

Abstract
Many anticancer and antiviral drugs are purine or pyrimidine analogs, which use membrane transporters to cross cellular membranes. Concentrative nucleoside transporters (CNTs) mediate the salvage of nucleosides and the transport of therapeutic nucleoside analogs across plasma membranes by coupling the transport of ligands to the sodium gradient. Of the three members of the human CNT family, CNT3 has the broadest selectivity and the widest expression profile. However, the molecular mechanisms of the transporter, including how it interacts with and translocates structurally diverse nucleosides and nucleoside analogs are unclear. Recently, the crystal structure of vcCNT showed that the prokaryotic homolog of CNT3 forms a homo-trimer. In this study, we successfully expressed and purified the wild type human homolog, hCNT3, demonstrating the homo-trimer by size exclusion profiles and glutaraldehyde cross-linking. Further, by creating a series of cysteine mutants at highly conserved positions guided by comparative structure models, we cross-linked hCNT3 protomers in a cell-based assay, thus showing the existence of hCNT3 homo-trimers in human cells. The presence and absence of cross-links at specific locations along TM9 informs us of important structural differences between vcCNT and hCNT3. Comparative modeling of the trimerization domain and sequence co-evolution analysis both indicate that oligomerization is critical to the stability and function of hCNT3. In particular, trimerization appears to shorten the translocation path for nucleosides across the plasma membrane and may allow modulation of the transport function via allostery.
Graphical abstract
Introduction
Nucleosides are metabolic precursors in nucleic acid synthesis with a wide variety of critical roles in cell homeostasis.1,2 For example, when converted to nucleotides (e.g., ATP and GTP), they represent units of cellular currency, while as components of NAD and FAD, they act as coenzymes critical in energy metabolism. Some nucleosides can act alone as signaling molecules; for example, adenosine can act as an autocrine and paracrine hormone by serving as a ligand for adenosine (P1) receptors.3 Although most cell types are capable of de novo synthesis of nucleosides, others, like cells of the brain, muscle, erythrocytes, leukocytes, and bone marrow, lack these biosynthetic pathways.4 In addition, de novo synthesis is energetically costly.2 Nucleoside salvage (i.e., the recycling of extracellular nucleosides to replenish the intracellular levels) via nucleoside transporters is therefore an attractive strategy to achieve nucleoside homeostasis. Being largely hydrophilic, nucleosides require membrane transporters to cross the plasma membrane. To date, six human nucleoside transporters have been identified from two SLC transporter families, the concentrative nucleoside transporters (CNT) (SLC28 family) and the equilibrative nucleoside transporters (ENT) (SLC29 family).5
Of the three human members of the SLC28 family, hCNT3 is characterized by the broadest tissue distribution and substrate specificity. High levels of hCNT3 have been found in the pancreas, bone marrow, and mammary gland; lower levels have been found in the intestine, lung, prostrate, testis, and liver.6 hCNT3 is a symporter that couples the transport of one nucleoside to the symport of two Na+ ions or one proton.4 Its pharmacological importance stems from its ability to transport a wide variety of nucleoside-derived drugs,7 including first-line therapeutic agents for viral infections, such as valacyclovir, or solid tumors, such as gemcitabine. As such, it is an important mediator of drug response and resistance to anti-cancer nucleoside analogs. For example, elevated CNT3 expression levels in chronic lymphocytic leukemia (CLL) is associated with lower complete response rate to fludarabine therapy.8 In addition, in pancreatic cancer, some uncharacterized hCNT3 polymorphisms, such as the nonsynonymous A25G mutation, have been associated with gemcitabine toxicity.9
Although the regulation and function of hCNT3 have been subject to extensive characterization through mutagenesis and uptake studies over the past two decades, there have been only a few insights into the structure and molecular mechanism of these transporters.10–15 The crystal structure of vcCNT provided the first breakthrough, by establishing a previously unknown protein-fold.16 The most striking feature, however, proved to be the unexpected quaternary structure. No previous study has suggested that CNTs form oligomeric structures. Subsequently, it was established that the functional form of vcCNT is indeed a trimer, with clearly defined ligand and ion binding sites on each of the protomers.16
With the increasing number of crystal structures of membrane transporters, their oligomerization and symmetry have recently become topics of intense study due to a variety of possible functional roles.17,18 For example, crystallographic studies of the glycine betaine transporter BetP from Corynebacterium glutamicum have identified a symmetric trimer, with coupled binding of betaine and sodium ions within the protomer during the transport process.19,20 Further, experimental evidence for an engineered functional/non-functional BetP heterotrimer indicates that functional crosstalk between protomers is not essential, neither for transport catalysis nor for regulation by cytoplasmic K+ concentration.21 Nevertheless, isolated monomers displayed a significantly reduced transport catalysis, and no regulation.22 Therefore, oligomerization has been proposed to influence transporter function via its impact on stability and structural assembly.21
As a trimer, BetP does not represent an outlier. Oligomerization is remarkably common among membrane proteins. A survey of the Protein Data Bank of Transmembrane Proteins shows that approximately 65% of membrane proteins form oligomers.17 Amongst more than 50 SLC transporter families, several, including SLC1, SLC13, and SLC42, are predicted or shown to have members that form oligomeric structures.5
Here, we report a series of cross-linking studies to examine two main questions. 1) Does hCNT3 also form homo-trimers? 2) Is there a functional role for the formation of this quaternary structure? By using a comparative structure model of hCNT3 based on the vcCNT structure (39% sequence identity) followed by cysteine mutagenesis, we determined that the trimer is preserved in hCNT3, using in vitro and cell-based assays. The cross-linking results can be parsimoniously rationalized through localized structural differences between hCNT3 and vcCNT in the trimerization domain. The model of the hCNT3 trimer also indicates how the translocation distance is significantly reduced in the trimer compared to that in the monomeric membrane transporters.
