

Abstract
We describe the synthesis and characterization of a new fluorescence probe whose emission spectra, anisotropies, and wavelength-dependent decay times are highly sensitive to the phase state of phospholipid vesicles. This probe is 6-palmitoyl-2-[[2-(trimethylammonio)ethyl]methylamino] naphthalene chloride (Patman). The emission maximum of Patman shifts from 425 to 470 nm at the bilayer transition temperatures. The spectral properties of Patman reveal nanosecond time-dependent spectral shifts, which are the result of membrane relaxation around the excited state of Patman. The apparent fluorescence lifetimes of Patman are strongly dependent upon the emission wavelength, and the fluorescence phase and modulation data prove that the spectral shifts are due to an excited-state process, and not ground-state heterogeneity. As expected, the fluorescence anisotropies reflect the phase transitions of the bilayers. In addition, the anisotropies are dependent upon the emission wavelength because the duration of the excited state varies across the emission spectrum. The different apparent lifetimes across the emission spectrum allow the relaxed and unrelaxed emission spectra to be resolved by phase-sensitive detection of fluorescence. Also, the emission spectra of Patman show marked shifts to longer wavelengths as the excitation wavelength is increased. These red-edge excitation shifts are sensitive to the temperature and phase state of the bilayers.
Registry No. Patman, 87393-54-2; Patman iodide, 87393-57-5; DMPC, 18194-24-6; DOPC, 4235-95-4; DPPG, 4537-77-3; DPPC, 63-89-8; CTABr, 57-09-0; SDS, 151-21-3; 2-methoxynaphthalene, 93-04-9; palmitoyl chloride, 112-67-4; 6-palmitoyl-2-methoxynaphthalene, 87393-55-3; trimethylethylenediamine, 142-25-6; 6-palmitoyl-2-[[2-(dimethylamino)ethyl]methylamino] naphthalene, 87393-56-4; propylene glycol, 57-55-6; dioxane, 123-91-1; methanol, 67-56-1; water, 7732-18-5; dimethylformamide, 68-12-2; acetone, 67-64-1; acetonitrile, 75-05-8; chloroform, 67-66-3; 1-butanol, 71-36-3
Fluorescence probes are widely used to study the physical properties of proteins and membranes. Generally, such probes are selected on the basis of their sensitivity to the phenomenon of interest. For instance, probes such as l,6-diphenyl-1,3,5-hexatriene (DPH)1 are highly sensitive to the phase state of lipid bilayers, as revealed by its fluorescence anisotropy (Shinitzky & Barenholz, 1974; Lentz et al., 1976a,b). This sensitivity originates with its rodlike structure, the motions of which are highly sensitive to the dynamic and order properties of the acyl side chain regions of membranes (Kawato et al., 1977; Lakowicz et al., 1979). Other probes, such as the widely used naphthylaminesulfonic acids, are sensitive to contact with water and to the polarity of the surrounding environment (Weber, 1952; Slivak, 1982).
In this paper, we describe the synthesis and characterization of a new fluorescence probe for biological membranes, Patman (Scheme I). The fluorescent moiety of Patman is based upon 2-(alkylamino)-6-acylnaphthalene. Weber and co-workers demonstrated that the emission spectra of these compounds are highly sensitive to solvent polarity and relaxation (Weber & Farris, 1979; Macgregor & Weber, 1981). This sensitivity is consistent with the separation of approximately a unit charge in the excited state, with the negative and positive charges being localized on the carbonyl oxygen and the amino group nitrogen, respectively (Scheme I). To localize and orient the chromophore in the membranes, we attached a palmitoyl side chain, which serves to anchor this end of Patman in the bilayer. A trimethylammonium group was used to orient the alkylamino end of the fluorophore toward the lipid–water interface.
Scheme I.

