

Abstract
Recent work demonstrated the propensity of dopamine transporters (DATs) to form trimers or higher oligomers, enhanced upon binding a furopyrimidine, AIM-100. AIM-100 binding promotes DAT endocytosis and thereby moderates dopaminergic transmission. Despite the neurobiological significance of these events, the molecular mechanisms that underlie the stabilization of DAT trimer and the key interactions that modulate the trimerization of DAT, and not serotonin transporter SERT, remain unclear. In the present study, we determined three structural models, termed trimer-W238, -C306 and -Y303, for possible trimerization of DATs using structural data resolved for DAT and its paralogs and structural homologs that share the LeuT fold in advanced computational modeling and simulations, site-directed mutagenesis experiments and live-cell imaging assays. The models are in accord with the versatility of LeuT fold to stabilize dimeric or higher order constructs. Selected residues show a high propensity to occupy interfacial regions. Among them, D231-W238 in the extracellular loop EL2, including the intersubunit salt-bridge forming pair D231/D232-R237 (not present in SERT) (in trimer-W238), the loop EL3 (trimers-C306 and -Y303), and W497 on the intracellularly exposed IL5 loop (trimer-C306) and its spatial neighbors (e.g. K525) near the C-terminus are computationally predicted and experimentally confirmed to play important roles in enabling the correct folding and/or oligomerization of DATs in the presence of AIM-100. The study suggests the possibility of controlling the effective transport of dopamine by altering the oligomerization state of DAT upon small molecule binding, as a possible intervention strategy to modulate dopaminergic signaling.
Keywords: Dopamine transporter, Oligomerization, in silico saturation mutagenesis, docking and molecular dynamics simulations, furopyrimidine AIM-100, structural dynamics
Graphical Abstract:
1. Introduction
Neurotransmitter:sodium symporters (NSSs) play a major role in clearing excess neurotransmitters from extra-neuronal regions, thus regulating synaptic transmission and preventing neurotoxicity. As essential regulators of neurotransmission, NSSs are important pharmacological targets (Amara and Sonders, 1998; Iversen, 2006; Kanner and Zomot, 2008; Kristensen et al., 2011; Pramod et al., 2013). Mutations, mis-localization and aberrant expression of the solute carrier 6 (SLC6) family of NSSs result in neurological and behavior disorders, drug abuse and neurodegenerative diseases (Gether et al.; Giros et al., 1996; Kristensen et al., 2011; Pramod et al., 2013; Sulzer, 2011).
SLC6 family members include dopamine (DA) transporter (DAT), serotonin and norepinephrine transporters (SERT and NET), and GABA and glycine transporters (Kanner and Zomot, 2008). DAT regulates dopaminergic signaling by reuptake of DA into presynaptic terminals, assisted by co-transport of two sodium ions and the permeation of a chloride ion. It is the principal target for psychostimulants and/or drugs of abuse such as cocaine and amphetamines (AMPH) (Sulzer, 2011); and understanding the molecular basis of its cellular actions has pharmacological and biomedical significance.
It is now widely established that proteins’ structural dynamics enable their activities in the cell and their adaptation to changes in environment or to mutations (for reviews see (Bahar et al., 2010; Haliloglu and Bahar, 2015)). In the case of NSS family members that share the so-called LeuT fold (Yamashita et al., 2005), an alternating access (Jardetzky, 1966) or rocking bundle mechanism (Forrest and Rudnick, 2009; Forrest et al., 2008) has been proposed. The latter enables the uptake and release of the cargo in the respective outward-facing (OF) and inward-facing (IF) states of the transporter. In addition to these global changes in structure, local changes in residue-residue associations modulate the opening/closure of the extracellular (EC) and intracellular (IC) gates in the two respective states.
Recent studies suggest that the tightly regulated structural changes of NSSs are not solely restricted to their substrate/ion translocation events, but also mediate their assembly into multimers. Growing data from radiation inactivation (Berger et al., 1994), crosslinking (Hastrup et al., 2001), mutagenesis (Torres et al., 2003), co-immuno-precipitation (Kilic and Rudnick, 2000; Sorkina et al., 2003), fluorescence resonance energy transfer (Schmid et al., 2001; Sorkina et al., 2003; Sorkina et al., 2018), electrophoresis (Baucum et al., 2004; Sorkina et al., 2018), single molecule imaging (Anderluh et al., 2014), and super-resolution microscopy (Rahbek-Clemmensen et al., 2017) suggest that NSSs may co-exist as monomers and oligomers. Oligomerization has been suggested to be a determinant of NSS distribution in the plasma membrane (PM) in addition to modulating transport efficiency (Li et al., 2010; Scholze et al., 2002; Sitte et al., 2004; Sorkina et al., 2003); and it can be promoted upon ligand binding to SERT (Anderluh et al., 2017) and DAT (Siciliano et al., 2018). A cooperativity between protomers has been reported for DAT dimers and/or oligomers (Cheng et al., 2017; Gur et al., 2017; Zhen et al., 2015); and an oligomer-based counter-transport model has been proposed for describing the AMPH-induced substrate efflux (Seidel et al., 2005; Sitte and Freissmuth, 2010; Sitte and Freissmuth, 2015). There is a compelling need to learn more about the biological mechanisms and significance of NSS oligomers.