Experimental Procedures
Expression and purification of hCNT3 in High Five insect cells hCNT3 was expressed in High Five insect cells using the Bac-to-Bac Baculovirus Expression System (Thermo Scientific). The gene encoding the full-length hCNT3 was cloned into the pFastBac1 vector, including a HRV 3C cleavable His10-tag at the N-terminus (Figure S1A). The protein was purified as described previously.23 Cells were spun down and solubilized in the lysis buffer (50 mM Tris pH 8.0, 300 mM NaCl, cOmplete Mini Protease Inhibitor Cocktail (Roche 11836153001)) before passing the solution through 5 cycles inside of Emulsiflex at 5,000–10,000 psi. The lysate was cleared with a 15 min spin at 15,000 × g at 4°C before pelleting the membranes with an 80 minute ultracentrifuge spin at 40,000 × g at 4°C. Membranes were resuspended in the resuspension buffer (50mM HEPES pH 7, 300 mM NaCl, 10% glycerol, 2 mM imidazole pH 7, 1% n-dodecyl-β-D-maltopyranoside (DDM) (Inalco 1758–1350), 1 mg/mL iodoacetamide, cOmplete Mini Protease Inhibitor Cocktail (Roche 11836153001), 0.05% cholesteryl hemisuccinate, 0.1 mg/mL E. coli polar lipid extract) and stirred for 1 hour at 4°C. Excess lipids were pelleted with a 30 min spin at 35,000 rpm at 4°C. Supernatants were applied to TALON/Co2+ affinity resin and stirred for 2 hours. Protein was eluted off the column with 300 mM imidazole, following a series of washes. The protein was concentrated with 100kD cutoff Amicon concentrators (Sigma Z648043) and applied to a Superdex 200 size-exclusion column along with the running buffer (50 mM HEPES pH 7, 300 mM NaCl, 10% glycerol, 0.1% DDM). Fractions were collected every 500 μL to be further analysis. For Coomassie gel stains, fractions were loaded onto 4–15% Criterion Tris-HCl gels (Bio-Rad 3450027) under reducing and denaturing conditions and stained using SimplyBlue SafeStain (Invitrogen LC6060) following manufacturers’ protocols. Protein size was estimated with Amersham ECL Full-Range Rainbow pre-stained protein ladder (GE RPN800E).
Glutaraldehyde cross-linking of purified hCNT3
Glutaraldehyde cross-linking was performed following an adapted protocol from Perez, et al.22 A volume of 100 μL of purified protein in running buffer was supplemented with 5 μL of 2.3% glutaraldehyde and incubated at 37°C for 5 min. The reaction was terminated by the addition of 10 μL of 1 M Tris-HCl (pH 8.0). Samples were loaded onto 4–15% Criterion Tris-HCl gels (Bio-Rad 3450027). Protein size was estimated with Precision Plus Protein™ All Blue pre-stained protein ladder (1610373). Polyclonal antibody Sigma HPA023311 against CNT3 was used for detection, with SuperSignal West Femto Chemiluminescent substrate (Thermo Scientific 34095) on the FluorChem E development system (proteinsimple).
Comparative structure modeling of hCNT3
Comparative protein structure models of wild type and mutant hCNT3 protomers were created with MODELLER 9.10 (http://salilab.org/modeller),24 using the 2.4 Å co-crystal structure of a concentrative nucleoside transporter from Vibrio cholerae, vcCNT, in complex with uridine (PDB ID 3TIJ)16 as a template (39% sequence identity to hCNT3). Sequence alignment was created by a manual refinement of gaps in the alignment from PROMALS3D.25 The alignment results in an 84% coverage of hCNT3. Predicted TM 1–3, intracellular loop between TM8 and TM9, and extracellular loop between HP2b and TM10 were not modeled. We generated 100 models using the automodel class with default settings. The models had acceptable normalized discrete optimized protein energy scores (zDOPE) in the range of −0.41 to −0.63.26 The top scoring models were selected for analysis. To construct a model of the trimer, three copies of the hCNT3 protomer were aligned to the crystal structure of the vcCNT trimer using PyMol.
Cell culture of PK15NTD cells
Porcine kidney tubular epithelium nucleoside transporter deficient cells (PK15NTD)27 were donated by Dr. Chung-Ming Tse (The Johns Hopkins University School of Medicine, Baltimore, MD, USA). The cells were cultured in Eagle’s minimal essential medium with Earle’s balanced salt solution with 1 mM sodium pyruvate, 0.1 mM non-essential amino acids, 10% FBS, 100 I.U./mL penicillin, 100 μg/mL streptomycin, and 200 μg/mL hygromycin B at 37°C and 5% CO2, as reported previously.28,29
Cloning and site-directed mutagenesis of hCNT3
Full-length human CNT3 cDNA was cloned into the pcDNA5/FRT mammalian expression vector (Invitrogen V601020). SLC28A3 (UniProt ID Q9HAS3) cDNA were obtained from GE Dharmacon MGC cDNAs collection (MHS6278–202857241). Locations of cysteine mutants were selected based on the comparative structure models. Standard protocols for QuikChange II site-directed mutagenesis were followed (Agilent 200523), using KOD Xtreme Hot Start DNA polymerase (Novagen 71975–3) instead of PfuUltra High-Fidelity DNA polymerase and MAX Efficiency DH5α competent cells (Invitrogen 18258–012) instead of XL1-Blue supercompetent cells.
Cross-linking of hCNT3 containing cysteine mutants in PK15NTD cells
The protocol was adapted from Hastrup et al.30 PK15NTD cells were seeded on non-treated polystyrene 60 mm dishes (Corning). Upon reaching 80–90% confluency, they were transfected with the appropriate construct and Lipofectamine LTX per the manufacturer’s protocol (Invitrogen 15338–030). For co-expressing studies, half of the DNA material added came from each of the single mutant constructs.
