

Abstract
Following the discovery of numerous long non-coding RNA (lncRNA) transcripts in the human genome, their important roles in biology and human disease are emerging. Recent progress in experimental methods has enabled the identification of structural features of lncRNAs. However, determining high-resolution structures is challenging as lncRNAs are expected to be dynamic and adopt multiple conformations, which may be modulated by interaction with protein binding partners. The X-inactive specific transcript (Xist) is necessary for X inactivation during dosage compensation in female placental mammals and one of the best-studied lncRNAs. Recent progress has provided new insights into the domain organization, molecular features, and RNA binding proteins that interact with distinct regions of Xist. The A-repeats located at the 5′ end of the transcript are of particular interest as they are essential for mediating silencing of the inactive X chromosome. Here, we discuss recent progress with elucidating structural features of the Xist lncRNA, focusing on the A-repeats. We discuss the experimental and computational approaches employed that have led to distinct structural models, likely reflecting the intrinsic dynamics of this RNA. The presence of multiple dynamic conformations may also play an important role in the formation of the associated RNPs, thus influencing the molecular mechanism underlying the biological function of the Xist A-repeats. We propose that integrative approaches that combine biochemical experiments and high-resolution structural biology in vitro with chemical probing and functional studies in vivo are required to unravel the molecular mechanisms of lncRNAs.
Keywords: lncRNA, structural biology, Xist, chemical probing, enzymatic footprinting, computational structure prediction
Introduction
Despite their low primary sequence conservation (Eddy, 2014) and abundance levels (Cabili et al., 2015), long non-coding RNAs (lncRNAs; non-protein encoding transcripts >200 nucleotides) are involved in a wide repertoire of biological processes that have been extensively reviewed (Ponting et al., 2009; Wapinski and Chang, 2011; Kung et al., 2013; Ulitsky and Bartel, 2013; Guo et al., 2016; Marchese et al., 2017). These processes include (i) interacting with chromatin complexes, (ii) serving as modulators of protein and enzyme cofactors, (iii) binding DNA/RNA-binding proteins to regulate transcriptional expression, (iv) regulating DNA stability through R-loop and triple helix formation, and (v) forming higher-order structure for purposes such as X-chromosome inactivation (XCI). It is expected that the function of an RNA is closely linked to its structural features in a fashion similar to the relationship between function and structure in proteins. This link between structure and function is what also enables the exploration of lncRNAs as potential therapeutic targets. Despite the promising prospect of developing lncRNA-targeted therapy techniques, <20 of the near 30000 existing (GENCODE v29) lncRNAs in humans have been studied on a structural level (Table 1). This is primarily due to the large size and dynamic conformations of lncRNAs, free and in complex with their cognate RNA binding proteins, which render common high-resolution techniques, such as nuclear magnetic resonance (NMR) and X-ray crystallography, challenging. Over the last decade, advancements in chemical probing techniques such as selective 2’ hydroxyl acylation analyzed by primer extension (SHAPE), the re-emergence of enzymatic footprinting and psoralen crosslinking approaches, and comparative sequence analysis have made it possible to explore the secondary structural motifs formed by lncRNAs both in vitro and in vivo. The structures of lncRNAs and methods used to determine them have been recently reviewed (Novikova et al., 2013b, c; Pintacuda et al., 2017b; Zampetaki et al., 2018).
Table 1.
Secondary structures of lncRNAs (supported by experimental data).
| Length | Method used to determine structure | Function | References | |
|---|---|---|---|---|
|
Steroid receptor RNA
activator (SRA) |
0.86 kb | SHAPE, enzymatic, and inline probing |
Coactivation of steroid nuclear
receptors |
Novikova et al. (2012b) |
| X-inactive specific transcript (Xist) | 17 kb | SHAPE, Targeted Structure-Seq, enzymatic, and inline probing |
X-chromosomal inactivation | Wutz et al. (2002); Maenner et al. (2010); Duszczyk et al. (2011); Fang et al. (2015); Cirillo et al. (2016); Lu et al. (2016); Smola et al. (2016a); Liu et al. (2017a) |
| Hox antisense intergenic RNA (HOTAIR) | 2.2 kb | SHAPE, dimethyl sulfate (DMS), and terbium structure probing in parallel with phylogenetic determination |
Protein ubiquitination | Somarowthu et al. (2015) |
| Metastasis associated lung adenocarcinoma transcript 1 (MALAT1) | 8 kb | SHAPE, chemical, and inline probing | Nuclear speckle formation | Zhang et al. (2017) |
| Nuclear enriched abundant transcript 1 (NEAT1) | 3.2 kb | SHAPE | A scaffolding factor for nuclear paraspeckle formation |
Lin et al. (2018) |
| RNA on the X1 and X2 (ROX1/ROX2) | 3.8 kb; 0.6 kb | SHAPE and parallel analysis of RNA structure (PARS) analysis |
Dosage compensation | Ilik et al. (2013) |
| Braveheart | 0.6 kb | SHAPE and inline probing | Regulation of cardiovascular lineage commitment |
Xue et al. (2016) |
| COOLAIR | 0.4 kb; 0.7 kb | SHAPE and inline probing | Downregulation of FLC flowering and expression |
Lee et al. (2017b) |
| SPRIGHTLY | 0.62 kb | SHAPE-seq | Intranuclear organization of pre-mRNA molecules |
Hawkes et al. (2016) |
| SRA-like non-coding RNA | 0.4 kb; 0.7 kb | SHAPE | Transcriptional repression of Sox9b |
Schmidt et al. (2016) |
| P21 | 3 kb | SHAPE | Regulation of p53-mediated stress response |
Chillón and Pyle (2016) |
|
Maternally expressed
gene 3 |
1.7 kb | mFold and deletion analysis | Transcriptional gene regulation of the transforming growth factor β pathway |
Zhang et al. (2010) |
|
Polyadenylated nuclear
RNA |
1.1 kb | SHAPE-MaP | Expression suppression of host genes involved in the antiviral response |
Sztuba-Solinska et al. (2017) |
Two important features of RNA molecules are their intrinsic flexibility and propensity for adopting multiple conformations. RNA molecules often sample multiple dynamic conformations and are best represented as an ensemble of structures (Dethoff et al., 2012). This renders structural studies challenging and requires that a combination of techniques be used to define the dynamic conformational space of lncRNAs. This problem is illustrated by the observation that eight major, distinct and incompatible secondary structural models have been proposed for the A-repeat section of the lncRNA X-inactive specific transcript (Xist) by researchers using different approaches. It is thus important to consider and experimentally address the possibility of multiple conformations of lncRNAs to understand the structural mechanisms underlying their biological function. In this review, we illustrate the challenges associated with understanding the structure and dynamics of lncRNAs with the example of the Xist A-repeats. We discuss the methodology used and secondary structures proposed for the Xist A-repeats, the cell-based and in vitro assays that have been used to characterize them, and the impact that protein binding partners may have on the structure and dynamics of this essential transcript. We propose that the combination of various complementary techniques will be important to overcome the difficulties in studying Xist and other lncRNAs and map their dynamic conformational landscape.
The A-repeat region of the lncRNA Xist
Xist, one of the most well-studied lncRNAs, is a 17 kb transcript responsible for dosage compensation in placental mammals; during early development, Xist coats the inactive X chromosome and represses transcription in a process known as XCI (Brown, 1992; Penny et al., 1996; Lucchesi et al., 2005). This occurs with the aid of several protein binding events that take place along the length of the transcript, and which have been well investigated and reviewed (Hasegawa et al., 2010; Chu et al., 2015; McHugh et al., 2015; Moindrot and Brockdorff, 2016). Xist comprises six interspersed repeat regions: A, B, C, D, E, and F (Brockdorff, 1992) (Figure 1A). Chromosomal silencing and localization are mediated by different domains of the Xist transcript. The A-repeats, which are located at the 5′ end of the transcript, are required for transcriptional silencing. In their absence, coating of the inactive X chromosome occurs; however, the effect of silencing is abolished (Wutz et al., 2002). The A-repeat region is also transcribed as a separate transcript, called Rep A (Zhao et al., 2008). Because the A-repeats serve such a vital role in XCI, much research has been done to determine the molecular mechanism underlying their role in silencing both through protein binding interactions and secondary structure.
Figure 1.
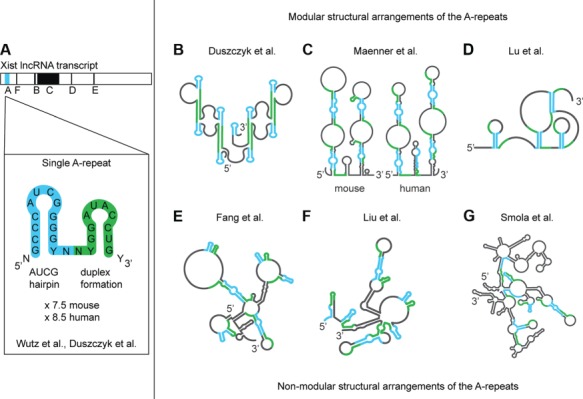
Structural arrangements of the Xist A-repeats. (A) The Xist A-repeats are located on the 5′ end of the Xist transcript. Each repeat (7.5 in mouse and 8.5 in human) is separated by a U/A-rich linker. Wutz et al. (2002) first predicted that each A-repeat formed two stable hairpins using free energy minimization; however, Duszczyk et al. (2011) showed that only the AUCG hairpin is stable while the latter drives duplex formation. (B–G) Distinct structural models for the A-repeats. Modular arrangements (where the A-repeats assemble in a modular fashion by inter-repeat duplex formation) (B–D) and non-modular arrangements (where the A-repeats are base paired in a variety of ways) (E–G) of the A-repeats. (B) Model based on NMR analysis of single and tandem repeats in vitro (see Figure 2). (C) Mouse (left) and human (right) models based on in vitro experiments: enzymatic cleavage (V1, T1, T2), chemical probing (DMS, CMCT), FRET, and comparative sequence analysis. (D) Mouse in vivo: icSHAPE (NAI-N3), PARIS (AMT), and comparative sequence analysis. (E) Mouse in vivo: Targeted Structure-Seq (DMS) and comparative sequence analysis. (F) Mouse in vitro: chemical probing using DMS and SHAPE (1 M7) and comparative sequence analysis. (G) Mouse in vivo: SHAPE-MaP (1M6, 1M7, NMIA).
