weight, and neurological distress was diminished. Three weeks post radiation, we started to observe side effects of radiation treatment, with hair loss localized to the radiation field and flaky, irritated skin. To minimize the pain and discomfort of side effects, these mice were treated with ibuprofen by our veterinary staff. Control mice that did not receive radiation rapidly declined and were euthanized within days of treated mice having completed radiation (Fig. 5C). A 10 Gy treated mouse initially showed significant improvement following completion of radiation but was found dead 1 week post treatment, likely due to lung lesions found during necropsy. MRI scans at 4 weeks post radiation treatment showed the remaining 10 Gy treated mouse had a small increase in posterior fossa disease and had developed a small metastasis in the olfactory bulb (Fig. 5A, right panel). Both 20 Gy treated mice recurred locally and developed large metastases in the olfactory bulb (Fig. 5B, right panel). Histological analysis shows both posterior fossa and metastatic disease maintained the histopathologic features characteristic of EPN, such as ependymal rosettes and pseudorosettes and finely granular/stippled chromatin (Supplementary Figure 9). Mitoses were readily seen. We did not observe significant radiation necrosis or vascular changes such as hyalinization. While the number of mice used in this study was not enough to achieve statistical significance, radiation treatment does extensively prolong survival of mice with MAF-928 tumors (Fig. 5D). Additionally, reducing the posterior fossa disease allows for mice to survive long enough to develop metastatic disease, modeling human 1q+ PFA1 disease that often recurs metastatically.
Fig. 5.

Fraction radiation treatment of MAF-928_XC. (A) Sagittal plane, T2 scans prior to 10 Gy fraction radiation (left); 2 weeks post 10 Gy fraction radiation (middle) and 4 weeks post 10 Gy fraction radiation (right). (B) Sagittal plane, T2 scans prior to 20 Gy fraction radiation (left); 2 weeks post 20 Gy fraction radiation (middle) and 4 weeks post 20 Gy fraction radiation (right). (C) Representative MRI of untreated mouse. Tumor outlined in yellow. (D) Survival of mice following radiation treatment. Blue denotes untreated mouse; red denotes 10 Gy treated mice; purple denotes 20 Gy treated mice.
Chemotherapy trials for relapsed PFA ependymoma have failed, most likely because, until recently, preclinical models for these tumors did not exist. We recently identified 5FU as a PFA-specific chemotherapy target from an FDA-approved chemotherapy drug screen on our 1q+ PFA cell lines.17 Interestingly, 5FU was identified as a top hit from the screen for candidate drugs for EPN by Atkinson et al.18 To test whether our PDX model is robust enough for preclinical chemotherapy trials, mice were injected with p4 MAF-928_XC into the fourth ventricle. Unfortunately, due to a temporary shutdown of animal imaging, starting tumor size was not feasible and mice were randomized into 2 groups based on tail identification number. Mice were given i.p. injection of 30 mg/kg 5FU or equivalent volume of PBS once weekly. Previous studies using a supratentorial RELA EPN model used 75 mg/kg/week,18 but we found this dose too toxic. Mice were given a total of 7 doses and MRI scans at 4 and 8 weeks (Fig. 6A). At 4 weeks, tumors were smaller in the 5FU treated mice compared with vehicle; however, the decrease was not statistically significant (Fig. 6B). All mice had increased local disease (blue arrows) at 8 weeks, and many developed metastatic diseases to the cortex (Fig. 6A, yellow arrows). Despite having an effect on tumor volume at 8 weeks, 5FU does not significantly increase survival (Fig. 6C); however, a 5FU treated mouse with minimal disease at 8 weeks is still alive, suggesting that repeat experiments with more mice may show a survival benefit for 5FU alone.
Fig. 6.

Fluorouracil treatment of MAF-928_XC. (A) Sagittal plane, T2 scans at 4 and 8 weeks post initiation of 5FU treatment. Mice received either vehicle or 30 mg/kg 5FU i.p. once weekly for 7 total doses. Blue arrow indicates posterior fossa disease; yellow arrow indicates metastases. (B) Quantification of tumor volume at each scan time. Blue denotes vehicle treated mice; red denotes 5FU treated mice. (C) Survival time of vehicle (blue), 5FU (red) treated mice.
Discussion
Children with 1q+ PFA tumors have particularly poor outcomes, and it is uncertain whether they benefit from radiation given their comparatively short time to recurrence after that modality.5 It is important to note that 1q+ is not a robust marker in PFB patients, and it is important that clinicians realize that the poor prognosis impact of 1q+ is confined to PFA patents.19 Patients with 1q+ PFA are particularly suitable for experimental trials as progression is rapid and usually fatal. Up to 50% of children with 1q+ PFA relapsed by 30 months in one study,20 and only 7% were progression free in another.21 In contrast, although outcomes for many other subtypes of EPN are poor, accounting for low disease-free rate at 10 years, many children with other subtypes of EPN relapse late. Given the unsatisfactory results of most experimental interventions at diagnosis in trials encompassing all subgroups of EPN, continuing to conduct trials at diagnosis with a 7- to 10-year timeline is impractical, but relapse studies have not been effective.16 Reirradiation at relapse is the exception, although even this gave an interval of freedom from progression of only just over 27 months in the large St Jude study and did not benefit as much children with 1q+ PFA.22 Children with 1q+ PFA seem to be a group in whom new therapies could be introduced at diagnosis with a reasonable timeline to completion, and the preclinical model described herein can directly inform such trials.