Following a 48-hour incubation, the cells were washed with PBS (1.54 mM KH2PO4, 155.17 mM NaCl, 2.71 mM Na2HPO4, pH 7.4) and subsequently incubated in 100 μM CuSO4 and 400 μM 1,10-phenanthroline for 10 minutes at room temperature. The cells were then washed twice with PBS and incubated in 10 mM N-ethylmaleimide (NEM) for 20 minutes at room temperature, before being scraped into PBS/protease inhibitor (PI) buffer (PBS, 10 mM NEM, cOmplete Mini Protease Inhibitor Cocktail (Roche 11836153001)). The suspension was pelleted at 800 x g for 5 min at 4°C. The pellet was resuspended in 0.2% digitonin in PBS/PI buffer for 20 min at 4°C. The suspension was pelleted at 2000 x g for 10 min. The pellet was resuspended in 100 μL of 1% Triton X-100 in PBS/PI at 4°C for 1 hour. The suspension was centrifuged at 14,000 x g for 30 min at 4°C.
The extract was then assayed for protein concentration with the BCA Protein Assay Kit (Pierce 23225) and 2 μg of protein was deglycosylated overnight with 1.5 μL of PNGase F (NEB P0704L) in a total volume of 20 μL, following the nonreducing manufacturer’s protocol. Western blots were performed under denaturing but nonreducing conditions by mixing 15 μL of the sample with 4X NuPAGE LDS Sample Buffer (Invitrogen NP0008) and loading it onto NuPAGE 3–8% Tris-Acetate mini gel (Invitrogen EA0375). Protein size was estimated with HiMark pre-stained protein ladder (Invitrogen LC5699). Polyclonal antibody Sigma HPA023311 antibody against CNT3 was used for detection, with SuperSignal West Femto Chemiluminescent substrate (Thermo Scientific 34095) on the FluorChem E development system (proteinsimple).
[3H]uridine uptake
Uptake studies were performed as described previously.31 Briefly, PK15NTD cells were seeded on non-coated polystyrene 24-well plates (Corning). Upon reaching 70–80% confluency, cells were transfected with the appropriate gene containing construct or an empty vector, and Lipofectamine LTX per the manufacturer’s protocol (Invitrogen 15338–030). Following a 48-hour incubation, the cells were incubated for 10 minutes in a 37°C sodium-free buffer (5 mM HEPES, 10 mM glucose, 1 mM CaCl2, 140 mM N-methyl-D-glucamine, 5 mM KH2PO4, 1 mM MgCl2, pH 7.4). The uptake was initiated by the addition of 33.3 nM [5,6-3H]-uridine (Moravek MT 799) in a sodium-containing buffer (5 mM HEPES, 10 mM glucose, 1 mM CaCl2, 5 mM KCl, 135 mM NaCl, 1 mM MgCl2, 0.8 mM Na2HPO4, 3.3 mM NaH2PO4, pH 7.4) and terminated by washing the cells twice with 4°C sodium-free buffer. Cells were lysed by the addition of lysis buffer (0.1% SDS vol/vol, 0.1 N NaOH). Intracellular radioactivity was measured by scintillation counting and normalized per well of protein content using the BCA Protein Assay Kit (Pierce 23225).
Results
Purification of wild type hCNT3
To determine whether or not hCNT3 forms a homo-oligomer, we first expressed and purified the wild type protein. The created construct was a truncated variant of the full-length hCNT3, missing the 75 C-terminal residues predicted to be extracellular and to lack both secondary structure and glycosylation sites (Figure S1A). Earlier work performed in our laboratory indicated that glycosylation most likely does not affect trafficking of CNTs.32
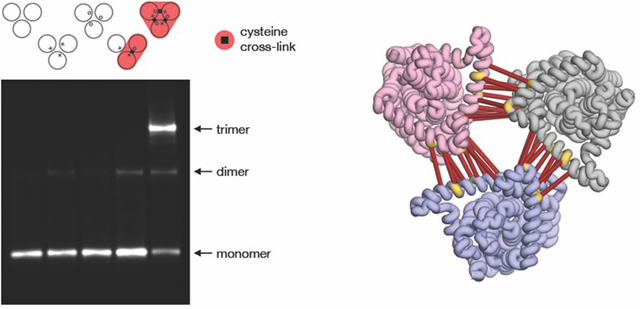
The size-exclusion chromatography profile of the sample showed that using a mild, nonionic detergent like DDM preserved CNT3 as an oligomer. The elution peak corresponded to the micelle size of approximately 300 kDa, as expected for a trimer/detergent micelle complex (Figure 1A). Further analysis by mass spectrometry validated the purity of the fractions as mostly containing hCNT3 (Figure S1B, Table S1). A Western blot of the sample indicated three potential species whose molecular weight was in agreement with the size of a CNT3 monomer, dimer, and trimer (Figure 1B). Nonspecific cross-linking of the detergent-solubilized sample using glutaraldehyde stabilized the highest molecular weight species (without cross-linking, any potential complex could dissociate during electrophoresis). This result is the first indication that hCNT3 forms a homo-trimer.
Figure 1.

Expression and purification of hCNT3 from High Five insect cells. A) Size exclusion chromatography (SEC) elution profile where the void peak is indicated with V0 and the collected fractions are represented by grey vertical lines. B) Western blot of the wild-type hCNT3 sample before and after glutaraldehyde cross-linking.