The Xist A-repeats consist of a highly conserved 26-nucleotide region that is repeated 8.5 times in human and 7.5 times in mouse. The repeats are separated by uracil/adenosine-rich linkers, which lack primary sequence conservation (Wutz et al., 2002). The A-repeats are believed to regulate transcriptional repression through an interaction with several proteins, including SHARP (Chu et al., 2015; McHugh et al., 2015; Monfort et al., 2015), RBM15 and RBM15B (Moindrot et al., 2015), WTAP (Moindrot et al., 2015), YTHDC1 (Patil et al., 2016), PRC2 (Zhao et al., 2008; Kanhere et al., 2010; Cerase et al., 2014; McHugh et al., 2015; Almeida et al., 2017), PTB (Maenner et al., 2010), HuR (Smola et al., 2016a), FUS (Smola et al., 2016b), ASF/SF2 (Royce-Tolland et al., 2010), LBR (Chen et al., 2016), ATRX (Sarma et al., 2015), and Rnf20 (Chu et al., 2015) (see below). The secondary structure of the A-repeats has been extensively investigated, but remains unresolved, as at least eight different structural models have been proposed with additional alternative models that have similar minimum free energies (Wutz et al., 2002; Duszczyk et al., 2008, 2011; Maenner et al., 2010; Fang et al., 2015; Delli Ponti et al., 2016; Lu et al., 2016; Smola et al., 2016a; Liu et al., 2017a; Rivas et al., 2017) (Figure 1). The structural models of the A-repeats have been recently reviewed by Pintacuda et al. (2017b). In the following, we first discuss the methods that are available to assess the secondary structures of RNA, and then discuss how different combinations of these methods were used to derive models of the A-repeat secondary structure.
Experimental and computational methods for RNA structure prediction and probing
A range of methods have been developed in recent years to evaluate the secondary structure of RNA. These include, but are not limited to, enzymatic footprinting, chemical probing, NMR, and comparative sequence analysis (Table 2). The information obtained by these methods can be incorporated as restraints for secondary structure predictions (Lorenz et al., 2016). In this section, we briefly describe each method and some of the limitations associated with them.
Table 2.
Experimental and computational methods to probe and predict RNA structure.
| Tools for determining secondary structure of RNAs* | Description of target | |
|---|---|---|
| Enzymatic cleavage, footprinting | RNAse A (in vitro) | Cleaves 3′ of single stranded C, U |
| RNAse T1 (in vitro) | Cleaves 3′ of single stranded G | |
| RNAse T2 (in vitro) | Cleaves 3′ of single stranded N (preference for A) | |
| S1 nuclease (in vitro) | Cleaves all single stranded nucleotides | |
| RNAse V1 (in vitro) | Cleaves double stranded RNA | |
| Hydroxyl radicals (Fe (II)-EDTA, H2O2) (in vitro) | Degradation of ribose backbone based on solvent accessibility of phosphodiester bonds | |
| In-line probing (in vitro) (Pb2+) | RNA allowed to degrade over time, single-stranded regions typically degrade before structured regions | |
| Chemical probing | Kethoxal (in vitro) | Modifies single stranded G (N1) |
| DEPC (in vitro) | Modifies single stranded A (N7) | |
| CMCT (in vitro) | Acylation of single stranded U (N3) and G (N1) | |
| DMS (in vitro, in vivo) | Methylation of single stranded A, C (N1, N3) | |
| SHAPE (in vitro, in vivo) | Acylation of flexible 2′OH ribose groups, performed with several different molecules such as 1M7 | |
| PARIS (in vitro, in vivo) | Reversible crosslinking of base paired nucleotides using AMT | |
| Computation | R-scape | Comparative sequence analysis |
| WAR | Comparative sequence analysis |
*Stern et al. (1988); Ziehler and Engelke (2001); Torarinsson and Lindgreen (2008); Weeks (2010); Kwok (2016); Rivas et al. (2017).
DEPC, diethyl pyrocarbonate; CMCT, 1-cyclohexyl-3-(2-morpholinoethyl) carbodiimide metho-p-toluenesulfonate; SHAPE, selective 2′ hydroxyl acylation analyzed by primer extension; PARIS, psoralen analysis of RNA interactions and structures; R-scape, RNA structural covariation above phylogenetic expectation; WAR, webserver for aligning structural RNAs; 1M7, 1-methyl-7-nitroisatoic anhydride; AMT, 4′-aminomethyltrioxsalen.
Enzymatic footprinting is an in vitro approach performed by treating a radioisotope-labeled RNA with an enzyme that recognizes and cleaves either single- or double-stranded nucleotides. There are several enzymes that each have specific targets (Table 2). The fragmented RNA products are typically run alongside a sequencing or an alkaline hydrolysis RNA ladder on a denaturing polyacrylamide gel. Next generation sequencing can also be used to evaluate RNA fragments in a method known as PARS, allowing for evaluation of up to 3000 transcripts in a single experiment (Kertesz et al., 2010). The biggest limitation of enzymatic footprinting is that it cannot be used in vivo, a state that is known to influence secondary structures of RNA (Leamy et al., 2016).
Chemical probing makes use of small reactive molecules that chemically modify atoms of single stranded or flexible nucleotides (Table 2). The versatility of available chemical probes allows this approach to be performed both in vitro and in vivo. For the latter, RNA structure should reflect its native state, as it is probed in the cellular context and in the presence of protein binding partners, followed by extraction of the RNA from cells.
SHAPE] is a more recently introduced versatile chemical probing method that can be used to indiscriminately identify flexible nucleotides in RNA transcripts both in vitro and in vivo (Wilkinson et al., 2006; Spitale et al., 2012). The C2’ hydroxyl group of a ribose that is exposed and/or flexible undergoes acylation by a SHAPE reagent (McGinnis et al., 2012; Mlynsky and Bussi, 2018). There are several reagents that can be used with SHAPE, including BzCN (benzoyl cyanide), 1M7 (1-methyl-7-nitro-isatoic anhydride), 1M6 (1-methyl-6-nitroisatoic anhydride), NMIA (N-methyl isatoic anhydride), NAI (2-methylnicotinic acid imidazolide), FAI (2-methyl-3-furoic acid imidazolide), and NAI-N3 (2-methylnicotinic acid imidazolide-azide) (Weeks and Mauger, 2011; Kwok, 2016; Lee et al., 2017a).
After treatment with a chemical probe, RNA is then reverse transcribed; depending on which reverse transcriptase is used, the modified nucleotides will either cause a mutation of the corresponding nucleotides in the final, full-length cDNA, or will halt elongation, resulting in a series of cDNA fragments. Much like enzymatic footprinting, the cDNA can be analyzed by denaturing polyacrylamide gel electrophoresis or capillary electrophoresis. Approximately 20 methods have been developed involving the evaluation of the cDNA using next generation sequencing, including SHAPE-MaP, Targeted Structure-Seq, and icSHAPE. These methods, among several others, have been recently reviewed (Kwok, 2016).
Incorporation of enzymatic footprinting and chemical probing data as restraints during folding can drastically reduce the number of possible structures and the amount of time needed to generate a secondary structure (Low and Weeks, 2010). Using a combinatorial approach provides a greater abundance of restraints that can be used to guide RNA folding programs like ‘MFOLD’ and ‘RNAstructure’ (Zuker, 2003; Reuter and Mathews, 2010). Most importantly, a combinatorial approach addresses the ambiguities that can arise due to experimental limitations. In the case of SHAPE, one such limitation involves the tendency of riboses to exhibit SHAPE reactivity when they are, in fact, not flexible (false positive), as well as the tendency of flexible riboses to evince a low or intermediate reactivity profile (false negative) (McGinnis et al., 2012; Kenyon et al., 2014). Noise and uncertainties in data and their analysis affect the accuracy of predicted RNA folds. False positives can be caused by several factors. These include (i) the effect of dimethyl sulfoxide (DMSO) (required to dissolve SHAPE reagents), which is known to cause denaturation and destabilization of RNA secondary structure in concentrations as low as 5% (Strauss et al., 1968; Lee et al., 2013), (ii) the dynamic and flexible nature of RNA, which can result in base paired nucleotides that are near bulges and loops showing higher reactivity than expected, and (iii) the mechanism of SHAPE chemical probes; some have a propensity to be reactive toward some nucleotides more than others; purines are typically 1.5 times more reactive than pyrimidines (McGinnis et al., 2012). Thus, complementing SHAPE with other structural probing techniques can clarify ambiguous SHAPE data, and vice versa.