Identifying new therapies without an in vivo model of 1q+ PFA where there is no known mutational driver is difficult and liable to error. This paper described the first in vivo model of 1q+ PFA and demonstrates its utility in the testing of therapies. A few other orthotopic models of EPN in childhood have been reported, but none of them have been reported as harboring 1q+ and have not had the extensive molecular characterization described here.23,24 This publication presents the possibility of confirming in vitro testing of therapy in 1q+ PFA cells in an appropriate orthotopic mouse model. Cell lines of 1q+ PFA established by our group have enhanced our understanding of tumor biology and provided a model to identify potential therapeutic targets.7–9,17 Passaging the cells through a mouse has also given us more proliferative cell lines and has extended the passaging time compared with our originally published MAF-811 and MAF-928 1q+ PFA cell lines.
In order to develop a robust preclinical model, it was essential to validate the fidelity of an orthotopic model to recapitulate human disease, especially given that tumors first had to be established subcutaneously before transplant to intracranial model. PDXs of 1q+ PFA were verified to grow in the fourth ventricle and to metastasize to lateral ventricle locations. Importantly, from MRI we were able to observe tumors growing in the fourth ventricle compressing the brainstem and cerebellum, which is a classic feature of PF EPN. Serial intracranial transplants decreased the time for tumor formation in MAF-928_XC, but this was not seen with MAF-811_XC. We confirmed that tumors in both the posterior fossa and metastatic lesions retain histological EPN features with Ki67, GFAP, and EMA staining similar to the original patient tumors. MAF-811_XC were classified as PFA using DNA methylation CNS tumor classification.16 While initially MAF-928_XC could not be classified as PFA using the online brain tumor classifier, our collaborator at Heidelberg was able predict the tumor falls in the PFA1d subgroup. Both PDX models retain the silenced LDOC1 phenotype and high levels of CXorf67 expression, which are both thought to drive PFA1 tumor biology.5,9 The major disadvantage of these models is that they fail to replicate the immune microenvironment, which we have shown to be critical in the biology of PFA EPN. We are currently working on developing an immune competent model.
We recently published the results of an FDA-approved oncology drug screen using the MAF-811 and MAF-928 cell lines. The results showed that 5FU and retinoids are effective in the 1q+ PFA1 cell lines,17 but we did not test the efficacy of using these drugs in an in vivo model, a step that is critical for developing future clinical trials. We show here that this novel in vivo model is capable of testing such novel agents and can be used to explore clinically relevant combination drug and radiation studies. In this model, we showed that 20 Gy fractionated radiation almost completely eliminated tumors but these subsequently relapsed. Using this radiation dose and allowing the tumors to progress would potentially simulate a relapsed model that could be utilized to determine toxicities of reirradiating with combination chemotherapy. There has been concern that adding therapy, either during or after reirradiation, might increase the risk of radionecrosis. We can now eloquently model the normal progression of disease and can therefore study these potential toxicities.
A number of papers have emphasized the importance of molecular subtyping in childhood EPN.2–5,19 It is possible that the different subtypes of EPN may need specific therapies. It is therefore important, in our view, not only to develop models corresponding to the different subtypes of EPN but also to characterize as fully as possible the molecular nature of these models so that their relationship to the human EPN subtype can be assessed. We have strived to do this with our model, which is now available to the brain tumor community by providing low passage number viably frozen single-cell flank tumors that can be propagated in the flank of NSG mice upon arrival before transferring to intracranial sites.
Funding
This work was funded by the Tanner Seebaum Foundation. The Genomics and Microarray Core and Histology Core receives direct funding support from the National Cancer Institute (P30CA046934). All imaging protocols were developed at the Animal Imaging Shared Resource (N.J.S.) supported by the University of Colorado Cancer Center (NCI P30 CA046934) and the Colorado Clinical and Translational Sciences Institute (National Institutes of Health/National Center for Advancing Translational Sciences, UL1 TR001082).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We would like to thank the Tanner Seebaum Foundation for generously providing the funding for this work. We would like to thank the University of Colorado Denver Cancer Center Genomics and Microarray Core, Histology Core, Small Animal Imaging Core, and UC Denver vivarium veterinarians and staff.
Conflict of interest statement.
No conflicts declared.
Authorship statement:
Conception and design: AMP, DAW, BVC, AO, SDK, NJS, RV, NKF, AMG. Acquisition of data: AMP, DAW, AG, BVC, JS, AMG. Analysis and interpretation of data: AMD, AG, BS, MS, EWP, ED, KLJ, SDK, NJS, AMG. Drafting or revising the article: AMP, DAW, AMD, RV, NKF, AMG. Tumor sample procurement: KD, TH, MHH.