Comparative structure modeling and selection of sites for cysteine mutagenesis
To further examine the oligomerization of hCNT3, we turned to cysteine cross-linking. By introducing cysteines at key residue positions, we aimed to stabilize the complexes suggested in Figure 1B for further analysis. Cysteine cross-linking has been successfully used in previous studies of transporter oligomerization.16,30,33,34 First, we created a comparative structure model of the hCNT3 spanning the predicted TM4-TM11 helices, using the structure of vcCNT as a template (Figure 2A). Cysteine mutagenesis sites were then chosen based on the proximity of interface residues and consideration of disulfide geometry.35 Three separate sites along the trimerization interface, representing 4 separate pairs of cysteine mutants, were selected to determine whether or not the hCNT3 forms a trimer similar to that of vcCNT (Figure 2B).
Figure 2.

Comparative structure model of hCNT3 and the location of the mutated sites. A) A cartoon representation of the comparative structure model of hCNT3 based on the structure of vcCNT (PDB ID 3TIJ)(ref. 16). The trimerization domain is shown in green, scaffold domain in pink, and the inverted repeats performing the transport catalysis function in blue and yellow. B) Cartoon representation of the putative hCNT3 homo-trimer shown in the plane of the membrane. Two separate protomers are shown in blue and pink. Three mutagenesis sites are numbered and shown in yellow. C) Cartoon representation of the first mutagenesis site, where the side chains of V274C and A285C are shown as sticks. D) Cartoon representation of the second mutagenesis site, where the side chains of Y266C and F463C are shown as sticks. E) Cartoon representation of the third mutagenesis site, where the side chains of two separate pairs of mutants, T297C/A456C and A440C/A456C are shown as sticks.
The first selected site is in the extracellular “crown” region of hCNT3, at the junction of the predicted IH1, EH, and TM6 helices (Figure 2C). Both V274 and A285 are flanked in sequence by phenylalanine residues, which participate in the inter-and intra-protomer π-π stacking interactions in the vcCNT crystal structure. The second site is also extracellular, positioned at the predicted interface of IH1 and the C-terminal end of TM9 (Figure 2D). The third site residing in the predicted transmembrane region was probed with two independent pairs of cysteines (Figure 2E). The first pair, A440 and A452, is equivalent to the pair shown to cross-link in vcCNT (A253 and A269, respectively).16 Due to the symmetry of the trimer, it represents a double mutation on the predicted TM9, where the N-terminal A440 of one protomer intersects the centrally positioned A456 of another protomer. The second pair probes the junction of A456 with T297 located on TM6.
All residues selected for mutagenesis are conserved among the three human members of the SLC28 family, except for T297, which is a cysteine in both hCNT1 and hCNT2. Endogenous cysteines of hCNT3 are present only on the predicted helices TM1, IH3, TM10a, and TM11, and thus are located away from the trimerization region and expected not to interfere with the cross-linking. A series of constructs was created, containing either single or double cysteine mutants. As shown in Figure 3A using the 274/285 pair as an example, due to the homo-trimer symmetry, it is possible to cross-link the vcCNT-like trimer only by expressing the hCNT3 construct containing a double mutation or co-expressing two single mutant constructs in a single cell.
Figure 3.

Functional validation and cross-linking of hCNT3 cysteine mutants. A) Schematic of the experimental design. Mutations are shown as X and O. Sites of potential cross-linking are indicated with a red highlight and a thick black line. B) Uridine uptake inhibition studies in PK15NTD cells transiently transfected with hCNT3 constructs. C) Western blot of the cysteine cross-linking experiment involving V274C/A285C mutants, D) Y266C/F463C mutants, E) T297C/A456C mutants, and F) A440C/A456C mutants.
Validation of hCNT3 homo-trimer formation with cysteine cross-linking
To test whether or not the mutants fold properly and still traffic to the cell membrane, we transiently expressed the mutant constructs in the PK15NTD nucleoside transporter deficient porcine kidney cell line. This cell line was specifically chosen to ensure the lack of background concentrative nucleoside transporter expression. All mutants retained uridine transport function, though to variable extents (Figure 3B), and were thus suitable for cell-based cross-linking tests.
To preserve the potential hCNT3 homo-trimer, we performed the cross-linking assay in PK15NTD cells transiently transfected with the mutant constructs. As hypothesized, no hCNT3 complex survived the solubilization with Triton X-100 and electrophoresis, as demonstrated by a single band corresponding to the monomeric molecular weight of the wild type hCNT3. For the 274/285 site, when co-expressing both of the single mutants, we observed a band corresponding to the molecular weight of the dimer, in line with a prediction that two protomers in a trimer are able to cross-link (Figure 3C). The sharp band that appeared for the double mutant at the molecular weight of three monomers thus indicates that hCNT3 forms homo-trimers in cells.
Equivalent cross-linking studies for the second site, 266/463, were in agreement with the first site (Figure 3D). Co-expressing the single mutant constructs led to a sharp band corresponding to a dimeric species. Interestingly, there were two bands near the molecular weight of a homo-trimer in the double mutant. It is conceivable that there were two populations of homo-trimers, one with three cross-links and the other with two cross-links, each with a slightly different electrophoretic mobility.
The third site, containing two separate cysteine pairs, surprisingly did not lead to the cross-linking of the trimer (Figure 3E,F). Because the equivalent residues in vcCNT do cross-link,16 the absence of a cross-link in hCNT3 indicates a structural difference in the position and/or conformation of TM9 in vcCNT and hCNT3. In addition, the band corresponding to a dimer in the 440 single mutant suggests that the 440 residues were sufficiently close for disulfide bond formation. We repeated the experiment several times, including the replacement of the O2/1,10-phenanthroline with molecular iodine, which is often a more effective oxidation agent for cross-linking transmembrane regions (Figure S2),36 but with no change in results.