Arguably, the largest drawback to enzymatic footprinting and chemical probing techniques is that they fail to identify specific base pairs, which can lead to incorrectly predicted secondary structures. The factors that can contribute to an inaccuracy of SHAPE-predicted helices have been investigated by both the Das (Kladwang et al., 2011) and Weeks groups (Leonard et al., 2013). Enzymatic or chemical probing assesses the flexibility or accessibility of nucleotides, thereby suggesting the presence flexible single- vs. more rigid double-stranded regions. Though this information can be quite useful for predicting secondary structures of short RNAs, longer RNAs, which have a tendency to fold both locally (base pairs are formed by nucleotides close in primary sequence) and globally (long distance base pairs), remain a challenge. Moreover, internal dynamics and flexibility of a structured region will also render it more reactive, and thus an unambiguous identification of single-stranded and structured RNA regions is difficult. The shotgun secondary structure (3S) method was recently developed by the Sanbonmatsu group to differentiate between local and global folding arrangements and involves performing chemical probing or enzymatic cleavage on subfragments of an RNA, and comparing the reactivity profiles to that of the full-length RNA transcript (Novikova et al., 2013a). Good agreement between a subfragment and its corresponding sequence in the full-length transcript supports local folding, whereas disagreements in data suggest more long-distance interactions.
NMR spectroscopy is a versatile biophysical and structural biology technique that can readily probe base pairing and secondary structure in structured regions of an RNA. It is also a powerful high-resolution solution method for high-resolution analysis of the structure and dynamics of RNAs (Varani et al., 1996; Mollova and Pardi, 2000; Fürtig et al., 2003; Al-Hashimi, 2013; Xue et al., 2015). The imino region of one-dimensional 1H and two-dimensional 1H-1H NOESY NMR experiments readily reveals base-paired nucleotides. The advantage of using NMR is that it can reveal specific base pairs and it is performed in solution, yielding information that can be used to predict RNA secondary structure (Chen et al., 2015). A limitation for using NMR is the molecular weight of the RNA. Spectral overlap and line broadening complicates the analysis of RNAs with >80–100 nucleotides, unless specific methods are used to focus on specific regions (Lukavsky and Puglisi, 2005; Lu et al., 2010; Barnwal et al., 2017).
Comparative sequence analysis (also referred to as phylogenetic analysis) is used to detect compensatory mutations, which allow an RNA to maintain its structure and function, despite evolutionary divergence in primary sequence (Chen et al., 1999; Parsch et al., 2000; Eddy, 2014). Comparative sequence analysis programs generate covariance models by aligning RNA sequences based on sequence conservation and single-sequence structure prediction. As a result, covarying base pairs (where a base pair switches from G-C to A-U, for example) and consistent mutations (where only one nucleotide changes, but base pairing is maintained: e.g., a G-C to a G-U base pair) are identified and a covariance model can be generated.
Secondary structure models of the Xist A-repeat RNA
Given the experimental uncertainties and often ambiguous information associated with the various methods to probe RNA secondary structure, it is not surprising that incongruent structural models are frequently proposed. A notable case is that of the HIV-1 encapsidation signal RNA, where in-gel SHAPE probing, supported by high resolution NMR analysis, culled the disparate proposed secondary structures by revealing that this RNA samples two conformations (Kenyon et al., 2013; Keane et al., 2015). All of the SHAPE structure-probing datasets published before had (unknowingly) studied a mixture of the two states, thus leading to conflicting and incorrect structural models. It is not unlikely that lncRNAs, including the Xist A-repeats, similar to the HIV-1 encapsidation signal RNA, sample multiple conformations. This possibility has to be considered in structural analysis. In the following, we describe the various structural models that have been proposed for the Xist A-repeats.
Wutz et al. (2002) were the first to propose a secondary structure for a single Xist A-repeat using a computational structure prediction algorithm. Their model suggested that a 26-mer repeat folds into two short stem loops, each adopting a hairpin conformation (Figure 1A). Duszczyk et al. (2008, 2011) have reported a high-resolution structure of the first hairpin, which was found to adopt a thermodynamically stable stem-loop (Figure 2). They also found that the second predicted hairpin adopts a dimeric duplex conformation, suggesting that this may be a building principle of higher order assemblies of the complete A-repeat region (Duszczyk et al., 2008, 2011). Since then, eight secondary structural models (based on various in vitro or in vivo approaches) (Figure 1) have been proposed. The proposed structures can be divided into two categories: modular assembly of individual repeats and non-modular structures with overall tertiary folds (Figure 1B–G).
Figure 2.
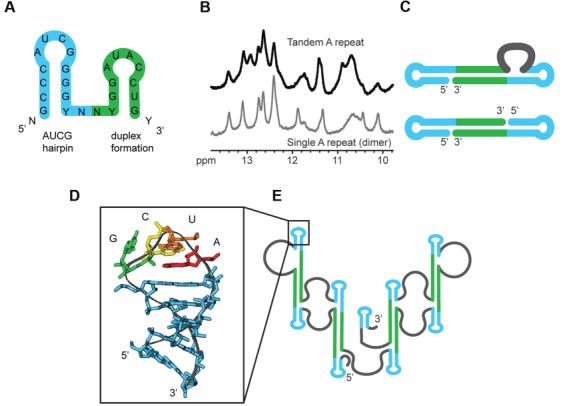
(A) A single 26-nucleotide A-repeat region comprising two predicted hairpins. (B and C) Similarity of one-dimensional imino spectra of the dimeric single and tandem A-repeats suggests the formation of inter-repeat dimers involving the theoretical ‘hairpin 2’. (D and E) NMR structure of the stable AUCG hairpin 1 suggests this a basic folding unit of the complete A-repeat (Duszczyk et al., 2008, 2011).
Modular assembly of the A-repeats
Duszczyk et al. (2008, 2011), Maenner et al. (2010), and Lu et al. (2016) (Figure 1B–D) have proposed secondary structural models for the assembly of the A-repeats, characterized by the modular arrangement of individual repeat elements. In the Duszczyk model, hairpin 1 of each repeat adopts a stable AUCG tetraloop fold. Modular assembly of the hairpin 1 fold in different repeats is established by inter-repeat duplex formation involving the region of the predicted hairpin 2 (Figure 2). The duplex formation involving two repeats then occurs four times along in the complete A-repeat RNA fold (Figure 1B). In the Maenner model, none of the predicted hairpins in the individual repeats are formed and instead the A-repeats are involved in inter-repeat duplexes forming overall extended stem-loop structures (Figure 1C). The third model, from Lu, is a hybrid of the first two duplex types: the major hairpins are extended and form a duplex with subsequent major hairpins, while the minor hairpin is single stranded, driving formation of both internal and terminal loops (Figure 1D).
Non-modular assembly of the A-repeats
Non-modular assembly has been proposed for the Xist A-repeats by Fang et al. (2015) (later supported by Delli Ponti et al., 2016), Liu et al. (2017a), and Smola et al. (2016a) (Figure 1E–G). These secondary structures possess a variety of distinct and non-modular structural arrangements of the A-repeats across the length of the transcript, including duplex formation and major and/or minor hairpin formation. Unlike all other previously reported structures, Smola et al. (2016a) proposed that the A-repeats interact with other regions of the Xist transcript, instead of folding independently. However, both Maenner et al. (2010) and the Pyle lab have provided experimental evidence that suggests that the A-repeat region is, indeed, an individual folding unit, consistent with the fact that functional Rep A transcripts exist, which comprise mainly the A-repeat regions (Zhao et al., 2008).
Addressing the limitations of methodology used to predict lncRNA secondary structure
Whether the Xist A-repeats adopt a non-modular or modular higher-order assembly is currently unknown. However, despite the disparity in proposed secondary structures, there is a consensus that the Xist A-repeats are highly structured. The propensity of the Xist A-repeats to form secondary structure was also determined computationally by both Fang et al. (2015) and Delli Ponti et al. (2016) Fang used thermodynamic z-scores to evaluate the secondary structure; Xist A-repeats were scanned in 150-nucleotide sliding windows and the folding free energy was predicted relative to a randomized version of the same window sequence. The Xist A-repeats had a lower minimal free energy of folding compared to a randomized Xist A-repeat sequence, and are therefore structured (Mathews et al., 1999; Fang et al., 2015). Delli Ponti developed and used an artificial neural network (CROSS: computational recognition of secondary structure), which was trained by SHAPE, PARS, and NMR structural restraints, to evaluate the propensity of the Xist A-repeats to form secondary structure. They concluded that where there is high primary sequence conservation (as occurs with the A-repeats), there is a strong tendency for secondary structure (Delli Ponti et al., 2016).
What is most interesting about these disparate secondary structures is that there are remarkable similarities in the experimental data, from which they are derived, despite the fact that different specific methods have been used. What, then, leads to the observed disparities in the structural models derived? The presence of multiple conformations will affect experiments that probe RNA secondary structure and render computational structure predication challenging. Also, experimental imperfections and limitations of structure prediction may contribute to distinct structural models.
Duszczyk et al. (2008, 2011), Maenner et al. (2010) and Liu et al. (2017a) used in vitro experimental approaches to propose secondary structures for the Xist A-repeats. Duszczyk determined the high-resolution structure of the major hairpin of a Xist A-repeat in solution (Figure 2) using NMR. Duszczyk et al. (2008, 2011) found that the NMR imino fingerprint of a dimeric single A-repeat and a single-chain tandem A-repeat (both ∼50 nucleotides long) are highly similar, indicating comparable base pairing and structural features. This suggests a higher modular arrangement of A-repeats (Figure 1B), although experimental evidence for the modularity has yet to be demonstrated for the complete A-repeat RNA.
