

Summary
During mammalian spermatogenesis, germ cell chromatin undergoes dramatic histone acetylation-mediated reorganization, whereby 90-99% of histones are evicted. Given the potential role of retained histones in fertility and embryonic development, the genomic location of retained nucleosomes is of great interest. However, the ultimate position and mechanisms underlying nucleosome eviction/retention are poorly understood, including several studies utilizing MNase-seq methodologies reporting remarkably dissimilar locations. We utilized ATAC-seq in mouse sperm and found nucleosome enrichment at promoters, but also retention at inter- and intragenic regions, and repetitive elements. We further generated germ cell specific, conditional knockout mice for the key histone acetyltransferase Gcn5, which resulted in abnormal chromatin dynamics leading to increased sperm histone retention and severe reproductive phenotypes. Our findings demonstrate Gcn5 mediated histone acetylation promotes chromatin accessibility and nucleosome eviction in spermiogenesis, and that loss of histone acetylation leads to defects that disrupt male fertility and potentially, early embryogenesis.
Keywords: Sperm, epigenetics, nucleosome retention, Gcn5, spermiogenesis, infertility
Graphical Abstract
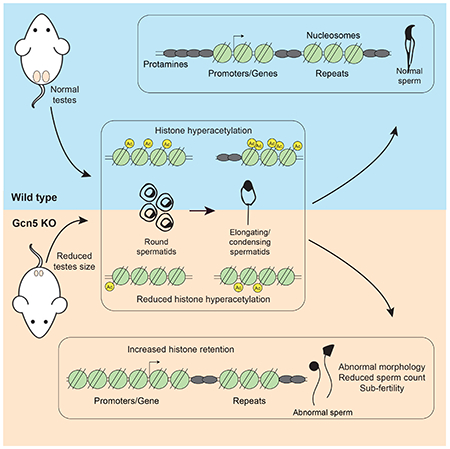
eTOC Blurb
Luense et. al. utilize ATAC-seq to track nucleosomes through male germ cell maturation and report retention at promoters and repetitive elements in sperm. They develop a mouse model, via conditional deletion of the acetyltransferase Gcn5, to study histone hyperacetylation/eviction resulting in abnormal sperm histone retention and male fertility defects.
Introduction
Mammalian sperm exhibit a unique chromatin structure, whereby ~90 to 99% of nuclear histones are removed and replaced by small, basic proteins termed protamines. Incomplete histone removal and/or protamine incorporation are linked to fertility defects, as mice lacking protamines are infertile (Cho et al., 2001) and multiple human studies demonstrate a correlation between reduced protamine levels and infertility (Carrell et al., 2007; Carrell and Liu, 2001; Chevaillier et al., 1987; de Mateo et al., 2010; X. Zhang, 2006). Hence, the process of histone eviction is crucial to generating a viable male gamete and the location of retained sperm nucleosomes may play a crucial role in post-fertilization transcriptional and epigenetic reprogramming events (Carrell and Hammoud, 2010; Schagdarsurengin et al., 2012). Advances in next-generation sequencing have allowed nucleosome mapping in sperm, and initial findings in human and mice indicate that retained nucleosomes are enriched at promoters, including loci important for developmental processes and master regulatory genes (Arpanahi et al., 2009; Brykczynska et al., 2010; Erkek et al., 2013; Hammoud et al., 2011; 2009). In stark contrast, subsequent studies utilizing similar micrococcal-nuclease digestion (MNase) methodologies, found nucleosomes localized to distal intergenic regions and repetitive element-rich regions in human and bovine sperm (Samans et al., 2014), or localized to gene deserts and depleted at promoters in mouse sperm (Carone et al., 2014). Variations in mapping techniques from different laboratories present inherent difficulties in carrying out nucleosome mapping within the compact sperm chromatin; thus alternative technical and genetic approaches are imperative to solve this controversy.
The process of nucleosome eviction occurs during spermiogenesis, which comprises the post-meiotic stages of germ cell maturation. During this process, cellular morphology and chromatin structure undergo dramatic reorganization, leading to a compact and condensed chromatin state and transcriptional quiescence in sperm. Chromatin destabilization is initiated by histone hyperacetylation in post-meiotic round spermatids, resulting in the loosening of chromatin structure, and promoting the brief incorporation of testes-specific variant histones and transition proteins, followed by their removal and replacement with protamines (Govin et al., 2004; Hazzouri et al., 2000; Oliva, 2006). The occurrence of this post-meiotic histone hyperacetylation has been well described (Govin et al., 2007; Hazzouri et al., 2000; Luense et al., 2016); however, the key enzymes involved in hyperacetylation have not been elucidated.
Histone acetyltransferases (HATs) are evolutionarily conserved enzymes that modify lysine (K) residues through post-translational, covalent attachment of acetyl groups (Lee and Workman, 2007). Histone acetylation regulates chromatin structure and promotes accessibility via decondensation and relaxation of chromatin fibers (Shogren-Knaak et al., 2006). In mammalian spermatogenesis there is poor understanding of the HATs that hyperacetylate nucleosomes for eviction. Conditional deletion in round spermatids of both CBP and p300, which acetylate histone H3 lysine 27(H3K27ac), altered the subsequent transcriptional program but did not change histone retention in sperm (Boussouar et al., 2014). Loss of Epc1 or Tip60, components of the NuA4 complex that acetylates H4 lysine residues, resulted in abnormal spermatid formation and development (Dong et al., 2017). Epc1 knock-out mice exhibited decreased histone acetylation and were infertile, however effects on hyperacetylation-mediated nucleosome eviction were not determined. This limited information warrants a deeper and systematic study of HATs during spermiogenesis and their influence on chromatin structure, nucleosome retention, and fertility.
Here, we investigated spermiogenesis and sperm nucleosome retention via ATAC-seq (assay for transposase accessible chromatin), as an orthogonal approach to map nucleosome positions and chromatin compaction through multiple stages of spermiogenesis and in sperm. This method reveals genome wide changes in chromatin accessibility throughout spermiogenesis. We also generated a pre-meiotic, conditional mutant mouse for the HAT Gcn5 to investigate its role in nucleosome eviction/retention in spermiogenesis. Gcn5 (general control of amino acid synthesis 5, also known as KAT2A) is a central enzyme for histone acetylation, transcriptional activation, cell survival, cell cycle progression and importantly, embryonic development (Bu et al., 2007; Lin et al., 2008a; 2008b; Xu et al., 2000). Gcn5 acetylates K9 and K14 on histone H3 (H3K9 and H3K14) (Bonnet et al., 2014; Grant et al., 1999) and additionally acetylates amino terminal lysines on histone H4, including K5, K8, K12, and K16 (Kuo et al., 1996; W. Zhang et al., 1998). Gcn5 null mice are embryonic lethal by e11.5 (Bu et al., 2007), therefore loss of Gcn5 has not been investigated in the testis. We show that Gcn5 promotes histone hyperacetylation and nucleosome eviction, accurate chromatin dynamics, and ultimately, sperm function and fertility. Combined, our results provide insight into mechanisms of histone acetylation in nucleosome eviction and into the location of sperm nucleosomes.
Results
ATAC-seq identifies changes to chromatin accessibility during spermiogenesis
Chromatin undergoes dramatic reorganization in the haploid germ cell, first with post-meiotic histone hyperacetylation to promote chromatin relaxation, followed by nucleosome eviction (Figure 1A). However, the specific genomic locations of these step-wise changes in nucleosome position and removal have not been elucidated. We employed ATAC-seq to interrogate genome-wide changes to chromatin accessibility during spermiogenesis. ATAC-seq utilizes transposase-mediated cutting and barcoding of open, accessible regions of the genome for next-generation sequencing (Buenrostro et al., 2013). This assay reveals open (accessible) or closed (inaccessible) regions of chromatin. Paired-end sequenced fragments partitioned by size into nucleosomal or subnucleosomal fractions determine, respectively, specific nucleosome locations or the presence/absence of regulatory elements and potential transcription factor (TF) binding sites (Buenrostro et al., 2013; Chen et al., 2013).
Figure 1.
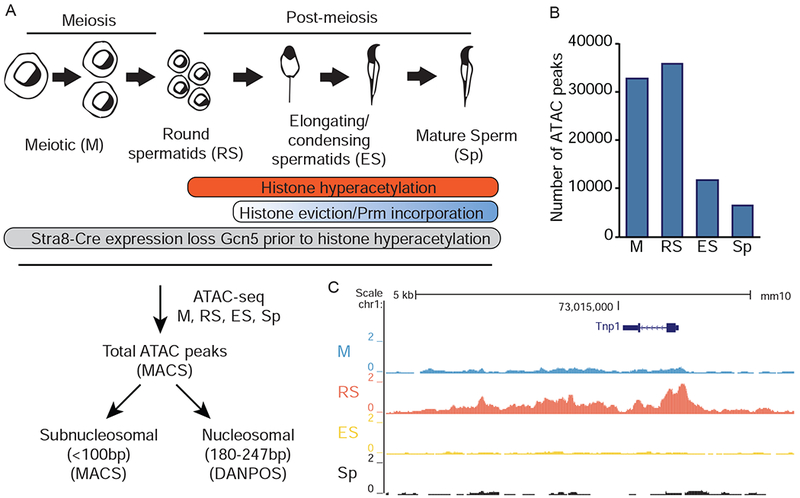
Assessment of chromatin dynamics during spermiogenesis by ATAC-seq. A) Schematic representation (top) of spermiogenesis beginning with meiotic spermatocytes (M) and progressing through round spermatid (RS), elongating/condensing spermatid (ES), and sperm (Sp). (Bottom) Experimental and computational pipeline for ATAC-seq experiments. B) Total ATAC-seq peaks in SV129 germ cells called by MACS peak calling algorithm. C) Representative UCSC Genome Browser tracks of total ATAC-seq signal at Tnp1 locus. See also Figure S1.
We fractionated adult SV129E mouse testes into cellular developmental stages that represent key time-points during germ cell maturation (Bryant et al., 2013). In parallel, mature sperm were collected from cauda epididymides (Figure 1A). We utilized meiotic spermatocytes (M, prior to histone hyperacetylation and nucleosome eviction) as a baseline to assess alterations in round spermatids (RS, undergoing histone hyperacetylation but no nucleosome eviction), elongating/condensing spermatids (ES, undergoing histone hyperacetylation, nucleosome eviction, and protamine incorporation), and ultimately, sperm (Sp, 95-99% nucleosomes are evicted in mouse) (Oliva, 2006). These distinct cell types allowed comparison of multiple chromatin states during spermiogenesis.
We first determined the total number of ATAC-seq peaks to assess the general state of chromatin during stages of spermiogenesis (sequencing and alignment statistics in Supplemental Table 1). Total ATAC-seq peak numbers decreased from M to Sp (Figure 1B), with signal present at expected loci, including, stage-specific chromatin opening of M (Figure S1) and RS (Tnp1, Figure 1C) specific genes. The decline in chromatin accessibility during spermiogenesis, coupled with anticipated locus-specific chromatin changes, provided validation of our ATAC-seq data.
Nucleosome retention in sperm
To determine the genomic location of nucleosomes, ATAC-seq fragments of nucleosomal length (180-247bp) were determined by the DANPOS computational pipeline (Buenrostro et al., 2013; Chen et al., 2013). To validate the ability of our ATAC-seq to reproducibly and accurately determine nucleosome position in multiple stages of germ cells, we compared our data to multiple genomic sequencing methods. First, we performed MNase-seq on M and RS cells to compare nucleosome location in germ cells prior to the profound chromatin compaction in ES and Sp. Nucleosome positions identified in our ATAC-seq datasets corresponded with stage-matched MNase-seq datasets (Figure S2A). Secondly, ATAC-seq-defined nucleosomes in RS cells also corresponded with signal in our previously published RS H3 ChIP-seq dataset, utilizing sonication to shear chromatin (Bryant et al., 2015) (Figure S2A). Thirdly, nucleosomes in our Sp ATAC-seq datasets corresponded with previously published MNase-seq (Erkek et al., 2013), H3 ChIP-seq, and ATAC-seq (Jung et al., 2017) for mouse sperm (Figures S2B, 2C), providing evidence that the use of ATAC-seq can confidently determine nucleosome position.
Figure 2.