While Drosophila DAT (dDAT) (Penmatsa et al., 2013) and human SERT (hSERT) (Coleman et al., 2016) have been resolved as monomers, the bacterial orthologue LeuT, which long served as a prototype for structure-based studies of NSS family members (Nyola et al., 2010), has been resolved as a dimer (Krishnamurthy and Gouaux, 2012; Yamashita et al., 2005); and single-molecule images of intact cells suggested constitutive DAT dimerization (Das et al., 2019). Among other transporters whose protomers share the LeuT fold, betaine transporter (BetP) and the L-carnitine/γ-butyrobetaine exchanger (CaiT) have been resolved as trimers (Ressl et al., 2009; Schulze et al., 2010); and BetP trimerization has been noted to be a requirement for regulating transporter activity (Perez et al., 2011).
Our recent experiments revealed that a cell-permeable furopyrimidine, AIM-100, promotes the oligomerization and endocytosis of DAT but not of other closely-related SLC6 transporters, NET and SERT (Sorkina et al., 2018). DAT oligomers were detected as high molecular weight DAT species by SDS-PAGE, and based on the mobility of these SDS-resistant species, it was proposed that a minimal oligomerization state of DAT could be a trimer (Sorkina et al., 2018). While the ability of AIM-100 to stabilize DAT trimers or oligomers appears to depend on the conformation, OF or IF, of DAT (Sorkina et al., 2018), the molecular mechanisms that underlie the stabilization of the trimers remain unknown.
The breakthrough in structural (Penmatsa et al., 2013, 2015; Wang et al., 2015), in silico (Cheng and Bahar, 2015; Cheng et al., 2015; Cheng et al., 2017; Cheng et al., 2018; Gur et al., 2017; Kaya et al., 2018; Khelashvili et al., 2015; LeVine et al., 2018) and live-cell (Adkins et al., 2007; Caltagarone et al., 2015; Sorkina et al., 2013; Sorkina et al., 2003) studies of DAT dynamics and function now permit us to gain a better understanding of the link between DAT structural dynamics, oligomerization, and function. Here we explore the propensity of DAT to trimerize in the absence and presence of AIM-100, as well as its implication on DAT structural dynamics using a combination of computational and biochemical methods, and live-cell imaging assays. Our in silico experiments pointed to specific residues likely to drive or stabilize intermolecular associations. Experiments with mutants conducted for investigating the effects of substitutions at those sites supported their potential role in DAT oligomerization and PM localization (enhanced by AIM-100). Application of a novel machine learning method for assessing the impact of single amino acid variants (SAVs) (Ponzoni and Bahar, 2018) further helped rationalize the experimental observations.
Overall these results invite attention to the versatility of DAT, intrinsically endowed by the LeuT fold, to assume various oligomerization states. Selective modulation of oligomerization state by small-molecule (e.g. AIM-100) binding emerges as a potential intervention method for altering DA reuptake or alleviating associated neurological disorders.
2. Materials and Methods
2.1. Molecular modeling of DAT and SERT trimers.
We generated in silico a series of trimeric models for human DAT and SERT using the trimer docking module of the software ClusPro (Comeau et al., 2004; Kozakov et al., 2017). Supplemental Fig S1 illustrates the top-ranking models obtained for DAT protomers in the OF and IF states. In the former case, we used as input an OF conformer equilibrated in the presence of DA and two Na+ and one Cl− ions; and for the IF state, a conformer sampled after releasing the substrate DA and ions to the IC region (Cheng and Bahar, 2015). Structural data for both conformers used as input are provided in PDB format as Supplemental Files. We obtained 19 and 22 clusters of conformations for the respective IF and OF trimers, upon clustering the ~1,000 trimeric models generated by ClusPro. The clusters were rank-ordered by cluster size, as recommended (Comeau et al., 2004; Kozakov et al., 2017). Representative members belonging to the top-ranking six clusters for IF and OF trimer models are shown in Fig S1, and the corresponding cluster sizes and weighted scores are listed in Table S1. Three trimers (highlighted in green and yellow boxes in Fig S1) were selected for further studies based on the NSS-specific criteria described below. OF SERT trimers were generated by applying the same protocol using the crystal structure of OF SERT monomer (PDB: 5I75) (Coleman et al., 2016).