Both Maenner et al. (2010) and Liu et al. (2017a) used enzymatic footprinting and/or chemical probing to obtain folding restraints that were used by Mfold and RNAstructure, respectively, to generate secondary structures for the A-repeats. Specifically, Maenner et al. used enzymatic footprinting (with RNAses T1, T2, and V1) and chemical probing (with small molecules DMS and CMCT) to propose three different models of the A-repeats for both mouse and human. Additional distance measurements based on fluorescence resonance energy transfer (FRET) and compatibility of binding with the polycomb repressive complex 2 (PRC2, see below) led them to propose one of three possible folds as a model of the A-repeat region. Liu et al. (2017a) on the other hand, used SHAPE and DMS chemical probing to generate their structural model. In their study, emphasis was placed on using non-denaturing purification techniques (Chillón et al., 2015) to ensure a monomeric, homogenous arrangement of the Xist Rep A lncRNA (Rep A includes both the Xist A and F repeats, but the authors showed that the A-repeat structure is not affected by the F repeats). Liu varied the magnesium concentrations of the buffer solution of Rep A RNA and performed size exclusion chromatography (SEC) and sedimentation velocity (SV) experiments on each sample. Evaluation of the SV-analytical ultracentrifugation profiles (SV-AUC) was performed to evaluate transcript compactness; it was determined that 15 mM magnesium chloride is sufficient for obtaining homogenous compaction of monomeric Rep A RNA, and that under these conditions, the RNA most likely reflects folding under native, cellular conditions. The buffer conditions used by Liu et al. (2017a), and those in studies of the A-repeats by other groups are listed in Table 3. Interestingly, under varying magnesium concentrations (0.01–50 mM), Liu et al. (2017a) observed that the compactness, and, essentially, structure, of the Rep A transcript varied significantly. While this could be attributable to any portion of the Rep A transcript, it is possible that the differences in compactness are attributable to the Xist A-repeats. Native gel analysis or SEC/SV-AUC experiments on the A-repeats alone could reveal the existence of multiple conformations, offering a solution to the disparity in secondary structures.
Table 3.
Experimental conditions used to probe Xist A-repeat secondary structure.
| References | RNA buffer probing conditions |
|---|---|
| Duszczyk et al. (2008, 2011) | 25°C/4°C; 10 mM NaH2PO4/Na2HPO4 pH 6.0, 100 mM NaCl, 0.02 mM EDTA, 0.02% azide |
| Maenner et al. (2010) | 25°C; 20 mM Hepes-KOH pH 7.9, 100 mM KCl, 0.2 mM EDTA pH 8.0, 0.5 mM DTT, 0.5 mM PMSF, 20% glycerol, 3.25 mM MgCl2 |
| Lu et al. (2016) | 37°C; HeLa/HEK293T/mES cells |
| Fang et al. (2015) | 37°C; MEF cells |
| Liu et al. (2017a) | 37°C/25°C; 25 mM K-Hepes pH 7.0, 0.1 mM Na-EDTA, 150 mM KCl, 15 mM MgCl2 |
| Smola et al. (2016a) | 37°C; mouse TSCs/100 mM Hepes pH 8.0, 100 mM NaCl, 10 mM MgCl2 |
Altogether, it is evident that incorporation of experimental data obtained in vitro in secondary structure predictions improves the accuracy of RNA folding programs. This provides insightful secondary structural models that can be used to further understand RNA structure. However, it is important to consider how cellular conditions such as salt concentration, pH, temperature and biological processes can influence RNA secondary structure and function in vivo, and thus, depending on conditions, RNA may adopt different structures in vitro and in vivo (Kwok et al., 2013; Leamy et al., 2016). Fang et al. (2015), Lu et al. (2016), and Smola et al. (2016a) each evaluated the secondary structure of the Xist A-repeats in cells.
Fang et al. (2015) developed and used Targeted Structure-Seq to probe specifically the Xist secondary structure. While the Xist RNA was probed with DMS before being extracted from the cells, thus reflecting its native state, Fang et al. (2015) did not discuss or perform crosslinking and pull-down experiments to demonstrate the effect that proteins may have on the secondary structure of the Xist A-repeats. Lu et al. (2016) and Smola et al. (2016a). on the other hand, evaluated the structure of the Xist A-repeats while taking into consideration some of the protein binding partners. Lu et al. (2016) developed and used PARIS (psoralen analysis of RNA interactions and structures), in combination with icSHAPE (in vivo click selective 2′-hydroxyl acylation and profiling experiment) (Spitale et al., 2015; Flynn et al., 2016) and CLIP (crosslinking and immunoprecipitation) (Lee and Ule, 2018) to predict the secondary structure of the A-repeats in the presence of their protein binding partner SHARP. The single stranded-ness of the uracil/adenosine-rich linkers is believed to serve as a platform for SHARP binding. Smola et al. (2016a,b) using SHAPE-MaP (selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling), proposed that differences observed between their ex vivo vs. in-cell SHAPE reactivity profiles are due to HuR and FUS protein binding. However, it was noted (due to reduced analysis stringency) that these proteins may interact transiently with a dynamic 5′ end or bind to double-stranded elements in such a way as to exhibit no SHAPE reactivity changes.
The differences in structures observed by Smola et al. (2016a) and Lu et al. (2016) could be due to the differences in their experimental approaches and the methods they used (i.e. they used different cell lines and probing molecules used for SHAPE). Smola et al. (2016a) studied the Xist A-repeats in the presence of HuR and FUS, while Lu et al. (2016) evaluated the A-repeat structure in the presence of SHARP. The different proteins may have affected the conformation of the RNA that was observed by each research group. This highlights the complications associated with studying intricately involved and dynamic processes such as XCI in cells (Spitale et al., 2012; Kubota et al., 2015), where various protein binding events may lead to a structure that reflects, in different areas, several conformations that happen to be adopted over time. Therefore, a more integrative approach ought to be taken; one that includes structural evaluation in the presence of the many known protein binding partners, and even rationalization of the results with functional assays (which we discuss in the next section) when validating secondary structure.
Maenner et al. (2010), Fang et al. (2015), Lu et al. (2016), Smola et al. (2016a), and Liu et al. (2017a) used comparative sequence analysis to bolster their respective proposed secondary structures. Delli Ponti et al. (2016) used their CROSS algorithm to show that where there is primary sequence conservation there is secondary structure; however, the phylogenetic analysis performed by different groups on the A-repeats rendered significantly different results. For example, Fang et al. (2015) used MAFFT (Katoh et al., 2002) and an iterative refinement method (Katoh et al., 2005) to identify eight covarying base pairs and 31 consistent mutations among the Xist A-repeats, whereas Liu et al. (2017a) used Infernal 1.1 (Nawrocki and Eddy, 2013) and reported only four covarying base pairs and 17 consistent mutations. Most interesting about these results is that the structural assembly of A-repeats five to eight are nearly identical between their two structures, but few of the identified covarying base pairs and consistent mutations agree.
The disparity between the identified compensatory mutations among the A-repeats is not satisfying. Thus, Rivas et al. (2017) developed a program, R-scape, specifically designed to statistically score the significance of observed covariation in RNA; a feature not yet fully developed in the programs used for phylogenetic analysis. Rivas et al. (2017) found no statistically significant support for secondary structure conservation in the Xist A-repeats. This does not mean that the Xist A-repeats do not harbor secondary structure; on the contrary, it may suggest that multiple conformations exist. In this case, it is important to consider the experimental approaches, whether it be in vitro or in vivo, that each research group used to predict and propose the Xist A-repeat RNA secondary structure.
Multiple and dynamic conformations of the Xist A-repeats
While it has been speculated that the Xist A-repeats may adopt multiple conformations (Lu et al., 2016; Smola et al., 2016a), this has not yet been further investigated. Furthermore, the lack of conserved secondary structure, as suggested by Rivas et al. (2017), may be due to the requirement of the A-repeats to adopt different conformations to facilitate particular protein binding events. One such example of this is supported by the presence of N6 methyladenosine (m6A) in the transcript (Patil et al., 2016). The Xist A-repeats are reported to harbor two sites with m6A modifications, one in the apical loop of A-repeat 2, and the other residing in the U/A rich linker between repeats four and five (Patil et al., 2016). These methylated adenosines interact with YTHDC1 to mediate transcriptional silencing. A recent study suggested that m6A sites occur only in structured regions of RNA (Spitale et al., 2015), causing destabilization of RNA secondary structure to potentially facilitate protein binding (Liu et al., 2017b). Interestingly, both adenosines, prior to methylation, occur at nucleotide positions that exhibit high chemical probing and enzymatic cleavage reactivity; these adenosines are single stranded. Given this observation, it is possible that the Xist A-repeats, especially in the areas where m6A are found, may exhibit a conformational change in secondary structure after methylation. Secondary structural probing of the methylated Xist A-repeats would provide valuable information regarding the effect that m6A has on Xist A-repeat secondary structure. Eventually, high-resolution methods will be needed to probe the effect of m6A on the dynamic conformational landscape.
Recognition of Xist A-repeat RNA by RNA binding proteins
Several proteins have been identified to interact with the A-repeats. The preferred RNA binding sequences and functional roles of these proteins with the Xist A-repeats are summarized in Table 4; the proposed binding sites are mapped onto the structure in Figure 3. In the following, we discuss protein interactions that have been more extensively studied with electrophoretic mobility binding shift assays (EMSA), pull down and crosslinking experiments. These include PRC2, ATRX, SHARP, and ASF/SF2 proteins, which may direct folding of the A-repeats (Table 2; Figure 3).
Table 4.