In this paper, we demonstrate that the emission maxima and anisotropies of Patman change dramatically at the phase transition temperature of lipid bilayers. In addition, because of the large spectral shifts caused by dipolar relaxation, the anisotropies and apparent lifetimes are strongly dependent upon the emission wavelength. As a result of its unique spectral properties, Patman may be a useful probe of the dynamic properties of biological membranes and for quantifying lateral phase separations or the extent of water penetration into membranes. Our objective is not to provide a complete study of Patman under all possible conditions. Rather, the presented results were chosen to illustrate the potential uses of this new probe.
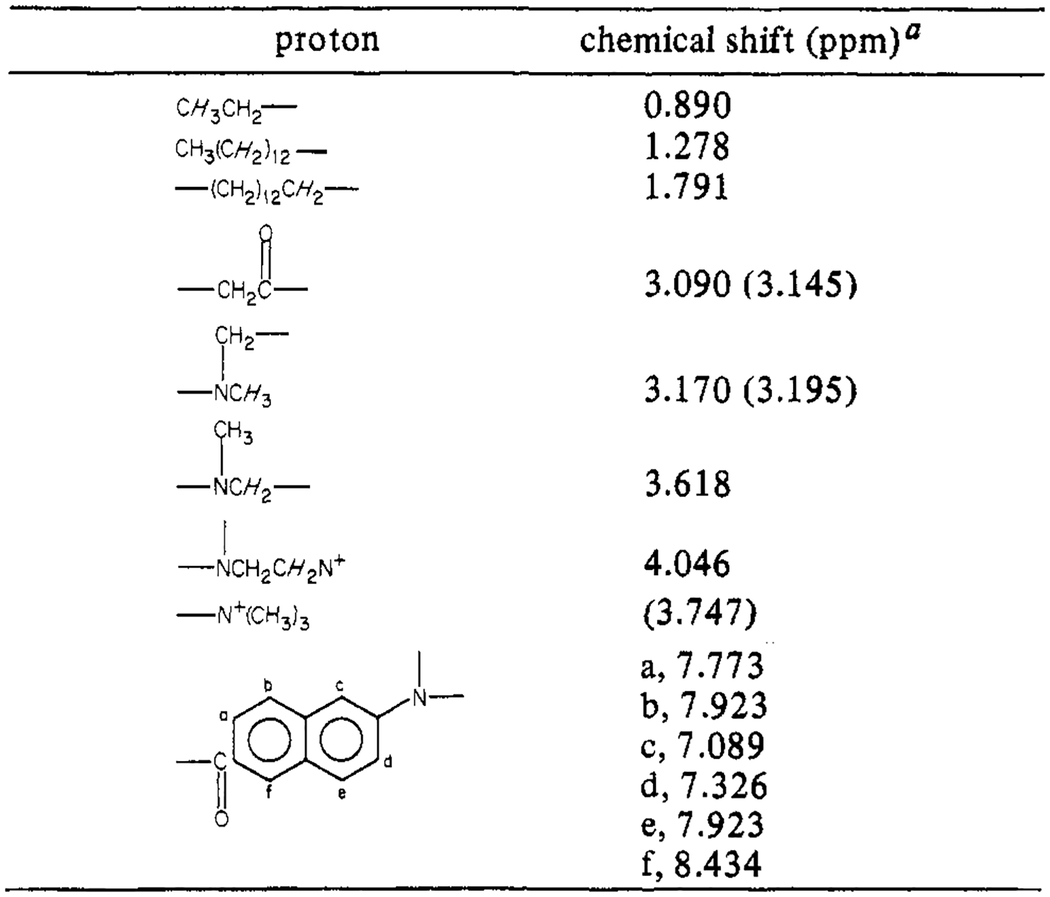
Synthesis of Patman
The synthesis of 6-palmitoyl-2-[[2-(trimethylammonio)-ethyl]methylamino]naphthalene chloride (Patman) requires several steps. First, 2-methoxynaphthalene is acylated with palmitoyl chloride, and then the methoxy group of the product, 6-palmitoyl-2-methoxynaphthalene, is substituted with trimethylethylenediamine. The product of this reaction, 6-palmitoyl-2-[[2-(dimethylamino)ethyl]methylamino]-naphthalene, is then N-methylated with methyl iodide to yield the iodide salt of Patman. The resulting product was found to yield a single spot on silica plates in chloroform/methanol/water (65/25/2 v/v). Also, the 270-MHz proton NMR spectrum was consistent with the structure shown in Scheme I, in terms of the chemical shifts, multiplet structure, and integrated peak areas. The resonances found for the Cl− form in CD3OD are summarized in Table I. A more detailed description of each step of the synthesis is given below. The 2-methoxynaphthalene, palmitoyl chloride, trimethylethylenediamine, and hexamethylphosphoramide (HMPA) were obtained from Aldrich. The Li wire was from Alfa, and the nitrobenzene was from Eastman.
Table I:
270-MHz Proton Magnetic Resonance Parameters of Patman
|
These values are relative to tetramethylsilane. The values in parentheses were measured in pyridine-d5,. All others were obtained in CD3OD.
Synthesis of 6-Palmitoyl-2-methoxynaphthalene.
The 2-methoxynaphthalene (50 g; 0.32 mol) was dissolved in 200 mL of nitrobenzene. Palmitoyl chloride (88 g; 0.32 mol) was slowly added to the solution with stirring at 0 °C. AlCl3 (40 g; 0.32 mol) was added in small portions, also while stirring at 0 °C (Buu-Hoi, 1949). The AlCl3 must be dry, and we found it advantageous to grind it to a fine powder immediately before use. After the AlCl3 was added, the mixture was allowed to warm to room temperature, and stirring was continued for approximately 24 h.
The reaction was terminated by pouring the reaction mixture into 300 mL of concentrated HCl in ice (150 mL of water and 150 mL of HCl). This produced a brown solid material which was filtered by vacuum filtration, washed with 0.1 N NaOH, and again isolated by filtration. The solid material was added to methanol, and the solvent was removed under vacuum with heating. The solid material was not completely soluble in the methanol, but since this step primarily served to remove the excess nitrobenzene, it was not important that all of the material did not dissolve. The solid remaining was recrystallized 3 times from benzene to which decolorizing carbon was added.
After the final crystallization, the product was washed with a small amount of cold methanol. A white powder (40.5 g; 32% yield) was obtained. The product was characterized by proton NMR and judged to be pure by TLC (50/50 petroleum ether/ethyl ether).
Synthesis of 6-Palmitoyl-2-[[2-(dimethylamino)ethyl]-methylamino]naphthalene.
The methoxy group of 6-palmitoyl-2-methoxynaphthalene was substituted with the desired amine as described by Cuvigny & Normant (1971). All reagents must be dry for this reaction. The benzene was distilled and then dried further by storage over 3-Å molecular sieves. Hexamethylphosphoramide and trimethylethylenediamine were also dried over molecular sieves. Benzene and hexamethylphosphoramide (10 mL of each) were mixed, and argon was bubbled through the solution for about 5 min. Trimethylethylenediamine (4.4 mL; 0.035 mol) was added, and argon was again bubbled through the solution for an additional 2–3 min. The argon flow was then adjusted to pass over the surface of this stirring solution. A 6-cm length of Li wire (Alfa) (0.24 g; 0.035 mol) was rinsed in ether to remove the mineral oil. The 6-cm piece was further cut into 1-mm pieces. To prevent oxidation, the cutting was done while the Li was in a pool of ether. The ether was quickly evaporated under argon, and the pieces of Li were dropped into the stirring solution. Prior to addition, the Li was stored in ether to prevent oxidation. This procedure was found to minimize air oxidation of the Li. As the Li dissolved, the solution first turned blue and then a deep red. This color change occurred before all the Li had been added. After all the Li was added, the argon flow was stopped, and the reaction vessel was tightly stoppered. After the Li was completely dissolved, the 6-palmitoyl-2-methoxynaphthalene (3 g; 0.0075 mol) was added, and the mixture was stirred for 48 h at room temperature. Samples were removed at various times and analyzed for product formation by TLC.
The reaction was terminated by pouring the mixture into 40 mL of ice water. The product was extracted with ethyl ether (5 × 60 mL). The ether was evaporated under vacuum. The resulting yellow oil was crystallized from absolute ethanol. The product was not completely pure as judged from TLC, but it was decided that a completely pure product was not necessary for the next step in the reaction sequence. Note that not all of the material crystallized from the ethanol. About 300 mg of a white solid was obtained, and an oil remained which also contained about 300 mg of product, as estimated from the UV absorbance. It was judged from thin-layer chromatography that both the solid and the oil contained the desired product.
Synthesis of 6-Palmitoyl-2-[[2-(trimethylammonio)-ethyl]methylamino]naphthalene Iodide.
The 6-palmitoyl-2-[[2-(dimethylamino)ethyl] methylamino] naphthalene (200 mg; 0.43 mmol) was dissolved in 30 mL of absolute ethanol; 50 μL (8.6 × 10−4 mol) of CH3I was added to 3 mL of ethanol, and this solution was added dropwise with stirring to the solution of the amine. The solution was refluxed gently for 30 min and then cooled to room temperature, during which time the product began to precipitate. Complete precipitation was effected by adding 30 mL of ether. The product was filtered under vacuum and washed with cold ethanol. Approximately 100 mg of product was obtained. The success of this reaction requires that the reagents and the reaction mixture be free of water. The methylation reaction was also carried out by using the yellow oil obtained in preparing 6-palmitoyl-2-[[2-(dimethylamino)ethyl] methylamino]-naphthalene. It was estimated that approximately 300 mg of the amine was present in the oil. Therefore, the oil containing approximately 0.64 mmol of the amine was taken up to 30 mL with absolute ethanol; 0.12 mL (2 × 10−3 mol) of CH3I was added to 3 mL of absolute ethanol and then added dropwise to the amine. The solution was refluxed for 30 min and the product isolated as described above. Approximately 200 mg of the product was obtained.
