

Abstract
The neuropeptides oxytocin (OT) and vasopressin (VP) and their G protein-coupled receptors OTR, V1aR, V1bR, and V2R form an important and widely-distributed neuroendocrine signaling system. In mammals, this signaling system regulates water homeostasis, blood pressure, reproduction, as well as social behaviors such as pair bonding, trust and aggression. There exists high demand for ligands with differing pharmacological profiles to study the physiological and pathological functions of the individual receptor subtypes. Here, we present the pharmacological characterization of an arthropod (Metaseiulus occidentalis) OT/VP-like nonapeptide across the human OT/VP receptors. I8-arachnotocin is a full agonist with respect to second messenger signaling at human V2R (EC50 34 nM) and V1bR (EC50 1.2 µM), a partial agonist at OTR (EC50 790 nM), and a competitive antagonist at V1aR [pA2 6.25 (558 nM)]. Intriguingly, I8-arachnotocin activated the Gαs pathway of V2R without recruiting either β-arrestin-1 or β-arrestin-2. I8-arachnotocin might thus be a novel pharmacological tool to study the (patho)physiological relevance of β-arrestin-1 or -2 recruitment to the V2R. These findings furthermore highlight arthropods as a novel, vast and untapped source for the discovery of novel pharmacological probes and potential drug leads targeting neurohormone receptors.
Subject terms: Receptor pharmacology, Peptides
Introduction
Oxytocin (OT) and vasopressin (VP) are prototypical neuropeptides that together with their G protein-coupled receptors (GPCRs), OTR, V1aR, V1bR, and V2R, form a versatile neuroendocrine signaling system in humans. Peripherally, OT is important in the regulation of labor1 and milk let-down2, while VP is crucial for water homeostasis and vasoconstriction3,4. Centrally, OT and VP influence different behaviors, such as empathy5, social recognition2,6–8, sexual arousal2,6–8, attachment9, parental care10, and anxiety-related behavior11. Consequently, its dysregulation is associated with a wide range of disorders, including post-partum complications, cardiovascular diseases, diabetes insipidus and dysmenorrhea as well as social anxiety disorders, autism, schizophrenia, Prader-Willi syndrome and depression2,12–18.
OT and VP differ only in two amino acids and their receptors share ~80% binding site sequence homology19, which results in OT and VP being unselective and able to activate all four receptors. Consequently, neuroscientists are looking for a repertoire of ligands with different pharmacological profiles to study the (patho)physiology of the individual receptor subtypes of this fundamental signaling system. In an attempt to provide such a pharmacological toolbox, we started to explore different animal species in the search for OT/VP-like ligands that are active on the human receptors, yet better at discriminating signaling between the four OT/VP receptors. Our strategy14 relies on the fact that OT/VP-like signaling system is highly conserved and widely distributed across vertebrates, including fish and amphibians, as well as in several invertebrate species such as mollusks, annelids, nematodes, insects, starfish, and hydra6,14,20. Its origin can be traced back ~600 million years to the closely-related ancestral vertebrate nonapeptide vasotocin and similar invertebrate nonapeptides (Fig. 1)20,21. Our strategy has already yielded several important pharmacological probes, including a selective human V1aR antagonist based on an OT/VP-like peptide isolated from the black garden ant Lasius niger19, a selective antagonist at the human V1aR antagonist, based on an OT/VP-like peptide from the venom of the marine predatory cone snail22, and a selective human OTR agonist based from an OT/VP-like sequence identified in a cyclotide from the plant Oldenlandia affinis23.
Figure 1.

Phylogenetic relationship and molecular sequence of OT/VP-like neuropeptides. Taxonomic groups and neuropeptide names (in brackets). Conserved cysteines are highlighted in yellow, and the disulfide bond between Cys1 and Cys6 is indicated. *C-terminus amidated; #Renamed to arachnotocin to simplify nomenclature. Analogous to the existing phylum nomenclature, we refer to the mite-derived peptide by I8-arachnotocin, to distinguish it from arachnotocin (also referred to as crustacean OT/VP-like peptide in the past)20,21.
In this work, we report the chemical synthesis and pharmacological characterization of I8-arachnotocin across the four human OT/VP receptors using second messenger quantification as well as β-arrestin-1 and -2 recruitment assays in cells heterologously expressing the individual receptor subtypes OTR, V1aR, V1bR, and V2R.
Materials and Methods
Peptide synthesis
I8-arachnotocin was produced by solid phase peptide synthesis (SPPS) using fluorenylmethyloxycarbonyl (Fmoc) chemistry, purified and analyzed using methods previously described19. Briefly, the peptide was synthesized on a Rink amide 4-methylbenzyhydrylamine (MBHA) resin (Auspep, Australia) using 4-fold excess of protected amino acids, 4 equivalents of hexafluorophosphate benzotriazole tetramethyl uranium (HBTU) (Iris Biotech, Germany) and N,N-diisopropylethylamine (DIPEA) (Auspep, Australia) in dimethylformamide (DMF) (Auspep, Australia) and a coupling time of 15 min. The peptide was cleaved from the resin and the side chain protecting groups were removed by treatment with trifluoroacetic acid (TFA) (Auspep, Australia): triisopropylsilane (TIPS) (Sigma Aldrich, Australia): ethandithiol (EDT) (Sigma Aldrich, Australia): H2O (90:2.5:2.5:5) for 2 h. The crude peptide was folded for 24 h at 100 μM at 25°C in 0.1 M NH4HCO3 (Auspep, Australia), pH 8.2, and purified using a Vydac Protein and Peptide C18 preparative column. Analytical HPLC was performed with column heating at 40 °C and detection at 214 nm. The final product was analyzed by LC-MS on an API QSTAR PULSAR from PE Sciex, used in series with Agilent 1100 series HPLC system with a Kromasil C18 column at a gradient of 0–40% eluent B (90% acetonitrile, 0.045% trifluoroacetic acid) in 20 min.