In addition, we constructed DAT (OF and IF) trimers by homology modeling using as template the crystal structure resolved for CaiT trimer (Schulze et al., 2010). Briefly, hDAT OF or IF monomers were first structurally aligned with each subunit of CaiT using the STAMP algorithm (Russell and Barton, 1992) from MultiSeq (Roberts et al., 2006) tool of VMD (Humphrey et al., 1996) and assembled to generate the trimer construct, followed by loop refinement using MODELLER (Fiser and Sali, 2003) and 20,000 steps of energy minimization using NAMD (Phillips et al., 2005).
2.2. Simulations of AIM-100 binding to DAT trimers.
AIM-100 binding to selected DAT trimers was modeled using two methods, AutoDock (Trott and Olson, 2010) and MD simulations in explicit membrane, as described in the Supplemental Method. The lipid composition was adopted from previous studies (Ingólfsson et al., 2017; Khelashvili et al., 2015) to mimic the asymmetric distribution of lipids in neuronal PMs (see Supplemental Method). Each simulation system contained ~ 235,000 atoms, including the DAT trimer, approximately 600 lipid molecules, and 46,000 water molecules. Eight to ten AIM-100 molecules were randomly distributed in the aqueous environment on both sides of the membrane to examine their diffusion and binding to the DAT trimer. Additionally, a series of MD runs were carried out with higher concentration of AIM-100s to investigate the binding to DAT monomers. The complete list of MD runs along with their specifications is presented in Table S2. The scripts for initiating NAMD runs and AutoDock ligand docking, and the AIM-100 topology parameters are available in the Appendices of the Supplemental Material.
2. 3. Mutagenesis.
Yellow fluorescent protein (YFP) and hemagglutinin epitope (HA)- tagged wild-type (wt) human DAT (YFP-HA-DAT, Addgene plasmid # 90244) and truncated human DAT mutant lacking the N-terminal 65 residues (YFP-ΔN-HA-DAT) were described previously (Sorkina et al., 2006). All mutations in the full-length YFP-HA-DAT construct were made using a Stratagene Quick-change mutagenesis kit according to the manufacture’s protocol using YFP-HA-DAT as a template. The mutations were verified by automatic dideoxynucleotide sequencing.
2.4. Cell culture, transient and stable expression.
Porcine aortic endothelial (PAE) cells were grown in F12 medium with 10% fetal bovine serum (FBS). Effectene method (Qiagen, Valencia, CA) was used for DNA transfection of PAE cells. Clonal pools of PAE cells stably expressing DAT mutants were generated by G418 selection for 2–3 weeks. These pools of G418-resistant cells contained cells not expressing YFP-HA-DAT and 10–20% cells expressing various levels of YFP-HA-DAT mutants. The cells were grown on glass coverslips for direct imaging of YFP in fixed cells, or on 12-well tissue culture plates for biochemical experiments.
2.5. Western blotting detection of YFP-HA-DAT.
Cells grown in 12-well plates were incubated with vehicle (DMSO) or AIM-100 for 2 hrs at 37°C, washed with ice-cold phosphate-buffer saline, and solubilized in TGH lysis buffer (1%Triton X-100, 10% glycerol, 20 mM HEPES, 50 mM NaCl) supplemented with 1% deoxycholate, 10 mM N-ethyl maleimide, iodacetamide, 10 mM DTT and protease (including MG132) and phosphatase inhibitors for 15 min at 4°C. Lysates were resolved by SDS-PAGE, transferred to nitrocellulose and probed by western blotting using antibodies to GFP as described previously (Sorkina et al., 2018).
2.6. Fluorescence microscopy imaging.
The cells transiently or stably expressing wild type (wt) or mutant DATs were fixed with freshly-prepared 4% papa-formaldehyde untreated or after 2-hour incubation with DMSO (vehicle) or AIM-100 (20 μM). To obtain three-dimensional (3D) images of the cells, a z-stack of confocal images was acquired using a spinning disk confocal imaging system (Intelligent Imaging Innovation, Denver, CO), as described previously (Sorkina et al., 2018). Typically, 20–25 serial 2D confocal images through a 515 nm channel were acquired at 200–400 nm intervals. Images were analyzed and prepared using SlideBook 6 software.