References
- 1. Marinoff AE, Ma C, Guo D, et al. Rethinking childhood ependymoma: a retrospective, multi-center analysis reveals poor long-term overall survival. J Neurooncol. 2017;135(1):201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hoffman LM, Donson AM, Nakachi I, et al. Molecular sub-group-specific immunophenotypic changes are associated with outcome in recurrent posterior fossa ependymoma. Acta Neuropathol. 2014;127(5):731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Witt H, Mack SC, Ryzhova M, et al. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell. 2011;20(2):143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pajtler KW, Witt H, Sill M, et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell. 2015;27(5):728–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pajtler KW, Wen J, Sill M, et al. Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. 2018;136(2):211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kilday JP, Mitra B, Domerg C, et al. Copy number gain of 1q25 predicts poor progression-free survival for pediatric intracranial ependymomas and enables patient risk stratification: a prospective European clinical trial cohort analysis on behalf of the Children’s Cancer Leukaemia Group (CCLG), Societe Francaise d’Oncologie Pediatrique (SFOP), and International Society for Pediatric Oncology (SIOP). Clin Cancer Res. 2012;18(7):2001–2011. [DOI] [PubMed] [Google Scholar]
- 7. Amani V, Donson AM, Lummus SC, et al. Characterization of 2 novel ependymoma cell lines with chromosome 1q gain derived from posterior fossa tumors of childhood. J Neuropathol Exp Neurol. 2017;76(7):595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Griesinger AM, Josephson RJ, Donson AM, et al. Interleukin-6/STAT3 pathway signaling drives an inflammatory phenotype in group A ependymoma. Cancer Immunol Res. 2015;3(10):1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Griesinger AM, Witt DA, Grob ST, et al. NF-κB upregulation through epigenetic silencing of LDOC1 drives tumor biology and specific immunophenotype in Group A ependymoma. Neuro Oncol. 2017;19(10):1350–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alimova I, Pierce A, Danis E, et al. Inhibition of MYC attenuates tumor cell self-renewal and promotes senescence in SMARCB1-deficient Group 2 atypical teratoid rhabdoid tumors to suppress tumor growth in vivo. Int J Cancer. 2019;144(8):1983–1995. [DOI] [PubMed] [Google Scholar]
- 11. Baird NL, Bowlin JL, Cohrs RJ, Gilden D, Jones KL. Comparison of varicella-zoster virus RNA sequences in human neurons and fibroblasts. J Virol. 2014;88(10):5877–5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bradford AP, Jones K, Kechris K, et al. Joint MiRNA/mRNA expression profiling reveals changes consistent with development of dysfunctional corpus luteum after weight gain. PLoS One. 2015;10(8):e0135163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Henderson HH, Timberlake KB, Austin ZA, et al. Occupancy of RNA polymerase II phosphorylated on serine 5 (RNAP S5P) and RNAP S2P on varicella-zoster virus genes 9, 51, and 66 is independent of transcript abundance and polymerase location within the gene. J Virol. 2016;90(3):1231–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maycotte P, Jones KL, Goodall ML, Thorburn J, Thorburn A. Autophagy supports breast cancer stem cell maintenance by regulating IL6 secretion. Mol Cancer Res. 2015;13(4):651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Panwalkar P, Clark J, Ramaswamy V, et al. Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol. 2017;134(5):705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555(7697):469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Donson AM, Amani V, Warner EA, et al. Identification of FDA-approved oncology drugs with selective potency in high-risk childhood ependymoma. Mol Cancer Ther. 2018;17(9):1984–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Atkinson JM, Shelat AA, Carcaboso AM, et al. An integrated in vitro and in vivo high-throughput screen identifies treatment leads for ependymoma. Cancer Cell. 2011;20(3):384–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cavalli FMG, Hübner JM, Sharma T, et al. Heterogeneity within the PF-EPN-B ependymoma subgroup. Acta Neuropathol. 2018;136(2): 227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Araki A, Chocholous M, Gojo J, et al. Chromosome 1q gain and tenascin-C expression are candidate markers to define different risk groups in pediatric posterior fossa ependymoma. Acta Neuropathol Commun. 2016;4(1):88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Korshunov A, Witt H, Hielscher T, et al. Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol. 2010;28(19):3182–3190. [DOI] [PubMed] [Google Scholar]
- 22. Tsang DS, Burghen E, Klimo P Jr, Boop FA, Ellison DW, Merchant TE. Outcomes after reirradiation for recurrent pediatric intracranial ependymoma. Int J Radiat Oncol Biol Phys. 2018;100(2):507–515. [DOI] [PubMed] [Google Scholar]
- 23. Yu L, Baxter PA, Voicu H, et al. A clinically relevant orthotopic xenograft model of ependymoma that maintains the genomic signature of the primary tumor and preserves cancer stem cells in vivo. Neuro Oncol. 2010;12(6):580–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pavon LF, Sibov TT, Caminada de Toledo SR, et al. Establishment of primary cell culture and an intracranial xenograft model of pediatric ependymoma: a prospect for therapy development and understanding of tumor biology. Oncotarget. 2018;9(31):21731–21743. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.