Cell culture and transient receptor expression
All cell culture work was performed with human embryonic kidney cells 293 (HEK293)24. Unless otherwise stated, cells were incubated at 37 °C and 5% CO2 and grown in Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo Fisher Scientific, Australia and Fisher Scientific, Austria) containing 10% fetal bovine serum (GE Life Sciences, Australia and Sigma-Aldrich, Germany), 50 U/mL penicillin and 50 U/mL streptomycin (Thermo Fisher Scientific, Australia and Sigma-Aldrich, Germany). Transient transfections were performed via Lipofectamine 2000 (Thermo Fisher Scientific, Australia) or jetPRIME (Polyplus transfection, France) using 2 µg of pEGFP-N1 plasmid DNA coding for EGFP-tagged human OT/VP receptors23 or a combination of 2 µg each of receptor-encoding plasmids and a plasmid coding for β-arrestin-1- or -2-Nluc (subcloned into pcDNA3, with NanoLuc provided under a Limited Use Label License from Promega, Madison, USA).
Second messenger quantification
Inositol-1-phosphate (IP1) accumulation in response to Gαq coupled activation of human OTR, V1aR, and V1bR were measured using the IP-One Gq assay kit (Cisbio, France). Cells were seeded 4 h after transfection onto 384-well plates at a density of 10,000 cells per well and incubated for 2 days. At the time of the assay, all media was removed and the cells equilibrated to the provided stimulation buffer for 15 min at 37 °C. The cells were stimulated with peptide ligands at varying concentration (10 pM – 30 µM) for 1 h at 37 °C. Cyclic adenosine monophosphate (cAMP) accumulation induced by Gαs coupling of V2R activation was determined using the LANCE Ultra cAMP detection kit (Perkin Elmer, Waltham, USA). Cells were re-passaged 4 h after transfection at a 1:2 ratio and incubated overnight. The next day, all media was removed, and cells were suspended with cAMP stimulation buffer (5 mM HEPES, 0.5 mM 3-isobutyl-1-methylxanthine, 0.1% bovine serum albumin in Hank’s balanced salt solution, HBSS, pH 7.4) and the cells transferred onto a 384-well plate at a density of 300 cells per well. Stimulation was carried out for 30 min at 25 °C. Second messenger levels were measured by homogenous time-resolved fluorescence resonance energy transfer via fluorescence measurement on a Flexstation 3 (Molecular Devices, San Jose, USA) using the ratios 665/620 nm (IP1) and 665/615 nm (cAMP) at an excitation wavelength of 340 nm. For antagonist screening, cells were stimulated with the endogenous ligand OT at OTR (50 nM), or VP for V1aR (1 nM), V1bR (3 nM) and V2R (0.5 pM), in presence (1 or 10 µM) and absence of I8-arachnotocin, as well as with 10 µM of the respective endogenous ligand.
Antagonism of I8-arachnotocin at V1aR was characterized by Schild regression analysis (as published earlier)19. Briefly, several concentration-response curves of the endogenous agonist VP (as described above) were measured in the presence (1 µM, 3 µM and 10 µM) and absence of I8-arachnotocin. The logarithm of the dose-ratio (A′/A-1) was plotted vs. the logarithm of the respective concentration of I8-arachnotocin (B) to obtain the pA2 value.
β-arrestin-1 and -2 recruitment
Recruitment of β-arrestin-1- and -2 upon receptor stimulation was measured via real-time measurement of bioluminescence resonance energy transfer (BRET) between β-arrestin-1/2-luciferase and EGFP-tagged receptors. Cells were co-transfected with β-arrestin-1/2-Nluc and OT/VP receptor encoding plasmids at a ratio of 1:10. At 6 h post-transfection, the cells were transferred onto a white, clear bottom 96-well plate at 50,000 cells/well in phenol-red free DMEM containing 10% fetal bovine serum. The following day, the cells were serum starved for 1 h in phenol-free DMEM. Furimazine (Promega, Madison, USA), diluted 1:50 in HBSS, was added to the cells 5 min prior to monitoring at a 1:1 ratio. Light emissions were measured at 460 nm (Nluc) and 510 nm (EGFP) on a Flexstation 3 (Molecular Devices, San Jose, USA). After establishment of a baseline for 5 min, peptides diluted in HBSS were added and the response measured for 35 min. The ligand-induced BRET signal was calculated as: (emission EGFPligand/emission Nlucligand) − (emission EGFPHBSS/emission NlucHBSS). Concentration-response curves were generated from the BRET signal at 5 min after addition of various peptide concentrations (10 pM – 30 µM).
Immunoblotting for ERK 1/2
Immunoblotting was performed as described previously25. Briefly, following an overnight incubation and 16 h starvation HEK293 cells transiently expressing human V2R were treated with 1 µM I8-arachnotocin or 1 µM VP prepared in DMEM. Cells were incubated with peptide ligands at 37 °C for indicated periods and 1 mL of chilled 1x phosphate-buffered saline were used to terminate the incubation. After freeze-thaw cycle in liquid nitrogen cells were solubilized in 100 µl of lysis buffer (50 mM HEPES, 0.5% NP-40 substitute, 50 mM glycerol-2-phosphate, 250 mM NaCl, 5 mM EDTA, 2 mM imidazole, 1 mM Na3VO4, 1 mM hepta-molybdate, pH adjusted to 7.0 with NaOH), freshly added 1 mM PMSF, one cOmplete Mini EDTA-free tablet (Roche) and one PhosSTOP tablet (Roche) and then centrifuged at 10,000 × g for 10 min. The bicinchoninic acid assay (Micro-BCA kit, Pierce) was used to measure total protein content. Phosphorylated and total ERK 1/2 were detected by immunoblotting on the same membrane using the same exposure method with an anti-phospho-p44/42 MAPK antibody (ERK 1/2) (1:1,000; Cell Signaling Technology) and an anti-p44/42 MAPK (ERK 1/2) (1:1,000; Cell Signaling Technology), respectively. Detection and quantification of resulting bands were executed by secondary antibodies (Donkey anti rabbit 680RD and 880RD) and Odyssey Clx (LiCor Biosciences) infrared fluorescent imaging system, respectively.
