

Abstract
β2-adrenergic receptor (β2AR) agonists are clinically used to elicit rapid bronchodilation for the treatment of bronchospasms in pulmonary diseases such as asthma and COPD, both of which exhibit characteristically high levels of reactive oxygen species (ROS); likely secondary to over-expression of ROS generating enzymes and chronically heightened inflammation. Interestingly, β2AR has long-been linked to ROS, yet the involvement of ROS in β2AR function has not been as vigorously studied as other aspects of β2AR signaling. Herein, we discuss the existing body of evidence linking β2AR activation to intracellular ROS generation and importantly, the role of ROS in regulating β2AR function. The reciprocal interplay of the β2AR and ROS appear to endow this receptor with the ability to self-regulate signaling efficacy and ligand binding, hereby unveiling a redox-axis that may be unfavorably altered in pathological states contributing to both disease progression and therapeutic drug responses.
Keywords: G protein-coupled receptors, β2AR, reactive oxygen species, redox, cysteine-S-sulfenation
Graphical Abstract
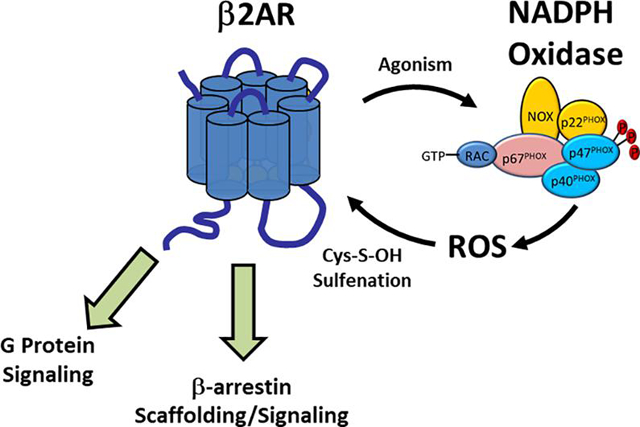
1. Introduction
The β2-adrenergic receptor (β2AR), a prototype of the G protein-coupled receptor (GPCR) superfamily, is ubiquitously expressed and involved in homeostasis of the cardiopulmonary, vascular, endocrine, digestive, and ocular systems, amongst others. Clinical use of β2AR-acting drugs is prevalent, perhaps most so within the setting of pulmonary pathologies such as asthma and chronic obstructive pulmonary disorder (COPD). Agonism of the β2AR in pulmonary tissue elicits rapid bronchodilation and is therefore the current gold standard for therapeutic treatment of acute bronchoconstriction and bronchospasms. In addition, pleotropic effects of β2-receptor agonism include decrease of mast cell degranulation, enhanced mucociliary transport, and reduced neutrophil recruitment, amongst others, and together, these outcomes have made short-acting β2-agonists (SABA) or long-acting β2-agonists (LABA) the mainstays for treatment of all stages of clinical severity of both asthma and COPD. Chronic inflammation is a hallmark of diseased pulmonary tissue due to perpetual activation of the immune system in response to either repeated exposure to an irritant or an inappropriate response of the immune system to a non-noxious stimuli [1]. Chronic inflammation of bronchial tissue results in resting bronchoconstriction and a hypersensitivity to irritants that cause episodes of severe bronchospasms [2]. Inhaled corticosteroids are used to reduce chronic inflammation in the lungs, however, inflamed asthmatic tissue is characterized by high levels of reactive oxygen species (ROS), and over-expression of the ROS generating enzyme NADPH oxidase (NOX) as well as the hydrogen peroxide generating enzyme superoxide dismutase (SOD) [3]. Interestingly, ROS can be generated by acute agonism of the β2AR, and these oxidants are also known to regulate receptor function. This review will summarize the growing body of literature linking this β2AR-ROS axis, and its potential role in human health and disease.
1.1. β2AR Signaling
As with other GPCRs, signaling of the β2AR is initiated upon agonist binding, promoting receptor GEF activity and guanine nucleotide exchange of the Gαs subunit, which facilitates downstream dissociation of the Gα and βγ subunits [4]. The GTP-bound Gαs subunit activates adenylyl cyclases, forming 3’,5’-cyclic monophosphate (cAMP), the second messenger that activates protein kinase A, which itself catalyzes phosphorylation of downstream targets leading to biological responses. Gαs signaling of agonist-occupied β2AR is effectively arrested upon the receptors phosphorylation by G protein-receptor kinases (GRK), notably GRK 2–3, leading to the high affinity recruitment of cytosolic β-arrestin partner proteins to the phosphorylated receptor [5]. β-arrestin-dependent desensitization of G protein signaling is followed by β-arrestin-dependent scaffolding, a well characterized cascade that can lead to further downstream physiological outcomes that include activation of other kinases, including mitogen-activated protein kinases (MAPK), transcription factors, and trafficking proteins [6–8], amongst others. While canonical G protein and β-arrestin signaling have been the subject of intense research efforts over the last three decades, β2AR has also been linked to ROS and redox cycles, yet this aspect has been largely overlooked.
1.2. Reactive Oxygen Species
ROS are transient and readily diffusible oxidants that are formed via metabolism of O2. ROS are ubiquitous throughout the human body and include hydrogen peroxide (H2O2), superoxide (O2−), hydroxide radical (OH•) and oxygen free radicals. Hydrogen peroxide is the most abundant and stable form of ROS [9–10], and given its pKa of 11.8, it is found predominately in the unionized state at physiological pH [11]. As a consequence, hydrogen peroxide can readily diffuse biological membranes [12], implying that the oxidative effects of these species are not limited to the cellular microdomain, or even the cell, of origin. While ROS are known to be formed from a variety of O2-metabolizing sources, including enzymes such as cyclooxygenase, xanthine oxidase, nitric oxide synthase and via mitochondrial respiration, within this review, we focus primarily on ROS generated via the membrane-bound NADPH oxidase (NOX) complex. In phagocytic cells, ROS are primarily generated via NOX, which is comprised of the membrane bound p22 and gp91phox subunits that function as O2-sensors, and the cytosolic p47phox and p67phox subunits, which catalyze electron transfer from NADPH to O2, forming superoxide (O2−). Due to its high reactivity and toxicity, superoxide is rapidly dismutated by superoxide dismutase (SOD), yielding hydrogen peroxide [13–16]. NOX activity is dependent on flavin cofactors, heme, and NADPH, while some catalytic NOX isoforms are also dependent on the small GTPase Rac1, which is recruited to the complex [13, 17]. Indeed, five distinct NOX isoforms (NOX1–5) have been characterized and shown to have variable distribution and regulation in non-phagocytic cells, where they play central roles in homeostatic ROS signaling as well as oxidative stress [18–19].
1.3. ROS mediated protein post-translational modification
While ROS have long been known to significantly affect nucleic acids [20–22], they also have underappreciated, yet pronounced, effects on protein structure and function. In this regard, ROS readily oxidize both methionine and cysteine residues, however, due to the ubiquitous expression of methionine sulfoxide reductase, methionine-ROS adducts are rapidly reduced back to the native thiol states [23]. On the contrary, no such specific reductase has been discovered for oxidized cysteine residues, endowing them with a more physiologically active function upon oxidation by ROS. Indeed, first order oxidation of cysteine residues has recently emerged as a transient form of post translational protein modification, much like phosphorylation and SUMOlation. Such oxidation has been termed cysteine-S-sulfenation, and converts the cysteine thiol (Cys-SH) to the oxidized cysteine sulfenic acid (Cys-S-OH) (Figure 1) [24]. Cysteine sulfenic acids can be reduced back into thiols or can react further to form other more stable species such as cysteine-cysteine disulfide bonds (Cys-S-S-Cys) (Figure 1). Additionally, in the presence of higher concentrations of or prolonged exposure to ROS, cysteine sulfenic acids can be further oxidized to form cysteine sulfinic (SO2H) or sulfonic (SO3H) acids, which unlike the transient and unstable sulfenic acid state, are stable and irreversible modifications typically associated with oxidative stress and damage [24]. Moreover, high order oxidation leaves the cysteine residues incapable of further interacting with ROS and undergoing transient, yet necessary, homeostatic redox reactions.