The NMR spectrum of the product was as expected, but careful analysis of this spectrum is not presented because it was still desired to convert the iodide salt obtained in this reaction to the chloride salt.
Formation of Patman.
A technique based on the classic Folch extraction mixture (CHCl3/CH3OH/H2O 1/1/0.9) was used to prepare the chloride salt (Folch et al., 1957; Bligh & Dyer, 1957; Kates, 1972). The basic method to convert the iodide to the chloride salt was to use a two-phase system, an organic phase containing the iodide salt of the product and an aqueous phase containing a large excess of NaCl. Exchange of the iodide with the chloride would occur upon mixing the phases.
The iodide salt of Patman (100 mg) was dissolved with heating in 200 mL of CHCl3. To this were added 200 mL of CH3OH and 190 mL of 2 M NaCl. Mixing and then separating the two phases gave the desired product which was isolated by evaporating the solvent to dryness under vacuum. The yellow solid was recrystallized from absolute ethanol. The product was judged to be pure by silica gel TLC (CHCl3/CH3OH/H2O 65/25/5) and 270-MHz NMR (Table I).
Materials and Methods
Fluorescence Spectral Measurements.
For recording of emission spectra, the excitation wavelength was 350 nm, with a band-pass of 8 nm. For lifetime measurements, the slit widths were 8 and 2 nm, for the entrance and exit slits of the excitation monochromator, respectively. These slit widths are required for the Debye–Sears modulator. Emission was generally isolated with a monochromator, with the emission band-pass at 4 nm. Occasionally, lifetime measurements were performed with interference filters (10-nm band-pass). For all measurements, the excitation was polarized vertically, and the emission at 54.7° from the vertical. This eliminates the bias caused by polarization on either the measured lifetimes or the yields (Kalantar, 1968; Spencer & Weber, 1970; Shinitzky, 1972). Fluorescence anisotropies were obtained in the usual manner (Lakowicz, 1983). For red-edge excitation, the excitation band-pass was 2 nm. Where necessary, blank spectra were obtained by using unlabeled lipids and were subtracted from the spectra obtained from the labeled lipids.
The full widths of the emission spectra, at the half-maximal intensity (fwhm), were obtained from the uncorrected steady-state spectra. The fwhm value is the spacing, in nanometers, between the wavelengths corresponding to half of the maximal intensity on the short- and long-wavelength sides of the emission. Of course, a more appropriate measure of the spectral width would be provided from the corrected spectra on the wavenumber scale. However, this is not needed for the comparisons used in this report.
Apparent fluorescence lifetimes were calculated by the phase shift and modulation methods (Spencer & Weber, 1969), using a modulation frequency of 30 MHz. The errors resulting from the use of scattered light as the phase reference were avoided by using reference fluorophores to determine the phase and modulation of the incident light (Lakowicz et al., 1981). These reference compounds were POPOP [l, 4-bis[2-(5-phenyloxazolyl)]benzene] or Me2POPOP [l, 4-bis(4-methyl-5-phenyloxazol-2-yl)benzene] in ethanol. The reference lifetimes were 1.35 and 1.45 ns, respectively.
Preparation of Phospholipid Vesicles.
Patman-labelea phospholipid vesicles were prepared as follows. Patman and lipid were dissolved in chloroform at the molar ratio of one probe molecule per 200 lipid molecules. The solvent was evaporated under a stream of argon while the container was gently warmed in a water bath. Buffer (0.01 M Tris/0.05 M KCl, pH 7.5) was added to yield the desired concentration of lipid, 0.2 mg/mL. The samples were sonicated with a probe-type sonicator, incubated for 30 min at 40 °C, and then centrifuged at 20 000 rpm for 30 min to remove the larger lipid aggregates. The optical density at 350 nm due to the absorbance of Patman was near 0.20. The extinction coefficient of the fluorophore in Patman is near 15 000 M−1 cm−1 at 350 nm (Weber & Farris, 1979). To prepare detergent micelles, an amount of Patman adequate to yield a final optical density of 0.5 at 350 nm was dissolved in chloroform. The solvent was evaporated with a stream of argon. Either 0.1 M SDS or 0.1 M CTABr was added, and the solution was stirred overnight.
Results and Discussion
Effects of Solvent Relaxation and Solvent Polarity on the Spectral Properties of Patman.
Upon excitation, Patman and other fluorophores undergo an instantaneous change in dipole moment. The magnitude of this change reflects the difference in the electronic distribution of charge between the ground and excited states. This change is substantial for Patman, probably near 20 D (Weber & Farris, 1979), which is equivalent to the separation of a unit electronic charge by about 4 Å. Following excitation, the polar solvent molecules can reorient around the excited state, yielding time-dependent spectral shifts [Bakhshiev et al. 1966; DeToma et al., 1976; see Lakowicz (1980, 1983) and references cited therein]. The effects of solvent reorientation or relaxation upon the emission spectra are generally revealed by the temperature-dependent spectra in vitrifying solvents. In such solvents, the rate of relaxation can be slowed by decreases in temperature. Such spectra for Patman in propylene glycol are shown in Figure 1. At low temperature (−63 °C), the emission maximum is 417 nm. As the temperature is increased, the spectra shift progressively to longer wavelengths. The spectral shift is about half completed at −30 °C, where the emission maximum is 442 nm. The red shift is largest at 20 °C, where the emission maximum is 472 nm (Figure 2). These results are similar to those observed by Macgregor & Weber (1981) for Prodan. It is interesting to note that the emission spectra shift to shorter wavelengths at temperatures above 20 °C. Such temperature-dependent blue shifts have been observed previously (Cherkasov & Dragneva, 1961; Bakhshiev & Peterskaya, 1965; Macgregor & Weber, 1981). At high temperature, thermal energy can prevent complete alignment of the solvent dipoles around the excited state.
FIGURE 1:

Temperature-dependent emission spectra of Patman in propylene glycol.
FIGURE 2:

Fluorescence spectral properties of Patman in propylene glycol. The fluorescence emission maxima (●) and approximate widths (○) of the emission spectra are shown.
The effects of solvent polarity on the emission spectra of Patman are summarized in Table II. The emission maxima shift dramatically to longer wavelengths with increasing solvent polarity. For example, in dioxane the maximum is at 404 nm, whereas the maximum is at 481 nm in methanol/water (1:1). The red shift in the emission from nonpolar to polar solvents is accompanied by an increase in the lifetime of the total emission from 1.3 to 3.5 ns.
Table II:
Emission Maxima, fwhm Values, and Lifetimes of Patman in Various Solventsa
| solvent | emission maximum (nm) | fwhm (nm) | τ (ns)b |
|---|---|---|---|
| dioxane | 404 | 52 | 1.3 |
| chloroform | 423 | 62 | 1.7 |
| acetone | 425 | 60 | 1.9 |
| acetonitrile | 440 | 60 | 3.0 |
| dimethylformamide | 440 | 57 | 3.1 |
| butanol | 459 | 60 | 3.2 |
| methanol | 472 | 63 | 3.3 |
| methanol/water (1:1) | 481 | 65 | 3.5 |
At 25 °C; excitation wavelength, 350 nm.
Phase lifetime, 30 MHz; emission filter, 1 M sodium nitrite, 2-mm thickness.
Comparison of the emission spectra of Patman in propylene glycol at various temperatures (Figure 1) with the spectra observed in fluid solvents of varying polarity (Table II) reveals an increase in the width of the emission spectrum in the case of propylene glycol. As a simple measure of the spectral width, we used the full width at the half-maximum intensities (fwhm) on the short- and long-wavelength sides of the emission. The increase in the fwhm is evident in the emission spectrum of Patman in propylene glycol at −30 °C (Figure 1). The width is maximal at this temperature and becomes smaller as the temperature is either increased or decreased (Figure 2). The increased spectral width is due to emission from a multitude of partially relaxed states, these being the result of time-dependent solvent relaxation. At the intermediate temperatures, the rate of spectral relaxation is comparable to the decay rate of the fluorescence intensity [see Lakowicz (1983) and references cited therein]. It is important to notice that the emission maxima of Patman in propylene glycol are most dependent upon temperature when the fwhm values are largest.
For comparative purposes, we note that the spectral shifts caused by increasing solvent polarity are not accompanied by a comparable increase in the spectral width (Table II). For example, even though the emission maximum in dimethylformamide is an intermediate value, the spectral width is similar to that seen in the less and more polar solvents. We stress that a rigorous comparison of spectral widths requires the use of corrected emission spectra and the use of wave-numbers.
A useful characteristic of Patman is the dependence of its apparent lifetime upon the rate of solvent relaxation. Wavelength-dependent lifetimes can be a result of either heterogeneity (a mixture of directly excited fluorophores with different lifetimes) or an excited-state reaction. The observation of apparent phase lifetimes (τp) which are greater than the apparent modulation lifetimes (τm) (i.e., τp > τm) proves that an excited-state process has occurred (Lakowicz & Balter, 1982b). For a single-exponential decay of fluorescence intensity, τp = τm, and for a mixture of fluorophores, each excited directly, τp< τm. Therefore, the data in Figure 3 clearly indicate that the temperature-dependent spectral shifts observed for Patman (Figure 1) are clearly due to time-dependent solvent relaxation. At an intermediate temperature (−20 °C), where the solvent relaxation time is expected to be comparable with the fluorescence lifetime, the apparent phase and modulation lifetimes are highly dependent upon the emission wavelength (Figure 3). At lower temperatures (−65 °C), the rate of solvent relaxation is slower than the decay rate of the fluorophore; as a result, the apparent lifetimes are less dependent upon emission wavelength.
FIGURE 3:

Apparent fluorescence lifetimes of Patman in propylene glycol. The modulation frequency was 30 MHz. τp and τm refer to the apparent phase and modulation lifetimes, respectively. For m/cos ϕ: (●) −20 °C; (○) −65 °C.
Also shown in Figure 3 are the values of m/cos ϕ, where m is the demodulation factor and ϕ is the phase angle. The ratio m/cos ϕ exceeds unity only in the presence of an excited-state reaction (Lakowicz & Balter, 1982b) and when the reacted species is observed. This ratio was found to increase with increases in the emission wavelength, with the largest values being observed on the long-wavelength side of the emission. Although the detailed temperature-dependent values of m/cos ϕ are not shown, this ratio is (at 520 nm) largest near −30 °C and decreases as the temperature is either increased or decreased. From the results presented in Figures 1–3, it is apparent that the spectral properties of Patman reflect both the overall polarity of its surrounding environment and the rate at which this environment relaxes around the increased dipole moment of the excited state.
Emission Spectra of Patman-Labeled Phospholipid Vesicles.
The fluorescence emission spectra of Patman-labeled phospholipid vesicles shift dramatically at the phase transition temperature of the vesicles. This is illustrated in Figure 4 for Patman-labeled DMPC vesicles. At 2 °C, which is below the phase transition temperature of about 23 °C, the emission spectrum is similar to that seen in a relatively nonpolar solvent such as acetone or acetonitrile. At 32 °C, the emission spectrum is red shifted and considerably broadened. As described above for Patman in propylene glycol when compared with various solvents, a broadened spectrum is characteristic of emission from both the relaxed and the unrelaxed states. At higher temperature (52 °C), the spectrum shifts further to the red and becomes more narrow, indicating that the relaxation process is almost complete. In addition to a red shift, increasing temperature results in a modest decrease in the peak intensity (Figure 4, bottom panel). Since the spectra are also broader, it is not clear to what extent the quantum yield decreases above the transition temperature, but any such decrease is less than 2-fold. Analogous results were obtained in vesicles composed of other saturated phosphatidylcholines. In DOPC vesicles at 2 °C, the spectra are already broadened (Figure 5). Further increases in temperature result in a narrowing of the emission spectrum. These results reflect the greater fluidity of the unsaturated phospholipids.
FIGURE 4:

Emission spectra of Patman-labeled DMPC vesicles. Spectra are shown normalized (top) and at the same instrumental gain (bottom). The temperatures were 2 (—), 22 (-·-·), and 52 °C (---).
FIGURE 5:

Emission spectra of Patman-labeled DOPC vesicles (top) and Patman-labeled micelles (bottom). Top panel: 2 (—), 22 (-·-·), and 52 °C (---). Bottom panel: CTABr (---); SDS (—), 25 °C.
The temperature-dependent emission maxima of Patman-labeled vesicles are summarized in Figure 6. The dependence upon the chemical composition of the vesicles is similar to that found by using other probes, such as diphenylhexatriene, and other techniques, such as calorimetry. However, the temperatures at which the spectral shifts are half complete are higher than those found by these other methods. This disparity is probably a result of the lower fluorescence intensity of the Patman in bilayers above their phase transition temperature, and hence a smaller contribution of these molecules to the emission spectrum. The emission maxima of Patman-labeled DPPG vesicles shifts at temperatures above 40 °C, as expected for the transition temperature of this lipid (Boggs et al., 1981). The surface charge of the vesicle does not seem to have a significant effect, as seen by the similar results for DPPC and DPPG. For all these lipids, the temperature-dependent spectra are similar to that seen in Figure 4 for DMPC. That is, the spectra shift to longer wavelengths above the transition temperature, and the peak intensity decreases about 2-fold. Also, an approximate isoemissive point is found near 470 nm. The effects of temperature on the widths of the emission spectra are summarized in Figure 7 for Patman-labeled vesicles. It is apparent that these widths are largest at temperatures near the phase transitions of the vesicles, and these widths become smaller at either higher or lower temperatures. The similarity of these results with those observed for Patman in propylene glycol (Figure 2) indicates that time-dependent spectral relaxation is the probable cause of the spectral shifts summarized in Figure 6.
FIGURE 6:

Effects of temperature on the emission maxima of Patman-labeled vesicles. The lipids were DOPC (○), DMPC (●), DPPC (Δ), and DPPG (□).
FIGURE 7:

Effects of temperature on full widths at half-maximum intensity (fwhm) of Patman-labeled vesicles. The lipids were DOPC (○), DMPC (●), DPPC (Δ), and DPPG (□).
The emission spectra of Patman-labeled micelles illustrate the differential sensitivity of this probe to polarity and dipolar relaxation. Emission spectra of Patman bound to positively (CTABr) and negatively (SDS) charged micelles are shown in the bottom panel of Figure 5. These spectra are essentially identical. Moreover, these spectra are red shifted relative to that found for Patman in vesicles, and the spectra are considerably narrower. This indicates a more polar environment surrounding Patman in the vesicles and rapid relaxation of this environment around the excited state. This is consistent with the known penetration of water into micelles (Menger, 1979) and the higher fluidity of micelles relative to lipid vesicles.
Fluorescence Lifetimes of Patman-Labeled Vesicles.
Typical wavelength profiles of the apparent lifetimes and the ratio m/cos ϕ are shown for Patman-labeled DMPC vesicles in Figure 8. At lower temperature, the value of m/cos ϕ is lower. Typically m/cos ϕ is maximal near the transition temperature of the vesicles. Similar results were obtained for DOPC, except that for this lipid the dependence of the apparent lifetimes on the emission wavelength (Figure 9) and the values of m/cos ϕ were maximal at the lower temperature. This is in agreement with the lower phase transition temperature of this unsaturated lipid. For these and other Patman-labeled lipids, the apparent phase and modulation lifetimes are highly dependent on the emission wavelength. The dependence on the emission wavelength is substantial, with the apparent lifetimes ranging from 2 ns at 380 nm to 6 ns at 540 nm. The effects of relaxation are most apparent at temperatures where the spectra are broadest, such as DMPC at 32 °C and DOPC at 2 °C. The increase of τp over τm is most dramatic for these lipids at these respective temperatures.
FIGURE 8:

Apparent fluorescence lifetimes for Patman-labeled DMPC vesicles.
FIGURE 9:

Apparent fluorescence lifetimes for Patman-labeled DOPC vesicles.
The observation of τp > τm, or m/cos ϕ > 1, on the long-wavelength side of the emission demonstrates the occurrence of membrane relaxation around the excited state of Patman. As the temperatures are increased above the transition temperatures, the values of m/cos ϕ decrease because the emission becomes dominated by the relaxed state, as this state forms quickly from the initially excited state.
Phase-Sensitive Fluorescence Spectra of Patman-Labeled Vesicles.
Using phase-sensitive detection of fluorescence, one can directly record, in an approximate fashion, the emission spectra of the initially excited (unrelaxed) and the relaxed states of fluorophores (Lakowicz & Balter, 1982a). Phase-sensitive fluorescence spectra of Patman-labeled DMPC are shown in Figure 10. To record these spectra, we adjusted the detector phase angle to suppress emission with the lifetime values indicated on the figure. At 52 °C (bottom panel), suppression of the 5.1-ns component at 530 nm results in a blue-shifted spectrum with an emission maximum near 430 nm. Conversely, suppression of the 0.9-ns component at 395 nm yields a red-shifted spectrum with an emission maximum near 480 nm. To a first approximation, these spectra may be regarded as characteristic of the unrelaxed and relaxed states of Patman, respectively. The substantial difference in lifetime between the blue and red sides of the emission makes Patman particularly useful for such analysis by the phase-sensitive spectra. However, we caution that the resolution of states by the phase-sensitive method, although informative, is not unique. This is illustrated by the phase-sensitive spectra of Patman-labeled DMPC observed at 2 °C. If the two spectra recorded at 52 °C were indeed characteristic of the initially excited and relaxed states, then the phase-sensitive spectra should be invariant with temperature, except for changes in the relative amplitudes. The phase-sensitive spectra obtained at 2 °C are shown in the top panel of Figure 10. Once again, blue- and red-shifted spectra were observed, but these were distinct from those found at 52 °C. In particular, the spectral separation of the presumed relaxed and unrelaxed states of Patman is less pronounced at 2 °C than at 52 °C. Spectral relaxation is probably a complex process involving a number of fluorophore–lipid interactions (Bakhshiev et al., 1966; DeToma et al., 1976; Easter et al., 1976; Macgregor & Weber, 1981). Hence, one should not expect a two-state model to precisely describe the phase-resolved spectra.
FIGURE 10:

Phase-sensitive fluorescence spectra of Patman-labeled DMPC vesicles. The arrows indicate the wavelengths used for phase suppression, and the numbers indicate the apparent lifetime suppressed at the chosen phase angle.
Dependence of the Fluorescence Anisotropy on the Emission Wavelength.
The spectral properties of Patman provide a unique opportunity to investigate the relationship between spectral relaxation and rotational diffusion. This is possible because the fluorescence anisotropies of Patman-labeled vesicles are dependent upon the observation wavelength. Typical data are shown in Figure 11. At both high and low temperatures, but especially at 52 °C, the anisotropy decreases substantially with increasing emission wavelength. At 52 °C, the anisotropy decreases about 4-fold from 380 to 560 nm. Similar behavior was seen in other vesicles. In the case of Patman, the average duration of the excited state increases with the emission wavelength. The longer lived fluorophores, which are also those which emit at longer wavelengths, have more time to rotate and hence lower fluorescence anisotropies. Similar observations were made for fluorophores in viscous solvents (Mazurenko & Bakhshiev, 1970; Koyava et al., 1980), but the magnitude of these effects was less than that seen for Patman.
FIGURE 11:

Fluorescence anisotropies at various emission wavelengths for Patman-labeled DMPC vesicles. The excitation wavelength was 360 nm.
For completeness, we note that the steady-state anisotropies of Patman are sensitive to the phase transition of the bilayers. These data are summarized in Figure 12, where the anisotropies were measured near the center of the emission spectrum (460 nm). Above the phase transition, the anisotropies decrease, but not to the extent found for other probes such as 1,6-diphenyl-1,3,5-hexatriene and perylene (Lakowicz et al., 1979). The palmitoyl and trimethylammonioethyl groups attached to the fluorophore probably restrict its rotational motions. The degree to which the anisotropies decrease at the phase transition is dependent upon the chosen emission wavelength. At both 420 and 500 nm, the phase transition is observable. However, the magnitude of the anisotropy changes is severalfold larger at 5Ó0 nm than at 420 nm (results not shown). This is because at longer wavelengths the duration of the excited state is also longer. Hence, there is more time for rotational diffusion. The opposite is true for an observation wavelength of 420 nm. At this temperature, the duration of the excited state is decreased by relaxation, resulting in less time for the decay of anisotropy.
FIGURE 12:

Temperature-dependent fluorescent anisotropies of Patman-labeled vesicles. The anisotropies were measured near the center of the emission spectrum, 460 nm. The lipids were DOPC (○), DMPC (●), DPPC (Δ), and DPPG (□).
Red-Edge Excitation Shifts of Patman-Labeled Vesicles.
For many fluorophores, including Patman (see below), excitation on the red edge of the absorption spectrum results in red-shifted emission spectra (Rubinov & Tomin, 1970; Rudik & Pikulik, 1971; Azumi et al., 1966; Macgregor & Weber, 1981). It appears that the use of red-edge excitation selectively excites those fluorophores which are interacting most strongly with the solvent. These are the fluorophores around which the solvent molecules are oriented in a configuration comparable to that found in the solvent-relaxed state. The ability to observe red-shifted spectra requires that a different average population be excited at each excitation wavelength and that this difference be maintained for a time comparable to the fluorescence lifetime. Hence, red-shifted spectra should be most apparent when the solvent relaxation times are long relative to the fluorescence lifetime (Demchenko, 1982), and for fluorophores which are sensitive to solvent polarity.
Figure 13 summarizes the dependence of the emission maximum of Patman-labeled DOPC vesicles on the excitation wavelength at different temperatures. This dependence is largest at 2 °C and is no longer apparent at 52 °C. At intermediate temperatures, the variation of the emission maximum is also intermediate. Note that with 420-nm excitation, the completely relaxed emission is observed, irrespective of temperature. Careful quantitation of and correction for background fluorescence has ensured that impurities were not the origin of these excitation red shifts. From these data, it is apparent that substantial excitation shifts occur with Patman and that the extent of these shifts can be used to estimate the dynamics of its environment.
FIGURE 13:

Dependence of the emission maximum of Patman-labeled DOPC upon the wavelength of excitation.
In summary, we demonstrated that the fluorescence spectral properties of Patman are sensitive to solvent polarity and to the phase states of the surrounding lipid. This probe may be useful for the study of lateral phase separations in mixed vesicles (Lentz et al., 1976a,b) or in the study of boundary lipid around membrane-bound proteins (Lee et al., 1982). If regions of lipid exist with distinct physical properties, then the emission spectra of Patman from such regions may be distinct, allowing the amount of each region to be quantified.
Acknowledgments
This work was supported by Grants PCM 80-41320 and PCM 81-06910 from the National Science Foundation and was performed during the tenure of an Established Investigatorship (to J.R.L.) from the American Heart Association.
Footnotes
Abbreviations: DPH, 1,6-diphenyl-1,3,5-hexatriene; Patman, 6-palmitoyl-2-[[2-(trimethylammonio)ethyl]methylamino]naphthalene chloride; DOPC, DMPC, and DPPC, dioleoyl-, dimyristoyl-, and dipalmitoyl-l-α-phosphatidylcholine, respectively; DPPG, dipalmitoyl-l-α-phosphatidylglycerol; CTABr, cetyltrimethylammonium bromide; SDS, sodium dodecyl sulfate; fwhm, full width at half-maximum intensity of a steady-state emission spectrum; Prodan, 6-propionyl-2-(dimethylamino)naphthalene; TLC, thin-layer chromatography; Tris, tris(hydroxymethyl)aminomethane.
References
- Azumi T, Itoh KI, & Shiraiski H (1976) J. Chem. Phys 65, 2550–2555. [Google Scholar]
- Bakhshiev NC, & Piterskaya IV (1965) Opt. Spectrosc. (Engl. Transl.) 19, 309–395. [Google Scholar]
- Bakhshiev NG, Mazurenko Yu. T., & Piterskaya IV (1966) Opt. Spectrosc. (Engl. Transl.) 21, 307–309. [Google Scholar]
- Bligh EG, & Dyer WJ (1959) Can. J. Biochem. Physiol 37, 91. [DOI] [PubMed] [Google Scholar]
- Boggs JM, Wood DD, & Mossarello MA (1981) Biochemistry 20, 1065–1073. [DOI] [PubMed] [Google Scholar]
- Buu-Hoi N. G. pH. (1949) Recl. Trav. Chim. Pays-Bas 68, 759–780. [Google Scholar]
- Cherkasov AS, & Dragneva GI (1961) Opt. Spectrosc. (Engl. Transl.) 10, 238–241. [Google Scholar]
- Cuvigny T, & Normant H (1971) C. R. Hebd. Seances Acad. Sci., Ser. C 272, 1475–1430. [Google Scholar]
- Demchenko AP (1982) Biophys. Chem 15, 101–109. [DOI] [PubMed] [Google Scholar]
- DeToma RP, Easter JH, & Brand L (1976) J. Am. Chem. Soc 98, 5001–5007. [DOI] [PubMed] [Google Scholar]
- Easter JH, DeToma RP, & Brand L (1976) Biophys. J 16, 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Lees M, & Stoane-Stanley GA (1957) J. Biol. Chem 226, 497. [PubMed] [Google Scholar]
- Fraenkd G, Ellis SH, & Dix DT (1965) J. Am. Chem. Soc 87, 1406–1407. [Google Scholar]
- Kalantar AH (1968) J. Phys. Chem 72, 2801–2805. [Google Scholar]
- Kates M (1972) Techniques of Lipodology, pp 349–350, North-Holland Publishing Co., Amsterdam. [Google Scholar]
- Kawato S, Kinosita K, & Ikegami A (1977) Biochemistry 16, 2319–2324. [DOI] [PubMed] [Google Scholar]
- Koyava VT, Popechits VI, & Sarzhevskii AM (1980) Opt. Spectrosc. (Engl. Transl.) 48, 493–497. [Google Scholar]
- Lakowicz JR (1980) J. Biochem. Biophys. Methods 2, 91–119. [DOI] [PubMed] [Google Scholar]
- Lakowicz JR (1983) Principles of Fluorescence Spectroscopy, Plenum Press, New York. [Google Scholar]
- Lakowicz JR, & Knutson JR (1980) Biochemistry 19, 905–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz JR, & Cherek H (1981) J. Biochem. Biophys. Methods 5, 19–35. [DOI] [PubMed] [Google Scholar]
- Lakowicz JR, & Balter A (1982a) Photochem. Photobiol 36, 125–132. [DOI] [PubMed] [Google Scholar]
- Lakowicz JR, & Balter A (1982b) Biophys. Chem 16, 99–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz JR, Prendergast FG, & Hogen D (1979) Biochemistry 18, 508–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz JR, Cherek H, & Balter A (1981) J. Biochem. Biophys. Methods 5, 131–146. [DOI] [PubMed] [Google Scholar]
- Lee AG, East JM, Jones OT, McWhirter JJ, Rooney EK, & Simmonds AC (1982) Biochemistry 21, 6441–6446. [DOI] [PubMed] [Google Scholar]
- Lentz B, Barenholz Y, & Thompson TE (1976a) Biochemistry 15, 4521–4528. [DOI] [PubMed] [Google Scholar]
- Lentz BR, Barenholz Y, & Thompson TE (1976b) Biochemistry 15, 4529–4536. [DOI] [PubMed] [Google Scholar]
- Macgregor RB, & Weber G (1981) Ann. N.Y. Acad. Sci 366, 140–154. [Google Scholar]
- Mazurenko Yu. T., & Bakhshiev NG (1970) Opt. Spectrosc. (Engl. Transl.) 28, 490–494. [Google Scholar]
- Menger FM (1979) Acc. Chem. Res 12, 111–117. [Google Scholar]
- Rubinov AN, & Tomin VI (1970) Opt. Spectrosc. (Engl. Transl.) 29, 578–579. [Google Scholar]
- Rudik KI, & Pikulik LG (1971) Opt. Spectrosc. (Engl. Transl.) 30, 147–148. [Google Scholar]
- Shinitzky M (1972) J. Chem. Phys 56, 5979–5981. [Google Scholar]
- Shinitzky M, & Barenholz Y (1974) J. Biol. Chem 249, 2652–2657. [PubMed] [Google Scholar]
- Slivak J (1982) Biochim. Biophys. Acta 694, 1–25. [DOI] [PubMed] [Google Scholar]
- Spencer RD, & Weber G (1969) Ann. N.Y. Acad. Sci 158, 361–376. [Google Scholar]
- Spencer RD, & Weber G (1970) J. Chem. Phys 52, 1654–1663. [Google Scholar]
- Weber G (1952) Biochem. J 51, 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber G, & Farris FJ (1979) Biochemistry 18, 3075–3078. [DOI] [PubMed] [Google Scholar]