Data analysis
All data were analyzed using GraphPad Prism (GraphPad Software, San Diego) and all graphs were normalized to the activity of OT/VP above baseline. Concentration response curves were fitted to three-parameter non-linear regression curves with a bottom constrained to zero, a slope of one and sigmoidal shape at logarithmic scale to derive estimates of potency (EC50) and maximum efficacy (Emax). Concentration response curves for Schild regression analysis26 were additionally constrained to a top value of one hundred. All data were presented as mean ± SEM of at least three independent experiments (unless otherwise stated) conducted in triplicate.
Ethics Statement
The study presented in this manuscript did not involve human or animal subjects.
Results
I8-arachnotocin is an agonist at human OTR, V1bR and V2R
Fmoc-SPPS, cleavage, oxidative folding, followed by preparative C18-HPLC purification yielded I8-arachnotocin in >95% purity (15% overall yield) (Fig. 2). The concentration-response curves of I8-arachnotocin at the human OT/VP receptors (Fig. 3) indicated discriminatory effects in terms of Emax and EC50. At both V1bR and V2R, it was a full agonist, yet with highly disparate potencies, namely potencies of 1.2 µM (logEC50 = − 5.93 ± 0.15) and 34 nM (logEC50 − 7.47 ± 0.09) respectively. At OTR, I8-arachnotocin was a partial agonist (Emax = 62%) with an EC50 of 790 nM (logEC50 = − 6.11 ± 0.20) (Fig. 3). At all three receptors, I8-arachnotocin was less potent compared to the respective endogenous peptide (OTR 65-fold, V1bR 750-fold, V2R 5,000-fold). No activation of V1aR by I8-arachnotocin was observed for concentrations up to 10 µM (Table 1).
Figure 2.
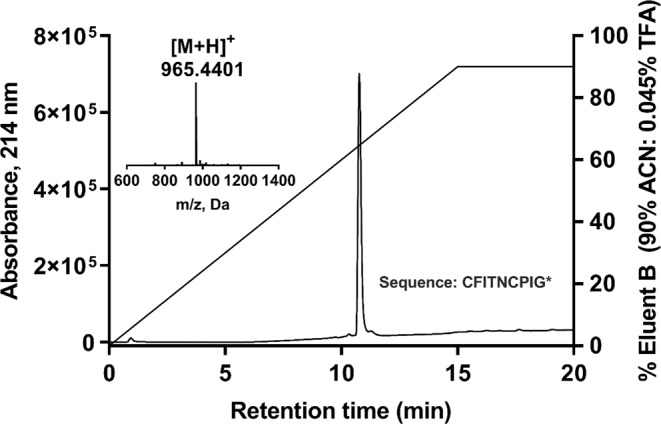
Quality of synthetic I8-arachnotocin. Analytical RP-HPLC of the purified I8-arachnotocin (purity > 95%). Inset: high resolution MS of the product with the observed molecular weight of 964.44 Da (theoretical: 964.44 Da). ACN = acetonitrile; TFA = trifluoroacetic acid; *C-terminus amidated.
Figure 3.

I8-arachnotocin is an agonist at human V2R, V1bR, and OTR. Concentration-dependent accumulation of second messengers (cAMP and IP1) after receptor stimulation of (a) OTR, (b) V1aR, (c) V1bR and (d) V2R with I8-arachnotocin (30 pM – 30 µM). Results were normalized to accumulation of IP1 and cAMP above baseline and maximal activation of the receptors by their endogenous ligands (OT for OTR and VP for VPRs). Data points were fitted by nonlinear regression curves (sigmoidal, slope = 1); error bars depict SEM; n = 3. For EC50 and Emax values refer to Table 1.
Table 1.
Potency and efficacy (G protein-mediated) of I8-arachnotocin at human receptors.
| I8-arachnotocin | VP/OT& | |||||
|---|---|---|---|---|---|---|
| EC50 | pEC50 | Emax# | EC50 | pEC50 | Emax# | |
| V2R | 34 nM | −7.47 ± 0.09 | 99 ± 3% | 6.7 pM | −11.17 ± 0.10 | 100% |
| V1bR | 1.2 µM | −5.93 ± 0.15 | 104 ± 8% | 1.6 nM | −8.80 ± 0.09 | 100% |
| V1aR | antagonist pA2 of 6.253 (~558 nM) | 0.56 nM | −9.25 ± 0.07 | 100% | ||
| OTR | 790 nM | −6.11 ± 0.20 | 62 ± 6% | 12 nM | −7.91 ± 0.09 | 100% |
#EC50 is given in nM or µM (as indicated) and as pEC50 as logEC50 ± SEM.
&controls were VP at V2R, V1aR, V1bR and OT at OTR.
I8-arachnotocin is a competitive antagonist at human V1aR
Based on the lack of V1aR activation, we performed an antagonist screen of I8-arachnotocin across all four receptors (Fig. 4a). Receptor stimulation with VP (0.55 nM) in the presence of 1 and 10 µM I8-arachnotocin, yielded lower IP1 levels than VP alone (Student’s t-test, p = 0.0226). The concentration-response curves of VP on V1aR in the absence and presence of I8-arachnotocin (1 µM, 3 µM, 10 µM) indicated an I8-arachnotocin-mediated dextral shift of the potency proportional to its concentration, without affecting Emax, typical for a competitive antagonist (Fig. 4b). The dextral shift was evaluated via Schild regression analysis26 yielding a linear regression slope of 1.28 ± 0.02 and a pA2 of 6.253 (~558 nM), thus demonstrating that I8-arachnotocin is a competitive antagonist of the V1aR.
Figure 4.