Figure 1. Redox dependent fates of cysteine residues.

Cysteine residues tonically exist in the thiol state with an oxidation state of −2. First order oxidation of cysteine residues via cysteine-S-sulfenation (A) is a transient oxidative modification leading to an oxidation state of 0. Sulfenic acids can be further oxidized to sulfinic (B) or sulfonic (C) acids, both of which are irreversible modifications, that lead to oxidation states of +2 and +4, respectively. Experimentally, a sulfenic acid can be selectively alkylated by cyclic diketones such as dimedone [11, 24, 67–68], which physically blocks subsequent redox reactions (D). A sulfenic acid can also react with a free cysteine thiol to form a disulfide bond with an oxidation state of −1 (E), yielding a stable modification that is experimentally labile to reaction with reducing agents (F).
1.4. ROS mediated signal transduction
ROS have historically been associated with cytotoxic and oxidative stress effects, yet, over the last two decades, a growing body of evidence has demonstrated that ROS play purposeful roles in intracellular signal transduction [25–26]. The compartmentalization of ROS generating enzymes and anti-oxidants (i.e. SOD, catalase, glutathione peroxidase, thioredoxin peroxidase) that metabolize cellular oxidants such as hydrogen peroxide [27–29], endows the cell with mechanisms to regulate ROS concentrations in specific cellular microdomains [27, 30]. The relative concentration of hydrogen peroxide associated with signal transduction is significantly lower than that associated with oxidative stress or cytotoxicity [31], however the physiological threshold for ROS signaling, versus oxidative stress, is tissue specific and variable [27, 32]. Regardless, ROS are now well established to play signaling roles in a wide variety of effector systems including mitogen-activated protein kinases (MAPK) [33–34], receptor tyrosine kinases, including EGFR and PDGF [35–38], transcription factors, including HIF-1 [39–42], as well as protein phosphatases [43–46], amongst others [25–26]. ROS have also been linked to GPCR function, with evidence of the angiotensin II-1R, dopamine D5R, and 5-HT2AR all being connected to ROS generation and/or signaling [39–42]. In addition, the literature is replete with data connecting β2AR and ROS, suggesting both the vitality of ROS for receptor function as well as a high propensity for permeant oxidative modifications, discussed below.
2. ROS are necessary for canonical β2AR function
The evidence indicating a functionally relevant interplay between ROS and the β2AR first surfaced in the 1980s when it was shown that a plethora of redox active substances could modulate β2AR function and binding of the non-selective β-adrenoreceptor agonist isoproterenol (ISO) [47], and that high affinity binding of related phenylethanolamine-backbone agonists to β2AR is dependent on redox [48]. Soon thereafter, it was shown that alkylation of β2AR cysteine residues resulted in a receptor that is locked in an agonist-receptor-G-protein complex [49], suggesting that normal receptor function requires redox capable cysteine residues. This line of research however appeared to have gone cold after these experiments and were not rekindled until the early 2000s. Using a pharmacological inhibitor-based approach that utilized the flavin-oxidase (i.e., NOX) inhibitor diphenyleneiodonium chloride (DPI), Rac1 inhibitor NSC23766 (NSC), and the ROS scavenger N-acetyl-L-cysteine (NAC) in HEK293 cells, which endogenously express β2, but not β1AR, Moniri and Daaka [50] demonstrated that reduction in intracellular ROS abolishes the receptors ability to activate Gαs proteins upon agonism with ISO [50], indicating ROS facilitate either the physical interaction of the receptor with Gαs or the ability of the receptor to successfully activate the G protein. Assessment of downstream Gαs protein mediated cAMP formation and cAMP phosphorylation activity were also significantly inhibited by NOX and Rac1 inhibition, and ROS scavenging, suggesting a broader requirement for ROS in canonical β2AR signaling [50].
Since G-protein mediated β2AR signaling is effectively desensitized by β-arrestin recruitment, which facilitates downstream β-arrestin scaffolding and subsequent signals, including receptor internalization, the requirement of ROS for β-arrestin-dependent effects have also been assessed. In this context, Shenoy and colleagues elegantly established that ISO-induced β2AR-signaling to phosphorylation of the extracellular-signal regulated kinases-1/2 (ERK1/2) MAPKs is mediated by Gαs-protein signaling at early time points (1–10 min) following agonism, whereas a more sustained phospho-ERK1/2 signal is mediated solely by β-arrestin scaffolding 10–60 min following agonism [7]. Based on these results, treatment of cells with DPI or NAC caused a significant decrease in ISO-induced phosphorylation of ERK1/2 during both the transient Gαs and the sustained β-arrestin-mediated time course following β2AR agonism [51], suggesting that ISO-induced β2AR-β-arrestin signaling is also regulated by ROS. Indeed, DPI also inhibited the ISO-induced physical interaction between β-arrestin and β2AR, as detected by both immunoprecipitation and BRET between β2AR-Rluc and β-arrestin-2-YFP [51]. These data are also in accordance with others that show that β2AR phosphorylation and internalization, which precede and follow β-arrestin recruitment respectively, are inhibited by ablation of ROS [50], and together, these results demonstrate that a degree of ROS are strictly required for canonical signaling of β2AR via both G-protein dependent and β-arrestin-dependent pathways.
3. β2AR Mediated ROS Generation
Given this apparent dependency of β2AR on ROS to execute all aspects of proper function, it is not surprising that agonism of the β2AR generates its own ROS supply. ROS generation appears to be a conserved, yet specialized function of cell-surface receptors, as several different receptors, including GPCRs, have been shown to generate ROS in response to agonism, including; the T cell receptor [52], α1A-adrenergic receptor [50], angiotensin II receptor [44], both 5HT-1A and −2A subclasses [45–46], as well as β2AR. Agonist mediated ROS generation through the β2AR has been demonstrated in a myriad of cells and tissues including; osteoclasts [53], aortic cells [54], microglia [55], cardiomyocytes [56–57], alveolar macrophages [58], human lung airway epithelial cells (CALU3) [59], RAW264.7 macrophages [53], and the clonal cell lines COS-7 [57] and HEK293 [50–51, 60], as discussed in detail below. These results demonstrate that the mechanisms of ROS generation that follow receptor agonism and the signaling outcomes of such ROS, are diverse and likely cell-type dependent. For example, in mature osteoclasts and precursor macrophages, including murine RAW264.7 cells, which express β2AR, but not β1- or β3- ARs, agonism by ISO significantly increased ROS generation and this effect directly mediated differentiation and osteoclastogenesis, which was blocked by propranolol, demonstrating β2AR-dependency in these cells [53].