A-repeat interactions with RNA binding proteins and complexes.
| Acronym | Protein name | RNA sequence motif | Xist A-repeat function | References |
|---|---|---|---|---|
| ASF/SF2 | Alternative splicing factor/splicing factor 2 | CAUCGGG, CUGCGGA | Required for spliced Xist RNA accumulation | Royce-Tolland et al. (2010) |
| ATRX | ATP dependent helicase; X-linked helicase II | UUAGGG | Promotes loading of PRC2 on the Xist A-repeats | Sarma et al. (2015); Chu et al. (2017) |
| FUS | Fused in sarcoma | CGCGC, GGUG, GUGGU | Not yet identified, but known to regulate transcription | Pérez et al. (1997); Smola et al. (2016a) |
| hnRNP U | Heterogeneous nuclear ribonucleoprotein U | GUGG | Localization of XIST to the X chromosome | Fackelmayer et al. (1994); Wang et al. (2015) |
| HuR | Human antigen R | poly U, poly A, AU rich elements | Not yet identified, but known to increase mRNA stability | Ma et al. (1996); Smola et al. (2016a) |
| LBR | Lamin B receptor | non-sequence specific | Repositioning of Xist-targeted loci to the lamina | Nikolakaki et al. (2008); Chen et al. (2016) |
| PRC2 | PRC2 | poly G | XIST upregulation/initiation and spread of XCI | Zhao et al. (2008) |
| PTB | Polypyrimidine tract binding protein | UCUU(C), UUCUCU, CUCUCU | Stabilization of A-repeat repeat structure | Maenner et al. (2010) |
| SHARP | SMART/HDAC1 associated repressor protein | GUGUG, ACACA | Recruitment of the SMRT co-repressor to the Xist A-repeat for activation of HDAC3 | Arieti et al. (2014); McHugh et al. (2015) |
| RBM15/RBM15B | RNA binding motif protein 15 | poly U | Required for transcriptional repression | Patil et al. (2016); Dominguez et al. (2018) |
| Rnf20 | Ring finger protein 20 | unknown | Not yet identified, but known to be an E3 ubiquitin ligase involved in H2BK120 ubiquitylation | Da Rocha and Heard (2017) |
| WTAP | Wilms’ tumor 1-associating protein | RRACH | m6A methylation-promoting effects | Ping et al. (2014); Patil et al. (2016) |
| YTHDC1 | YT521-B homology domain containing 1 | GG(m6A)C | Promotes XIST-mediated gene silencing | Xu et al. (2014); Patil et al. (2016) |
Figure 3.
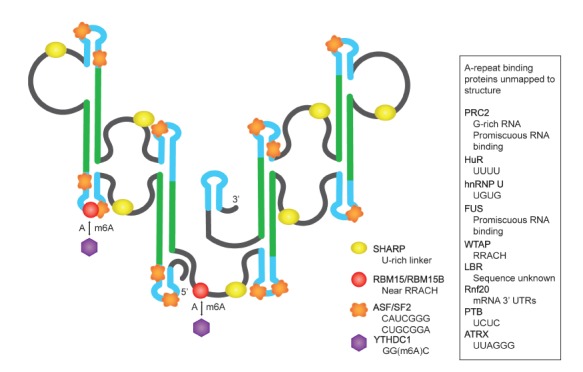
RNA binding proteins that have been reported to bind to the Xist A-repeats by CLIP and binding shift assays.
PRC2, has been identified as a promiscuous RNA binding partner, with a strong preference for G-rich RNA sequences (Long et al., 2017). While the role of PRC2 in silencing is still being investigated, in vitro binding assays reveal PRC2 has a low-nanomolar binding affinity with the Xist A-repeats (Maenner et al., 2010; Cifuentes-Rojas et al., 2014; Davidovich et al., 2015). Increasing the number of Xist A-repeats from two to eight resulted in an increase in PRC2 binding affinity, as observed by each research group. Maenner et al. concluded that their structural model supported the binding activity of PRC2; two A-repeats are unable to form a duplex, resulting in an incomplete and unstable structure, whereas four repeats are enough to facilitate duplex formation. These four repeats form one of the two large stem loops in their proposed structure (Figure 1C). This large stem loop is repeated twice, each being a binding domain for PRC2. While PRC2 interacts with high affinity to the Xist A-repeats in vitro, the relevance of this interaction in vivo appears unclear; some in vivo studies show that PRC2 subunits are associated with the Xist A-repeats, whereas other experiments show that Xist transcripts missing the A-repeats are still able to interact with PRC2 (Zhao et al., 2008; Kanhere et al., 2010; Maenner et al., 2010; Cerase et al., 2014; McHugh et al., 2015; Almeida et al., 2017; Da Rocha and Heard, 2017; Pintacuda et al., 2017a).
ATRX was identified by Sarma et al. (2015) as a binding partner of the Xist A-repeats. Sarma used both UV-crosslink RNA immunoprecipitation (UV-RIP) and EMSA to validate the interaction of ATRX with the A-repeats. Much like with PRC2, it was determined that increasing the length of the Xist A-repeat transcript (thus increasing the number of A-repeats in the sequence) resulted in an increased binding affinity of ATRX. Sarma et al. (2015) report the higher-order modular structure that was proposed by Maenner et al. (2010) as a supporting platform for ATRX binding.
Several research groups independently identified SHARP as a binding partner to the Xist A-repeats (Chu et al., 2015; McHugh et al., 2015; Moindrot et al., 2015; Monfort et al., 2015; Lu et al., 2016). This is one of the most extensively studied binding partners of the A-repeats. SHARP, which possesses four RRMs, is suggested to interact with both single-stranded (RRM3) and structured (xRRM4) RNA (Arieti et al., 2014). Monfort et al. (2015) used in vitro binding shift assays to show that SHARP can interact with a dimer of the Xist A-repeat RNA. Lu et al. (2016) showed that SHARP was crosslinked at least four times across the length of the Xist A-repeat transcript, typically three to five nucleotides upstream of each duplex unit in their model. A binding shift assay performed by Lu with the full-length mouse transcript of the A-repeats revealed multiple molecules of SHARP binding the Xist A-repeats as determined by the observance of a supershift in complex migration upon increasing the concentration of SHARP protein. The results obtained by Lu et al. (2016) supported a higher-order modular arrangement of the A-repeats.
ASF/SF2, a well-known alternative splicing factor, has been reported to interact with the Xist A-repeats both in vitro and in vivo. This interaction is believed to be a necessary component for normal spliced Xist RNA accumulation. The consensus sequences, which are listed in Table 2 and Figure 3, appear 13 times, overlapping both the major and minor hairpins of each A-repeat. Using the structural model proposed by Wutz et al. (2002), mutations that (i) disrupted the secondary structure and (ii) disrupted primary sequence but maintained major and minor hairpin conformations were made. Disruption of the major hairpin structure did not abolish binding of ASF/SF2; however, mutations to both binding site sequences prevented formation of the ASF/SF2 Xist A-repeat complex. ASF/SF2 is thus proposed to recognize primary sequence as opposed to secondary structure.
Together, these results reveal that proteins bind both primary sequence and structured elements in the Xist A-repeats. Furthermore, these results demonstrate that increasing the number of A-repeats increases the binding affinity for several protein interaction partners, interactions that may influence the structural arrangements of the A-repeats. Independent research has shown that increasing the number of A-repeats results in an increase of transcriptional silencing. This was observed after performing mutation, deletion, and insertion experiments. Wutz et al. (2002) determined that at least four repeats are necessary for silencing, while Minks et al. (2013) reported that as few as two A-repeats are sufficient, provided a longer period of time is allowed to pass to observe silencing effects. Both groups report that there is a near linear increase in silencing with an increase in the number of repeats. Mutations disrupting the stability of the major and minor hairpin stems completely abrogated silencing activity, whereas non-destabilizing major hairpin mutations had no significant effect. Scrambling the nucleotides in the apical loop of the major hairpin reduced the effect of silencing by two-fold in studies by both Wutz et al. (2002) and Minks et al. (2013). Thus, modifying the primary sequence not only disrupts binding of proteins that bind by recognizing sequence motifs (such as ASF/SF2) but also results in a disruption of secondary structural motifs, which are necessary for some proteins to bind.
Considering each of the in vitro and in vivo protein binding assays alongside the NMR data by Duszcyk and the functional assays discussed above, it is possible that increasing the number of A-repeats provides more binding sites for protein binding partners that are known to be involved in silencing. This could be the cause of the increased silencing observed in studies that evaluated the relationship between Xist A-repeat length and transcription repression. Formation of intermediate structures or multiple conformations are possible; as has been demonstrated with mRNA, rRNA and a riboswitch that RNA can fold co-transcriptionally, followed by rapid exchange of secondary structure due to binding events (Mahen et al., 2010; Lai et al., 2013; Watters et al., 2016). Similarly, the Xist A-repeats could initially adopt a non-modular structure during transcription, and subsequently refold with the use of RNA chaperones or helicases (Rajkowitsch et al., 2007; Li et al., 2008; Jarmoskaite and Russell, 2011, 2014) to assume physiologically relevant folds in the presence of protein interacting partners (Novikova et al., 2012a). Alternatively, given the numerous proteins that have been identified to interact with the Xist A-repeats, it is possible that distinct conformations of the A-repeats may be stabilized or modulated upon binding to distinct interacting proteins and complexes (e.g. SHARP and PRC2), thus regulating the function. Clearly, much work is needed to understand the structure and dynamics of lncRNAs and how protein interactions affect them in a functional context.
Integrative approaches to study the structure and dynamics of lncRNAs
As illustrated with the example of the Xist A-repeats, there are several limitations and complications associated with determining the structural features of lncRNAs. These include the inability of chemical probing and enzymatic cleavage techniques to identify specific base pairs, experimental uncertainties and ambiguities in structural interpretation of these data, and the intrinsic propensity of RNA to adopt dynamic conformations that may be differentially stabilized by interactions with RNA binding proteins or complexes. Structure probing methods fail to efficiently capture the dynamic nature and intrinsic flexibility of RNA. Thus, an integrated approach that combines various complementary methods and includes high-resolution structural biology techniques is necessary for investigating the structure and dynamics of lncRNAs both alone and in complex with protein binding partners.