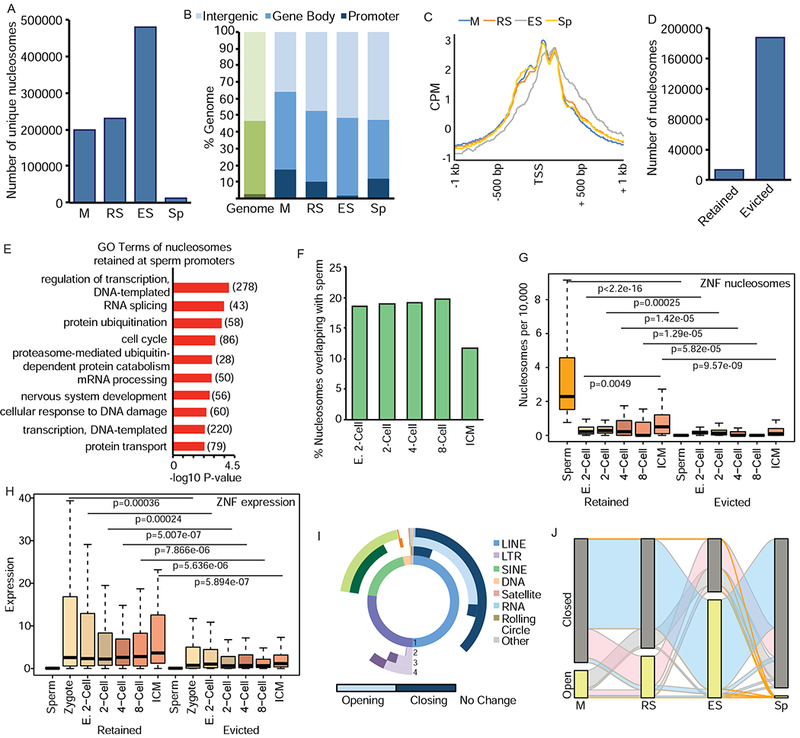
Nucleosome retention is mediated by dynamic changes in chromatin during spermiogenesis. A) Number of unique nucleosomes in germ cells following partitioning of ATAC-seq (180-247bp) and analysis by DANPOS. B) Genomic compartment analysis of nucleosome location in germ cells (blue) compared to genome (green). C) Meta-analysis of nucleosome position at TSS of genes with accessible nucleosomes. CPM=count per million reads. D) Number of nucleosomes evicted or retained in Sp previously present in M stage. E) Top enriched GO terms for retained Sp nucleosomes. Number of genes in parentheses. F) Percentage of nucleosomes retained in Sp overlapping with nucleosomes in pre-implantation embryos. E. 2-cell: early 2-cell, ICM: inner cell mass. G) Comparison of nucleosomes retained (left) or evicted (right) at ZNFs in Sp and pre-implantation embryos. H) Comparison of RNA expression at retained (left) or evicted (right) ZNFs in gametes and pre-implantation embryos. I) Donut plot depicting changes in REs from total ATAC-seq signal. (1) represents proportion of nucleotides for each class of REs in genome. (2) M to RS transition (3) RS to ES (4) ES to Sp. Light shades represent increased enrichment of RE, dark shades represent depletion. Lack of color (white) represents no change. J) Alluvial plot of nucleosomes located in open (detected by DANPOS, yellow bar) or closed (not detected, grey) chromatin compartments. Orange ribbons depict nucleosomes that are ultimately retained (present) in Sp, blue are present in ES but absent in Sp, pink are present in RS, but absent in ES and Sp, grey ribbons present only in M. See also Figure S2.
The total number of unique accessible nucleosomes increased during RS to ES, followed by a marked reduction in Sp (Figure 2A). The increase in accessible nucleosomes during the ES stage corresponds with chromatin relaxation induced by histone hyperacetylation prior to eviction. In addition, the cell heterogeneity of the ES population – consisting of multiple stages of elongating and condensing spermatids – may contribute to the increase in unique nucleosomes. Finally, there was a striking decrease in nucleosomes between ES and Sp (Figure 2A), corresponding to nucleosome eviction of most nucleosomes.
Nucleosomes were enriched at promoter regions in M and RS (6.7- and 4-fold, respectively, Figure 2B), whereas in ES, the proportion of nucleosomes at promoters declines (Figure 2B), likely due to transcriptional quiescence and eviction of nucleosomes throughout the genome. Similar to previous reports (Arpanahi et al., 2009; Brykczynska et al., 2010; Erkek et al., 2013; Hammoud et al., 2009), Sp nucleosomes were enriched at promoters (4.5-fold), but also present throughout the genome, including inter- and intragenic regions (Figure 2B, Figure S2D). Examination of transcription start sites (TSS) found well-positioned nucleosomes at the −1, +1, and +2 promoter positions in M, RS, and Sp (Figure 2C). ES cells also exhibited +1 and +2 nucleosomes at the TSS, but not −1 nucleosome; further, they exhibited non-specific positioning of nucleosomes downstream of the TSS (Figure 2C). This pattern suggests reduced nucleosome positioning in late stage spermiogenesis (ES) prior to re-stabilization and final positioning of nucleosomes in Sp. Importantly, as noted by others (van der Heijden et al., 2006; Ward, 2009), the presence of well-positioned promoter nucleosomes in quiescent sperm suggests paternally inherited nucleosomes may play an important gene regulatory role following fertilization.
Previous approaches focused primarily on retained Sp nucleosomes (Arpanahi et al., 2009; Brykczynska et al., 2010; Carone et al., 2014; Erkek et al., 2013; Hammoud et al., 2009; Samans et al., 2014); in contrast, our approach follows the fate of evicted nucleosomes. We compared nucleosomes in M to Sp, and as expected, the majority were evicted, with only 6.7% retained in Sp (Figure 2D). Gene Ontology (GO) analysis revealed retained nucleosomes enriched at genes involved in regulation of transcription, RNA splicing, protein ubiquitination, and the cell cycle (Figure 2E). Given the transcriptional quiescence of Sp, enrichment of these processes suggests retained nucleosomes are primed specifically for early pre-implantation developmental events (i.e. DNA transcription: 278 genes) as well as later in development (i.e. nervous system development: 56 genes). Nucleosomes were retained at a significant number of zinc-finger proteins (ZNF, total=75, of which 70 are KRAB-ZNFs, p=1.7E-10, Figure S2E). KRAB-ZNF TFs silence repetitive elements (RE) in the embryo (Ecco et al., 2017), hence, retention at these genes further indicates a specific and critical embryonic role for sperm nucleosomes.
Taken together, the use of ATAC-seq as an orthologonal approach to MNase-seq corroborates aspects of several previous reports of nucleosome enrichment at promoters in mouse and human sperm (Arpanahi et al., 2009; Brykczynska et al., 2010; Erkek et al., 2013; Hammoud et al., 2009). However, we note that the vast majority of unique nucleosomes sites mapped to distal and intragenic regions of the genome (Figure 2B), an observation that supports previous reports of nucleosome retention at genic locations (Arpanahi et al., 2009; Brykczynska et al., 2010; Erkek et al., 2013).
Retained sperm nucleosomes and pre-implantation embryos
Following fertilization the paternal pronucleus undergoes decondensation, and maternally provided histones are loaded onto paternal chromatin (Ecklund, 1975). A key question is if retained paternal nucleosomes play a role in embryo development. We compared our ATAC-seq with published ATAC-seq data from mouse pre-implantation embryos [(Wu et al., 2016), Supplemental Table 2] subjected to DANPOS. Nucleosomes retained in Sp overlapped with ~20% of nucleosomes present in early 2-cell embryos through the 8-cell stage, before exhibiting a reduction in the inner cell mass (ICM, Figure 2F).
Specifically, the 75 ZNFs which retain nucleosomes in Sp (Figure S2E) are also enriched in pre-implantation embryos (Figure 2G, left) compared to ZNFs where nucleosomes were evicted (Figure 2G, right). Nucleosome abundance at the 75 ZNFs increased during embryo development (Figure 2G, left), corresponding with increased expression following zygotic genome activation (ZGA) in the 4- and 8-cell embryo (Figure 2H). Notably, expression of the ZNFs was not detected in Sp (Figure 2H, left), suggesting that the paternal contribution derives from nucleosome position and not prior transcription.
Chromatin changes at repetitive elements
Given the role of KRAB-ZNFs in silencing REs (Ecco et al., 2017), and previous localization of retained nucleosomes at repetitive regions in human and bovine sperm (Samans et al., 2014), total ATAC-seq reads were analyzed for enrichment at REs using REPENRICH (Criscione et al., 2014). We also used REPENRICH on two different datasets for human sperm digested by MNase (Samans et. al. 2014, Hammoud et. al., 2009) to provide direct computational comparison of our ATAC-seq with MNase-seq in human sperm. We found a strong correlation between nucleosome retention at REs in mouse (ATAC) and human (MNase) sperm (R=0.846, Figure S2F).
As previous studies assessed nucleosome position at specific REs in sperm only, we utilized our mouse system to analyze RE chromatin at multiple stages of spermiogenesis. Total ATAC-seq reads from M, RS, ES, and Sp were analyzed for enrichment at REs. During spermiogenesis, REs undergo dynamic changes in chromatin state in all cellular transitions, with many classes exhibiting unique chromatin changes (Figure 2I). SINEs primarily close in ES (79%, dark green) before pronounced opening in Sp (99%, light green). LINEs undergo an opposite transition, opening in the RS to ES transition (51%, light blue), before a pervasive closing of 73% LINEs in Sp (dark blue). Interestingly, this same class-specific behavior was reported in human sperm analyzed by MNase-seq, with SINEs retaining nucleosomes and LINE2s exhibiting nucleosome depletion (Samans et al., 2014), thus suggesting the retention of nucleosomes at specific classes of REs.
Dynamic changes in nucleosome accessibility during spermiogenesis
ATAC-seq data from each cell type was next merged to track the fate of uniquely positioned nucleosomes during spermiogenesis, classifying them as either open (present in cell type, yellow bars, Figure 2J) or closed (not present, grey bars), thus determining their ultimate trajectory of being evicted or retained. Waves of nucleosomes exhibiting increased chromatin accessibility were present in RS (pink wave) and ES (blue wave), prior to profound nucleosome loss in Sp (Figure 2J). The increased accessibility in RS and ES stages is likely due to corresponding histone hyperacetylation (see below). Importantly, the majority of nucleosomes observed in ES are accessible only in ES, suggesting relaxation of chromatin at these loci occurs specifically in late spermiogenesis. In summary, ATAC-seq provides a technical approach to track specific nucleosomes, thus defining dynamic locus-specific chromatin changes during spermiogenesis.
Gcn5 conditional knock-out results in reproductive defects
We next investigated mechanisms underlying chromatin changes during spermiogenesis, in particular to explore whether histone acetylation drives nucleosome accessibility in RS and ES (Figure 2J). We compared ATAC-seq with our published ChIP-seq data for acetylated histone lysine residues in RS (Bryant et al., 2015). H3K9ac and H4K5/8/12ac, which are both typically associated with transcriptional activation, corresponded with nucleosome eviction in Sp; in contrast, H4K16ac, which does not correlate as closely with transcriptional activation, was not as correspondent with nucleosome eviction (Figure S3A). We detected expression of the HAT Gcn5 in M and RS, along with acetylation of its substrate H3K9 (Figure 3A, left), suggesting it may be involved in spermiogenesis. Because genome-wide knock-out of Gcn5 is embryonic lethal (Bu et al., 2007; Xu et al., 2000; Yamauchi et al., 2000), conditional male germ cell knock-out mice were generated by deleting Gcn5fl/fl alleles (Lin et al., 2008b) pre-meiotically with Stra8-Cre (Sadate-Ngatchou et al., 2008) to disrupt Gcn5-mediated histone acetylation during spermiogenesis, prior to initiation of histone eviction (Figure 1A). Efficiency of Gcn5 deletion was confirmed via ATAC-seq (gray box, Figure 3B), western blot, (Figure 3A, right), and a strong reduction of H3K9ac (Figure 3A, right).
Figure 3.
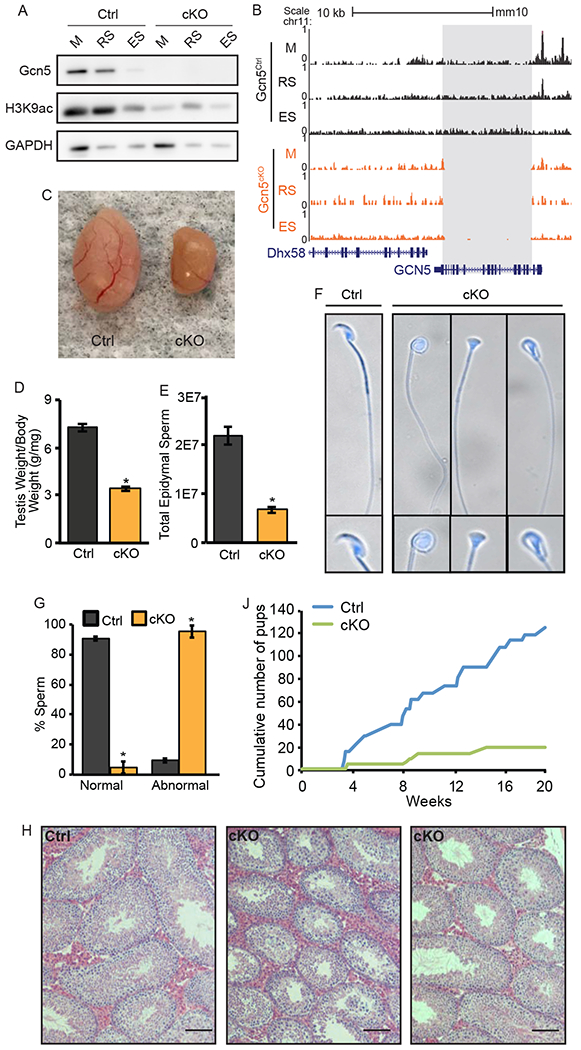
Pre-meiotic loss of Gcn5 leads to abnormal spermiogenesis and fertility defects. A) Protein expression of Gcn5 and H3K9ac in germ cells of Gcn5Ctrl (left) and Gcn5cKO (right) mice. B) UCSC Genome Browser track of total ATAC-seq for Gcn5Ctrl and Gcn5cKO cells depicting deletion of Gcn5 in Gcn5cKO (grey box). Loss of Gcn5 leads to decreased testes size (C), testis/body weight ratio (D), and total epididymal sperm (E). Data presented as means+/−SEM (p<0.001). F) Dapi stained Gcn5cKO sperm exhibit abnormal nuclear morphology. G) Abnormal morphology is increased in Gcn5cKO animals (p<0.01). J) Cumulative number of pups born during 5-month breeding trial. H) Representative H&E staining of adult testes. See also Figure S3.