In primary microglia, the β2-selective and LABA salmeterol dose-dependently increased intracellular ROS as well as extracellular O2− via activation of NOX in a manner dependent on downstream ERK1/2, but not PKA, activity [55]. These findings suggested that β2AR-mediated ROS generation contributes to dopaminergic neurotoxicity and reveal that the lower-efficacy partial agonist salmeterol, as opposed to a fully efficacious agonist like ISO, could also induce ROS generation. However, though salmeterol is highly selective for β2AR over β1- and β3-receptors, and the effect of salmeterol on the downstream neurotoxicity was fully blocked by the selective β2AR-antagonist ICI-118,551, the study did not examine the effect of the antagonist on ROS generation, which is proximal to neurotoxicity.
In rabbit ventricular cardiomyocytes, ISO was shown to facilitate significant intracellular mitochondrially produced ROS generation in a manner dependent on electrical conductance, as no effect was seen in quiescent myoctes [56]. However, no distinction was made regarding whether this effect was due to agonism of β1- or β2AR. Importantly, in primary rat cardiomyocytes, ISO was also shown to increase both mitochondrial oxygen and intracellular ROS, and in a manner that was blocked by ICI-118,551, but not by the β1AR-selective antagonist CGP20712A. These effects were also reproduced with the selective β2AR partial agonist zinterol [57] and in the clonal COS-7 cell line transfected with β2AR [57]. Similarly, ROS generation was heightened upon treatment of mouse thoracic aortic slices, which express both β1- and β2AR, with ISO, and this effect was significantly decreased, but in this case, not fully abolished in slices derived from β2AR-knockout animals [54], suggesting involvement of both subtypes.
In addition to COS-7 clonal cell lines, studies in HEK293 cells, which endogenously express β2- but not β1- or β3-AR, as well as in HEK293 cells that transiently overexpress β2AR reveal similar levels of ROS generation upon agonism with ISO, an effect that is blocked by propranolol, used in this instance due to the lack of expression of other beta-adrenoreceptors [50–51].
Consistent amongst these studies, short term agonism (up to 5 minutes) of βAR elicits a relatively modest increase in ROS, typically on the order of a 30–50% over control [50–51, 53, 57, 60], an extent equivalent to approximately 10 μM exogenous hydrogen peroxide [50–51]. However, longer agonist exposures elicit higher levels of ROS generation, a 250% increase was shown in microglial cells following a 30 minute stimulation with salmeterol [55], and a time dependent ISO-induced ROS response was seen in cardiomyocytes [56]. This is an important context as it infers that β2AR-mediated ROS generation serves a purposeful signaling role, rather than an oxidative-stress inducing role, which would be expected to yield ROS concentrations equivalent to millimolar levels of H2O2. On the contrary, agonism of β2AR in human neutrophils with ISO or terbutaline was shown to negatively modulate the inhibitory effects of fMLP and platelet activating factor-mediated ROS generation [61–62], but these inhibitory effects are likely due to the known regulatory role PKA on neutrophil priming and function, as others have shown that agonism of β2AR with epinephrine in neutrophils decreases extracellular ROS, while increasing intracellular ROS generation [63].
β2AR mediated ROS generation is overwhelmingly believed to be NADPH oxidase (NOX) mediated, as several groups have shown that inhibition of NOX abolishes ISO-induced ROS generation [50–51, 55–56, 60]. Though the definitive mechanism remains to be elucidated, current evidence suggests that the β2AR-NOX linkage may be facilitated by β2AR-β-arrestin signaling to activation of Rac1 [51, 60, 64], which is essential to the activity of some NOX isoforms (Figure 2) [60]. This hypothesis is in agreement with data demonstrating that NSC, a Rac1-GEF inhibitor, decreases both β2AR ROS generation and β2AR signaling [50–51]. Additionally, β2AR activation of ERK1/2, via G-protein signaling or β-arrestin scaffolding, can induce p47PHOX phosphorylation, a modification that initiates NOX complexation [13–14], ultimately acting as another route of β2AR mediated NOX activation and therefore ROS production [55]. Other, more speculative mechanisms may involve EGFR-1 transactivation, tyrosine protein kinase c-Src activation, or cross-talk to PKC, all of which are known to regulate NOX activity (figure 2). As β2AR signaling is studied in additional cell lines and primary cells, the exact nature of the ROS generation signal will likely be found to be variable and cell-type specific.
Figure 2. The β2AR-ROS feedback loop is dependent upon cysteine redox activity.

Known or hypothesized mechanisms whereby β2AR agonism may induce NADPH oxidase-based ROS generation include β-arrestin scaffolding of Rac-1, c-Src, or ERK1/2, all of which are known to activate NOX. β2AR-mediated transactivation of EGFR via c-Src may also presumably activate NOX via PI3K activity, while activation of Gαs signaling following β2AR agonism can also facilitate ERK1/2 activity. Once generated by NOX, superoxide can be dismutated to H2O2, which can oxidize β2AR cysteine residues forming Cys-S-sulfenic acids, which are seemingly required to uphold β2AR function, including G protein-signaling and β-arrestin scaffolding.
4. Oxidative modification to the β2AR
Marches and Bicho first presented the hypothesis that cysteine residues lying at the interface of the β2AR-Gαs protein were critical to agonist-induced signals, which were likely redox-dependent [49]. Since superoxide and hydrogen peroxide have a high propensity to oxidize cysteine residues, these ROS can also greatly influence protein structure and function via post-translational modification of cysteines yielding sulfenic, sulfinic, and sulfonic acids as described above, as well as disulfide bonds, which are indispensable for protein function. Using a novel biotin-switch methodology that was selective for detection of S-sulfenic acids, it has been shown that treatment of cells transiently expressing β2AR with H2O2 leads to significant β2AR S-sulfenation in a time- and H2O2-concentration dependent manner [65]. Importantly, S-sulfenation of β2AR was also induced in a dose-dependent manner by agonism with ISO, an effect which was blocked by non-selective β-antagonist propranolol [65], and which suggests that β2AR-mediated ROS generation alone is sufficient to stimulate receptor S-sulfenation, as other β-AR isoforms are not expressed in the model used. Furthermore, β2AR S-sulfenation was inhibited by scavenging of ROS with NAC, and was also decreased in the presence of the selective Cys-sulfenic acid alkylator dimedone [65], demonstrating selectivity of the effect. The use of this probe also allows for selective labeling of Cys-Sulfenic acids (Fig. 1), which can subsequently be detected by antibody-based approaches [24]. More recently, dimedone-based derivatives that are biotin-conjugated (e.g., DCP-Bio1) or can be subsequently biotinylated by Click-chemistry (e.g., DYn-2) have been introduced that allow for easier labeling and detection [66–68]. Using these probes, we were able to demonstrate cysteine-S-sulfenation of β2AR in situ in both clonal cell lines and the human airway epithelial cell model CALU3, which endogenously expresses β2AR [59]. Furthermore, cysteine S-sulfenic acid labeling was increased by the addition of exogenous hydrogen peroxide demonstrating that β2AR S-sulfenation is sensitive to local oxidant concentration of the cellular milieu. The realization that β2AR can be modified by hydrogen peroxide from external sources highlights the strong likelihood that the overwhelmingly oxidative conditions exhibited in pulmonary disease state lung tissue are probably oxidatively modifying the resident β2ARs.
Importantly, in situ cysteine-S-sulfenation and subsequent ‘trapping’ of Cys-S-OH groups with dimedone appears to modify either receptor conformation or the ligand binding pocket itself, as redox modifying constituents significantly alter ligand binding [47, 59]. Interestingly ROS scavenging did not alter agonist or antagonist binding to the β2AR in isolated plasma membranes [50], but the addition of ROS (e.g. hydrogen peroxide) to membrane preparations significantly increased the available number of ligand binding sites (i.e., Bmax) [59]. Furthermore, alkylation of redox active cysteine residues, which prevent normal receptor cysteine redox conductance, not only reversed the oxidation induced increases in ligand binding, but completely abolished the ability of endogenous β2AR on human lung tissue to bind ligand [59]. Redox mediated effects on ligand binding were also seen live whole cells [59], indicating this is a physiologically relevant phenomenon that plausibly may alter binding of clinically used β2AR agents.