To address this, both ‘divide-and-conquer’ approaches and studies of holo lncRNA structures using integrative structural biology approaches will be required (Figure 4). On one side enzymatic and chemical probing combined with structure prediction can be used to assess the presence of modular substructures. Similarity of SHAPE reactivities in vivo and in vitro, with the presence of candidate proteins for in vitro SHAPE can provide evidence for substructures. NMR can be used for the unambiguous identification of base pairs and can be combined with small angle X-ray scattering (SAXS) and small angle neutron scattering (SANS) to provide low-resolution information about substructures. Crystallography and NMR can be used for high-resolution structural analysis. On the other side, given the recent advances in cryo-electron microscopy (EM) techniques direct structural analysis of the holo lncRNA can be performed. To assess and determine the presence of multiple, dynamic conformations, FRET experiments can be performed on the holo RNA and RNA/protein complexes. Depending on the extent of dynamics present, single-particle cryo-EM analysis may be able to provide high-resolution structural information about the lncRNA and lncRNA/protein complexes. In this respect, the divide-and-conquer and holistic approaches are expected to be very complementary.
Figure 4.
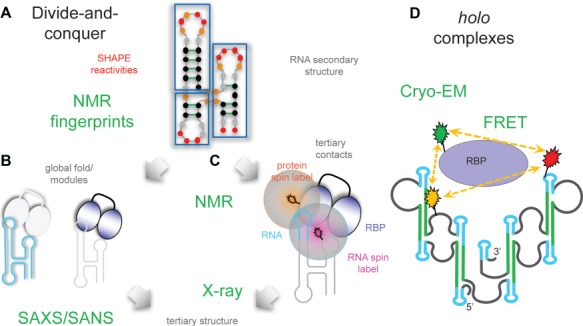
Integrated approach for tertiary structure determination of lncRNAs and their RNPs. (A) SHAPE chemical probing and NMR define RNA secondary structure. (B) SAXS/SANS provide global and subdomain shapes. (C) NMR PREs yield long-range distance restraints. Crystallography can be performed in parallel. (D) Structural analysis of holo lncRNA and RNPs can be performed using cryo-EM, and dynamics and spatial arrangements can be obtained from FRET.
In the case of the A-repeats, utilizing the existing in vitro and in vivo structural probing data (and possibly employing the 3S shotgun approach to differentiate between local and global base pair interactions) can be used to identify smaller subfragments of the A-repeats that can more feasibly be analyzed by NMR. These subfragments can also be used to study both protein binding mechanisms and conformational changes induced by protein binding partners. Upon resolving the high-resolution structure of RNA subfragments and their protein complexes, the full Xist A-repeat structure can be reconstructed with the help of SAXS, which can map overall structural arrangement, and SANS, which can be used to discriminate protein and RNA substructures in the overall arrangement (Yang et al., 2010; Madl et al., 2011; Burke and Butcher, 2012; Sonntag et al., 2017). To obtain distance restraints between identified and resolved subdomains, both FRET and NMR spin labeling techniques can be utilized (Walter, 2003; Simon et al., 2010; Wunderlich et al., 2013; Goebl et al., 2014; Hennig et al., 2015). This involves strategic placement of pairs of fluorophores (for FRET) or spin labels (for NMR) and subsequent analyzation of the resulting data. Cryo-EM can be used to map overall and subdomain shapes (Garmann et al., 2015). It will be worthwhile and presumably crucial to take such an integrated approach to characterizing the structure and dynamics of lncRNAs and their complexes in order to understand the molecular mechanisms underlying their biological function.
Conclusion
The combination of multiple techniques is required to investigate the secondary structures of lncRNAs. The presence of multiple dynamic conformations may need to be considered, including structural changes induced by protein interactions. For large RNAs, it is difficult to differentiate between local and global base pair interactions with structure probing methods, posing significant challenges to accuracy of structures derived. We propose that the Xist A-repeats adopt a higher-order modular structural arrangement, but may undergo conformational changes to accommodate protein binding partners. Taking the case of the Xist A-repeats, with their length, repetitive nature and variety of binding partners, it is evident that integrative and high-resolution approaches are required to unravel the molecular mechanisms of XCI and cull the disparate potential secondary structures. The development of integrative approaches that addresses the structures, dynamics, and protein interactions will be applicable to other lncRNAs. Given that nearly 30000 lncRNAs have been identified in the human genome, it will be crucial to develop efficient approaches for determining the structure and dynamics of large RNAs.
Acknowledgements
We thank Andrea Cerase, Santiago Martinez Lumbreras, and Hyun-Seo Kang for discussions and critical reading of the manuscript. We also thank Gregory Patrick Wolfe for assistance with figures and proofreading. We apologize to colleagues whose work we could not discuss due to space constraints.
Funding
M.S. is supported by the Deutsche Forschungsgemeinschaft, grants SFB1035, SFB1309, SPP1935, and GRK1721.
Conflict of interest: none declared.
References
- Al-Hashimi H.M. (2013). NMR studies of nucleic acid dynamics. J. Magn. Reson. 237, 191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida M., Pintacuda G., Masui O., et al. (2017). PCGF3/5-PRC1 initiates polycomb recruitment in X chromosome inactivation. Science 356, 1081–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arieti F., Gabus C., Tambalo M., et al. (2014). The crystal structure of the split end protein SHARP adds a new layer of complexity to proteins containing RNA recognition motifs. Nucleic Acids Res. 42, 6742–6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnwal R.P., Yang F., and Varani G. (2017). Applications of NMR to structure determination of RNAs large and small. Arch. Biochem. Biophys. 628, 42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockdorff N. (1992). The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell 71, 515–526. [DOI] [PubMed] [Google Scholar]
- Brown C.J. (1992). The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 71, 527–542. [DOI] [PubMed] [Google Scholar]
- Burke J.E., and Butcher S.E. (2012). Nucleic acid structure characterization by small angle X-ray scattering (SAXS). Curr. Protoc. Nucleic Acid Chem. Chapter 7, Unit 7.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabili M.N., Dunagin M.C., McClanahan P.D., et al. (2015). Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol. 16, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerase A., Smeets D., Tang Y.A., et al. (2014). Spatial separation of Xist RNA and polycomb proteins revealed by superresolution microscopy. Proc. Natl Acad. Sci. USA 111, 2235–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.-K., Blanco M., Jackson C., et al. (2016). Xist recruits the X chromosome to the nuclear lamina to enable chromosome-wide silencing. Science 354, 468–472. [DOI] [PubMed] [Google Scholar]
- Chen J.L., Bellaousov S., Tubbs J.D., et al. (2015). Nuclear magnetic resonance-assisted prediction of secondary structure for RNA: incorporation of direction-dependent chemical shift constraints. Biochemistry 54, 6769–6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Carlini D.B., Baines J.F., et al. (1999). RNA secondary structure and compensatory evolution. Genes Genet. Syst. 74, 271–286. [DOI] [PubMed] [Google Scholar]
- Chillón I., Marcia M., Legiewicz M., et al. (2015). Native purification and analysis of long RNAs. Methods Enzymol. 558, 3–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chillón I., and Pyle A.M. (2016). Inverted repeat Alu elements in the human lincRNA-p21 adopt a conserved secondary structure that regulates RNA function. Nucleic Acids Res. 44, 9462–9471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C., Zhang Q., da Rocha S., et al. (2015). Systematic discovery of Xist RNA binding proteins. Cell 161, 404–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu H.P., Cifuentes-Rojas C., Kesner B., et al. (2017). TERRA RNA antagonizes ATRX and protects telomeres. Cell 170, 86–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Rojas C., Hernandez A.J., Sarma K., et al. (2014). Regulatory interactions between RNA and polycomb repressive complex 2. Mol. Cell 55, 171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo D., Blanco M., Armaos A., et al. (2016). Quantitative predictions of protein interactions with long noncoding RNAs: to the editor. Nat. Methods 14, 5–6. [DOI] [PubMed] [Google Scholar]
- Da Rocha S.T., and Heard E. (2017). Novel players in X inactivation: insights into Xist-mediated gene silencing and chromosome conformation. Nat. Struct. Mol. Biol. 24, 197–204. [DOI] [PubMed] [Google Scholar]
- Davidovich C., Wang X., Cifuentes-rojas C., et al. (2015). Towards a consensus on the binding specificity and promiscuity of PRC2 for RNA. Mol. Cell 57, 552–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delli Ponti R., Marti S., Armaos A., et al. (2016). A high-throughput approach to profile RNA structure. Nucleic Acids Res. 45, e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethoff E.A., Chugh J., Mustoe A.M., et al. (2012). Functional complexity and regulation through RNA dynamics. Nature 482, 322–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez D., Freese P., Alexis M.S., et al. (2018). Sequence, structure, and context preferences of human RNA binding proteins. Mol. Cell 70, 854–867.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duszczyk M.M., Wutz A., Rybin V., et al. (2011). The Xist RNA A-repeat comprises a novel AUCG tetraloop fold and a platform for multimerization. RNA 17, 1973–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duszczyk M.M., Zanier K., and Sattler M. (2008). A NMR strategy to unambiguously distinguish nucleic acid hairpin and duplex conformations applied to a Xist RNA A-repeat. Nucleic Acids Res. 36, 7068–7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy S.R. (2014). Computational analysis of conserved RNA secondary structure in transcriptomes and genomes. Annu. Rev. Biophys. 43, 433–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fackelmayer F., Dahm K., Renz A., et al. (1994). Nucleic-acid-binding properties of hnRNP-U/SAF-A. Eur. J. Biochem. 221, 749–757. [DOI] [PubMed] [Google Scholar]