I8-arachnotocin is a competitive antagonist at V1aR. (a) Accumulation of second messengers (IP1 and cAMP) with partial activating concentrations of endogenous ligands (50 nM OT at OTR; 1 nM VP at V1aR; 3 nM VP at V1bR; 0.5 pM at V2R) in the absence and presence of 1 or 10 µM I8-arachnotocin in comparison to a saturating concentration of endogenous ligand (10 µM). All data were normalized to second messenger accumulation above baseline (0%) and maximum (100%) activity of the endogenous ligand. The dashed line depicts IP1/cAMP accumulation in the absence of I8-arachnotocin. Error bars depict SEM. n = 3, except n = 2 for V1aR (error bars depict SD). The asterisk (*) indicates significance in Student’s t-test (p = 0.0226). (b) Accumulation of IP1 by stimulation of human V1aR with VP (10 pM – 10 µM) alone or in the presence of 1, 3 or 10 µM I8-arachnotocin. Receptor activation was normalized to the accumulation of IP1 above baseline. Data points were normalized to the maximum (100%) and minimum (0%) response generated by VP. Error bars depict SEM; n = 3. Insert: Schild regression analysis: A = EC50 of VP in presence of I8-arachnotocin; A′ = EC50 of VP; B = logarithm of I8-arachnotocin concentration; Schild slope 1.28 ± 0.02 (SEM), R2 = 0.9998. The dotted line represents a reference line with a slope of 1; n = 3.
I8-arachnotocin does not induce β-arrestin-1 or -2 recruitment at V2R
With ligand bias becoming more relevant in understanding (patho)physiological responses, we also measured I8-arachnotocin-induced β-arrestin-2 recruitment. A concentration of 10 µM of the endogenous ligand induced rapid recruitment of β-arrestin-2 across all human OT/VP receptors, as judged by the increasing BRET signal from basal to maximum in 50–100 s. However, this effect was absent upon stimulation with 10 µM of I8-arachnotocin at OTR-, V1aR- and V2R-expressing cells (Fig. 5).
Figure 5.

Kinetics of OT/VP- and I8-arachnotocin-induced β-arrestin-2 recruitment at human OT/VP receptors. BRET was monitored between Nano-luciferase (Nluc) and EGFP introduced at the C-terminus of (a) OTR, (b) V1aR, (c) V1bR and (d) V2R (EGFP-OT/VP receptors) and the β-arrestin-2 (β-arrestin-2-Nluc). HEK293 cells co-expressing EGFP-OT/VP receptors and β-arrestin-2-Nluc were stimulated by 10 µM of VP, OT, and I8-arachnotocin respectively, 5 min after addition of the luciferase substrate (furimazine). The results are shown as differences in the BRET signals in the presence of ligands and are expressed as the mean value ± SD; n = 2.
To further explore this effect, we measured concentration-response curves of β-arrestin-2 recruitment across all four receptors (Fig. 6, Table 2). At the V2R, I8-arachnotocin did not recruit β-arrestin-2 up to a concentration of 100 µM, in contrast to VP which recruited β-arrestin-2 with an EC50 of 60 nM (logEC50 = − 7.22 ± 0.07). At the V1bR, I8-arachnotocin recruited β-arrestin-2 (Emax = 65%) with an EC50 of 1.2 µM (logEC50 = − 5.92 ± 0.14), compared to VP, which recruited β-arrestin-2 with an EC50 of 6.9 nM (logEC50 = − 8.16 ± 0.09). At the OTR, I8-arachnotocin recruited β-arrestin-2 with low efficacy (Emax = 32%) with an EC50 of 5.7 µM (logEC50 = − 5.24 ± 0.26), compared to OT which recruited β-arrestin-2 with an EC50 of 170 nM (logEC50 = − 6.77 ± 0.15). At the V1aR, no I8-arachnotocin-induced β-arrestin-2 recruitment was detected at a concentration up to 100 µM, in contrast to VP-induced β-arrestin-2 recruitment with an EC50 of 28 nM (logEC50 = –7.55 ± 0.12); this aligned with our findings of I8-arachnotocin being a competitive antagonist at this receptor subtype. We furthermore quantified bias on OTR and V1bR following the method outlined by Kenakin27, but did not detect differences compared to OT or VP.
Figure 6.

Differences between I8-arachnotocin- vs. VP-induced β-arrestin-2 recruitment at the human V2R. Concentration response curves of I8-arachnotocin and OT/VP at (a) OTR, (b) V1aR, (c) V1bR and (d) V2R using HEK 293 cells co-expressing EGFP-tagged receptors and β-arrestin-2-Nluc. Cells were pretreated with furimazine and measurements were taken 5 min after addition of ligands. Ligand-induced BRET was calculated as: (emission EGFPligand/emission NLucligand) − (emission EGFPHBSS/emission NLucHBSS). Results were normalized to β-arrestin-2 recruitment in response to OT/VP. Data points were fitted by nonlinear regression curves (sigmoidal, slope = 1); error bars indicate SEM; n = 3.
Table 2.
Potency and efficacy (β-arrestin recruitment) of I8-arachnotocin at human receptors.
| I8-arachnotocin | OT/VP& | |||||
|---|---|---|---|---|---|---|
| EC50 | pEC50 | Emax# | EC50 | pEC50 | Emax# | |
| V2Rβ-arr1 | n.d. | 56 nM | −7.25 ± 0.07 | 100% | ||
| V2Rβ-arr2 | n.d. | 60 nM | −7.22 ± 0.07 | 100% | ||
| V1bR | 1.2 µM | −5.92 ± 0.14 | 65 ± 5% | 6.9 nM | −8.16 ± 0.09 | 100% |
| V1aR | n.d. | 28 nM | −7.55 ± 0.12 | 100% | ||
| OTR | 5.7 µM | −5.24 ± 0.26 | 32 ± 5% | 170 nM | −6.77 ± 0.15 | 100% |
#EC50 is given in nM or µM (as indicated) and as pEC50 as logEC50 ± SEM.
&controls were VP at V2R, V1aR, V1bR and OT at OTR; n.d., not detectable; β-arr, β-arrestin.