Discussion
The main goal of this study was to determine whether or not hCNT3, like its bacterial homologue vcCNT, forms homo-trimers. By constructing a comparative structure model, we leveraged structural information to introduce cysteine residues at key points along the hypothesized protomer-protomer interface. Two key results emerge from the study. First, hCNT3 has a similar fold to its homologue, vcCNT, and second, hCNT3 trimerizes, a finding based on in vitro and cell-based experiments (Figure 1 and 3). We now discuss two major points in turn. First, we compare the crystallographic structure of vcCNT with a structural model of hCNT3 based on the cross-linking data. Second, we discuss the functional implications of trimer formation in hCNT3.
Comparison between structural model of hCNT3 and vcCNT
Unlike chemical cross-linking using agents such as DSS or BMB, the formation of a disulfide bond between two residues on separate protomers requires close proximity and specific geometry.37 Thus, the method of cysteine cross-linking limits false-positives resulting from transient protein-protein interactions and provides residue-level insight into the configuration of the protomers in the trimer. Both selected extracellular mutated sites along IH1 led to a cross-linked trimer, validating the relative placement of helices IH1, EH, and TM6 in the hCNT3 comparative structure model. In addition, these cross-links indicate that the fold of hCNT3 is similar to that of its homolog vcCNT, because interactions between IH1 and TMs 5–8 are some of its defining features.16 The results are in agreement with and further complement the substituted cysteine accessibility method (SCAM) analysis of hCNT3, which was published during the preparation of this manuscript.38
Though the homologues share the fold, some differences are likely. These differences may include the placement and/or conformation of the TM9 helix, which contributes to the trimerization domain (defined in Figure 2A). TM9 is the longest helix in the bacterial structure, (63 Å) and spans almost the entire thickness of the plasma membrane. None of the cross-links probing the cytoplasm-facing N-terminus of TM9 (440/456 and 297/456) led to the stabilization of the trimer. Instead, a dimer band surprisingly appeared for the 440 single mutant. To explain this result, we hypothesize that the N-terminal residues 440 in the unknown structure of hCNT3 are closer to each other than in the current hCNT3 model (Cβ-Cβ distance of 22 Å), while residue 456 (TM9) is farther from both 297 (TM6) and 440 (TM9) (456, 297, and 440 are all in the trimerization domain) than in the current model (approximately 4 Å). This hypothesis is consistent with an elevator-like model,16,39 in which the trimerization domain forms a stable, largely immobile scaffold, maintaining the overall architecture of the trimer. The motions necessary for the transport cycle states are confined within the monomer to the transport domain helices that undergo conformational changes, thus leaving the trimerization domain static throughout the cycle. First proposed in the context of inverted-topology repeat identification and “repeat-swap modeling”,16,40 the crystal structures of the inward-facing, intermediate, and outward-facing states of a CNT from Neisseria wadsworthii (CNTNW) provide experimental evidence for this mechanism of transport.41
The GREMLIN score, based on an alignment 1629 sequences, shows that 8 and 14 of the 43 TM9 residues involved in respectively intra-and inter-protomer interactions co-vary (Figure 4A).42 Because co-varying pairs of residues tend to be close in space, it is often possible to refine a structural model by minimizing distances between co-varying pairs of residues.42 Combining the co-evolutionary data, the C-terminal TM9 266/463 and the TM9 440 cross-links, and the lack of cross-links for sites involving the N-terminal TM9 456 residue, has led us to propose that TM9 is rotated in the plane of the membrane in hCNT3 as compared to TM6 in vcCNT (Figure 4B). This parsimonious model satisfies the proximity requirements needed for the experimentally observed cross-link between A440C of adjacent protomers and the lack of a cross-link between the 440/456 and 297/456 residue pairs. The implications of these potential local structural differences are significant, because TM9 is predicted to interact directly with the nucleoside substrates. By creating a portion of the “thin” intracellular gate in the inward occluded conformation,39,43 even slight positional changes in TM9 could affect the binding site volume, the specificity, and kinetics of transport.
Figure 4.

Residue covariation between different protomers and a schematic of the proposed differences between hCNT3 and vcCNT. A top) Cartoon representation of the hCNT3 comparative structure models is shown from the extracellular side. Three protomers are shown in different colors. Residue pairs with a high GREMLIN covariation score across the dimerization interface are shown in thick red bars. Location of mutated cysteines is shown in yellow. A bottom) Cartoon representation of the hCNT3 comparative structure model is shown with a simulated membrane aligned from the MemProtMD entry 3TIJ (ref. 44). Residue pairs involving TM9 residues with a high GREMLIN covariation score are shown in red. Red bars indicate the location of the other intra-or inter-protomer residue in a covariation pair. Location of mutated cysteines is shown in yellow. Black lines are drawn between pairs of mutated cysteines. B) Cartoon representation of the comparative structure model of hCNT3 homo-trimer shown in the plane of the membrane, where TM9 on each of the protomers is highlighted. We propose a model where TM9 in the human structure is rotated in the plane of the membrane compared to the equivalent TM6 in vcCNT.