Conditional Gcn5 knock-out mice (Gcn5cKO) exhibit normal body weight and appear healthy, however, distinct reproductive phenotypes were observed. Gcn5cKO mice exhibit approximately 50% reduction in testes/body weight ratio (Ctrl 7.24 mg/g, n=30; cKO 3.42 mg/g, n=70, p<0.001, Figures 3C, 3D), and reduced sperm count (Ctrl 2.2x107; cKO 0.68x107; p<0.001, Figure 3E). Gcn5cKO Sp exhibited distinct morphological abnormalities, with a rounded, or blunted, inverted-triangular shaped heads in approximately 95.2% of sperm (Figures 3F and 3G). Histological analysis of Gcn5cKO testes revealed the presence of spermatogonia, M, RS, and ES, although some seminiferous tubules appeared disorganized (Figure 3H). Mean tubule diameter was reduced in Gcn5cKO mice (Figure S3B), and combined cell counts of Stage II-III and Stage IIV-IIIV tubules exhibited decreased RS (Figure S3C), thus suggesting that while loss of Gcn5 leads to altered spermatogenesis, no stage-specific block occurs, allowing sperm production.
Male fertility was tested during a five-month breeding trial (n=5/genotype). Gcn5cKO male mice exhibited normal breeding behavior, as evidenced by the presence of seminal plugs in females. However, only 2 of 5 Gcn5cKO male mice produced litters (Figure S3D), resulting in a reduction of total number of litters born and pups per litter (Figure S3D). Of the 19 Gcn5cKO sired pups (compared to 124 Gcn5Ctrl, Figure 3J), 78% carried the Gcn5del allele, suggesting that, although Gcn5 is deleted, some Gcn5del sperm are able to fertilize oocytes and generate viable embryos. In the remaining 22% of Gcn5cKO sired offspring, the Gcn5del allele was not transmitted, indicating incomplete penetrance of the Stra8-Cre recombinase. Given the low, ~5% normal morphological sperm in Gcn5cKO mice (Figure 3G), it is possible that morphologically normal sperm that carry the Gcn5del allele had residual Gcn5 protein from pre-recombination, providing sufficient histone acetylation for normal nucleosome eviction, and/or a small percentage of sperm carry a Gcn5Ctrl allele due to incomplete cre-recombination.
Gcn5cKO sperm exhibit increased histone retention
To investigate the histone content of Gcn5cKO Sp, protein lysates were loaded at equal cell number and in a dilution series to compare histone abundance. Western blot analysis revealed a striking (~5-fold) increase in H3, H4, H2A, and H2B histones in Gcn5cKO compared to Gcn5Ctrl Sp (Figure 4A, 4B). Correspondingly, levels of Prm1 and Prm2 were reduced in Gcn5cKO Sp (Prm1: 2-fold, Prm2: ~2.3-fold reduction, Figure 4C, 4D). These results demonstrate that nucleoprotein content is altered in Gcn5cKO Sp, resulting in dramatically elevated core histone retention.
Figure 4.
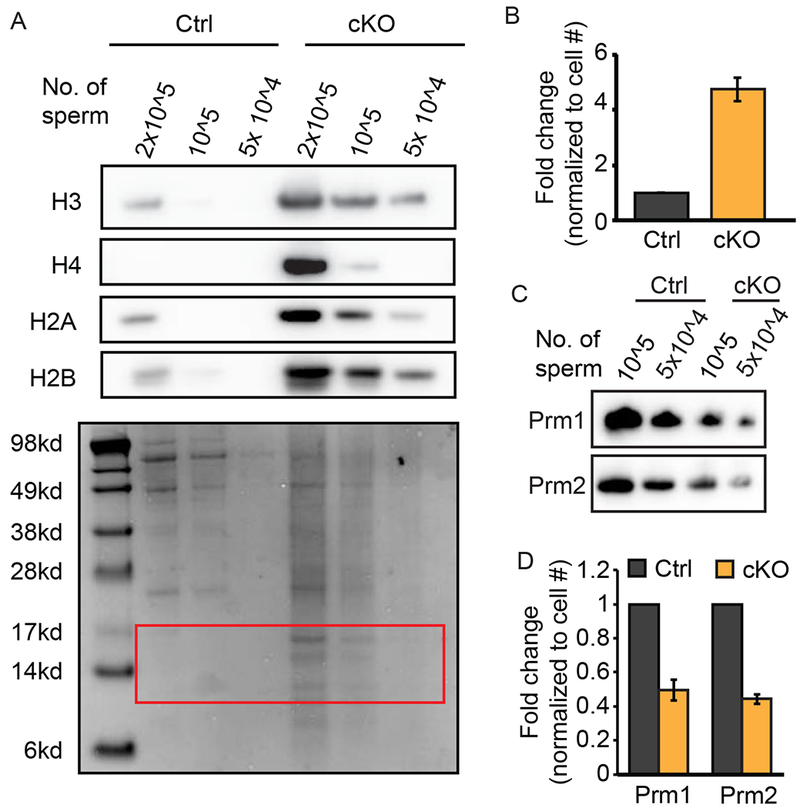
Sperm lacking Gcn5 exhibit increased histone retention. A) Western blot for histone proteins (top) of titrated Gcn5Ctrl or Gcn5cKO sperm. (Bottom) Coomassie stained membrane, red box, denotes histones. B) Relative quantification of protein bands from (B). Mean fold-change of all canonical histones normalized to cell number (n=3 experiments). C) Western blot analysis of titrated sperm for Prm1 or Prm2. D) Relative quantification of protein bands in (C). Mean fold-change normalized to cell number (n=2 experiments).
Loss of Gcn5 alters transcription and chromatin accessibility during spermiogenesis
To investigate the underlying changes to chromatin dynamics that result in increased histone retention, we utilized ATAC-seq (as described above) in Gcn5Ctrl and Gcn5cKO M, RS, ES, and Sp (representative tracks in Figure S4). ATAC-seq signal was first partitioned into subnucleosomal (<100bp) fragments to measure accessible promoters, which are typically associated with active transcription (Ruiz et al., 2018; Xiang et al., 2017) and compared to stage-matched RNA-seq expression. In Gcn5Ctrl cells, RNA expression corresponded with open chromatin at subnucleosomal promoters in transcriptionally active M and RS, but not ES (genes divided into deciles based on expression, Figure 5A). In Gcn5cKO M and RS, gene expression correlated with open chromatin, but was reduced compared to Gcn5Ctrl (Figure 5B, left and center panels). Also in contrast to Gcn5Ctrl, Gcn5cKO ES cells had an increase – although slight compared to M and RS cells – in open chromatin in the top 10% of expressed genes (Figure 5B right), thus indicating an overall change to chromatin accessibility in Gcn5cKO germ cells.
Figure 5.
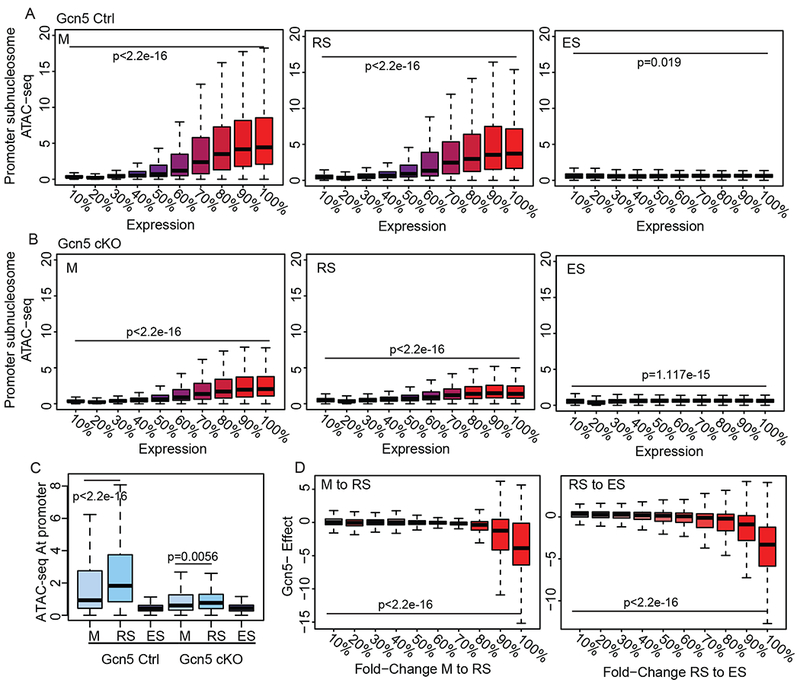
Loss of Gcn5 leads to altered chromatin dynamics and gene expression. Subnucleosomal ATAC-seq at promoters of all genes divided into deciles of RNA expression (0% non-expressed, 100% highest expressed) for M, RS, and ES in (A) Gcn5Ctrl and (B) Gcn5cKO. Statistical analysis performed on first vs last decile by Mann-Whitney rank-sum test. C) Subnucleosomal ATAC-seq signal at promoters of genes increasing in expression between M to RS in Gcn5Ctrl (Mann-Whitney rank-sum test). D) Genes with increased expression in Gcn5Ctrl during M to RS (left) or RS to ES (right) transitions, divided into deciles, exhibit dampened expression in Gcn5cKO cells (y-axis). Gcn5cKO effect is defined as (Gcn5cKO RS/M)/(Gcn5Ctrl RS/M) for M to RS transition (likewise RS to ES). Statistical analysis performed on first vs last decile by Mann-Whitney rank-sum test. See also Figure S4.
Genes with increased expression during the M to RS transition in Gcn5Ctrl cells exhibited the expected opening of chromatin, before closing in ES (Figure 5C, left). However, at the same genes in Gcn5cKO cells, chromatin failed to open (Figure 5C, right). Overall, differentially expressed genes upregulated during M to RS and RS to ES transitions in Gcn5Ctrl displayed a dampened increase in expression in Gcn5cKO (Figure 5D). This diminished increase in expression was most pronounced in genes exhibiting the greatest increase in expression in Gcn5Ctrl cells (90-100% decile, Figure 5D). This dampened opening of chromatin and subsequent attenuated gene expression of normally expressed genes, suggests loss of Gcn5 prevents chromatin accessibility for normal transcriptional programs in transcriptionally active cells.
Increased nucleosome retention in Gcn5cKO germ cells
To determine the effect of Gcn5 loss specifically on nucleosome eviction/retention, we mapped accessible nucleosomes using ATAC-seq combined with DANPOS in Gcn5Ctrl and Gcn5cKO germ cells. Total numbers of mapped nucleosomes were similar between Gcn5Ctrl and Gcn5cKO in M, RS, and ES cells (Figure S5A), however, as observed in western blot analysis (Figure 4A,B), retained nucleosomes increased dramatically in Gcn5cKO Sp (blue and orange bars, Figure 6A). Comparison of Sp nucleosomes to M, RS, and ES found a decreased number of evicted nucleosomes in Gcn5cKO (grey bars, Figure 6A), with a strikingly large number of retained nucleosomes present only in Gcn5cKO Sp (blue bars, Figure 6A). Genomic compartment analysis revealed that based on percentage, nucleosomes were depleted at promoters in Gcn5cKO , and enriched at distal intergenic regions (Figure 6B). However, due to the substantial increase in nucleosomes in Gcn5cKO Sp, the total number retained at promoters, and other genomic compartments, was increased compared to Gcn5Ctrl (Figure S5A). However, in Gcn5cKO Sp, the majority of these promoters retained only 1 or 2 nucleosomes; in contrast, in Gcn5Ctrl Sp, promoters retaining nucleosomes were nucleosome dense (5 or more nucleosomes, Figure S5B). Promoter-bound nucleosomes remained well-positioned at the TSS, with distinct +1 and +2 nucleosomes present in Gcn5cKO (Figure S5C), suggesting that, although depleted in promoters, nucleosome positioning is not affected by loss of Gcn5.
Figure 6.
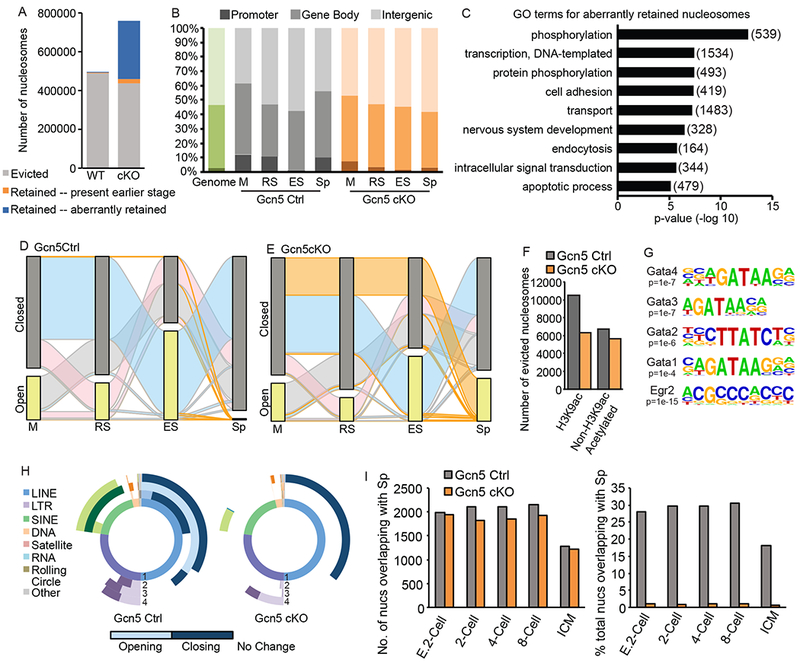
Aberrant nucleosome retention in Gcn5cKO sperm. A) Number of evicted (grey), retained from any previous stage (orange), or newly accessible (blue) nucleosomes in Gcn5Ctrl or Gcn5cKO Sp. B) Compartment analysis of nucleosome location based on percent of total nucleosomes (green bar, genomic background). C) Top enriched GO terms for aberrantly retained Gcn5cKO nucleosomes (number of genes in parentheses). Alluvial plot of nucleosomes in open (detected in specific cell type, yellow bar) or closed (not detected, grey) nucleosomes in (D) Gcn5Ctrl or (E) Gcn5cKO. Ribbon colors as described above. F) Number of evicted nucleosome dense regions with H3K9ac (defined by ChIP-seq, Bryant et. al., 2015) in RS. G) Enriched TF binding motifs present in newly open subnucleosomal regions in Gcn5cKO Sp. H) Donut plot depicting changes in REs from total ATAC-seq signal for Gcn5Ctrl (top) or Gcn5cKO (bottom). Plots as described above. F) Percentage of nucleosomes retained in Sp overlapping with nucleosomes in pre-implantation embryos. E. 2-cell: early 2-cell, ICM: inner cell mass. See also Figure S5.