The consequences of cysteine S-sulfenation on cell signaling outcomes have also been assessed using the ‘dimedone-trapping’ based approach subsequent to oxidation (figure 2). Dimedone alkylation significantly impairs the maximal ISO-induced efficacy (i.e., Emax) towards cAMP formation, an effect that was observed both in clonal cell lines and physiologically relevant human airway epithelial cells [59]. This effect was also demonstrated to yield significantly decreased phosphorylation, and hence activation, of the downstream transcription factor cAMP response element-binding protein (CREB). Furthermore, dimedone alkylation of β2AR cysteine-S-sulfenic acids also significantly decreased both G-protein and β-arrestin mediated phosphorylation, and hence activation, of ERK1/2, confirming earlier results demonstrating that ROS/redox cysteine chemistry are critical towards all aspects of canonical β2AR signaling.
4.1. Implication of specific cysteine residues on redox effects
Identification of the exact cysteine residues on the β2AR that are S-sulfenated would have important implications for the understanding of the mechanisms of ROS mediated alterations to receptor function, however some speculation can be made based upon functional and mutational data already in the literature. The β2AR contains 13 cysteine residues, with five embedded in the transmembrane regions, four within the extracellular domains, one in the third intracellular loop, and three on the C-terminus [69]. Each cysteine residue has its own propensity for oxidation due to the local microenvironment, and further, a cysteine residue may be involved in multiple redox reactions resulting in various forms of cysteine modifications. However, once the residue is oxidized to a sulfinic or sulfonic acid, it is no longer redox active as these are generally considered irreversible modifications [24]. Within the current body of literature, it is not yet determined if this “loss of function” oxidative modification would be mimicked by mutation or alkylation. Some researchers mutate cysteine residues to serine/alanine, each of which would have its own unique chemical properties and plausibly mimicking one or more various states of cysteine oxidation. Trapping of sulfenic acids by selective alkylators, such as dimedone (Fig. 1), has been shown to induce loss of protein function [67] and therefore currently provides the only means to inactivate redox active cysteines on wild type proteins in oxidative environments.
Of the β2AR cysteine residues, Cys341 is unquestionably the most well characterized, as receptor function is heavily dependent upon its palmitoylation to the plasma membrane, forming a fourth intracellular pseudoloop [70–71]. The actual chemical reaction resulting in palmitoyl-modification of a cysteine residue requires a reduced thiolic cysteine [72], and although the formation of a cysteine S-sulfenic acid and subsequent reduction prior to palmitoylation is plausible in the case of this residue, the formation of an S-sulfinic or S-sulfonic acid would block further palmitoylation and yield a predominantly non-functional receptor in this case [71]. However, given that these modifications would need to occur prior to membrane insertion, as Cys341 palmitoylation occurs in the Golgi [73], it is extremely unlikely that Cys341 redox chemistry or S-sulfenation is a major player in the functional changes seen by impairing ROS modification. Conversely, receptor fate has been correlated to Cys265 intracellular palmitoylation [74], which occurs after receptor membrane localization and agonist-mediated internalization, suggesting that S-sulfenation or higher order oxidation of this residue may interfere with palmitoylation and could alter receptor fate.
Evaluation of data using thiol-reactive agents does shed light on the suitability of various cysteine residues towards oxidation and indicates that reactive residues are accessible by intracellular constituents (e.g., intracellular-facing or transmembrane but inward-facing) and exist, at least in some period of time, as free thiols. Cys77, Cys125, Cys265 and Cys327 have been shown to be available for such reactivity with thiol-reactive N-ethylmaleimide (NEM) [75], indicating these residues are susceptible to modification in situ. Moreover, the reactivity of Cys77 and Cys327 with alkylating agents depended upon agonist occupancy and correlated directly to agonist efficacy in generating cAMP [75]. Therefore, the redox state of these cysteines may alter the propensity of the receptor to achieve various states of activation and plausibly these residues could contribute to ROS sensitive G protein signaling. Interestingly, a Cys77Ser mutation does not alter cAMP formation despite this residues functional correlation to receptor activation [76], further implicating the specific chemistry of Cys77 plays an important role in receptor activation.
Cys285 resides in at the base of transmembrane 6, a region that moves 11–14Å upon agonist binding [77]. Moreover, mutation of Cys285 results in nearly a 40% decrease in the efficacy of ISO-stimulated cAMP formation [76], indicating Cys285 may play a prominent role in the ability of β2AR to achieve an activated conformation or to confer G protein activation. Interestingly, Cys327 also has been indicated as a functionally relevant residue, whereby Cys327Ser mutation demonstrated no impact on cAMP formation or ligand binding [76], but a Cys327Arg mutation at this residue causes a significant decrease in both of these measures [78]. Given a serine residue has similar physical properties to that of a cysteine thiol or sulfenic acid, whereas an arginine substitution causes more drastic changes in the residue’s ionization, solubility, and reactivity, there is strong evidence that alterations to the surrounding microenvironment at this loci may impact receptor function. Finally, Cys378 and Cys406 reside on the C-terminus, a region known to interact with effector proteins including β-arrestin. Cys406 lies adjacent to a known GRK2 phosphorylation site that comprises the β-arrestin sensor [5], and therefore oxidation of Cys406 may impact Ser407 phosphorylation, and subsequent signaling outcomes. While Cys378 and Cys406 have been shown to form an intramolecular disulfide bond during protein purification, it is thus far unclear if this disulfide bond exists in endogenous conditions in situ [79], and the likelihood of this may be low due to the intrinsic reducing nature of the cytosol. Nonetheless, the intracellular accessibility of these residues and their importance in mediating β2AR signaling will certainly warrant further examination of these residues as possible foci for Cys-S-sulfenation. Given the breadth of functional consequences ROS-β2AR interplay has on all stages of canonical receptor pharmacology, it is likely that more than one residue is participating in this phenomenon. Elucidation of the involved residues followed by redox sensitive functional analysis of each loci would shed light as to which residues implore a physiologically relevant role in receptor function. Furthermore, detailing how these specific involved residues may differ in healthy verses disease state pulmonary tissues, may mechanistically explain changes in the clinical presentation of patients with various respiratory dysfunction and/or those using β2AR agonists.
5. Clinical significance of the ROS-β2AR connection
All human cells are likely exposed to some level of hydrogen peroxide [32], though the homeostatic concentration and/or the concentration needed to elicit specific physiological responses is largely tissue specific [27]. A growing body of evidence suggests that hydrogen peroxide is necessary for normal cell physiology [50–52], however higher levels of ROS are correlated to pathological states of oxidative stress [31]. Traditionally, hydrogen peroxide was typically thought of as a byproduct of cellular respiration [10], however, given receptor agonism generates ROS production as described above, and that hydrogen peroxide is directly or otherwise generated by enzymes such as NOX [80], SOD [12, 27] and members of the electron transport chain [32], it is more likely that generation of hydrogen peroxide serves purposeful roles in cells, and indeed, it is now generally well-accepted that this ROS species serves as an oxidative signal transducer.