Since recent studies uncovered an overlapping role of β-arrestin-1 and β-arrestin-2 with regard to V2R-dependent agonist-induced endocytosis and ERK activation28, we probed whether I8-arachnotocin is capable of recruiting β-arrestin-1 in a BRET-based assay. In comparison to VP (10 µM), which robustly induced β-arrestin-1 recruitment, I8-arachnotocin (10 µM) also failed to recruit β-arrestin-1 in a time-dependent manner (Fig. 7a). Moreover, VP treatment resulted in a concentration-dependent β-arrestin-1 recruitment with an EC50 of 56 nM (logEC50 = –7.25 ± 0.07), but receptor stimulation with I8-arachnotocin exhibited no β-arrestin-1 recruitment up to 100 µM (Fig. 7b). Overall, data obtained by BRET studies clearly demonstrate that modulation of the V2R with I8-arachnotocin results in neither recruitment of β-arrestin-1 nor β-arrestin-2, thereby confirming bias of this ligand towards G protein-coupling.
Figure 7.

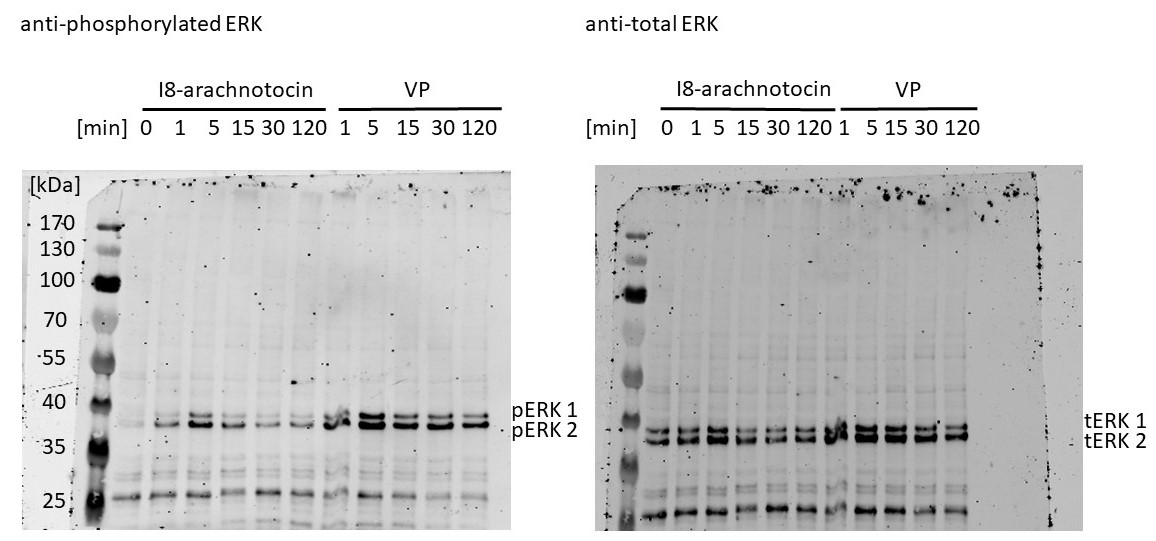
Characterization of β-arrestin-1 recruitment and ERK 1/2 phosphorylation induced by I8-arachnotocin vs. VP at the human V2R. (a) Kinetic profile of VP- and I8-arachnotocin-mediated β-arrestin-1 recruitment in HEK293 cells co-expressing EGFP-V2R and β-arrestin-1-Nluc. Cells were stimulated by 10 µM of VP or I8-arachnotocin, respectively, 5 min after addition of the luciferase substrate (furimazine). The results are shown as differences in the BRET signals in the presence of ligands and are expressed as the mean value ± SD; n = 2. (b) Concentration-response curves of VP and I8-arachnotocin at V2R using HEK 293 cells co-expressing EGFP-tagged V2R and β-arrestin-1-Nluc. Cells were pretreated with furimazine and measurements were taken 5 min after addition of ligands. Ligand-induced BRET was calculated as: (emission EGFPligand/emission NLucligand) − (emission EGFPHBSS/emission NLucHBSS). Results were normalized to β-arrestin-1 recruitment in response to VP. Data points were fitted by nonlinear regression curves (sigmoidal, slope = 1); error bars indicate SEM; n = 3. (c) Representative Western blot images of ERK 1/2 phosphorylation by stimulation of V2R with I8-arachnotocin vs. VP and (d) quantification of I8-arachnotocin- and VP-induced phosphorylated ERK 1/2 (pERK) relative to total ERK 1/2 (tERK) from four independent experiments (±SEM). Cells were transiently transfected with EGFP-V2R encoding plasmid and treated with 1 µM I8-arachnotocin or 1 µM VP at 37 °C for indicated time periods. Immunoblots were prepared from the same membranes using the same exposure method. Regions of interest were cropped from the full image (see Supplementary Information). Statistical significance was determined by Student’s t test (*P < 0.05; **P < 0.01; ns non-significant).
I8-arachnotocin modulates V2R-mediated ERK signaling
Having established that I8-arachnotocin is a G protein-biased peptide ligand, we examined its ability to modulate V2R-mediated ERK 1/2 phosphorylation. Previous studies reported that V2R activates ERK by two different pathways hypothesized to be dependent on either Gαs or β-arrestin signaling with distinct temporal patterns; the early phase (<5 min) being mediated by the G protein-dependent pathway, while the later phase (>10 min) being β-arrestin-2 dependent29. More recently, a study utilizing a combination of CRISPR/Cas9 genome-editing and pharmaceutical inhibition to generate HEK293 cells with ‘zero functional G’ indicated that ERK signaling mediated by V2R may still be dependent upon G protein even at later time points at which it may also be β-arrestin-dependent30. HEK293 cells transiently transfected to express the human V2R were stimulated with 1 µM I8-arachnotocin and 1 µM vasopressin for time periods between 1 min and 2 h. The immunoblotting data demonstrate that the late phase of V2R-dependent ERK 1/2 activation induced by I8-arachnotocin is significantly different from the late phase of VP-stimulated ERK 1/2 activation (30–120 min; Fig. 7c,d). While both peptides provoked an early and rapid ERK 1/2 phosphorylation peaking at 5 min, I8-arachnotocin-stimulated ERK 1/2 phosphorylation decreased over time in contrast to VP-elicited ERK 1/2 activation that resulted in a more sustained and prolonged ERK 1/2 activation (Fig. 7c,d). These data further support that I8-arachnotocin is a biased peptide ligand that modulates ERK 1/2 activity by preferentially activating the early phase G protein-dependent pathway at the plasma membrane over the late phase pathway that is β-arrestin-dependent.