- Fang R., Moss W.N., Rutenberg-Schoenberg M., et al. (2015). Probing Xist RNA structure in cells using Targeted Structure-Seq. PLoS Genet. 11, e1005668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn R.A., Zhang Q.C., Spitale R.C., et al. (2016). Transcriptome-wide interrogation of RNA secondary structure in living cells with icSHAPE. Nat. Protoc. 11, 273–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fürtig B., Richter C., Wöhnert J., et al. (2003). NMR spectroscopy of RNA. ChemBioChem 4, 936–962. [DOI] [PubMed] [Google Scholar]
- Garmann R.F., Gopal A., Athavale S.S., et al. (2015). Visualizing the global secondary structure of a viral RNA genome with cryo-electron microscopy. RNA 21, 877–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebl C., Madl T., Simon B., et al. (2014). NMR approaches for structural analysis of multidomain proteins and complexes in solution. Prog. Nucl. Magn. Reson. Spectrosc. 80, 26–63. [DOI] [PubMed] [Google Scholar]
- Guo X., Gao L., Wang Y., et al. (2016). Advances in long noncoding RNAs: identification, structure prediction and function annotation. Brief. Funct. Genomics 15, 38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa Y., Brockdorff N., Kawano S., et al. (2010). The matrix protein hnRNP U is required for chromosomal localization of Xist RNA. Dev. Cell 19, 469–476. [DOI] [PubMed] [Google Scholar]
- Hawkes E.J., Hennelly S.P., Novikova I.V., et al. (2016). COOLAIR antisense RNAs form evolutionarily conserved elaborate secondary structures. Cell Rep. 16, 3087–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig J., Warner L.R., Simon B., et al. (2015). Structural analysis of protein-RNA complexes in solution using NMR paramagnetic relaxation enhancements. Methods Enzymol. 558, 333–362. [DOI] [PubMed] [Google Scholar]
- Ilik I.A., Quinn J.J., Georgiev P., et al. (2013). Tandem stem-loops in roX RNAs act together to mediate X chromosome dosage compensation in Drosophila. Mol. Cell 51, 156–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarmoskaite I., and Russell R. (2011). DEAD-box proteins as RNA helicases and chaperones. Wiley Interdiscip. Rev. RNA 2, 135–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarmoskaite I., and Russell R. (2014). RNA helicase proteins as chaperones and remodelers. Annu. Rev. Biochem. 83, 697–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanhere A., Viiri K., Araújo C.C., et al. (2010). Short RNAs are transcribed from repressed polycomb target genes and interact with polycomb repressive complex-2. Mol. Cell 38, 675–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Kuma K.I., Toh H., et al. (2005). MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33,511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Misawa K., Kuma K., et al. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane S.C., Heng X., Lu K., et al. (2015). RNA structure. Structure of the HIV-1 RNA packaging signal. Science 348, 917–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon J., Prestwood L., and Lever A. (2014). Current perspectives on RNA secondary structure probing. Biochem. Soc. Trans. 42, 1251–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon J.C., Prestwood L.J., Le Grice S.F.J., et al. (2013). In-gel probing of individual RNA conformers within a mixed population reveals a dimerization structural switch in the HIV-1 leader. Nucleic Acids Res. 41, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz M., Wan Y., Mazor E., et al. (2010). Genome-wide measurement of RNA secondary structure in yeast. Nature 467, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kladwang W., Vanlang C.C., Cordero P., et al. (2011). Understanding the errors of SHAPE-directed RNA structure modeling. Biochemistry 50, 8049–8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota M., Tran C., and Spitale R.C. (2015). Progress and challenges for chemical probing of RNA structure inside living cells. Nat. Chem. Biol. 11, 933–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung J.T.Y., Colognori D., and Lee J.T. (2013). Long noncoding RNAs: past, present, and future. Genetics 193, 651–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok C.K. (2016). Dawn of the in vivo RNA structurome and interactome. Biochem. Soc. Trans. 44, 1395–1410. [DOI] [PubMed] [Google Scholar]
- Kwok C.K., Ding Y., Tang Y., et al. (2013). Determination of in vivo RNA structure in low-abundance transcripts. Nat. Commun. 4, 1–12. [DOI] [PubMed] [Google Scholar]
- Lai D., Proctor J.R., and Meyer I.M. (2013). On the importance of cotranscriptional RNA structure formation. RNA 19, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leamy K.A., Assmann S.M., Mathews D.H., et al. (2016). Bridging the gap between in vitro and in vivo RNA folding. Q. Rev. Biophys. 49, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B., Flynn R.A., Kadina A., et al. (2017a). Comparison of SHAPE reagents for mapping RNA structures inside living cells. RNA 23, 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B., Sahoo A., Marchica J., et al. (2017b). The long noncoding RNA SPRIGHTLY acts as an intranuclear organizing hub for pre-mRNA molecules. Sci. Adv. 3, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee F., and Ule J. (2018). Advances in CLIP technologies for studies of protein-RNA interactions. Mol. Cell 69, 354–369. [DOI] [PubMed] [Google Scholar]
- Lee J., Vogt C.E., McBrairty M., et al. (2013). Influence of dimethylsulfoxide on RNA structure and ligand binding. Anal. Chem. 85,9692–9698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard C.W., Hajdin C.E., Karabiber F., et al. (2013). Principles for understanding the accuracy of SHAPE-directed RNA structure modeling. Biochemistry 52, 588–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P.T.X., Vieregg J., and Tinoco I. (2008). How RNA unfolds and refolds. Annu. Rev. Biochem. 77, 77–100. [DOI] [PubMed] [Google Scholar]
- Lin Y., Schmidt B.F., Bruchez M.P., et al. (2018). Structural analyses of NEAT1 lncRNAs suggest long-range RNA interactions that may contribute to paraspeckle architecture. Nucleic Acids Res. 46, 3742–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Somarowthu S., and Marie Pyle A. (2017a). Visualizing the secondary and tertiary architectural domains of lncRNA RepA. Nat. Chem. Biol. 13, 282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N., Zhou K.I., Parisien M., et al. (2017b). N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 45, 6051–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Y., Bolanos B., Gong L., et al. (2017). Conserved RNA-binding specificity of polycomb repressive complex 2 is achieved by dispersed amino acid patches in EZH2. eLife 6, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R., Hofacker I.L., and Stadler P.F. (2016). RNA folding with hard and soft constraints. Algorithms Mol. Biol. 11, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low J.T., and Weeks K.M. (2010). SHAPE-directed RNA secondary structure prediction. Methods 52, 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K., Miyazaki Y., and Summers M.F. (2010). Isotope labeling strategies for NMR studies of RNA. J. Biomol. NMR 46, 113–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z., Zhang Q.C., Lee B., et al. (2016). RNA duplex map in living cells reveals higher-order transcriptome structure. Cell 165, 1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchesi J., Kelly W., and Panning B. (2005). Chromatin remodeling in dosage compensation. Annu. Rev. Genet. 39, 615–651. [DOI] [PubMed] [Google Scholar]
- Lukavsky P.J., and Puglisi J.D. (2005). Structure determination of large biological RNAs. Methods Enzymol. 394, 399–416. [DOI] [PubMed] [Google Scholar]
- Ma W.J., Cheng S., Campbell C., et al. (1996). Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J. Biol. Chem. 271, 8144–8151. [DOI] [PubMed] [Google Scholar]
- Madl T., Gabel F., and Sattler M. (2011). NMR and small-angle scattering-based structural analysis of protein complexes in solution. J. Struct. Biol. 173, 472–482. [DOI] [PubMed] [Google Scholar]
- Maenner S., Blaud M., Fouillen L., et al. (2010). 2-D structure of the a region of Xist RNA and its implication for PRC2 association. PLoS Biol. 8, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahen E.M., Watson P.Y., Cottrell J.W., et al. (2010). mRNA secondary structures fold sequentially but exchange rapidly in vivo. PLoS Biol. 8, e1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese F.P., Raimondi I., and Huarte M. (2017). The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 18, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews D.H., Sabina J., Zuker M., et al. (1999). Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 288, 911–940. [DOI] [PubMed] [Google Scholar]
- McGinnis J.L., Dunkle J.A., Cate J.H.D., et al. (2012). The mechanisms of RNA SHAPE chemistry. J. Am. Chem. Soc. 134, 6617–6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh C.A., Chen C.-K., Chow A., et al. (2015). The Xist lncRNA directly interacts with SHARP to silence transcription through HDAC3. Nature 521, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minks J., El Baldry S., Yang C., et al. (2013). XIST-induced silencing of flanking genes is achieved by additive action of repeat a monomers in human somatic cells. Epigentics Chromatin 6, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlynsky V., and Bussi G. (2018). Molecular simulations reveal an interplay between SHAPE reagent binding and RNA flexibilty. J. Phys. Chem. Lett. 9, 313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moindrot B., and Brockdorff N. (2016). RNA binding proteins implicated in Xist-mediated chromosome silencing. Semin. Cell Dev. Biol. 56, 58–70. [DOI] [PubMed] [Google Scholar]
- Moindrot B., Cerase A., Coker H., et al. (2015). A pooled shRNA screen identifies Rbm15, Spen, and Wtap as factors required for Xist RNA-mediated silencing. Cell Rep. 12, 562–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollova E.T., and Pardi A. (2000). NMR solution structure determination of RNAs. Curr. Opin. Struct. Biol. 10, 298–302. [DOI] [PubMed] [Google Scholar]