Functional implications of trimerization
Now that we have established that hCNT3 exists as a trimer, we address the question of its functional significance. The alignment of 883 sequences homologous to hCNT3 shows that the trimerization domain is as conserved in evolution as the inverted repeat domains, which perform the actual transport catalysis, indicating that the trimerization domain is indeed functionally important (Figure S3).45
As predicted by the structure of vcCNT, the translocation pathway of hCNT3 is confined to the monomer (i.e., each monomer contains an independent nucleoside and sodium binding sites). However, the possibility that trimerization may be needed for translocation, was considered. In particular, the helices in the trimerization and scaffold domains in the three protomers (Figure 2A) form one large aqueous basin within the membrane (Figure 5). This basin decreases the translocation distance for the ligand from approximately 40 Å to 25 Å.44 An aqueous basin of similar depth has been reported within the trimeric structure of GltPh, a eukaryotic glutamate transporter homologue from Pyrococcus horikoshii,46 that is not related in sequence or fold to CNTs. The quaternary structure of the trimer ensures that the translocation pathway is not exposed to the membrane while also minimizing the size of the hCNT3 monomer, compared to translocation in a monomeric transporter. On its own, without the other two protomers, and thus without the aqueous basin, each protomer is likely unstable in the membrane due to the exposure of hydrophilic regions (e.g., parts of TM9) to the lipid membrane. Based on these data, we hypothesize that hCNT3 is an obligate trimer, although we cannot rule out an engineered functional monomeric species; indeed, functional monomers of other SLC family members that appear to function as oligomers, such as OCT1 and BetP, have been engineered.22,47
Figure 5.

Cross-section of the hCNT3 comparative structure model. Three protomers are shown in different colors. Membrane is shown in yellow-green, colored according to local membrane deformations (MemProtMD)(ref. 44). Aqueous basin created in the center of the homo-trimer is shown in blue.
Symmetry is a common feature among protein complexes found in the PDB (approximately 85%).48 Potential benefits of symmetrical homo-complexes include increased coding efficiency, reduced aggregation, enhanced error control, and enhancement to sensitivity of selection; the reason is that symmetrical interfaces contain duplicates of pairwise contacts, and thus any favorable/unfavorable mutations are amplified.17,49–51 Although the sum of the three protomer-protomer interfaces is large (3 × 1250 Å2)52, it has been shown that only a small subset of residues, known as hotspots, contributes most of the free energy of binding.53 Because oligomerization has the potential to create new binding sites,17 it might be possible to disrupt the formation of the hCNT3 homo-trimer by targeting the dimeric interface. Such a binding site could be exploited for selectively modulating hCNT3, via allostery. Selective inhibition in particular might be appealing because hCNT3 substrate specificity overlaps with that of several other transporter families (e.g., ENTs).7 Potential applications involve cases such as chronic lymphocytic leukemia patients characterized by up-regulated cytoplasmic hCNT3 levels, who experience a lower response rate to fludarabine therapy. Targeting the cytoplasmic hCNT3 with allosteric inhibitors would result in destabilization of hCNT3 while leaving the required ENT-mediated drug uptake on the plasma membrane unaffected.8
In conclusion, we have shown that hCNT3 forms a homo-trimer in cells. By creating a comparative structure model, we leveraged structural data to introduce cysteines at various points along the dimerization interface. Cross-linking results indicate local structural differences between hCNT3 and vcCNT in TM9 of the trimerization domain. The quaternary structure creates an aqueous basin that significantly shortens the substrate translocation distance, but further exploration is needed to understand the full scope of its functional consequences.
Supplementary Material
Acknowledgements
We thank Dr. Robert M. Stroud and Dr. Thomas Tomasiak for sharing their facility and expertise concerning human SLC transporter expression and purification; Dr. Sook Wah Yee for valuable discussions regarding transporter uptake assays. The project was supported by grants from the National Institutes of Health (U19 GM61390 to K.M.G., R01 GM083960, P41 GM109824, P01 GM111126 to A. Sali, T32 GM007175 to A. Stecula), PhRMA Foundation Pre-Doctoral Fellowship in Pharmacology/Toxicology to A. Stecula, and APFE Pre-Doctoral Award in Pharmaceutical Sciences to A. Stecula.
Abbreviations
- CNT
concentrative nucleoside transporter
- ENT
equilibrative nucleoside transporter
- hCNT
human concentrative nucleoside transporter
- ENT
equilibrative nucleoside transporter
- SLC
solute carrier family of transport proteins
- TM
transmembrane helix
- IH
interfacial helix
- HP
helix–turn–helix hairpins
- EH
extracellular helix
References
- (1).Pastor-Anglada M, Cano-Soldado P, Errasti-Murugarren E, and Casado FJ (2008) SLC28 genes and concentrative nucleoside transporter (CNT) proteins. Xenobiotica 38, 972–994. [DOI] [PubMed] [Google Scholar]
- (2).Aymerich I, Duflot S, Fernández-Veledo S, Guillén-Gómez E, Huber-Ruano I, Casado FJ, and Pastor-Anglada M. (2005) The concentrative nucleoside transporter family (SLC28): new roles beyond salvage? Biochem. Soc. Trans 33, 216–219. [DOI] [PubMed] [Google Scholar]
- (3).Santos-Rodrigues A, Grañé-Boladeras N, Bicket A, and Coe IR (2014) Nucleoside transporters in the purinome. Neurochem. Int 73, 229–237. [DOI] [PubMed] [Google Scholar]
- (4).Young JD, Yao SYM, Baldwin JM, Cass CE, and Baldwin SA (2013) The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol. Aspects Med 34, 529–547. [DOI] [PubMed] [Google Scholar]