Discussion
Peptides are gaining momentum in the drug development field, due to their (i) ability to interact with proteins on a large surface and (ii) structural and chiral complexity, which allows for improved discrimination between highly homologous targets as compared to small molecules14. This is particularly the case for the OT/VP signaling system, where multiple small molecule drugs failed due to selectivity issues and the majority of approved therapeutics are peptide drugs15,31. The study of the complex signaling pathways of the widely-distributed and fundamental OT/VP signaling system remains however challenging due to the limited availability of pharmacological probes14,15,18. Differentiating between signaling events that occur pre or post β-arrestin recruitment has become an important focus32–34 for studies looking at understanding the (patho)physiological roles of β-arrestin-mediated receptor internalization, desensitization and trafficking35,36. In addition, β-arrestin-dependent signaling has been linked to chronic stress-evoked melanoma metastasis via OTR37, increased neonatal rat cardiac fibroblast proliferation via V1aR38, morphine tolerance via V1bR39 and sustained non-canonical signaling after receptor internalization via V2R, resulting in strong antidiuretic and anti-natriuretic effects40. Biased ligands such as I8-arachnotocin discovered in this work are thus important tools to advance our understanding in these areas of interest.
By utilizing a drug discovery strategy14,41 on the synthesis of evolutionarily-conserved, yet distinct, peptides, we were able to bypass the time- and resource-consuming fractionation, isolation and identification steps associated with the discovery of plant- or venom-derived compounds such as kalata B723, inotocin19 and conopressin T22. This strategy led us to explore the vast and untapped arthropod kingdom and resulted in the discovery and pharmacological characterization of I8-arachnotocin.
I8-arachnotocin activated the Gαs (cAMP) pathway, inducing ERK 1/2 phosphorylation without detectable recruitment of β-arrestin-1 or -2 at V2R, despite the capability of this receptor to form stable and strong interactions with β-arrestins42. These findings are consistent with the observation that I8-arachnotocin induced substantially lower levels of ERK 1/2 phosphorylation at later time points compared to VP, leading us to conclude that I8-arachnotocin displays a clear bias away from β-arrestin-dependent signaling at V2R. We are not aware of another V2R ligand capable of selectively activating the non-β-arrestin-dependent (EC50 = 50 nM) vs. the β-arrestin-dependent (EC50 > 100 µM) signaling pathway (>2,000-fold difference). Such biased ligands are highly sought-after for research tools that allow for the discrimination between multiple active conformations of GPCRs32,33. Since the pharmacology of the β-arrestin-1 and -2 pathways in respect to the OT and VP receptors is not fully elucidated yet43,44, I8-arachnotocin represents a valuable first probe to advance our understanding of this pathway at the human V2R.
Our data suggest that I8-arachnotocin can only effectively recruit β-arrestin-2 at the V1bR (Emax = 65%). To try to understand the structural differences resulting in bias between the four receptors, we compared the binding site residues of the V1bR vs. the OTR, V1aR and V2R; there are two positions that differ in these receptors, i.e. position 7.30 (Thr vs. Glu) and 7.42 (Asn vs. Ser)19. Although there are only a limited number of reports dealing with structural changes in GPCRs responsible for arrestin recruitment, it has been suggested for instance that residues in TM6 and TM7 are important for pathway selectivity45,46. Hence, the identified residues in positions 7.30 and 7.42 of the ligand binding pocket of OT/VP receptors, could contribute to the observed bias of the bound I8-arachnotocin ligand, by altering the interaction of the receptor C-tail with the N-terminal domain of arrestin47. However, this remains speculative until future studies reveal further details.
Overall, GPCRs are prime drug targets48 and the vast chemical diversity of nature will continue to deliver novel pharmacological and therapeutic leads, particularly with technological advances that accelerate the drug discovery pipeline49. This work follows this innovative trend by exploiting the evolutionary conservation and ubiquity of neuropeptides across the animal kingdom50,51. In particular, it highlights the abundance of neuropeptides in arthropods: e.g., there are >50–150 neuropeptides reported in the model species Tribolium castaneum52, Nasonia vitripennis53, Apis mellifera54 and Drosophila melanogaster55. We thus argue that arthropods represent a novel, vast and untapped source for the discovery of pharmacological probes and potential therapeutic leads for a broad range of signaling systems.
Supplementary information
{kind=link}
Acknowledgements
This research has been supported by the Austrian Science Fund (FWF) through projects I3243 and P32109 and by an Australian Research Council Future Fellowship (FT140100730) awarded to CWG. MM was supported by the European Research Council under the European Union’s Horizon 2020 research and innovation program (714366), by the Australian Research Council (DE150100784, DP190101667), and by the Vienna Science and Technology Fund (WWTF; LS18–053).
Author contributions
C.W.G. designed research. L.D., J.G., E.M., P.K., H.C.M. and M.M. performed research. L.D., J.G., E.M., P.K., K.D.G.P., M.M. and C.W.G. analyzed data. All authors wrote the paper and approved the final version.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Leopold Duerrauer and Edin Muratspahić.
Supplementary information
is available for this paper at 10.1038/s41598-019-55675-w.