GO analysis of nucleosomes aberrantly retained in Gcn5cKO Sp found enrichment at promoters of genes involved in several processes critical for pre-implantation embryo development including phosphorylation, DNA-templated transcription, cell adhesion, intracellular signal transduction, and apoptotic processes (Figure 6C). While many of these categories also retain nucleosomes in wildtype Sp (Figure 2E), excess nucleosomes at these genes potentially alter cellular processes in embryogenesis. Importantly, nucleosomes aberrantly retained in Gcn5cKO Sp were not present in published wildtype mouse sperm MNase-seq (right, Figure S5D), further validating the presence of aberrant nucleosomes resulting from Gcn5 loss.
Tracking of individual nucleosomes in open or closed chromatin compartments through spermiogenesis reveals striking differences in chromatin dynamics between Gcn5Ctrl and Gcn5cKO germ cells. In Gcn5Ctrl, stage-specific waves of nucleosomes become accessible (yellow bars, Figure 6D) similar to SV129E (Figure 2J). However, in Gcn5cKO cells, smaller waves of accessible nucleosomes are present in RS and ES (pink and blue ribbons, respectively, Figure 6E, Figure S5A), while a larger proportion of nucleosomes remain inaccessible throughout spermiogenesis, leaving them accessible only in Sp (Figure 6E, top orange ribbon).
To determine if histone acetylation predicts nucleosome eviction in the Gcn5cKO mouse, we identified H3K9ac peaks in RS (Bryant et al., 2015) and compared their locations to retained or evicted nucleosome regions (5+ nucleosomes within 1kb). Of the 11248 nucleosome dense H3K9ac regions in wildtype RS, 10522 are evicted from Gcn5Ctrl Sp (93.5%, grey bar, Figure 6F). However, only 6305 (59.9%) are evicted in Gcn5cKO Sp (Figure 6F), suggesting that loss of Gcn5 leads to loss of histone acetylation, which in turn leads to loss of nucleosome eviction. Interestingly, an additional 6747 nucleosome dense regions that are unacetylated at H3K9ac in RS are evicted in Gcn5Ctrl Sp (Figure 6F, orange bars), suggesting that additional histone residues are hyperacetylated to induce eviction.
Next, to determine potential regulatory functions of aberrant Gcn5cKO chromatin elements, newly open subnucleosomal regions in Gcn5cKO Sp (1kb window) were analyzed for enriched sequence motifs. TF binding sites associated with development including Smad3, p63, multiple Gata family members, and Znf263 and Zfp187 were identified (Figure 6G, Figure S5E, Supplemental Table 3). Additionally, the AC acceptor splice site was enriched at aberrantly open subnucleosomal regions, suggesting the potential for increased or altered splice variants in Gcn5cKO Sp or derived embryos.
While the aberrant TF motifs may point to potential altered embryogenesis, they may also serve as a signature to identify genes that are regulated by nucleosome eviction in preceding stages of spermiogenesis. To investigate if TFs with corresponding motifs are expressed during spermatogenesis, and more importantly, have altered expression in Gcn5cKO germ cells, the expression fold change of genes with at least 0.3 normalized FPKM were calculated (Figure S5F). Gata3 was the highest expressed RNA of the enriched motifs in our, and other, wildtype sperm (Hammoud et al., 2014), but was reduced in all stages of Gcn5cKO cells (Figure S5F), suggesting that this TF is involved in normal nucleosome eviction. In contrast, we found that Egr2 expression was robustly increased in Gcn5cKO M, RS, and ES cells, suggesting that aberrant expression of Egr2 may protect nucleosomes from eviction during spermiogenesis in Gcn5cKO. Hence, altered expression of these TF suggests roles in spermiogenesis and/or embryogenesis.
Unlike wildtype sperm (Figure 2I, Figure 6H, left panel), Gcn5cKO cells exhibit a distinct lack of alterations in chromatin accessibility in most classes of REs, including LINEs and SINEs (Figure 6H, right panel). No changes occur during M to RS or RS to ES transitions (Figure 6H, bottom panel, white regions of second and third most inner circles), but in ES to Sp depletion of accessible chromatin occurs at LINEs (dark purple, left and right). However, only a small fraction of SINEs become enriched in Gcn5cKO Sp (29.6%, right panel, light teal) compared to Gcn5Ctrl (85.2%, left panel, light teal). These differences in LINEs and SINEs suggest that eviction of nucleosomes at LINEs occurs irrespective of histone acetylation, while SINEs are dependent upon acetylation mediated eviction. Furthermore, lack of nucleosome retention at SINEs of paternal chromatin may lead to adverse retro element silencing in early embryonic development.
Finally, to determine whether aberrant nucleosome retention may impair early embryonic transcription and/or development, we compared locations of retained nucleosomes in Gcn5Ctrl and Gcn5cKO Sp to nucleosomes in pre-implantation embryos. We observed a similar number of Gcn5Ctrl and Gcn5cKO Sp nucleosomes present in the early embryo (Figure 6I, left), however when considering the increased number of retained nucleosomes in Gcn5cKO Sp, the percentage of total nucleosomes sharing similar positions in pre-implantation embryos is dramatically reduced (Figure 6I, right), thus suggesting a large number of newly retained Gcn5cKO nucleosomes at aberrant locations in the embryo. To assess how these changes in nucleosome position may alter RNA expression in pre-implantation embryos, we performed cluster analysis on the median expression levels of embryos and identified five clusters of genes. Similar percentages of Gcn5Ctrl and Gcn5cKO nucleosomes were retained at all clusters except cluster 2 (expressed in 2-, 4-, and 8-cell embryos), where Gcn5cKO nucleosomes were reduced. Given the abundance of newly retained nucleosomes in Gcn5cKO sperm, this further suggests the aberrant placement of nucleosomes at genes that are not normally expressed in early embryos.
Discussion
The need for eviction of nucleosomes to facilitate compaction of sperm chromatin has been documented for several decades. However, mechanisms underpinning this eviction are still being elucidated, including the involved enzymes, and the ultimate locations and potential function(s) of retained nucleosomes in sperm. Here, we report key advances in revealing the dynamic chromatin processes necessary for nucleosome eviction/retention.
We first address the location of retained nucleosomes in sperm by utilizing ATAC-seq as an orthogonal approach to MNase-seq, which was previously used in numerous studies but with different findings (Brykczynska et al., 2010; Carone et al., 2014; Erkek et al., 2013; Hammoud et al., 2009; Samans et al., 2014). We find nucleosomes are enriched specifically at promoters and are also present throughout the genome (Figure 2B). Tracking of nucleosomes throughout spermiogenesis identified waves of nucleosomes that become accessible during early or late spermiogenesis prior to eviction, including a large increase in nucleosome accessibility in late stage spermatids (Figure 2J). We show that retained nucleosomes in sperm correlate with nucleosome positions in pre-implantation embryos (Figure 2F–H), thus supporting the hypothesis that paternal nucleosomes transmit epigenetic information to the next generation (van der Heijden et al., 2006; Ward, 2009).
Following detection of these dynamic changes in chromatin and ultimate nucleosome positioning in sperm, we developed a mouse model with reduced histone acetylation to investigate the molecular mechanisms involved in chromatin changes during spermiogenesis and to perturb nucleosome retention in sperm. Our conditional knock-out of Gcn5 resulted in a severe reproductive phenotype (Figure 3). Sperm from Gcn5cKO mice exhibited a dramatic increase in histone retention resulting from abnormal chromatin dynamics during spermiogenesis (Figure 4). Furthermore, sperm lacking Gcn5 have reduced nucleosome retention specifically at SINE, but not LINE elements, suggesting a Gcn5-mediated mechanism that results in the eviction of nucleosomes at specific repetitive DNA elements (Figure 6H). Taken together, these findings address the location of nucleosomes in sperm and provide a model to study the mechanism and function of histone acetylation and eviction in spermiogenesis.
The use of ATAC-seq to evaluate chromatin accessibility is valuable to investigate the highly dynamic process of spermiogenesis, as ATAC-seq enables assessment of the open or closed state of chromatin at specific genomic locations. Importantly, the ability to determine nucleosome locations in multiple cell types provides a means to track nucleosomes prior to eviction. Previously, MNase-seq has been primarily used to map nucleosome position in sperm, although, notably, to varying results and interpretations (Brykczynska et al., 2010; Carone et al., 2014; Erkek et al., 2013; Hammoud et al., 2009; Samans et al., 2014). These widely differing results used similar MNase-seq methodology, thus pointing to difficulty in replication of the method across laboratories, possibly due to the sensitivity of MNase digestion (Carone et al., 2014). Other differences between approaches confound models, including cross-species comparisons in the studies (i.e. human to mouse), as the final histone content of sperm varies across species. Further, computational methods, particularly in alignment of repetitive DNA elements, may lead to divergent results (Dansranjavin and Schagdarsurengin, 2016; Royo et al., 2016). A recent study utilizing nucleoplasmin to displace protamines in sperm mapped the retained unmodified histones to distal intragenic regions, the retained H3K4me3-modified histones to promoters, and the retained H3K9me3-modified histones to satellite repeats (Yamaguchi et al., 2018), thus supporting diverse locations. Our results utilizing ATAC-seq reconcile these findings, in that we validate nucleosome retention at promoters, but also map retained nucleosomes throughout the genome, including inter- and intragenic regions, and REs.
The chief interest in the location of retained sperm nucleosomes is their function in the embryo; indeed, our findings suggest several potential roles for paternally inherited nucleosomes. Firstly, nucleosomes were enriched at promoters of genes involved in transcriptional regulation, including ZNF TFs, that may be critical for ZGA and repression of REs (Figure 2G–H). Previous studies in human sperm mapped retained nucleosomes to promoters critical for early embryonic development and master regulatory genes (i.e. Hox, miRNA clusters) (Hammoud et al., 2009). While we did not observe this specific group of factors enriched in sperm, this divergence may be due to species differences. Human sperm retain approximately 10% of histones, compared to the 1-5% estimated retained in mouse. This may lead to enrichment at additional promoters in human sperm. Secondly, we find enrichment of nucleosomes at SINEs and depletion at LINEs, corresponding with previous observations in human and bovine sperm (Samans et al., 2014). We also found a strong correlation between mouse and human sperm in enrichment of open chromatin at similar classes of REs. We see the strongest correlation with the Samans et. al. 2014 study that reports nucleosomes retained at REs, however we also observe similar clustering of REs with the Hammoud et. al., 2009 study, albeit with a lower correlation (Figure S2F). We believe the correlation between these cross-species datasets is indicative of broader agreement between the published MNase-seq studies than has so far been evident, and further validates the use of ATAC-seq data and underscores the importance of our ability to analyze changes to REs in the preceding stages of spermiogenesis in the mouse model.
Our data suggests that LINEs, as active retrotransposons, require strong repression via protamine-compacted chromatin lacking retained nucleosomes to protect the paternal germline in sperm. In contrast, because SINEs lack self-autonomy and require LINEs for their mobilization (Garcia-Perez et al., 2015), complete genomic compaction of SINEs may not be needed. Thus, our findings that nucleosomes are enriched at promoters for ZNF genes that are essential for repression of REs in the early embryo, contributes to mounting evidence that sperm nucleosomes have vital roles in embryogenesis. Analysis of embryos generated from Gcn5cKO sperm will reveal the consequences of abnormal nucleosome locations on pre-implantation development, chromatin dynamics, and transcription. Hence, this mouse model will be highly valuable in unraveling the role of paternal retained nucleosomes.
Previous studies have utilized ATAC-seq in early spermatogenesis including spermatogonial stem cells, spermatocytes, round spermatids (Maezawa et al., 2017), and in sperm (Jung et al., 2017). Similar to our results, previous ATAC-seq performed on mouse sperm reports the retention of well-positioned nucleosomes at TSSs, with a subset of these nucleosomes retained at genes, including RNA processing, which are expressed in early embryo development (Jung et al., 2017). Our study examines the developmental progression of late stage spermiogenesis when chromatin is undergoing dramatic reorganization to form the sperm cell. Thus, we have tracked specific nucleosomes through multiple stages of spermiogenesis prior to eviction, providing a model depicting chromatin dynamics of male germ cell maturation (Figure 7). We find two distinct waves of chromatin opening leading to nucleosome eviction. There is an initial “minor” wave when nucleosomes become accessible during early spermiogenesis (RS, pink nucleosomes, top panel, Figure 7), followed by a “major” wave of nucleosome accessibility occurring in late spermiogenesis (ES, yellow nucleosomes, top panel, Figure 7). One potential factor driving these minor and major waves may be specific histone PTMs driving accessibility. Numerous histone lysine residues undergo hyperacetylation during spermiogenesis (Govin et al., 2007; Hazzouri et al., 2000; Luense et al., 2016), however direct links between specific acetylation sites and eviction require further study.