In the context of the β2AR, the ROS-receptor relationship presents as an interesting example of a full circle positive feedback loop, whereby agonism of the β2AR produces ROS, which feedback to oxidize β2AR and uphold canonical signaling. Conversely, higher order oxidation of Cys-S-sulfenic acids to S-sulfinic or S-sulfonic acids would be expected to eliminate this positive feedback loop, suggesting that the oxidant-dependent modulation of β2AR requires and creates a delicate redox balance. Though hydrogen peroxide is detectible in exhaled air from healthy individuals [32], the level of hydrogen peroxide exhaled is increased in individuals with lung disease such as asthma and COPD [32], likely secondary to the increase in inflammation and the upregulation of ROS generating machinery [3, 81]. These results suggest that the β2AR-ROS axis, specifically oxidation of β2AR, may already be inclined towards higher-order oxidation states in diseased lung tissue.
Clinically, the β2AR is primarily used as the drug target for short- or long- acting bronchodilators in patients with an existing pulmonary pathology. Given the results discussed here, treatment of bronchoconstriction with β2AR agonists stimulates a degree of ROS generation in tissue that is pathologically high in oxidant levels, likely increasing the oxidant load to levels correlated with oxidative damage and out of the range of physiologically necessary ROS signaling. Though agonism of β2AR on airway smooth muscle cells (ASMC) is predominately responsible for bronchodilation, inhalation of β2AR agonists first interacts with the airway epithelial cells (AEC), which also express a high density of β2AR [64, 82]. Moreover, the β2AR signaling specifically in the epithelium is sufficient to mediate the asthmatic phenotype, including characteristics such as airway hyper-responsiveness and inflammation [64]. While β2AR agonists can penetrate AEC to act directly on the underlying ASMC [83], activation of AEC β2AR also mediates therapeutic benefits through indirect pathways such as the opening of Cl− ion channels [84], release of nitric oxide and activation of guanylyl cyclase mediated opening of additional ion channels [85], or by attenuating cholinergic mediated airway narrowing [86]. Evidence is mounting that β2AR agonists may actually be increasing disease progression and mortality [86], and that use of these agonists further increase inflammation [64]. With the recent findings that human airway epithelial cells generate ROS following β2AR agonism [59], it is plausible that this symptomatic treatment is advancing the pathological states. Furthermore, given the β2AR is itself modified by ROS, and that proper β2AR function is dependent upon the receptor’s ability to transiently interact with ROS, agonist mediated ROS generation adding to the already oxidative environment of pulmonary disease state tissue is likely causing oxidative stress and irreversible modification of the β2AR itself, disabling the receptors crucial ability to transiently interact with local ROS, and importantly, decreasing downstream receptor signaling that facilitate clinical benefits. In this context, higher-order oxidation of β2AR may also putatively explain, at least in part, the mechanisms of β2AR-agonist tachyphylaxis, or tolerance to the bronchodilatory effects, that are seen in patients upon chronic use of these agents. Under high oxidative stress induced by underlying inflammatory lung pathology and hypothetically, by chronic β2-agonist/β2AR-induced ROS generation, irreversible oxidation of ROS-sensitive β2AR cysteines to Cys-Sulfenic or Cys-Sulfonic acids would ablate receptor function leading to tachyphylaxis of the responses.
Of important note, β-receptor antagonists are widely used clinically for treatment of cardiovascular diseases, and many of these agents are in fact non-selective partial agonists with intrinsic sympathomimetic activity at β1- and β2-ARs, although some are cardioselective and exhibit strong β1AR selectivity. Interestingly, although historically, β-blocker use has been avoided in patients with obstructive pulmonary disorders, observational studies have demonstrated that β-blocker use in cardiovascular disease patients with comorbid COPD leads to a 38% decreased risk of COPD exacerbation [87]. This somewhat paradoxical observation has led to a controversy regarding the use of these agents in COPD patients, with or without CV, particularly given the lack of randomized, controlled clinical trials. Meanwhile, cardioselective β1-blockers such as metoprolol have been used in such patients [88], yet, a recent double-blind, placebo-controlled, prospective and randomized clinical trial in patients with moderate or severe COPD, but without comorbid CV disease, revealed that metoprolol significantly increased the rate of severe COPD exacerbation requiring hospitalization, as well as those that required mechanical ventilation [89]. Interesting, while β-blocking agents have been utilized in a variety of in vitro and animal studies to reverse agonist-mediated ROS generation and downstream signals, there are no known studies on what, if any, role β-blockers may have on the β2AR-ROS axis, and if modulation of ROS accounts for any of the clinical benefits seen with these agents. Indeed, several β-blockers including carvedilol [90–91], nadolol [92–93], and atenolol [93] have been shown to directly modulate ROS levels, while both carvedilol and the cardioselective agent nebivolol are also capable as acting as ROS scavengers [94]. Whether ROS modulating properties contribute to the beneficial clinical effects of these agents in cardiovascular and pulmonary diseases is currently not known, but the deleterious effects of ROS in these systems is well described [3, 95], and may be suggestive of a therapeutic role. Needless to say, much more research is needed to determine what role, if any, ROS modulation plays in the benefits of β-blocker therapy, and since many of these agents are partial agonists, it is conceivable that they elicit some degree of ROS generation, as do the β2-partial agonists albuterol (also referred to as salbutamol), salmeterol [55], and zinterol [57].
While these hypothesis are based on observations described in the literature using cysteine-reactive probes, the site(s) of β2AR S-sulfenation, and whether or not these residues do in fact become over-oxidized to higher-order S-sulfenic/sulfonic states are not currently know. Furthermore, the oxidation states of β2AR cysteine residues in airway cells of individuals with chronic pulmonary dysfunction (e.g., asthma or COPD) compared to those of healthy individuals would unveil the role of disease state in oxidative modifications of β2AR. Investigation of oxidative changes associated in these cells in patients on β2AR-agonist therapies, compared to corticosteroid or anti-cholinergic therapies would also shed light on the clinical importance of the role of β2AR-oxidation. Moreover, since most studies in the field make use of the fully-efficacious agonist ISO, yet patients on β2-agonist therapies commonly utilize partial agonists such as albuterol or salmeterol, the clinical importance of agonist efficacy and antagonism of the more efficacious endogenous agonist (i.e., epinephrine) in the presence of these clinically used drugs, and their relationship to the ROS axis remains elusive and will require much further study. On a similar note, the clinical use of short-acting β2-agonists (e.g., albuterol) versus long-acting β2-agonists (e.g., salmeterol) may very well have differential influence on the β2AR-ROS axis, yet remains completely unexamined, and will also require investigation. Together, the results reviewed here suggest important roles of ROS in β2AR function; yet, the study of this axis is in its infancy and much work remains to be done in order to fully resolve its role in physiological and pathophysiological tissues, particularly within the pulmonary system.
Acknowledgments
This work was supported by NIH grant HL138603 to N.H.M.