References
- 1.Gruber CW, O’Brien M. Uterotonic plants and their bioactive constituents. Planta Med. 2011;77:207–220. doi: 10.1055/s-0030-1250317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jurek B, Neumann ID. The Oxytocin Receptor: From Intracellular Signaling to Behavior. Physiological Reviews. 2018;98:1805–1908. doi: 10.1152/physrev.00031.2017. [DOI] [PubMed] [Google Scholar]
- 3.Henderson KK, Byron KL. Vasopressin-induced vasoconstriction: two concentration-dependent signaling pathways. J Appl Physiol. 2007;102:1402–1409. doi: 10.1152/japplphysiol.00825.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCormick SD, Bradshaw D. Hormonal control of salt and water balance in vertebrates. Gen Comp Endocrinol. 2006;147:3–8. doi: 10.1016/j.ygcen.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 5.Marsh AA. The Caring Continuum: Evolved Hormonal and Proximal Mechanisms Explain Prosocial and Antisocial Extremes. Annu Rev Psychol. 2019;70:347–371. doi: 10.1146/annurev-psych-010418-103010. [DOI] [PubMed] [Google Scholar]
- 6.Donaldson ZR, Young LJ. Oxytocin, Vasopressin, and the Neurogenetics of Sociality. Science. 2008;322:900–904. doi: 10.1126/science.1158668. [DOI] [PubMed] [Google Scholar]
- 7.Liu Y, et al. Oxytocin modulates social value representations in the amygdala. Nature Neuroscience. 2019;22:633–641. doi: 10.1038/s41593-019-0351-1. [DOI] [PubMed] [Google Scholar]
- 8.Stoop R. Sniffing and Oxytocin: Effects on Olfactory Memories. Neuron. 2016;90:431–433. doi: 10.1016/j.neuron.2016.04.033. [DOI] [PubMed] [Google Scholar]
- 9.Insel TR, Young LJ. The neurobiology of attachment. Nat Rev Neurosci. 2001;2:129–136. doi: 10.1038/35053579. [DOI] [PubMed] [Google Scholar]
- 10.Parreiras ESLT, et al. Functional New World monkey oxytocin forms elicit an altered signaling profile and promotes parental care in rats. Proc Natl Acad Sci USA. 2017;114:9044–9049. doi: 10.1073/pnas.1711687114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carter CS. The Oxytocin-Vasopressin Pathway in the Context of Love and Fear. Frontiers in Endocrinology. 2017;8:356. doi: 10.3389/fendo.2017.00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeMayo MM, Song YJC, Hickie IB, Guastella AJ. A Review of the Safety, Efficacy and Mechanisms of Delivery of Nasal Oxytocin in Children: Therapeutic Potential for Autism and Prader-Willi Syndrome, and Recommendations for Future Research. Paediatric Drugs. 2017;19:391–410. doi: 10.1007/s40272-017-0248-y. [DOI] [PubMed] [Google Scholar]
- 13.Goldman M, Marlow-O’Connor M, Torres I, Carter CS. Diminished plasma oxytocin in schizophrenic patients with neuroendocrine dysfunction and emotional deficits. Schizophr Res. 2008;98:247–255. doi: 10.1016/j.schres.2007.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gruber CW, Koehbach J, Muttenthaler M. Exploring bioactive peptides from natural sources for oxytocin and vasopressin drug discovery. Future Med Chem. 2012;4:1791–1798. doi: 10.4155/fmc.12.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manning M, et al. Oxytocin and vasopressin agonists and antagonists as research tools and potential therapeutics. J Neuroendocrinol. 2012;24:609–628. doi: 10.1111/j.1365-2826.2012.02303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McQuaid RJ, McInnis OA, Abizaid A, Anisman H. Making room for oxytocin in understanding depression. Neurosci Biobehav Rev. 2014;45:305–322. doi: 10.1016/j.neubiorev.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 17.Meyer-Lindenberg A, Domes G, Kirsch P, Heinrichs M. Oxytocin and vasopressin in the human brain: social neuropeptides for translational medicine. Nat Rev Neurosci. 2011;12:524–538. doi: 10.1038/nrn3044. [DOI] [PubMed] [Google Scholar]
- 18.Muttenthaler Markus, Andersson Åsa, Vetter Irina, Menon Rohit, Busnelli Marta, Ragnarsson Lotten, Bergmayr Christian, Arrowsmith Sarah, Deuis Jennifer R., Chiu Han Sheng, Palpant Nathan J., O’Brien Margaret, Smith Terry J., Wray Susan, Neumann Inga D., Gruber Christian W., Lewis Richard J., Alewood Paul F. Subtle modifications to oxytocin produce ligands that retain potency and improved selectivity across species. Science Signaling. 2017;10(508):eaan3398. doi: 10.1126/scisignal.aan3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Giglio MG, et al. Development of a human vasopressin V1a-receptor antagonist from an evolutionary-related insect neuropeptide. Sci Rep. 2017;7:41002. doi: 10.1038/srep41002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gruber CW. Physiology of invertebrate oxytocin and vasopressin neuropeptides. Exp Physiol. 2014;99:55–61. doi: 10.1113/expphysiol.2013.072561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stafflinger E, et al. Cloning and identification of an oxytocin/vasopressin-like receptor and its ligand from insects. Proc Natl Acad Sci USA. 2008;105:3262–3267. doi: 10.1073/pnas.0710897105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dutertre S, et al. Conopressin-T from Conus tulipa reveals an antagonist switch in vasopressin-like peptides. Journal of Biological Chemistry. 2008;283:7100–7108. doi: 10.1074/jbc.M706477200. [DOI] [PubMed] [Google Scholar]
- 23.Koehbach J, et al. Oxytocic plant cyclotides as templates for peptide G protein-coupled receptor ligand design. Proc Natl Acad Sci USA. 2013;110:21183–21188. doi: 10.1073/pnas.1311183110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keov P, Liutkeviciute Z, Hellinger R, Clark RJ, Gruber CW. Discovery of peptide probes to modulate oxytocin-type receptors of insects. Sci Rep. 2018;8:10020. doi: 10.1038/s41598-018-28380-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nasrollahi-Shirazi S, Sucic S, Yang Q, Freissmuth M, Nanoff C. Comparison of the beta-Adrenergic Receptor Antagonists Landiolol and Esmolol: Receptor Selectivity, Partial Agonism, and Pharmacochaperoning Actions. J Pharmacol Exp Ther. 2016;359:73–81. doi: 10.1124/jpet.116.232884. [DOI] [PubMed] [Google Scholar]