Figure 7.

Model of nucleosome accessibility during spermiogenesis: Transcriptionally active genes are acetylated in M and RS, prior to replacement by protamines during the transcriptionally quiescent stages of ES and Sp. Nucleosomes evicted in the minor-wave of chromatin accessibility are acetylated in RS (pink) or in the major-wave in ES (yellow) and evicted and replaced by protamines. In the Gcn5cKO mutant model, lack of acetylation leads to loss of chromatin accessibility and diminished minor and major waves, ultimately resulting in increased histone retention in sperm.
The Gcn5cKO mouse provides clear genomic evidence that loss of a specific HAT results in altered chromatin accessibility leading to increased nucleosome retention in sperm (bottom panel, Figure 7). Interestingly, mutant mouse models haploinsufficient for Prm1 or Prm2 do not show increased histone retention (Cho et al., 2001), with a recent finding showing, surprisingly, reduced levels of histone H3 in Prm2 KO mouse sperm (Schneider et al., 2016). Previous mutant mouse models for HATs targeting lysines on H4 (Tip60 and NuA4 complex member Epc1) show lowered fertility (Dong et al., 2017), thus suggesting that multiple acetylated lysine residues promote chromatin accessibility and subsequent nucleosome eviction. However, these mutants do not show increased histone retention in sperm, suggesting distinct roles for specific HATs during spermatogenesis (Dong et al., 2017). This is supported by our observation that some evicted nucleosomes are not marked with H3K9ac in RS. Interestingly, sperm from mice lacking the testes specific BET family protein Brdt also exhibit misshapen head morphology and infertility (Shang et al., 2007). Brdt binds acetylated histones and mediates the incorporation of transition proteins during spermiogenesis (Gaucher et al., 2012), suggesting a similar phenotype when a ‘reader’ of histone acetylation is altered in sperm development as in this Gcn5cKO model mutating a ‘writer’ of acetylation.
Abnormal chromatin composition in human sperm has been linked with fertility defects and poor embryogenesis (Carrell et al., 2007; Carrell and Liu, 2001; Chevaillier et al., 1987; de Mateo et al., 2010; X. Zhang, 2006). However, the causes of altered protamine/histone ratios and the detrimental effects on the embryo remain unknown. One potential cause is altered PTM signatures on paternally inherited nucleosomes, as evidenced by a recent mouse model with reduced H3K4me2 in sperm resulting in severe developmental transgenerational abnormalities (Siklenka et al., 2015). Limitations on human embryo studies have led to a lack of research on this important topic. The development of the Gcn5cKO mouse model that mimics an understudied human condition will provide an important tool in understanding the role of paternally inherited nucleosome on male fertility and embryo development.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
This study did not generate new unique reagents. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shelley Berger (bergers@upenn.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All experiments were approved and conducted according to guidelines, standards, and regulations from the University of Pennsylvania Institutional Animal Care and Use Committee. All mice were housed in the University of Pennsylvania animal vivarium with standard 12hr light/dark cycles with ad libitum access to standard laboratory chow and water. All experimental animals were adult male mice (8-36wks old), with no previous performed procedures, and were considered healthy at the time of experimental use.
Adult male SV129E mice (Charles River Laboratories) were utilized for initial ATAC-seq and RNA-seq experiments.
Mice with pre-meiotic deletion of Gcn5 in male germ cells were generated by mating Gcn5fl/fl mice (C57BL6), graciously provided by Dr. Sharon Dent (Baylor College of Medicine) (Lin et al., 2008b), with mice expressing Cre recombinase under the control of the Stra8 promoter (FVB/NJ, Jackson Labs, (Sadate-Ngatchou et al., 2008)). Expression of Stra8-Cre begins at post-natal d3 in early stage spermatogonia, continues through preleptotene stage spermatocytes (Sadate-Ngatchou et al., 2008) and is specific to the male germ line.
Subsequent mixed background F1 Gcn5fl/+; Stra8-Cre+ mice were backcrossed to Gcn5fl/fl mice to generate control (Gcn5fl/+, Gcn5Ctrl) or conditional knock-out mutant animals (Gcn5fl/Δ;Stra8-Cre+, Gcn5cKO) for subsequent experiments.
METHODS DETAILS
ATAC-seq, RNA-seq, and MNase-seq
Collection of mouse germ cells
Sexually mature, adult male mice (8-36wks old, Charles River Laboratories) were humanely euthanized by CO2 asphyxiation according to University of Pennsylvania Institutional Animal Care and Use Committee guidelines. Immature male germ cells were collected from testes by STAPUT velocity sedimentation as previously optimized, described, and validated by our laboratory (Bryant et al., 2013). Briefly, for each STAPUT collection, testes from 11 adult SV129E or Gcn5Ctrl mice were dissociated to a single cell suspension with sequential incubation of collagenase (0.9 mg/ml) and trypsin (0.6 mg/ml) with DNase (0.1 μg/ml) in KREBs buffer (1.2mM KH2PO4, 119mM NaCl, 1.2mM MgSO4-7H2O, 1.2mM CaCl2-2H2O, 4.8mM KCl, 25.2mM NaHCO3) supplemented with 11mM glucose. For Gcn5cKO testes fractionation, 24 mice were used per collection. Testes cells were subsequently resuspended in 0.5% BSA and slowly loaded onto a 2-4% BSA gradient. Cells were allowed to sediment for 105 min prior to fraction collection. Cell aliquots were stained with DAPI and assessed by fluorescent microscopy for meiotic (M), round spermatid (RS), and elongating/condensing spermatid (ES) fractions. Cell fractions with at least 85% purity for each stage were pooled, used immediately for ATAC-seq or snap frozen, and stored at −80°C for further processing. Mature sperm were collected from the cauda epididymis and vas deferens and incubated in somatic cell lysis buffer (0.1% SDS, 0.5% Triton-X-100) for 15min to remove any non-sperm contaminants. For each collection the sperm were counted, and subsequently snap frozen and stored at −80°C until further processing.
Assay for transposase-accessible chromatin
ATAC-seq was performed as previously described (Buenrostro et al., 2013), with slight modifications (Sammons et al., 2015). Following STAPUT isolation, cells were immediately washed with cold PBS, aliquoted (M and Sp: 100k cells/rxn, RS and ES: 200k cells), and centrifuged at 500xg for 10min at 4°C in a swinging bucket centrifuge. Cells were lysed in standard ATAC lysis buffer (10mM Tris-HCl, pH 7.4, 10mM NaCl, 3mM MgCl2, and 0.1% IGEPAL CA-630) and immediately centrifuged at 500xg for 10min at 4°C. Recovered nuclei were tagmented at 37°C for 30min with 2.5U and 5.0U of Tn5 (Nextera XT Kit, Illumina) and purified with Qiagen MinElute Kit in parallel reactions. Libraries were amplified for 12 PCR cycles (primer sequences listed in Table S4), purified by Qiagen PCR Cleanup Kit, tested for quality and correct size distribution by DNA 1000 Bioanalyzer (Agilent) and quantified by qPCR (KAPA Biosystems). Libraries were prepared from two independent STAPUT collections for SV129E cells and 1 STAPUT for Gcn5Ctrl and Gcn5cKO. Sperm libraries were prepared from individual animals. All libraries were sequenced on the Illumina NextSeq with paired-end sequencing for 150 cycles.
MNase-seq library preparation
SV129 M (100k) and RS (200k) cells were digested with MNase as previously described (Brind’Amour et al., 2015) with slight modifications. Frozen cell aliquots were first lysed in nuclear isolation buffer (Sigma) with protease inhibitors (Roche). MNase digestions (0U, 0.5U, 1U, or 2U, MNase (NEB)) were subsequently carried out in MNase Buffer (NEB) supplemented with 200mM DTT for 7.5min at 37°C. Reactions were stopped by adding EDTA. The resulting digested DNA was phenol:chloroform purified, quantified (Qubit), and tested for quality and correct nucleosomal size distribution with Bioanalzyer DNA 1000 analysis (Agilent). Sequencing libraries were generated from 37.5ng of DNA with the NEB Next Ultra II DNA Library Prep Kit for Illumina (5 amplification cycles) as described by manufacture protocol. Libraries were sequenced on the Illumina NextSeq with paired-end sequencing for 75 cycles.
RNA-seq library preparation
RNA was extracted from all cell types using the Qiagen RNAeasy kit per manufacturer instructions. RNA-seq libraries were prepared using the Script-Seq V2 RNA-seq kit (Illumina) with ribodepletion per manufacturing instructions. Following ribodepletion, sequencing libraries (20ng/sample) were prepared as described by the manufacturer (15 amplification cycles), tested for quality and size distribution (Bioanalyzer High Sensitivity DNA, Agilent), and quantified by qPCR (KAPA). All RNA-seq libraries were sequenced on the Illumina NextSeq with single end sequencing for 75 cycles.
Gcn5 pre-meiotic conditional mutant mice
Mice were generated as described above in animal model section. Body and testis weights and sperm count were measured following CO2 asphyxiation as approved by the University of Pennsylvania Institutional Animal Care and Use Committee. Statistical analysis of testes weights and sperm counts were conducted by t-test (Prism). A p-value of 0.05 was considered significant.
Histological analysis
For histological analysis, testes were fixed overnight in Bouin’s fixative or 4% PFA. Tissues were paraffin embedded, sectioned at 5um, and stained with hemotoxylyn and eiosin. To determine round spermatid numbers, paraffin embedded sections were dehydrated and incubated with lectin PNA (Invitrogen, L21409) for 1hr to detect the acrosome and DAPI to detect DNA. Total number of round spermatids (PNA positive cells with a distinct DAPI chromocenter) in Stage II-III and IIV-IIIV seminiferous tubules (>10 sections/stage, n=2 per genotype) were scored by two independent reviewers blind to the genotype.
Western blot
Cell pellets were resuspended in lysis buffer with benzonase (20 mM Tris pH 7.5, 1 mM MgCl2, 1 mM CaCl2, 137 mM NaCl, 10% Glycerol, 1% NP-40, 300 nM TSA, benzonase (12.5 U/ml), 1X protease inhibitors) and rotated for 1 hour at 4°C. For immature germ cells, 10ug of protein was separated by polyacrylamide gel electrophoresis and transferred to a PVDF membrane. For histone sperm titration experiments, protein from the designated number of cells were loaded in each lane. Following an 1hr block in 5%BSA in TBST buffer, membranes were incubated with primary antibody overnight, followed by horseradish-peroxidase-conjugated secondary antibodies. Proteins of interest were visualized following detection with ECL substrate (Pierce) and visualized by Amersham Gel Imager. Membranes were imaged at 10sec intervals and bands quantified by Fiji. Primary antibodies used: H3 1:1000 (Abcam 1791, lot# 63242682-2), H4 1:500 (Abcam 7311), H2A 1:500 (Abcam 18255), H2B 1:1000 (Abcam 1790), H3K9Ac (Abcam 4441, lot# GR252894-1) 1:1000, GCN5 (Cell Signaling (C26A10) 3305S, lot #4) 1:200 and GAPDH (Fitzgerald 10R-G109a, Batch # 1021) 1:10000. Secondary antibodies: Goat anti-rabbit IgG (H+L)-HRP conjugate (170-6515 Biorad), Goat Anti-Mouse IgG (H+L)-HRP (172-1011 Biorad), both secondary antibodies at 1:5000.
Protamine extraction and analysis
Protamines extracted from caudal epididymal sperm were pooled from Gcn5cKO (2-3 animals per pool, n=2 pools) or Gcn5cKO (6-8 animals per pool, n= 2 pools) mice as previously described (de Yebra and Oliva, 1993). Briefly, following somatic cell lysis, 107 sperm from each pool were washed with RNAase/DNAse-free H20 with 1mM PMSF and centrifuged at 4,000xg for 5min at room temperature. The resulting supernatant was resuspended in 100ul of 20mM EDTA, 1mM PMSF, and 100nM Tris pH 8.0, followed by the addition of 100ul 6M Guanidine with 575mM DTT. After thoroughly mixing, 200ul of 522mM sodium iodoacetate was added, mixed, and incubated for 30min in the dark. 1ml of ice-cold ethanol was subsequently added, mixed, and incubated for 10min at −20°C. Following centrifugation at 12,000xg for 8 min, the supernatant was removed and resulting precipitate was washed with 1ml of ice-cold ethanol. The precipitate was resuspended in 0.8ml of 0.5M HCl with 50mM DTT, mixed, and incubated at 37°C for 10min. Samples were centrifuged at 12,000xg for 5min, supernatants transferred to a new tube and protamines were precipitated with 200ul of TCA (100%) by vortexing and incubating at −20°C overnight. Following centrifugation, the supernatant was removed and the resultant precipitate was washed twice with 1% 2-mercaptoethanol in acetone, vortexed, and centrifuged for 12,000xg for 8min. After removal of the supernatant, the resulting protamines were air-dried and resuspended in loading buffer (0.375 M potassium acetate, pH 4.0, 15% sucrose, 0.05% Pyronin Y).
15% acid-urea gels were freshly poured as previously described (Liu et al., 2013), and equal numbers of sperm, normalized by DNA concentration from equal aliquots of pooled sperm, were loaded into wells and subjected to electrophoresis in 0.9 N acetic acid buffer. Gels were equilibrated and transferred to 0.45um PVDF membranes for 45min at 100V as previously described (Hazzalin and Mahadevan, 2016). Membranes were blocked in TBST with 5% BSA for 1hr and incubated overnight with antibodies to Prm1 (1:4000 in TBST with 2.5% BSA, BriarPatch) or Prm2 (1:5000 in TBST w/2.5% BSA, BriarPatch) at 4C. Following TBST washes, membranes were incubated with secondary anti-mouse antibody (1:10,000 in TBST with 2.5% BSA, Biorad) for 30min, washed in TBST, and developed with ECL substrate (WestPico, Pierce). Membranes were imaged at 10sec intervals and bands quantified by Fiji.