Footnotes
Neither author has any conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Murdoch J, Lloys C, Chronic inflammation in asthma, Mutat Res 690(1–2) (2010) 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bush A Pathophysiological Mechanisms of Asthma. Front Pediatr. 7 (2019) 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sutcliffe A, Hollins F, Gomez E, Saunders R, Doe C, Cooke M, Challiss RA, Brightling CE, Increased nicotinamide adenine dinucleotide phosphate oxidase 4 expression mediates intrinsic airway smooth muscle hypercontractility in asthma, Am J Respir Crit Care Med 185(3) (2012) 267–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pierce KL, Premont RT, Lefkowitz RJ, Seven-transmembrane receptors, Nature reviews. Molecular cell biology 3(9) (2002) 639–50. [DOI] [PubMed] [Google Scholar]
- [5].Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, Strachan RT, Huang TY, Bressler EA, Hara MR, Shenoy SK, Gygi SP, Lefkowitz RJ, Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin, Science Signal 4(185) (2011) ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shenoy SK, Lefkowitz RJ, β-arrestin-mediated receptor trafficking and signal transduction, Trends in pharmacological sciences 32(9) (2011) 521–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ, β-arrestin-dependent G protein-independent ERK1/2 activation by the beta2 adrenergic receptor, J Biol Chem 281(2) (2006) 1261–73. [DOI] [PubMed] [Google Scholar]
- [8].Ma L, Pei G, β-arrestin signaling and regulation of transcription, J Cell Sci 120(Pt 2) (2007) 213–8. [DOI] [PubMed] [Google Scholar]
- [9].Reddie KG, Carroll KS, Expanding the functional diversity of proteins through cysteine oxidation, Curr Opin Chem Biol 12(6) (2008) 746–54. [DOI] [PubMed] [Google Scholar]
- [10].Paulsen CE, Carroll KS, Orchestrating redox signaling networks through regulatory cysteine switches, ACS Chem Biol 5(1) (2010) 47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gupta V, Carroll KS, Sulfenic acid chemistry, detection and cellular lifetime, Biochim Biophys Acta 1840(2) (2014) 847–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mumbengegwi DR, Li Q, Li C, Bear CE, Engelhardt JF, Evidence for a superoxide permeability pathway in endosomal membranes, Mol Cell Biol 28(11) (2008) 3700–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hordijk PL, Regulation of NADPH oxidases: the role of Rac proteins, Circ Res 98(4) (2006) 453–62. [DOI] [PubMed] [Google Scholar]
- [14].Bedard K, Krause KH, The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology, Physiol Rev 87(1) (2007) 245–313. [DOI] [PubMed] [Google Scholar]
- [15].Sumimoto H, Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species, Febs j 275(13) (2008) 3249–77. [DOI] [PubMed] [Google Scholar]
- [16].Nisimoto Y, Motalebi S, Han C, Lambeth JD, The p67phox activation domain regulates electron flow from NADPH to flavin in flavocytochrome b558, The Journal of Biochemistry 274(33) (1999) 22999–23005. [DOI] [PubMed] [Google Scholar]
- [17].Koga H, Terasawa H, Nunoi H, Takeshige K, Inagaki F, Sumimoto H, Tetratricopeptide repeat (TPR) motifs of p67(phox) participate in interaction with the small GTPase Rac and activation of the phagocyte NADPH oxidase, J Biol Chem 274(35) (1999) 25051–60. [DOI] [PubMed] [Google Scholar]
- [18].Bardaweel SK, Gul M, Alzweiri M, Ishaqat A, HA AL, Bashatwah RM, Reactive Oxygen Species: the Dual Role in Physiological and Pathological Conditions of the Human Body, The Eurasian journal of medicine 50(3) (2018) 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Schieber M, Chandel NS, ROS function in redox signaling and oxidative stress, Current biology : CB 24(10) (2014) R453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cadet J, Wagner JR, DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation, Cold Spring Harbor perspectives in biology 5(2) (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jena NR, DNA damage by reactive species: Mechanisms, mutation and repair, Journal of biosciences 37(3) (2012) 503–17. [DOI] [PubMed] [Google Scholar]
- [22].Srinivas US, Tan BWQ, Vellayappan BA, Jeyasekharan AD, ROS and the DNA damage response in cancer, Redox biology (2018) 101084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Moskovitz J, Methionine sulfoxide reductases: ubiquitous enzymes involved in antioxidant defense, protein regulation, and prevention of aging-associated diseases, Biochim Biophys Acta 1703(2) (2005) 213–9. [DOI] [PubMed] [Google Scholar]
- [24].Gupta V, Carroll KS, Profiling the Reactivity of Cyclic C-Nucleophiles towards Electrophilic Sulfur in Cysteine Sulfenic Acid, Chem Sci 7(1) (2016) 400–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Finkel T, Signal transduction by reactive oxygen species, J Cell Biol 194(1) (2011) 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Reczek CR, Chandel NS, ROS-dependent signal transduction, Curr Opin Cell Biol 33 (2015) 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Veal EA, Day AM, Morgan BA, Hydrogen peroxide sensing and signaling, Mol Cell 26(1) (2007) 1–14. [DOI] [PubMed] [Google Scholar]
- [28].Lo Conte M, Carroll KS, The redox biochemistry of protein sulfenylation and sulfinylation, Journal of Biochemistry 288(37) (2013) 26480–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Choi SE, Min SH, Shin HC, Kim HE, Jung MW, Kang Y, Involvement of calcium-mediated apoptotic signals in H2O2-induced MIN6N8a cell death, Eur J Pharmacol 547(1–3) (2006) 1–9. [DOI] [PubMed] [Google Scholar]
- [30].Choi MH, Lee IK, Kim GW, Kim BU, Han YH, Yu DY, Park HS, Kim KY, Lee JS, Choi C, Bae YS, Lee BI, Rhee SG, Kang SW, Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II, Nature 435(7040) (2005) 347–53. [DOI] [PubMed] [Google Scholar]
- [31].Wouters MA, Fan SW, Haworth NL, Disulfides as redox switches: from molecular mechanisms to functional significance, Antioxidants & Redox Signaling 12(1) (2010) 53–93. [DOI] [PubMed] [Google Scholar]
- [32].Halliwell B, M.B. C, Longa LH, Hydrogen peroxide in the human body, FEBS Letters 486(2000) (2000) 10–13. [DOI] [PubMed] [Google Scholar]
- [33].Millar TM, Phan V, Tibbles LA, ROS generation in endothelial hypoxia and reoxygenation stimulates MAP kinase signaling and kinase-dependent neutrophil recruitment, Free Radic Biol Med 42(8) (2007) 1165–77. [DOI] [PubMed] [Google Scholar]
- [34].Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Abe Y, Mitochondria-derived reactive oxygen species and vascular MAP kinases: comparison of angiotensin II and diazoxide, Hypertension 45(3) (2005) 438–44. [DOI] [PubMed] [Google Scholar]
- [35].Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG, Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation, J Biol Chem 272(1) (1997) 217–21. [PubMed] [Google Scholar]
- [36].Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T, Requirement for generation of H2O2 for platelet-derived growth factor signal transduction, Science 270(5234) (1995) 296–9. [DOI] [PubMed] [Google Scholar]