- (5).Hediger MA, Clémençon B, Burrier RE, and Bruford EA (2013) The ABCs of membrane transporters in health and disease (SLC series): introduction. Mol. Aspects Med 34, 95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ritzel MW, Ng AM, Yao SY, Graham K, Loewen SK, Smith KM, Ritzel RG, Mowles DA, Carpenter P, Chen XZ, Karpinski E, Hyde RJ, Baldwin SA, Cass CE, and Young JD (2001) Molecular identification and characterization of novel human and mouse concentrative Na+-nucleoside cotransporter proteins (hCNT3 and mCNT3) broadly selective for purine and pyrimidine nucleosides (system cib). J. Biol. Chem 276, 2914–2927. [DOI] [PubMed] [Google Scholar]
- (7).Errasti-Murugarren E, and Pastor-Anglada M. (2010) Drug transporter pharmacogenetics in nucleoside-based therapies. Pharmacogenomics 11, 809–841. [DOI] [PubMed] [Google Scholar]
- (8).Mackey JR, Galmarini CM, Graham KA, Joy AA, Delmer A, Dabbagh L, Glubrecht D, Jewell LD, Lai R, Lang T, Hanson J, Young JD, Merle-Béral H, Binet JL, Cass CE, and Dumontet C. (2005) Quantitative analysis of nucleoside transporter and metabolism gene expression in chronic lymphocytic leukemia (CLL): identification of fludarabine-sensitive and -insensitive populations. Blood 105, 767–774. [DOI] [PubMed] [Google Scholar]
- (9).Okazaki T, Javle M, Tanaka M, Abbruzzese JL, and Li D. (2010) Single nucleotide polymorphisms of gemcitabine metabolic genes and pancreatic cancer survival and drug toxicity. Clin. Cancer Res 16, 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wang J, and Giacomini KM (1997) Molecular determinants of substrate selectivity in Na+-dependent nucleoside transporters. J. Biol. Chem 272, 28845–28848. [DOI] [PubMed] [Google Scholar]
- (11).Slugoski MD, Smith KM, Mulinta R, Ng AML, Yao SYM, Morrison EL, Lee QOT, Zhang J, Karpinski E, Cass CE, Baldwin SA, and Young JD (2008) A conformationally mobile cysteine residue (Cys-561) modulates Na+ and H+ activation of human CNT3. J. Biol. Chem 283, 24922–24934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Slugoski MD, Smith KM, Ng AML, Yao SYM, Karpinski E, Cass CE, Baldwin SA, and Young JD (2009) Conserved glutamate residues Glu-343 and Glu-519 provide mechanistic insights into cation/nucleoside cotransport by human concentrative nucleoside transporter hCNT3. J. Biol. Chem 284, 17266–17280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Slugoski MD, Ng AML, Yao SYM, Lin CC, Mulinta R, Cass CE, Baldwin SA, and Young JD (2009) Substituted cysteine accessibility method analysis of human concentrative nucleoside transporter hCNT3 reveals a novel discontinuous region of functional importance within the CNT family motif (G/A)XKX3NEFVA(Y/M/F). J. Biol. Chem 284, 17281–17292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Slugoski MD, Ng AML, Yao SYM, Smith KM, Lin CC, Zhang J, Karpinski E, Cass CE, Baldwin SA, and Young JD (2008) A proton-mediated conformational shift identifies a mobile pore-lining cysteine residue (Cys-561) in human concentrative nucleoside transporter 3. J. Biol. Chem 283, 8496–8507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Slugoski MD, Loewen SK, Ng AML, Smith KM, Yao SYM, Karpinski E, Cass CE, Baldwin SA, and Young JD (2007) Specific mutations in transmembrane helix 8 of human concentrative Na+/nucleoside cotransporter hCNT1 affect permeant selectivity and cation coupling. Biochemistry 46, 1684–1693. [DOI] [PubMed] [Google Scholar]
- (16).Johnson ZL, Cheong C-G, and Lee S-Y (2012) Crystal structure of a concentrative nucleoside transporter from Vibrio cholerae at 2.4 Å. Nature 483, 489–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Forrest LR (2015) Structural symmetry in membrane proteins. Annu. Rev. Biophys 44, 311–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Veenhoff LM, Heuberger EHML, and Poolman B. (2002) Quaternary structure and function of transport proteins. Trends Biochem. Sci 27, 242–249. [DOI] [PubMed] [Google Scholar]
- (19).Perez C, Koshy C, Yildiz Ö, and Ziegler C. (2012) Alternating-access mechanism in conformationally asymmetric trimers of the betaine transporter BetP. Nature 490, 126–130. [DOI] [PubMed] [Google Scholar]
- (20).Perez C, Faust B, Mehdipour AR, Francesconi KA, Forrest LR, and Ziegler C. (2014) Substrate-bound outward-open state of the betaine transporter BetP provides insights into Na+ coupling. Nat. Commun 5, 4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Becker M, Maximov S, Becker M, Meyer U, Wittmann A, and Krämer R. (2014) Analysis of putative protomer crosstalk in the trimeric transporter BetP: The heterotrimer approach. Biochim. Biophys. Acta 1837, 888–898. [DOI] [PubMed] [Google Scholar]
- (22).Perez C, Khafizov K, Forrest LR, Krämer R, and Ziegler C. (2011) The role of trimerization in the osmoregulated betaine transporter BetP. EMBO Rep. 12, 804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kim J, Wu S, Tomasiak TM, Mergel C, Winter MB, Stiller SB, Robles-Colmanares Y, Stroud RM, Tampé R, Craik CS, and Cheng Y. (2015) Subnanometre-resolution electron cryomicroscopy structure of a heterodimeric ABC exporter. Nature 517, 396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Sali A, and Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol 234, 779–815. [DOI] [PubMed] [Google Scholar]
- (25).Pei J, Kim B-H, and Grishin NV (2008) PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 36, 2295–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Shen M-Y, and Sali A. (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Aran JM, and Plagemann PG (1992) High-affinity, equilibrative nucleoside transporter of pig kidney cell line (PK-15). Biochim. Biophys. Acta 1108, 67–74. [DOI] [PubMed] [Google Scholar]
- (28).Ward JL, Sherali A, Mo ZP, and Tse CM (2000) Kinetic and pharmacological properties of cloned human equilibrative nucleoside transporters, ENT1 and ENT2, stably expressed in nucleoside transporter-deficient PK15 cells. J. Biol. Chem 275, 8375–8381. [DOI] [PubMed] [Google Scholar]