Breeding trial
Sexually mature, 8-wk old male Gcn5Ctrl and Gcn5cKO mice (n=5/genotype) were singly housed with 6-wk old female C57Bl/6 mice for five months. Initially, female mice were checked daily for presence of a seminal plug to determine if mating occurred and if breeding behavior was normal. Cages were checked daily for the presence of pups and litter size was recorded. If after 6 weeks, male mice did not sire any litters, dams were replaced with new 6-wk old female mice to eliminate potential female fertility defects.
QUANTIFICATION AND STATISTICAL ANALYSIS
ATAC-seq analysis
All ATAC-seq paired-end data were aligned to mouse genome assembly GRCm38/mm10 using STAR v2.5.2a with the following parameters: --outFilterMultimapNmax 20 –outFilterMismatchNmax 999 –alignMatesGapMax 1000000. SAM files were preprocessed with samtools to convert to BED format, merge across the four NextSeq lanes, and to filter out mitochondrial aligned tags as well as mate pairs with a fragment size in excess of 1kb.
To make UCSC Genome Browser tracks, filtered BED files were subjected to BEDtools genomeCoverageBed -bg, and the tag counts in the resulting bedGraphs were normalized per million reads to adjust for library size, then the normalized bedGraphs were converted to bigWigs using the UCSC Genome Browser software bedGraphToBigWig.
Peaks were called using MACS2 v2.1.0.20140616 with default parameters (no control sample was specified), except that the tag size and the genome size were provided and a q-value threshold was set at 0.1%. To identify peaks in the sub-nucleosome-sized fragments, an additional preprocessing step was performed to filter out any fragments with mate pairs on separate chromosomes and any unmatched mates; additionally, mate pairs with multiple alignments were collapsed to one locus. Sub-nucleosome sized fragments were defined as those with a total size of 100 bp or smaller, and the same MACS invocation was performed on this filtered fragment library.
Compartment analysis of peak enrichment was performed using CEAS v0.9.9.7 with the -b input option (to accept the peak BED file) and setting the upstream / downstream size to 1kb or 10kb (--sizes= 1000,10000,10000).
Nucleosome positions were identified using DANPOS v2.1.3 per the previously described settings (Buenrostro et al., 2013). Briefly, fragments were curated by size after employing the same preprocessing and filtering technique used to produce the sub-nucleosome fragments. In this case, fragments ranging from 180 bp to 247 bp were deemed to be nucleosome sized and those ranging from 0 bp to 100 bp were deemed to be sub-nucleosome-sized. DANPOS was then run to detect nucleosome enrichment vs a background of sub-nucleosome enrichment. All nucleosome-sized fragments from replicate samples (e.g., SV129 meiotic) were put into a folder “Nuc” and all sub-nucleosome-sized fragments from the same replicate samples were put into a folder “Sub”; DANPOS was then run using the following command-line:
$> python danpos.py Nuc:Sub -p 1 -a 10 -d 20 --clonalcut 0
Resulting spreadsheets were then processed to extract all positions with SMT_Diff_FDR < 0.005 (FDR < 0.5%); loci were centered on the provided locus plus and minus 75 bases to yield the nucleosome position.
Repeats were examined for ATAC-seq signal using RepEnrich v1.0. Briefly, data were realigned to mouse genome assembly NCBI37/mm9 using bowtie v0.12.7 and parameters -m 1 -S --max to extract multiple-mapping tags. Human MNase-seq datasets from Hammoud et. al., 2009 and Samans et. al., 2014 were aligned to GRCh27/hg19 with precisely the same parameters. Resulting SAM files were converted to BAM and indexed using samtools v0.1.18. RepEnrich was then run on the re-aligned data using default parameters. Data from the *_fraction_counts.txt files was collated and analyzed using EdgeR with a univariable linear model (the spermatogenesis stage) and contrasts for each successive pair of stages (meiotic to round, round to elongating, and elongating to mature sperm). Wheel plots were constructed by first counting the number of nucleotides occupied by members of each broad class of repeat, then loading EdgeR results to count the number of nucleotides occupied by members of that class with a statistically significant increase or decrease in each stage transition. Correlation scatter plots were assessed at the level of repeat classes using counts per million in RPM-adjusted *_fraction_counts.txt files, averaging over several replicates for the Luense et. al. ATAC-seq data, and correlations was calculated using the Spearman rank correlation.
Alluvial plots were constructed in the following way. Nucleosome positions (or sub-nucleosome peaks) from the entire time-course were merged into a single pan-stage locus set, with overlapping positions collapsed into one. The pan-stage regions were then examined for overlap with any locus at each stage using BEDtools intersect, so that each pan-stage region could be assigned a profile describing whether it was accessible (overlapping) or inaccessible (non-overlapping) at each stage (e.g., a pan-stage nucleosome region might have a profile of 1.0.0.1, meaning it is present in meiotic, absent in round and elongating spermatid, and reappears in mature sperm). A table summing the number of regions with each profile was then constructed and used as input to the alluvial library in R.
Topographical plots were constructed by taking tags from the sub-nucleosome-sized fragments or the nucleosome-sized fragments defined in the DANPOS run and binning them into 10bp bins at a distance of +/− 2 kilobases around all annotated RefSeq TSS’s. Fragment pile-ups were adjusted for library size using an RPM coefficient derived from the sum of all nucleosome mate pairs and sub-nucleosome mate pairs. Curves were then standardized (z-scores) to better visualize nucleosome stability.
Gene Ontology characterization of a peak set at a given spermatogenesis stage was performed by using DAVID on the nearest RefSeq transcript to each peak, filtering the FDR at a threshold of 10%.
Pre-implantation embryo ATAC-seq data were downloaded from GEO GSE66390 (Wu et al., 2016) and processed as described above to acquire nucleosome positions. Nucleosome positions from the published data and those generated by this study were then examined for pairwise overlap using the MTL approach (joining adjacent nucleosomes if the centers are within 100 bp). Genes retaining or evicting nucleosomes in mature sperm were then examined for nucleosome density at each stage. Each nucleosome was assigned by nearest TSS to a single gene, and that gene’s score was the number of assigned nucleosomes divided by the total nucleosomes in that stage (per 10k). Genes retaining or evicting nucleosomes in mature sperm were also examined for expression values, using the ERCC-adjusted RNA-seq scores computed for mature sperm in this study as well as the reported scores in the published work (GSE66582).
Zinc fingers and KRAB ZNFs were curated from a list created by Trono et al, Table S2 (Imbeault et al., 2017)
To perform motif analysis, nucleosomes present only in Gcn5cKO sperm (not present in earlier stages in Gcn5cKO or present in Gcn5ctrl sperm) were divided into those in promoters (1kb TSS upstream regions), gene bodies, or intergenic non-promoter. Sub-nucleosome peaks in Gcn5cKO mature sperm were then curated by their proximity to aberrant Gcn5cKO nucleosomes (within 1kb) to identify putative transcription factors that might influence nucleosome deposition. Motif analysis was then performed in one of two ways: using HOMER (-size given –mask against a randomly selected background of the same size) or by intersecting with ENCODE ChIP-seq peaks for 119 different transcription factors.
MNase-seq analysis
MNase-seq from Erkek et. al. 2013 and from this study, along with ATAC-seq and H3 ChIP-seq from Jung et. al., 2017, were aligned to mouse genome assembly mm10 using STAR v2.5.2a and ENCODE parameters (--outFilterMultimapNmax 20 --outFilterMismatchNmax 999 – align MatesGapMax 1000000). Heatmaps were generated by making standardized topographies of RPM-adjusted tag counts over 1kb windows centered on nucleosome positions identified from this study’s ATAC-seq data by DANPOS.
RNA-seq analysis
All RNA-seq data were aligned to mouse genome assembly NCBI37/mm9 using tophat2 v2.0.11 and parameters --library-type fr-secondstrand --b2-very-sensitive. Because we expected a dramatic and global decline in transcription in the later stages of spermatogenesis, the usual RNA-seq quantitative assessments (library size adjustment, or FPKM) were deemed to be inaccurate, as these measurements rely on the assumption that most of the transcriptome does not change with experimental conditions. To obtain a true measurement of expression per gene, ERCC spike-in probes were included in the sequencing reaction, and the alignment index was therefore constructed to contain ERCC spike-in sequences in addition to the assembled mouse chromosomes. Tag counts per gene per sample (and per spike-in probe per sample) were assessed using HTSeq v0.6.1p1 with parameters -r pos -s yes -t exon -i gene_id. Library-size and gene-length adjusted FPKM scores for genes were then corrected by leveraging spike-in data in the following way: for each spike-in probe, the tag ratio at each stage post-meiotic spermatid to meiotic spermatid was computed. This was then compared to the expected spike-in tag ratio, and the “deviation” was defined as the ratio of ratios observed:expected. The average deviation among all ERCC probes in a given stage was called the deviation of the stage. The reciprocal of this coefficient was multiplied by the FPKM scores at that stage to correct for large-scale alterations in the transcriptional landscape. In the event that transcription globally decreases, ERCC spike-in tags become more prominent in the sequencing reaction, thus inflating the observed:expected ratio; multiplying the tag counts by expected/observed therefore deflates the transcriptional read-out to an appropriate level.
To determine the effect of the Gcn5cKO on transcription, up-regulated genes in the meiotic to round spermatid transition or the round to elongating spermatid transition were assessed for the ratio of ratios (Gcn5cKO S1/S0) / (Gcn5Ctrl S1/S0), where S1 is the second stage of the transition and SO is the first.
ChIP-seq analysis
All histone acetylation ChIP-seq data were previously published; analysis was performed unless otherwise noted as described in (Bryant et al., 2015). Briefly, bowtie v0.12.7 was used to align to mouse genome assembly NCBI37/mm9 with --best and all other parameters were default. Peaks were called using MACS2 against an input sample to control for sonication efficiency bias; the genome size and tag size were specified and a q-value threshold of 0.1% was used. Peaks were lifted from mm9 to mm10 using UCSC Genome Browser software liftOver, and overlaps of acetylation sites with nucleosomes was performed using BEDtools intersect.
DATA AND CODE AVAILABILITY
All computational tools and software utilized in this study are listed in the Key Resources Table. Additional dedicated scripts developed for this work are available upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| H3 | Abcam | Cat#:1791 |
| H3K9ac | Abcam | Cat#:4441 |
| H4 | Abcam | Cat#:7311 |
| H2A | Abcam | Cat#:18255 |
| H2B | Abcam | Cat#:1790 |
| Gcn5 | Cell Signaling | Cat#:3305S |
| GAPDH | Fitzgerald | Cat#:10R-G109a |
| Prm1 | BriarPatch | Cat#:MAb-Hup1N |
| Prm2 | BriarPatch | Cat#:MAb-Hup2B |
| Lectin PNA | Invitrogen | Cat#:L21409 |
| Goat anti-rabbit IgG (H+L) HRP conjugated | Biorad | Cat#:170-6515 |
| Goat anti-mouse IgG (H+L) HRP conjugated | Biorad | Cat#:172-1011 |
| Biological Samples | ||
| Mouse: 129SVE | Taconic | Taconic#:129SVE-M |
| Mouse: Gcn5fl/fl | Lin et. al., 2008b | NA |
| Mouse: Tg(Stra8-icre)1Reb/J | Jackson Labs | Jax#:008208 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BSA | Affymetrix | Cat#10857 |
| Bouin’s fixative | Sigma | Cat#:HT10132 |
| Benzonase nuclease | Millipore | Cat#:70746 |
| Protease inhibitor cocktail | Roche | Cat#:4693132001 |
| Trichostatin A | Sigma | Cat#:T1952 |
| Pyronin Y | Sigma | Cat#:213519 |
| Collagenase, Type 1A | Sigma | Cat#:C9891 |
| Trypsin | Sigma | Cat#:T9201 |
| MNase | NEB | Cat#:M0247S |
| DNase | Sigma | Cat#:DNEP |
| Nuclei EZ isolsation buffer | Sigma | Cat#:N3408 |
| PMSF | Sigma | P7626 |
| DTT | Sigma | D9779 |
| Critical Commercial Assays | ||
| KAPA Library Quant Kit | KAPA Biosystems | Cat#:KK4835 |
| Ultra II DNA Library prep kit for Illumina | NEB | Cat#:E7645L |
| RNeasy Mini Kit | Qiagen | Cat#:74106 |
| Script-Seq Complete Gold RNA-seq Kit | Illumina | Cat#:SCL24G |
| Nextera XT Kit | Illumina | Cat#:15028212 |
| NextSeqTM 500 High Output Kit (75 cycles) v2 kit | Illumina | Cat#:FC-404-2005 |
| NextSeqTM 500 High Output Kit (150 cycles) v2 kit | Illumina | Cat#:FC-404-2002 |
| Min Elute PCR Purification Kit | Qiagen | Cat#:28006 |
| PCR Purification Kit | Qiagen | Cat#:28106 |
| DNA 1000 Kit | Agilent | Cat#:5067-1504 |
| DNA High Sensitivity Kit | Agilent | Cat#:5067-4626 |
| SuperSignal West Pico Chemiluminescent Substrate | Thermo | Cat#:34080 |
| Deposited Data | ||
| ATAC-seq | GEO | GEO: GSE134561 |
| RNA-seq | GEO | GEO: GSE134561 |
| MNase-seq | GEO | GEO: GSE134561 |
| MNase-seq | Hammoud et al., 2009 | GEO: GSE15594 |
| MNase-seq | Erkek et al., 2013 | GEO: GSE42629 |
| MNase-seq | Samans et al., 2014 | GEO: GSE47843 |
| ChIP-seq | Bryant et al., 2015 | GEO: GSE56526 |
| ATAC-seq/ChIP-seq | Jung et al., 2017 | GEO: GSE79225 |
| RNA-seq | Hammoud et al., 2014 | GEO: GSE49624 |
| MNase-seq | Wu et al., 2016 | GEO: GSE669390 |
| Oligonucleotides | ||
| ATAC-seq indices, see Table S4 | Buenrostro et. al., 2013 | N/A |
| Software and Algorithms | ||
| Gene ontology analysis | Dennis et. al., 2003 | https://david.ncifcrf.gov/summary.jsp |
| STAR v2.5.2a | Dobin et. al., 2013 | https://github.com/alexdobin/STAR |
| SAMtools | Li et. al., 2009 | http://samtools.sourceforge.net/ |
| BEDtools | Quinlan et. al., 2010 | https://bedtools.readthedocs.io/en/latest/ |
| MACS2 | Zhang et. al., 2008 | https://github.com/taoliu/MACS |
| CEAS | Shin et. al., 2009 | http://liulab.dfci.harvard.edu/CEAS/ |
| DANPOS v2.1.3 | Chen et. al., 2013 | https://sites.google.com/site/danposdoc/ |
| RepEnrich v1.0 | Criscione et. al., 2014 | https://github.com/nskvir/RepEnrich |
| EdgeR | Robinson et. al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| R | R Core Team, 2017 | https://www.r-project.org/ |
| tophat2 v2.0.11 | Kim et. al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| HTseq v0.6.1p1 | Anders et. al., 2015 | https://htseq.readthedocs.io/en/release_0.11.1/ |
| bowtie v0.12.7 | Langmead et. al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| HOMER | Heinz et. al., 2010 | http://homer.ucsd.edu/homer/ |
All sequencing data reported in this study (ATAC-seq, RNA-seq, and MNase-seq) is available at NCBI Accession #GSE134561.