- [37].Heppner DE, Hristova M, Dustin CM, Danyal K, Habibovic A, van der Vliet A, The NADPH Oxidases DUOX1 and NOX2 Play Distinct Roles in Redox Regulation of Epidermal Growth Factor Receptor Signaling, J Biol Chem 291(44) (2016) 23282–23293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Maziere C, Floret S, Santus R, Morliere P, Marcheux V, Maziere JC, Impairment of the EGF signaling pathway by the oxidative stress generated with UVA, Free Radic Biol Med 34(6) (2003) 629–36. [DOI] [PubMed] [Google Scholar]
- [39].Lee SR, Kwon KS, Kim SR, Rhee SG, Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor, J Biol Chem 273(25) (1998) 15366–72. [DOI] [PubMed] [Google Scholar]
- [40].Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG, Reversible inactivation of the tumor suppressor PTEN by H2O2, J Biol Chem 277(23) (2002) 20336–42. [DOI] [PubMed] [Google Scholar]
- [41].Meng TC, Fukada T, Tonks NK, Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo, Mol Cell 9(2) (2002) 387–99. [DOI] [PubMed] [Google Scholar]
- [42].Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M, Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases, Cell 120(5) (2005) 649–61. [DOI] [PubMed] [Google Scholar]
- [43].Yang Z, Asico LD, Yu P, Wang Z, Jones JE, Escano CS, Wang X, Quinn MT, Sibley DR, Romero GG, Felder RA, Jose PA, D5 dopamine receptor regulation of reactive oxygen species production, NADPH oxidase, and blood pressure, Am J Physiol Regul Integr Comp Physiol 290(1) (2006) R96–R104. [DOI] [PubMed] [Google Scholar]
- [44].Privratsky JR, Wold LE, Sowers JR, Quinn MT, Ren J, AT1 blockade prevents glucose-induced cardiac dysfunction in ventricular myocytes: role of the AT1 receptor and NADPH oxidase, Hypertension 42(2) (2003) 206–12. [DOI] [PubMed] [Google Scholar]
- [45].Munkin Y, Garnovskaya MN, Collinsworth G, Grewal JS, Pendergrass D, Nagai T, Pinckney S, Greene EL, Raymond JR, 5-Hydroxytryptamine1A receptor/Gi βγ stimulates mitogen-activated protein kinase via NAD(P)H oxidase and reactive oxygen species upstream of Src in Chinese hamster ovary fibroblasts, Biochem J 347(2000) (2000) 61–67. [PMC free article] [PubMed] [Google Scholar]
- [46].Greene, 5-HT2A receptors stimulate mitogen-activated protein kinase via H2O2 generation in rat renal mesangial cells, Am J Physiol Renal Physiol 278( 2000) (2000) 650. [DOI] [PubMed] [Google Scholar]
- [47].Davies AO, Coupling of human β2-adrenergic receptors: relationship to redox potential, J Endocrinol Invest 11(4) (1988) 239–245. [DOI] [PubMed] [Google Scholar]
- [48].Wong A, Hwang SM, Cheng HY, Crooke ST, Structure-activity relationships of beta-adrenergic receptor-coupled adenylate cyclase: implications of a redox mechanism for the action of agonists at beta-adrenergic receptors, Mol Pharmacol 31(4) (1987) 368–76. [PubMed] [Google Scholar]
- [49].Marques F, Bicho MP, Activation of a NADH dehydrogenase in the human erythrocyte by β-adrenergic agonists: possible involvement of a G protein in enzyme activation, Neurosignals 6(2) (1997) 52–61. [DOI] [PubMed] [Google Scholar]
- [50].Moniri NH, Daaka Y, Agonist-stimulated reactive oxygen species formation regulates β2-adrenergic receptor signal transduction, Biochem Pharmacol 74(1) (2007) 64–73. [DOI] [PubMed] [Google Scholar]
- [51].Singh M, Moniri NH, Reactive oxygen species are required for β2-adrenergic receptor-β-arrestin interactions and signaling to ERK1/2, Biochem Pharmacol 84(5) (2012) 661–9. [DOI] [PubMed] [Google Scholar]
- [52].Devadas S, Zaritskaya L, Rhee SG, Oberley L, Williams, Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression, Journal of Exp Methods 195(1) (2002) 59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kondo H, Takeuchi S, Togari A, β-adrenergic signaling stimulates osteoclastogenesis via reactive oxygen species, Am J Physiol Endocrinol Metab 304(5) (2013) E507–15. [DOI] [PubMed] [Google Scholar]
- [54].Davel AP, Brum PC, Rossoni LV, Isoproterenol induces vascular oxidative stress and endothelial dysfunction via a Gialpha-coupled β2-adrenoceptor signaling pathway, PLoS One 9(3) (2014) e91877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Qian L, Hu X, Zhang D, Snyder A, Wu HM, Li Y, Wilson B, Lu RB, Hong JS, Flood PM, β2-adrenergic receptor activation induces microglial NADPH oxidase activation and dopaminergic neurotoxicity through an ERK-dependent/protein kinase A-independent pathway, Glia 57(15) (2009) 1600–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bovo E, Lipsius SL, Zima AV, Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during β-adrenergic receptor stimulation in rabbit cardiomyocytes, J Physiol 590(14) (2012) 3291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Li J, Yan B, Huo Z, Liu Y, Xu J, Sun Y, Liu Y, Liang D, Peng L, Zhang Y, Zhou ZN, Shi J, Cui J, Chen YH, β2- but not β1-adrenoceptor activation modulates intracellular oxygen availability, J Physiol 588(Pt 16) (2010) 2987–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chiarella SE, Soberanes S, Urich D, Morales-Nebreda L, Nigdelioglu R, Green D, Young JB, Gonzalez A, Rosario C, Misharin AV, Ghio AJ, Wunderink RG, Donnelly HK, Radigan KA, Perlman H, Chandel NS, Budinger GR, Mutlu GM, β2-adrenergic agonists augment air pollution-induced IL-6 release and thrombosis, J Clin Invest 124(7) (2014) 2935–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Rambacher KM, Moniri NH, Redox Deficient Cysteine Residues Impair β2-Adrenergic Receptor Function, FASEB J. 33(1) (2019) 668.3.30024789 [Google Scholar]
- [60].Gong K, Li Z, Xu M, Du J, Lv Z, Zhang Y, A novel protein kinase A-independent, beta-arrestin-1-dependent signaling pathway for p38 mitogen-activated protein kinase activation by β2-adrenergic receptors, J Biol Chem 283(43) (2008) 29028–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Barnett CC Jr., Moore EE, Partrick DA, Silliman CC, Beta-adrenergic stimulation down-regulates neutrophil priming for superoxide generation, but not elastase release, The Journal of surgical research 70(2) (1997) 166–70. [DOI] [PubMed] [Google Scholar]
- [62].Opdahl H, Benestad HB, Nicolaysen G, Effect of beta-adrenergic agents on human neutrophil granulocyte activation with N-formyl-methionyl-leucyl-phenylalanine and phorbol myristate acetate, Pharmacology & toxicology 72(4–5) (1993) 221–8. [DOI] [PubMed] [Google Scholar]
- [63].Kopprasch S, Gatzweiler A, Graessler J, Schroder HE, Beta-adrenergic modulation of FMLP- and zymosan-induced intracellular and extracellular oxidant production by polymorphonuclear leukocytes, Molecular and cellular biochemistry 168(1–2) (1997) 133–9. [DOI] [PubMed] [Google Scholar]
- [64].Nguyen LP, Al-Sawalha NA, Parra S, Pokkunuri I, Omoluabi O, Okulate AA, Windham Li E, Hazen M, Gonzalez-Granado JM, Daly CJ, McGrath JC, Tuvim MJ, Knoll BJ, Dickey BF, Bond RA, beta2-Adrenoceptor signaling in airway epithelial cells promotes eosinophilic inflammation, mucous metaplasia, and airway contractility, Proc Natl Acad Sci U S A 114(43) (2017) E9163–E9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Burns RN, Moniri NH, Agonist- and hydrogen peroxide-mediated oxidation of the β2-adrenergic receptor: evidence of receptor s-sulfenation as detected by a modified biotin-switch assay, J Pharmacol Exp Ther 339(3) (2011) 914–921. [DOI] [PubMed] [Google Scholar]