- Monfort A., Di Minin G., Postlmayr A., et al. (2015). Identification of Spen as a crucial factor for Xist function through forward genetic screening in haploid embryonic stem cells. Cell Rep. 12, 554–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki E.P., and Eddy S.R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolakaki E., Drosou V., Sanidas I., et al. (2008). RNA association or phosphorylation of the RS domain prevents aggregation of RS domain-containing proteins. Biochim. Biophys. Acta 1780, 214–225. [DOI] [PubMed] [Google Scholar]
- Novikova I., Dharap A., Hennelly S.P., et al. (2013a). 3S: shotgun secondary structure determination of long non-coding RNAs. Methods 63, 170–177. [DOI] [PubMed] [Google Scholar]
- Novikova I., Hennelly S.P., and Sanbonmatsu K.Y. (2012a). Sizing up long non-coding RNAs: do lncRNAs have secondary and tertiary structure. BioArchitecture 2, 189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novikova I., Hennelly S.P., and Sanbonmatsu K.Y. (2012b). Structural architecture of the human long non-coding RNA, steroid receptor RNA activator. Nucleic Acids Res. 40, 5034–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novikova I., Hennelly S.P., and Sanbonmatsu K.Y. (2013b). Tackling structures of long noncoding RNAs. Int. J. Mol. Sci. 14, 23672–23684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novikova I., Hennelly S.P., Tung C.S., et al. (2013c). Rise of the RNA machines: exploring the structure of long non-coding RNAs. J. Mol. Biol. 425, 3731–3746. [DOI] [PubMed] [Google Scholar]
- Parsch J., Braverman J.M., and Stephan W. (2000). Comparative sequence analysis and patterns of covariation in RNA secondary structures. Genetics 154, 909–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil D.P., Chen C.K., Pickering B.F., et al. (2016). M6A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537,369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penny G., Kay G.F., Sheardown S.A., et al. (1996). Requirement for Xist in X-chromosome inactivation. Nature 379, 131–137. [DOI] [PubMed] [Google Scholar]
- Pérez I., McAfee J.G., and Patton J.G. (1997). Multiple RRMs contribute to RNA binding specificity and affinity for polypyrimidine tract binding protein. Biochemistry 36, 11881–11890. [DOI] [PubMed] [Google Scholar]
- Ping X.L., Sun B.F., Wang L., et al. (2014). Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24, 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintacuda G., Wei G., Roustan C., et al. (2017a). hnRNPK recruits PCGF3/5-PRC1 to the Xist RNA B-repeat to establish polycomb-mediated chromosomal silencing. Mol. Cell 68, 955–969.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintacuda G., Young A.N., and Cerase A. (2017b). Function by structure: spotlights on Xist long non-coding RNA. Front. Mol. Biosci. 4, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting C.P., Oliver P.L., and Reik W. (2009). Evolution and functions of long noncoding RNAs. Cell 136, 629–641. [DOI] [PubMed] [Google Scholar]
- Rajkowitsch L., Chen D., Stampfl S., et al. (2007). RNA chaperones, RNA annealers and RNA helicases. RNA Biol. 4, 118–130. [DOI] [PubMed] [Google Scholar]
- Reuter J.S., and Mathews D.H. (2010). RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas E., Clements J., and Eddy S.R. (2017). Lack of evidence for conserved secondary structure in long noncoding RNAs. Nat. Methods 14,45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royce-Tolland M.E., Andersen A.A., Koyfman H.R., et al. (2010). The A-repeat links ASF/SF2-dependent Xist RNA processing with random choice during X inactivation. Nat. Struct. Mol. Biol. 17, 948–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma K., Cifuentes-rojas C., Ergun A., et al. (2015). ATRX directs binding of PRC2 to Xist RNA and polycomb targets. Cell 159, 869–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt K., Joyce C.E., Buquicchio F., et al. (2016). The lncRNA SLNCR1 mediates melanoma invasion through a conserved SRA1-like region. Cell Rep. 15, 2025–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon B., Madl T., Mackereth C.D., et al. (2010). An efficient protocol for NMR-spectroscopy-based structure determination of protein complexes in solution. Angew. Chem. Int. Ed. 49, 1967–1970. [DOI] [PubMed] [Google Scholar]
- Smola M.J., Cristy T.W., Inoue K., et al. (2016a). SHAPE reveals transcript-wide interactions, complex structural domains, and protein interactions across the Xist lncRNA in living cells. Proc. Natl Acad. Sci. USA 113, 10322–10327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smola M.J., Rice G.M., Busan S., et al. (2016b). Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile, and accurate RNA structure analysis. Nat. Protoc. 10, 1643–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somarowthu S., Legiewicz M., Liu F., et al. (2015). HOTAIR forms an intricate and modular secondary structure. Mol. Cell 58, 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonntag M., Jagtap P.K.A., Simon B., et al. (2017). Segmental, domain-selective perdeuteration and small-angle neutron scattering for structural analysis of multi-domain proteins. Angew. Chem. Int. Ed. 56, 9322–9325. [DOI] [PubMed] [Google Scholar]
- Spitale R.C., Crisalli P., Flynn R.A., et al. (2012). RNA SHAPE analysis in living cells. Nat. Chem. Biol. 9, 18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitale R.C., Flynn R.A., Zhang Q.C., et al. (2015). Structural imprints in vivo decode RNA regulatory mechanisms. Nature 519, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern S., Moazed D., and Noller H.F. (1988). Structural analysis of RNA using chemical and enzymatic probing monitored by primer extension. Methods Enzymol. 164, 481–489. [DOI] [PubMed] [Google Scholar]
- Strauss J.H., Kelly R.B., and Sinsheimer R.L. (1968). Denaturation of RNA with dimethyl sulfoxide. Biopolymers 6, 793–807. [DOI] [PubMed] [Google Scholar]
- Sztuba-Solinska J., Rausch J.W., Smith R., et al. (2017). Kaposi’s sarcoma-associated herpesvirus polyadenylated nuclear RNA: a structural scaffold for nuclear, cytoplasmic and viral proteins. Nucleic Acids Res. 45, 6805–6821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torarinsson E., and Lindgreen S. (2008). WAR: webserver for aligning structural RNAs. Nucleic Acids Res. 36, W79–W84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I., and Bartel D.P. (2013). LincRNAs: genomics, evolution, and mechanisms. Cell 154, 26–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varani G., Aboul-ela F., and Allain F.H.T. (1996). NMR investigation of RNA structure. Prog. Nucl. Magn. Reson. Spectrosc. 29, 51–127. [Google Scholar]
- Walter N.G. (2003). Probing RNA structural dynamics and function by fluorescence resonance energy transfer (FRET). Curr. Protoc. Nucleic Acid Chem. 11, 11.10.1–11.10.23. [DOI] [PubMed] [Google Scholar]
- Wang X., Schwartz J.C., and Cech T.R. (2015). Nucleic acid-binding specificity of human FUS protein. Nucleic Acids Res. 43, 7535–7543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski O., and Chang H.Y. (2011). Long noncoding RNAs and human disease. Trends Cell Biol. 21, 354–361. [DOI] [PubMed] [Google Scholar]
- Watters K.E., Strobel E.J., Yu A.M., et al. (2016). Cotranscriptional folding of a riboswitch at nucleotide resolution. Nat. Struct. Mol. Biol. 23, 1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks K.M. (2010). Advances in RNA secondary and tertiary structure analysis by chemical probing. Curr. Opin. Struct. Biol. 20, 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks K.M., and Mauger D.M. (2011). Exploring RNA structural codes with SHAPE chemistry. Acc. Chem. Res. 44, 1280–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson K.A., Merino E.J., and Weeks K.M. (2006). Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 1, 1610–1616. [DOI] [PubMed] [Google Scholar]
- Wunderlich C.H., Huber R.G., Spitzer R., et al. (2013). A novel paramagnetic relaxation enhancement tag for nucleic acids: a tool to study structure and dynamics of RNA. ACS Chem. Biol. 8, 2697–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wutz A., Rasmussen T.P., and Jaenisch R. (2002). Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat. Genet. 30, 167–174. [DOI] [PubMed] [Google Scholar]
- Xu C., Wang X., Liu K., et al. (2014). Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat. Chem. Biol. 10, 927–929. [DOI] [PubMed] [Google Scholar]
- Xue Y., Kellogg D., Kimsey I.J., et al. (2015). Characterizing RNA excited states using NMR relaxation dispersion. Methods Enzymol. 558, 39–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Z., Hennelly S., Doyle B., et al. (2016). A G-rich motif in the lncRNA Braveheart interacts with a zinc-finger transcription factor to specify the cardiovascular lineage. Mol. Cell 64, 37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S., Parisien M., Major F., et al. (2010). RNA structure determination using SAXS data. J. Phys. Chem. B 114, 10039–10048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampetaki A., Albrecht A., and Steinhofel K. (2018). Long non-coding RNA structure and function: is there a link? Front. Physiol. 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Mao Y.S., Diermeier S.D., et al. (2017). Identification and characterization of a class of MALAT1-like genomic loci. Cell Rep. 19, 1723–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Rice K., Wang Y., et al. (2010). Maternally expressed gene 3 (MEG3) noncoding ribonucleic acid: isoform structure, expression, and functions. Endocrinology 151, 939–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Sun B.K., Erwin J.A., et al. (2008). Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322, 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziehler W.A., and Engelke D.R. (2001). Probing RNA structure with chemical reagents and enzymes. Curr. Protoc. Nucleic Acid Chem. Chapter 6, Unit 6.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]