Supplementary Material
Highlights.
ATAC-seq localizes retained nucleosomes to promoters and repetitive DNA in sperm
Gcn5-mediated histone acetylation is necessary for proper spermiogenesis
Gcn5 loss alters chromatin dynamics leading to increased histone retention in sperm
Gcn5 is necessary for normal sperm formation and male fertility in mice
Acknowledgements
The authors thank Dr. Sharon Dent for generously providing Gcn5 floxed mice. Funding for this study was provided by the National Institutes of Health: R01 GM055360 and HD06817 to SLB, F32HD086939 to LJL, P50HD06817 to SLB, LJL, MSB, and F31GM123744 to ELS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Anders S, Pyl PT, Huber W, 2015. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpanahi A, Brinkworth M, Iles D, Krawetz SA, Paradowska A, Platts AE, Saida M, Steger K, Tedder P, Miller D, 2009. Endonuclease-sensitive regions of human spermatozoal chromatin are highly enriched in promoter and CTCF binding sequences. Genome Research 19, 1338–1349. doi: 10.1101/gr.094953.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet J, Wang C-Y, Baptista T, Vincent SD, Hsiao W-C, Stierle M, Kao C-F, Tora, , Devys D., 2014. The SAGA coactivator complex acts on the whole transcribed genome and is required for RNA polymerase II transcription. Genes & Development 28, 1999–2012. doi: 10.1101/gad.250225.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussouar F, Goudarzi A, Buchou T, Shiota H, Barral S, Debernardi A, Guardiola P, Brindle P, Martinez G, Arnoult C, Khochbin S, Rousseaux S, 2014. A specific CBP/p300-dependent gene expression programme drives the metabolic remodelling in late stages of spermatogenesis. Andrology 2, 351–359. doi: 10.1111/j.2047-2927.2014.00184.x [DOI] [PubMed] [Google Scholar]
- Brind’Amour J, Liu S, Hudson M, Chen C, Karimi MM, Lorincz MC, 2015. An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nature Communications 6, 6033. doi: 10.1038/ncomms7033 [DOI] [PubMed] [Google Scholar]
- Bryant JM, Donahue G, Wang X, Meyer-Ficca M, Luense LJ, Weller AH, Bartolomei MS, Blobel GA, Meyer RG, Garcia BA, Berger SL, 2015. Characterization of BRD4 during mammalian post-meiotic sperm development. Molecular and Cellular Biology MCB.01328–14−56. doi: 10.1128/MCB.01328-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant JM, Meyer-Ficca ML, Dang VM, Berger SL, Meyer RG, 2013. Separation of spermatogenic cell types using STA-PUT velocity sedimentation. J Vis Exp e50648–e50648. doi: 10.3791/50648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brykczynska U, Hisano M, Erkek S, Ramos L, Oakeley EJ, Roloff TC, Beisel C, Schubeler D, Stadler MB, Peters AHFM, 2010. Repressive and active histone methylation mark distinct promoters in human and mouse spermatozoa. Nat Struct Mol Biol 17, 679–687. doi: 10.1038/nsmb.1821 [DOI] [PubMed] [Google Scholar]
- Bu P, Evrard YA, Lozano G, Dent SYR, 2007. Loss of Gcn5 Acetyltransferase Activity Leads to Neural Tube Closure Defects and Exencephaly in Mouse Embryos. Molecular and Cellular Biology 27, 3405–3416. doi: 10.1128/MCB.00066-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ, 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Meth 10, 1213–1218. doi: 10.1038/nmeth.2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carone BR, Hung J-H, Hainer SJ, Chou M-T, Carone DM, Weng Z, Fazzio TG, Rando OJ, 2014. High-Resolution Mapping of Chromatin Packaging in Mouse Embryonic Stem Cells and Sperm. Developmental Cell 1–20. doi: 10.1016/j.devcel.2014.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrell DT, Emery BR, Hammoud S, 2007. Altered protamine expression and diminished spermatogenesis: what is the link? Human Reproduction Update 13, 313–327. doi: 10.1093/humupd/dml057 [DOI] [PubMed] [Google Scholar]
- Carrell DT, Hammoud SS, 2010. The human sperm epigenome and its potential role in embryonic development. Molecular Human Reproduction 16, 37–47. doi: 10.1093/molehr/gap090 [DOI] [PubMed] [Google Scholar]
- Carrell DT, Liu L, 2001. Altered protamine 2 expression is uncommon in donors of known fertility, but common among men with poor fertilizing capacity, and may reflect other abnormalities of spermiogenesis. Journal of Andrology 22, 604–610. [PubMed] [Google Scholar]
- Chen K, Xi Y, Pan X, Li Z, Kaestner K, Tyler J, Dent S, He X, Li W, 2013. DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 23, 341–351. doi: 10.1101/gr.142067.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaillier P, Mauro N, Feneux D, Jouannet P, David G, 1987. Anomalous protein complement of sperm nuclei in some infertile men. Lancet 2, 806–807. [DOI] [PubMed] [Google Scholar]
- Cho C, Willis WD, Goulding EH, Jung-Ha H, Choi YC, Hecht NB, Eddy EM, 2001. Haploinsufficiency of protamine-1 or −2 causes infertility in mice. Nat Genet 28, 82–86. doi: 10.1038/88313 [DOI] [PubMed] [Google Scholar]
- Criscione SW, Zhang Y, Thompson W, Sedivy JM, Neretti N, 2014. Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics 15, 860–17. doi: 10.1186/1471-2164-15-583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dansranjavin T, Schagdarsurengin U, 2016. The Rationale of the Inevitable, or Why Is the Consideration of Repetitive DNA Elements Indispensable in Studies of Sperm Nucleosomes. Developmental Cell 37, 13–14. doi: 10.1016/j.devcel.2016.03.020 [DOI] [PubMed] [Google Scholar]
- de Mateo S, Ramos L, van der Vlag J, de Boer P, Oliva R, 2010. Improvement in chromatin maturity of human spermatozoa selected through density gradient centrifugation. International Journal of Andrology 34, 256–267. doi: 10.1111/j.1365-2605.2010.01080.x [DOI] [PubMed] [Google Scholar]
- de Yebra L, Oliva R, 1993. Rapid analysis of mammalian sperm nuclear proteins. Anal. Biochem 209, 201–203. [DOI] [PubMed] [Google Scholar]
- Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA, 2003. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4, P3. [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Isono K-I, Ohbo K, Endo TA, Ohara O, Maekawa M, Toyama Y, Ito C, Toshimori K, Helin K, Ogonuki N, Inoue K, Ogura A, Yamagata K, Kitabayashi I, Koseki H, 2017. EPC1/TIP60-Mediated Histone Acetylation Facilitates Spermiogenesis in Mice. Mol. Cell. Biol 37, e00082–17−16. doi: 10.1128/MCB.00082-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecco G, Imbeault M, Trono D, 2017. KRAB zinc finger proteins. Development 144, 2719–2729. doi : 10.1242/dev.132605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecklund PS, 1975. Mouse sperm basic nuclear protein. Electrophoretic characterization and fate after fertilization. The Journal of Cell Biology 66, 251–262. doi: 10.1083/jcb.66.2.251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkek S, Hisano M, Liang C-Y, Gill M, Murr R, Dieker J, Schubeler D, Vlag JVD, Stadler MB, Peters AHFM, 2013. Molecular determinants of nucleosome retention at CpG-rich sequences in mouse spermatozoa. Nat Struct Mol Biol 20, 868–875. doi: 10.1038/nsmb.2599 [DOI] [PubMed] [Google Scholar]
- Garcia-Perez JL, Doucet AJ, Kopera HC, Richardson SR, Moldovan JB, Moran JV, 2015. The Influence of LINE-1 and SINE Retrotransposons on Mammalian Genomes. Microbiology Spectrum 3, 1–40. doi: 10.1128/microbiolspec.MDNA3-0061-2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaucher J, Boussouar, al FC, Montellier E, Curtet S, Buchou T, Bertrand S, Hery P, Jounier S, Depaux A, Vitte A-L, Guardiola P, Pernet K, Debernardi A, Lopez F, Holota HELEN, Imbert J, Wolgemuth DJ, rard MGE, Rousseaux S, Khochbin S, 2012. Bromodomain-dependent stage-specific male genome programming by Brdt. The EMBO Journal 31, 3809–3820. doi: 10.1038/emboj.2012.233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govin J, Caron C, Lestrat C, Rousseaux S, Khochbin S, 2004. The role of histones in chromatin remodelling during mammalian spermiogenesis. European Journal of Biochemistry 271, 3459–3469. doi: 10.1111/j.1432-1033.2004.04266.x [DOI] [PubMed] [Google Scholar]
- Govin J, Escoffier E, Rousseaux S, Kuhn L, Ferro M, Thevenon J, Catena R, Davidson I, Garin J, Khochbin S, Caron C, 2007. Pericentric heterochromatin reprogramming by new histone variants during mouse spermiogenesis. The Journal of Cell Biology 176, 283–294. doi: 10.1083/jcb.200604141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant PA, Eberharter A, John S, Cook RG, Turner BM, Workman JL, 1999. Expanded lysine acetylation specificity of Gcn5 in native complexes. Journal of Biological Chemistry 274, 5895–5900. [DOI] [PubMed] [Google Scholar]
- Hammoud SS, Low DHP, Yi C, Carrell DT, Guccione E, Cairns BR, 2014. Chromatin and Transcription Transitions of Mammalian Adult Germline Stem Cells and Spermatogenesis. Stem Cell 1–15. doi: 10.1016/j.stem.2014.04.006 [DOI] [PubMed] [Google Scholar]
- Hammoud SS, Nix DA, Hammoud AO, Gibson M, Cairns BR, Carrell DT, 2011. Genome-wide analysis identifies changes in histone retention and epigenetic modifications at developmental and imprinted gene loci in the sperm of infertile men. Human Reproduction 26, 2558–2569. doi: 10.1093/humrep/der192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammoud SS, Nix DA, Zhang H, Purwar J, Carrell DT, Cairns BR, 2009. Distinctive chromatin in human sperm packages genes for embryo development. Nature 460, 473–478. doi: 10.1038/nature08l62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazzalin CA, Mahadevan LC, 2016. Acid-Urea Gel Electrophoresis and Western Blotting of Histones, in: Carrell DT, Aston KI (Eds.), Spermatogenesis, Methods in Molecular Biology. Springer New York, New York, NY, pp. 173–198. doi: 10.1007/978-1-4939-6630-1_11 [DOI] [PubMed] [Google Scholar]
- Hazzouri M, Pivot-Pajot C, Faure AK, Usson Y, Pelletier R, Sele B, Khochbin S, Rousseaux S, 2000. Regulated hyperacetylation of core histones during mouse spermatogenesis: involvement of histone deacetylases. Eur. J. Cell Biol 79, 950–960. doi: 10.1078/0171-9335-00123 [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK, 2010. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Molecular Cell 38, 576–589. doi: 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbeault M, Helleboid P-Y, Trono D, 2017. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 543, 550–554. doi: 10.1038/nature21683 [DOI] [PubMed] [Google Scholar]
- Jung YH, Sauria MEG, Lyu X, Cheema MS, Ausio J, Taylor J, Corces VG, 2017. Chromatin States in Mouse Sperm Correlate with Embryonic and Adult Regulatory Landscapes. CellReports 18, 1366–1382. doi: 10.1016/j.celrep.2017.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL, 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. doi: 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo MH, Brownell JE, Sobel RE, Ranalli TA, Cook RG, Edmondson DG, Roth SY, Allis CD, 1996. Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Nature 383, 269–272. doi: 10.1038/383269a0 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL, 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. doi: 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KK, Workman JL, 2007. Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol 8, 284–295. doi: 10.1038/nrm2145 [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup, 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Zhang Z, Chen C-H, Behringer RR, Dent SYR, 2008a. Proper Gcn5 histone acetyltransferase expression is required for normal anteroposterior patterning of the mouse skeleton. Dev. Growth Differ 50, 321–330. doi: 10.1111/j.1440-169X.2008.01041.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Zhang Z, Srajer G, Chen YC, Huang M, Phan HM, Dent SYR, 2008b. Proper expression of the Gcn5 histone acetyltransferase is required for neural tube closure in mouse embryos. Dev. Dyn 237, 928–940. doi: 10.1002/dvdy.21479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Aston KI, Carrell DT, 2013. Protamine extraction and analysis of human sperm protamine 1/protamine 2 ratio using Acid gel electrophoresis. Methods Mol. Biol 927, 445–450. doi : 10.1007/978-1-62703-038-0_38 [DOI] [PubMed] [Google Scholar]