- [66].Poole LB, Klomsiri C, Knaggs SA, Furdui CM, Nelson KJ, Thomas MJ, Fetrow JS, Daniel LW, King SB, Fluorescent and affinity-based tools to detect cysteine sulfenic acid formation in proteins, Bioconjug Chem 18(6) (2007) 2004–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS, Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity, Nat Chem Biol 8(1) (2011) 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Benitez LV, Allison WS, The inactivation of the acyl phosphatase activity catalyzed by the sulfenic acid form of glyceraldehyde 3-phosphoate dehydrogenase by dimedone and olefins, J Biol Chem 249(19) (1974) 6234–6243. [PubMed] [Google Scholar]
- [69].Kobilka BK, Dixon RAF, Frielle T, Dohlman HG, Bolanowski MA, Sigal IS, Yang-Feng TL, Francke U, Caron MG, Lefkowitz RJ, cDNA for the human β2-adrenergic receptor: A protein with multiple membrane-spanning domains and encoded by a gene whose chromosomal location is shared with that of the receptor for platelet-derived growth factor, Proceedings from the National Academy of Science 84 (1987) 46–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hannawacker A, Krasel C, Lohse MJ, Mutation of Asn293 to Asp in transmembrane helix VI abolishes agonist-induced but not constitutive activity of the β2-adrenergic receptor, Molecular Pharmacology 62(6) (2002) 1431–1438. [DOI] [PubMed] [Google Scholar]
- [71].O’Dowd BF, Hnatowich M, Caron MG, Lefkowitz RJ, Bouvier M, Palmitoylation of the human β2-adrenergic receptor, J Biol Chem 264(13) (1989) 7564–7569. [PubMed] [Google Scholar]
- [72].Dietrich LE, Ungermann C, On the mechanism of protein palmitoylation, EMBO Rep 5(11) (2004) 1053–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Tabaczar S, Czogalla A, Podkalicka J, Biernatowska A, Sikorski AF, Protein palmitoylation: Palmitoyltransferases and their specificity, Experimental biology and medicine (Maywood, N.J.) 242(11) (2017) 1150–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Adachi N, Hess DT, McLaughlin P, Stamler JS, S-palmitoylation of a novel site in the beta2-adrenergic receptor associated with a novel intracellular itinerary, The Journal of Biochemistry 291(38) (2016) 20232–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kahsai AW, Xiao K, Rajagopal S, Ahn S, Shukla AK, Sun J, Oas TG, Lefkowitz RJ, Multiple ligand-specific conformations of the β2-adrenergic receptor, Nature Chemical Biology 7(10) (2011) 692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Fraser CM, Site-directed mutagenesis of β-adrenergic receptors, The Journal of Biochemistry 264(16) (1989) 9266–9270. [PubMed] [Google Scholar]
- [77].Bang I, Choi HJ, Structural features of β2-adrenergic receptor: crystal structures and beyond, Mol Cells 38(2) (2015) 105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].O’Dowd BF HM, Regan JW, Leader WM, Caron MG, Lefkowitz RJ, Site-directed mutagenesis of the cytoplasmic domains of the human β-adrenergic receptor, The Journal of Biological Chemistry 263(31) (1988) 15985–15992. [PubMed] [Google Scholar]
- [79].Pejman Ghanouni JJS, Farrens† David L., Kobilka Brian K., Agonist-induced conformational changes in the G-protein-coupling domain of the β2 adrenergic receptor, Proceedings from the National Academy of Science 98(11) (2001) 5997–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Nisimoto Y, Diebold BA, Cosentino-Gomes D, Lambeth JD, Nox4: a hydrogen peroxide-generating oxygen sensor, Biochemistry 53(31) (2014) 5111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Hollins F, Sutcliffe A, Gomez E, Berair R, Russell R, Szyndralewiez C, Saunders R, Brightling C, Airway smooth muscle NOX4 is upregulated and modulates ROS generation in COPD, Respir Res 17(1) (2016) 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Abraham G, Kneuer C, Ehrhardt C, Honscha W, Ungemach FR, Expression of functional β2-adrenergic receptors in the lung epithelial cell lines 16HBE14o-, Calu-3 and A549, Biochem Biophys Res Commun 1691(2004) (2004) 169–179. [DOI] [PubMed] [Google Scholar]
- [83].Salomon JJ, Hagos Y, Petzke S, Kuhne A, Gausterer JC, Hosoya K, Ehrhardt C, Beta-2 adrenergic agonists are substrates and inhibitors of human organic cation transporter 1, Mol Pharm 12(8) (2015) 2633–41. [DOI] [PubMed] [Google Scholar]
- [84].Banga A, Flaig S, Lewis S, Winfree S, Blazer-Yost BL, Epinephrine stimulation of anion secretion in the Calu-3 serous cell model, American journal of physiology. Lung cellular and molecular physiology 306(10) (2014) L937–L946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Chen L, Bosworth CA, Pico T, Collawn JF, Varga K, Gao Z, Clancy JP, Fortenberry JA, Lancaster JR Jr., Matalon S, DETANO and nitrated lipids increase chloride secretion across lung airway cells, Am J Respir Cell Mol Biol 39(2) (2008) 150–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Barisione G, Baroffio M, Crimi E, Brusasco V, Beta-Adrenergic Agonists, Pharmaceuticals (Basel) 3(4) (2010) 1016–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Du Q, Sun Y, Ding N, Lu L, Chen Y. Beta-blockers reduced the risk of mortality and exacerbation in patients with COPD: a meta-analysis of observational studies. PLoS One 9(11) (2014) e113048–e113048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Camsari A, Arikan S, Avan C, Kaya D, Pekdemir H, Ciçek D, et al. Metoprolol, a beta-1 selective blocker, can be used safely in coronary artery disease patients with chronic obstructive pulmonary disease. Heart Vessels. 18(4) (2003) 188–92. [DOI] [PubMed] [Google Scholar]
- [89].Dransfield MT, Voelker H, Bhatt SP, Brenner K, Casaburi R, Come CE, et al. Metoprolol for the Prevention of Acute Exacerbations of COPD. N Engl J Med. (2019), in press, doi: 10.1056/NEJMoa1908142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Dandona P, Karne R, Ghanim H, Hamouda W, Aljada A, Magsino CH. Carvedilol inhibits reactive oxygen species generation by leukocytes and oxidative damage to amino acids. Circulation. 101(2) (2000) 122–4. [DOI] [PubMed] [Google Scholar]
- [91].Dandona P, Ghanim H, Brooks DP. Antioxidant activity of carvedilol in cardiovascular disease. J Hypertens. 25(4) (2007) 731–41. [DOI] [PubMed] [Google Scholar]
- [92].Magsino CH, Hamouda W, Bapna V, Ghanim H, Abu-Reish IA, Aljada A, Dandona P. Am J Cardiol. Nadolol inhibits reactive oxygen species generation by leukocytes and linoleic acid oxidation. 15;86(4) (2000) 443–8. [DOI] [PubMed] [Google Scholar]
- [93].Laurens C, Abot A, Delarue A, Knauf C. Central Effects of Beta-Blockers May Be Due to Nitric Oxide and Hydrogen Peroxide Release Independently of Their Ability to Cross the Blood-Brain Barrier. Front Neurosci. 13:33 (2019) doi: 10.3389/fnins.2019.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Coats A, Jain S. Protective effects of nebivolol from oxidative stress to prevent hypertension-related target organ damage. J Hum Hypertens. 31(6) (2017) 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Nakamura K, Murakami M, Miura D, Yunoki K, Enko K, Tanaka M, et al. Beta-Blockers and Oxidative Stress in Patients with Heart Failure. Pharmaceuticals (Basel). 4(8) (2011) 1088–100. [DOI] [PMC free article] [PubMed] [Google Scholar]