- 26.Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. Br J Pharmacol Chemother. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S. A simple method for quantifying functional selectivity and agonist bias. ACS Chem Neurosci. 2012;3:193–203. doi: 10.1021/cn200111m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghosh E, et al. Conformational Sensors and Domain Swapping Reveal Structural and Functional Differences between β-Arrestin Isoforms. Cell Reports. 2019;28:3287–3299.e3286. doi: 10.1016/j.celrep.2019.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ren X-R, et al. Different G protein-coupled receptor kinases govern G protein and β-arrestin-mediated signaling of V2 vasopressin receptor. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:1448–1453. doi: 10.1073/pnas.0409534102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grundmann M, et al. Lack of beta-arrestin signaling in the absence of active G proteins. Nature Communications. 2018;9:341. doi: 10.1038/s41467-017-02661-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manning M, et al. Peptide and non-peptide agonists and antagonists for the vasopressin and oxytocin V1a, V1b, V2 and OT receptors: research tools and potential therapeutic agents. Progress in Brain Research. 2008;170:473–512. doi: 10.1016/S0079-6123(08)00437-8. [DOI] [PubMed] [Google Scholar]
- 32.Rahmeh R, et al. Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc Natl Acad Sci USA. 2012;109:6733–6738. doi: 10.1073/pnas.1201093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rankovic Z, Brust TF, Bohn LM. Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorganic and Medicinal Chemistry Letters. 2016;26:241–250. doi: 10.1016/j.bmcl.2015.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Porter-Stransky KA, Weinshenker D. Arresting the Development of Addiction: The Role of beta-Arrestin 2 in Drug Abuse. J Pharmacol Exp Ther. 2017;361:341–348. doi: 10.1124/jpet.117.240622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lefkowitz RJ. Seven transmembrane receptors: something old, something new. Acta Physiol (Oxf) 2007;190:9–19. doi: 10.1111/j.1365-201X.2007.01693.x. [DOI] [PubMed] [Google Scholar]
- 36.Terrillon, S., Barberis, C. & Bouvier, M. In Proc Natl Acad Sci U S A 101 1548–1553 (2004). [DOI] [PMC free article] [PubMed]
- 37.Ji H, et al. Oxytocin involves in chronic stress-evoked melanoma metastasis via beta-arrestin 2-mediated ERK signaling pathway. Carcinogenesis. 2019 doi: 10.1093/carcin/bgz064. [DOI] [PubMed] [Google Scholar]
- 38.Chen Y, et al. GRK2/beta-arrestin mediates arginine vasopressin-induced cardiac fibroblast proliferation. Clin Exp Pharmacol Physiol. 2017;44:285–293. doi: 10.1111/1440-1681.12696. [DOI] [PubMed] [Google Scholar]
- 39.Koshimizu TA, et al. Complex formation between the vasopressin 1b receptor, beta-arrestin-2, and the mu-opioid receptor underlies morphine tolerance. Nat Neurosci. 2018;21:820–833. doi: 10.1038/s41593-018-0144-y. [DOI] [PubMed] [Google Scholar]
- 40.Feinstein TN, et al. Noncanonical control of vasopressin receptor type 2 signaling by retromer and arrestin. J Biol Chem. 2013;288:27849–27860. doi: 10.1074/jbc.M112.445098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liutkeviciute Z, Koehbach J, Eder T, Gil-Mansilla E, Gruber CW. Global map of oxytocin/vasopressin-like neuropeptide signalling in insects. Sci Rep. 2016;6:39177. doi: 10.1038/srep39177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem. 2000;275:17201–17210. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- 43.Passoni, I., Leonzino, M., Gigliucci, V., Chini, B. & Busnelli, M. Carbetocin is a Functional Selective Gq Agonist That Does Not Promote Oxytocin Receptor Recycling After Inducing beta-Arrestin-Independent Internalisation. J Neuroendocrinol28, 10.1111/jne.12363 (2016). [DOI] [PMC free article] [PubMed]
- 44.Reversi A, et al. The oxytocin receptor antagonist atosiban inhibits cell growth via a “biased agonist” mechanism. J Biol Chem. 2005;280:16311–16318. doi: 10.1074/jbc.M409945200. [DOI] [PubMed] [Google Scholar]
- 45.Che T, et al. Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell. 2018;172:55–67.e15. doi: 10.1016/j.cell.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wingler LM, et al. Angiotensin Analogs with Divergent Bias Stabilize Distinct Receptor Conformations. Cell. 2019;176:468–478.e411. doi: 10.1016/j.cell.2018.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shukla AK, et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature. 2014;512:218–222. doi: 10.1038/nature13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hauser AS, Attwood MM, Rask-Andersen M, Schioth HB, Gloriam DE. Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 2017;16:829–842. doi: 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cvijic ME, Sum CS, Alt A, Zhang L. GPCR profiling: from hits to leads and from genotype to phenotype. Drug Discovery Today Technologies. 2015;18:30–37. doi: 10.1016/j.ddtec.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 50.Elphick Maurice R., Mirabeau Olivier, Larhammar Dan. Evolution of neuropeptide signalling systems. The Journal of Experimental Biology. 2018;221(3):jeb151092. doi: 10.1242/jeb.151092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoyle CH. Neuropeptide families and their receptors: evolutionary perspectives. Brain Res. 1999;848:1–25. doi: 10.1016/s0006-8993(99)01975-7. [DOI] [PubMed] [Google Scholar]
- 52.Li B, et al. Genomics, transcriptomics, and peptidomics of neuropeptides and protein hormones in the red flour beetle Tribolium castaneum. Genome Res. 2008;18:113–122. doi: 10.1101/gr.6714008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hauser F, et al. Genomics and peptidomics of neuropeptides and protein hormones present in the parasitic wasp Nasonia vitripennis. J Proteome Res. 2010;9:5296–5310. doi: 10.1021/pr100570j. [DOI] [PubMed] [Google Scholar]
- 54.Han B, et al. Quantitative Neuropeptidome Analysis Reveals Neuropeptides Are Correlated with Social Behavior Regulation of the Honeybee Workers. J Proteome Res. 2015;14:4382–4393. doi: 10.1021/acs.jproteome.5b00632. [DOI] [PubMed] [Google Scholar]
- 55.Wegener C, Gorbashov A. Molecular evolution of neuropeptides in the genus Drosophila. Genome Biol. 2008;9:R131. doi: 10.1186/gb-2008-9-8-r131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.