- Luense LJ, Wang X, Schon SB, Weller AH, Shiao EL, Bryant JM, Bartolomei MS, Coutifaris C, Garcia BA, Berger SL, 2016. Comprehensive analysis of histone post-translational modifications in mouse and human male germ cells. Epigenetics Chromatin 9, 1–15. doi: 10.1186/s13072-016-0072-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa S, Yukawa M, Alavattam KG, Barski A, Namekawa SH, 2017. Dynamic reorganization of open chromatin underlies diverse transcriptomes during spermatogenesis. Nucleic Acids Research 1–16. doi: 10.1093/nar/gkx1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva R, 2006. Protamines and male infertility. Human Reproduction Update 12, 417–435. doi: 10.1093/humupd/dml009 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM, 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK, 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royo H, Stadler MB, Peters AHFM, 2016. Alternative Computational Analysis Shows No Evidence for Nucleosome Enrichment at Repetitive Sequences in Mammalian Spermatozoa. Developmental Cell 37, 98–104. doi: 10.1016/j.devcel.2016.03.010 [DOI] [PubMed] [Google Scholar]
- Ruiz JL, Tena JJ, Bancells C, Cortes A, Gomez-Skarmeta JL, Gomez-Diaz E, 2018. Characterization of the accessible genome in the human malaria parasite Plasmodium falciparum. Nucleic Acids Research 46, 9414–9431. doi: 10.1093/nar/gky643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadate-Ngatchou PI, Payne CJ, Dearth AT, Braun RE, 2008. Cre recombinase activity specific to postnatal, premeiotic male germ cells in transgenic mice. genesis 46, 738–742. doi: 10.1002/dvg.20437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samans B, Yang Y, Krebs S, Sarode GV, Blum H, Reichenbach M, Wolf E, Steger K, Dansranjavin T, Schagdarsurengin U, 2014. Uniformity of nucleosome preservation pattern in Mammalian sperm and its connection to repetitive DNA elements. Developmental Cell 30, 23–35. doi: 10.1016/j.devcel.2014.05.023 [DOI] [PubMed] [Google Scholar]
- Sammons MA, Zhu J, Drake AM, Berger SL, 2015. TP53 engagement with the genome occurs in distinct local chromatin environments via pioneer factor activity. Genome Research 25, 179–188. doi: 10.1101/gr.181883.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schagdarsurengin U, Paradowska A, Steger K, 2012. Analysing the sperm epigenome: roles in early embryogenesis and assisted reproduction. Nat Rev Urol 9, 609–619. doi: 10.1038/nrurol.2012.183 [DOI] [PubMed] [Google Scholar]
- Schneider S, Balbach M, Jikeli JF, Fietz D, Nettersheim D, Jostes S, Schmidt R, Kressin M, Bergmann M, Wachten D, Steger K, Schorle H, 2016. Re-visiting the Protamine-2 locus: deletion, but not haploinsufficiency, renders male mice infertile. Sci Rep 1–13. doi: 10.1038/srep36764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang E, Nickerson HD, Wen D, Wang X, Wolgemuth DJ, 2007. The first bromodomain of Brdt, a testis-specific member of the BET sub-family of double-bromodomain-containing proteins, is essential for male germ cell differentiation. Development 134, 3507–3515. doi: 10.1242/dev.004481 [DOI] [PubMed] [Google Scholar]
- Shin H, Liu T, Manrai AK, Liu XS, 2009. CEAS: cis-regulatory element annotation system. Bioinformatics 25, 2605–2606. doi: 10.1093/bioinformatics/btp479 [DOI] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishii H, Sun J-M, Pazin MJ, Davie JR, Peterson CL, 2006. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847. doi: 10.1126/science.1124000 [DOI] [PubMed] [Google Scholar]
- Siklenka K, Erkek S, Godmann M, Lambrot R, McGraw S, Lafleur C, Cohen T, Xia J, Suderman M, Hallett M, Trasler J, Peters AHFM, Kimmins S, 2015. Disruption of histone methylation in developing sperm impairs offspring health transgenerationally. Science aab2006. doi: 10.1126/science.aab2006 [DOI] [PubMed] [Google Scholar]
- van der Heijden GW, Derijck AAHA, Ramos L, Giele M, van der Vlag J, de Boer P, 2006. Transmission of modified nucleosomes from the mouse male germline to the zygote and subsequent remodeling of paternal chromatin. Developmental Biology 298, 458–469. doi: 10.1016/j.ydbio.2006.06.051 [DOI] [PubMed] [Google Scholar]
- Ward WS, 2009. Function of sperm chromatin structural elements in fertilization and development. Molecular Human Reproduction 16, 30–36. doi: 10.1093/molehr/gap080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Huang B, Chen H, Yin Q, Liu Y, Xiang Y, Zhang B, Liu B, Wang Q, Xia W, Li W, Li Y, Ma J, Peng X, Zheng H, Ming J, Zhang W, Zhang J, Tian G, Xu F, Chang Z, Na J, Yang X, Xie W, 2016. The landscape of accessible chromatin in mammalian preimplantation embryos. Nature Publishing Group 534, 652–657. doi: 10.1038/nature18606 [DOI] [PubMed] [Google Scholar]
- Xiang Y, Tanaka Y, Patterson B, Kang Y-J, Govindaiah G, Roselaar N, Cakir B, Kim K-Y, Lombroso AP, Hwang S-M, Zhong M, Stanley EG, Elefanty AG, Naegele JR, Lee S-H, Weissman SM, Park I-H, 2017. Fusion of Regionally Specified hPSC-Derived Organoids Models Human Brain Development and Interneuron Migration 1–24. doi: 10.1016/j.stem.2017.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Edmondson DG, Evrard YA, Wakamiya M, Behringer RR, Roth SY, 2000. Loss of Gcn5l2 leads to increased apoptosis and mesodermal defects during mouse development. Nat Genet 26, 229–232. doi: 10.1038/79973 [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Hada M, Fukuda Y, Inoue E, Makino Y, Katou Y, Shirahige K, Okada Y, 2018. Re-evaluating the Localization of Sperm-Retained Histones Revealed the Modification-Dependent Accumulation in Specific Genome Regions. CellReports 23, 3920–3932. doi: 10.1016/j.celrep.2018.05.094 [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Yamauchi J, Kuwata T, Tamura T, Yamashita T, Bae N, Westphal H, Ozato K, Nakatani Y, 2000. Distinct but overlapping roles of histone acetylase PCAF and of the closely related PCAF-B/GCN5 in mouse embryogenesis. Proceedings of the National Academy of Sciences 97, 11303–11306. doi: 10.1073/pnas.97.21.11303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Bone JR, Edmondson DG, Turner BM, Roth SY, 1998. Essential and redundant functions of histone acetylation revealed by mutation of target lysines and loss of the Gcn5p acetyltransferase. The EMBO Journal 17, 3155–3167. doi: 10.1093/emboj/17.11.3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, 2006. Sperm Nuclear Histone to Protamine Ratio in Fertile and Infertile Men: Evidence of Heterogeneous Subpopulations of Spermatozoa in the Ejaculate. Journal of Andrology 27, 414–420. doi: 10.2164/jandrol.05171 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS, 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. doi: 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All computational tools and software utilized in this study are listed in the Key Resources Table. Additional dedicated scripts developed for this work are available upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| H3 | Abcam | Cat#:1791 |
| H3K9ac | Abcam | Cat#:4441 |
| H4 | Abcam | Cat#:7311 |
| H2A | Abcam | Cat#:18255 |
| H2B | Abcam | Cat#:1790 |
| Gcn5 | Cell Signaling | Cat#:3305S |
| GAPDH | Fitzgerald | Cat#:10R-G109a |
| Prm1 | BriarPatch | Cat#:MAb-Hup1N |
| Prm2 | BriarPatch | Cat#:MAb-Hup2B |
| Lectin PNA | Invitrogen | Cat#:L21409 |
| Goat anti-rabbit IgG (H+L) HRP conjugated | Biorad | Cat#:170-6515 |
| Goat anti-mouse IgG (H+L) HRP conjugated | Biorad | Cat#:172-1011 |
| Biological Samples | ||
| Mouse: 129SVE | Taconic | Taconic#:129SVE-M |
| Mouse: Gcn5fl/fl | Lin et. al., 2008b | NA |
| Mouse: Tg(Stra8-icre)1Reb/J | Jackson Labs | Jax#:008208 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BSA | Affymetrix | Cat#10857 |
| Bouin’s fixative | Sigma | Cat#:HT10132 |
| Benzonase nuclease | Millipore | Cat#:70746 |
| Protease inhibitor cocktail | Roche | Cat#:4693132001 |
| Trichostatin A | Sigma | Cat#:T1952 |
| Pyronin Y | Sigma | Cat#:213519 |
| Collagenase, Type 1A | Sigma | Cat#:C9891 |
| Trypsin | Sigma | Cat#:T9201 |
| MNase | NEB | Cat#:M0247S |
| DNase | Sigma | Cat#:DNEP |
| Nuclei EZ isolsation buffer | Sigma | Cat#:N3408 |
| PMSF | Sigma | P7626 |
| DTT | Sigma | D9779 |
| Critical Commercial Assays | ||
| KAPA Library Quant Kit | KAPA Biosystems | Cat#:KK4835 |
| Ultra II DNA Library prep kit for Illumina | NEB | Cat#:E7645L |
| RNeasy Mini Kit | Qiagen | Cat#:74106 |
| Script-Seq Complete Gold RNA-seq Kit | Illumina | Cat#:SCL24G |
| Nextera XT Kit | Illumina | Cat#:15028212 |
| NextSeqTM 500 High Output Kit (75 cycles) v2 kit | Illumina | Cat#:FC-404-2005 |
| NextSeqTM 500 High Output Kit (150 cycles) v2 kit | Illumina | Cat#:FC-404-2002 |
| Min Elute PCR Purification Kit | Qiagen | Cat#:28006 |
| PCR Purification Kit | Qiagen | Cat#:28106 |
| DNA 1000 Kit | Agilent | Cat#:5067-1504 |
| DNA High Sensitivity Kit | Agilent | Cat#:5067-4626 |
| SuperSignal West Pico Chemiluminescent Substrate | Thermo | Cat#:34080 |
| Deposited Data | ||
| ATAC-seq | GEO | GEO: GSE134561 |
| RNA-seq | GEO | GEO: GSE134561 |
| MNase-seq | GEO | GEO: GSE134561 |
| MNase-seq | Hammoud et al., 2009 | GEO: GSE15594 |
| MNase-seq | Erkek et al., 2013 | GEO: GSE42629 |
| MNase-seq | Samans et al., 2014 | GEO: GSE47843 |
| ChIP-seq | Bryant et al., 2015 | GEO: GSE56526 |
| ATAC-seq/ChIP-seq | Jung et al., 2017 | GEO: GSE79225 |
| RNA-seq | Hammoud et al., 2014 | GEO: GSE49624 |
| MNase-seq | Wu et al., 2016 | GEO: GSE669390 |
| Oligonucleotides | ||
| ATAC-seq indices, see Table S4 | Buenrostro et. al., 2013 | N/A |
| Software and Algorithms | ||
| Gene ontology analysis | Dennis et. al., 2003 | https://david.ncifcrf.gov/summary.jsp |
| STAR v2.5.2a | Dobin et. al., 2013 | https://github.com/alexdobin/STAR |
| SAMtools | Li et. al., 2009 | http://samtools.sourceforge.net/ |
| BEDtools | Quinlan et. al., 2010 | https://bedtools.readthedocs.io/en/latest/ |
| MACS2 | Zhang et. al., 2008 | https://github.com/taoliu/MACS |
| CEAS | Shin et. al., 2009 | http://liulab.dfci.harvard.edu/CEAS/ |
| DANPOS v2.1.3 | Chen et. al., 2013 | https://sites.google.com/site/danposdoc/ |
| RepEnrich v1.0 | Criscione et. al., 2014 | https://github.com/nskvir/RepEnrich |
| EdgeR | Robinson et. al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| R | R Core Team, 2017 | https://www.r-project.org/ |
| tophat2 v2.0.11 | Kim et. al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| HTseq v0.6.1p1 | Anders et. al., 2015 | https://htseq.readthedocs.io/en/release_0.11.1/ |
| bowtie v0.12.7 | Langmead et. al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| HOMER | Heinz et. al., 2010 | http://homer.ucsd.edu/homer/ |
All sequencing data reported in this study (ATAC-seq, RNA-seq, and MNase-seq) is available at NCBI Accession #GSE134561.