- (29).Wang C, Pimple S, and Buolamwini JK (2011) Interaction of fused-pyrimidine nucleoside analogs with human concentrative nucleoside transporters: High-affinity inhibitors of human concentrative nucleoside transporter 1. Cell 81, 82–90. [DOI] [PubMed] [Google Scholar]
- (30).Hastrup H, Karlin A, and Javitch JA (2001) Symmetrical dimer of the human dopamine transporter revealed by cross-linking Cys-306 at the extracellular end of the sixth transmembrane segment. Proc. Natl. Acad. Sci. U. S. A 98, 10055–10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hoque KM, Chen L, Leung GPH, and Tse C-M (2008) A purine-selective nucleobase/nucleoside transporter in PK15NTD cells. Am. J. Physiol. Regul. Integr. Comp. Physiol 294, R1988–95. [DOI] [PubMed] [Google Scholar]
- (32).Mangravite LM, and Giacomini KM (2003) Sorting of rat SPNT in renal epithelium is independent of n-glycosylation. Pharm. Res 20, 319–323. [DOI] [PubMed] [Google Scholar]
- (33).Keller T, Egenberger B, Gorboulev V, Bernhard F, Uzelac Z, Gorbunov D, Wirth C, Koppatz S, Dötsch V, Hunte C, Sitte HH, and Koepsell H. (2011) The large extracellular loop of organic cation transporter 1 influences substrate affinity and is pivotal for oligomerization. J. Biol. Chem 286, 37874–37886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hughson AG, Lee GF, and Hazelbauer GL (1997) Analysis of protein structure in intact cells: Crosslinking in vivo between introduced cysteines in the transmembrane domain of a bacterial chemoreceptor. Protein Sci. 6, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Careaga CL, and Falke JJ (1992) Thermal motions of surface alpha-helices in the D-galactose chemosensory receptor. Detection by disulfide trapping. J. Mol. Biol 226, 1219–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bass RB, Butler SL, Chervitz SA, Gloor SL, and Falke JJ (2007) Use of site-directed cysteine and disulfide chemistry to probe protein structure and dynamics: applications to soluble and transmembrane receptors of bacterial chemotaxis. Methods Enzymol. 423, 25–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Careaga CL, and Falke JJ (1992) Structure and dynamics of Escherichia coli chemosensory receptors. Engineered sulfhydryl studies. Biophys. J 62, 209–16– discussion 217–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Mulinta R, Yao SYM, Ng AML, Cass CE, and Young JD (2017) Substituted cysteine accessibility method (SCAM) analysis of the transport domain of human concentrative nucleoside transporter 3 (hCNT3) and other family members reveals features of structural and functional importance. J. Biol. Chem jbc.M116.743997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Johnson ZL, Lee J-H, Lee K, Lee M, Kwon D-Y, Hong J, and Lee S-Y (2014) Structural basis of nucleoside and nucleoside drug selectivity by concentrative nucleoside transporters. Elife e03604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Vergara-Jaque A, Fenollar-Ferrer C, Kaufmann D, and Forrest LR (2015) Repeat-swap homology modeling of secondary active transporters: updated protocol and prediction of elevator-type mechanisms. Front. Pharmacol 6, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Hirschi M, Johnson ZL, and Lee S-Y (2017) Visualizing multistep elevator-like transitions of a nucleoside transporter. Nature 545, 66–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ovchinnikov S, Kamisetty H, and Baker D. (2014) Robust and accurate prediction of residue-residue interactions across protein interfaces using evolutionary information. Elife 3, e02030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Krishnamurthy H, Piscitelli CL, and Gouaux E. (2009) Unlocking the molecular secrets of sodium-coupled transporters. Nature 459, 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Stansfeld PJ, Goose JE, Caffrey M, Carpenter EP, Parker JL, Newstead S, and Sansom MSP (2015) MemProtMD: automated insertion of membrane protein structures into explicit lipid membranes. Structure 23, 1350–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Capra JA, and Singh M. (2007) Predicting functionally important residues from sequence conservation. Bioinformatics 23, 1875–1882. [DOI] [PubMed] [Google Scholar]
- (46).Yernool D, Boudker O, Jin Y, and Gouaux E. (2004) Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 431, 811–818. [DOI] [PubMed] [Google Scholar]
- (47).Keller T, Egenberger B, Gorboulev V, Bernhard F, Uzelac Z, Gorbunov D, Wirth C, Koppatz S, Dötsch V, Hunte C, Sitte HH, and Koepsell H. (2011) The large extracellular loop of organic cation transporter 1 influences substrate affinity and is pivotal for oligomerization. J. Biol. Chem 286, 37874–37886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Levy ED, Pereira-Leal JB, Chothia C, and Teichmann SA (2006) 3D complex: a structural classification of protein complexes. PLoS Comput. Biol 2, e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).André I, Strauss CEM, Kaplan DB, Bradley P, and Baker D. (2008) Emergence of symmetry in homooligomeric biological assemblies. Proc. Natl. Acad. Sci. U. S. A 105, 16148–16152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Goodsell DS, and Olson AJ (2000) Structural symmetry and protein function. Annu. Rev. Biophys. Biomol. Struct 29, 105–153. [DOI] [PubMed] [Google Scholar]
- (51).Monod J, Wyman J, and Changeux J-P (1965) On the nature of allosteric transitions: a plausible model. J. Mol. Biol 12, 88–118. [DOI] [PubMed] [Google Scholar]
- (52).Vangone A, Spinelli R, Scarano V, Cavallo L, and Oliva R. (2011) COCOMAPS: a web application to analyze and visualize contacts at the interface of biomolecular complexes. Bioinformatics 27, 2915–2916. [DOI] [PubMed] [Google Scholar]
- (53).Wells JA, and McClendon CL (2007) Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature 450, 1001–1009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.