2.7. In silico saturation mutagenesis experiments.
In silico saturation mutagenesis experiments evaluate the effect of all 19 amino acid substitutions at all sequence positions, using the machine-learning-based method we recently introduced (Ponzoni and Bahar, 2018) for predicting the pathogenicity of SAVs. The corresponding software, Rhapsody v2, has been upgraded to incorporate features derived from Pfam sequence alignments (El-Gebali et al., 2019), namely the Shannon entropy as a metric of conservation, and mutual information as a metric of coevolution propensity (Bakan et al., 2014; Dunn et al., 2008; Gloor et al., 2005); and benchmarked against a dataset of ~ 19,000 annotated mutants to yield a prediction accuracy of 0.85 (measured by area under ROC curve). We also computed the predictions from PolyPhen-2 (Adzhubei et al., 2010) and EVmutation (Hopf et al., 2017) for comparison. EVmutation estimates the fitness of the mutant with respect to the wild-type (wt) protein (whose score is 0). EVmutation scores were normalized by computing an optimal threshold that maximized the classification accuracy on our dataset of variants (based on Youden’s index (Youden, 1950)), and then rescaling the cutoff to 0.5 (decision threshold). The resulting rescaled values fall in the range [−0.3, 1.16], with the lower and upper limits corresponding to neutral (or even beneficial) and strongly pathogenic substitutions, respectively.
2.8. Elastic network model (ENM) analysis.
We analyzed an ensemble of 49 structures, composed of 18 DATs (five hDAT models and 13 structures resolved for dDAT), 25 LeuTs, and 6 hSERTs. See Supplementary Methods for the dataset selection procedure and Table S3 for the PDB codes of the structures included in our ensemble. The collective dynamics of each member was analyzed using the anisotropic network model (ANM) (Eyal et al., 2015) implemented in ProDy (Bakan et al., 2014). Residues with occupancies below 70% in the multiple sequence alignment (MSA) were considered as environment in a system-environment formalism. We computed the softest 50 modes for each structure, reordered them using a linear algorithm (Kuhn, 1955) to match the order of modes in hDAT and identified shared (generic) and distinctive (specific) features of their mode spectra using the SignDy tool developed for evaluating the signature dynamics of protein families (Zhang et al., 2019).
3. Results
3.1. Intersubunit contacts made by EL2b or EL3 loops stabilize alternative trimers.
Among structural models generated for DAT trimers in the OF and IF states (Fig S1), only three were found to satisfy the criteria specific to NSS multimers (Cheng et al., 2017; Gur et al., 2017), mainly: (1) symmetric or pseudo-symmetric organization of the protomers; (2) exposure of the N- and C- termini to the IC region; (3) suitable orientation of the aromatic residues (i.e., Trp) on the EC and IC sides to enable anchoring to the membrane. These models are highlighted in green and yellow boxes in Fig S1.
Fig 1 panels A-B and D-E describe in more detail these two trimeric models. The former, termed trimer-W238, was stabilized in both OF and IF states of the protomers, driven by strongly favorable entropic effects (as accounted for by the large sizes of the clusters in Table S1) in addition to favorable interfacial interactions involving W238 residues from the three protomers; whereas the latter, trimer-C306, was selected in the OF state only, with three C306 residues located at the trimerization interface, and IL5 W497 and TM11 K595 forming close contacts near the IC-exposed region. Members of the corresponding clusters retained the interfacial contacts exemplified by these representative models, apart from slight fluctuations in the size of the central hydrophobic cavity. Notably, 60% of top-ranking DAT trimers generated by ClusPro showed interfacial contacts at the EL2b portion (S227-W238) of the EC loop 2 (EL2), complemented by TM4/TM9 contacts typical of trimer-W238; and 40%, at the EL3 loop (P288-S309) with contributions from TM2, 10, 11 or 12 typical of trimer-C306.
Fig. 1. Structural models for DAT trimer, and corresponding AIM-100 binding sites.

Top view, side view and AIM-100 binding site of three structural models (trimers-W38, -C306 and –Y303) are illustrated in the respective panels A-C, D-F and G-I. (A-C) Trimer-W238 is stabilized by interfacial contacts between EL2b (green), TM4 and TM9. AIM-100 (cyan van der Waals (vdW) representation) binds to the trimeric cavity with an estimated affinity of −7.6 kcal/mol. W238 and W256 (purple sticks) line the EC and IC entrances to the cavity. Inset in A displays that interfacial salt bridge R237(blue)-D231(red). (D-F) Trimer-C306 constructed from OF DAT monomer. Three C306 residues (yellow, vdW) from EL3 (magenta) cluster in the center. Inter-subunit cation-π interactions are detected between W497 and K525. Three AIM-100 binding sites are detected near: C306 (occluded state), W497 (entropically favorable; shown in panel F), and D79 at the substrate binding site (S1), all with comparable (~ −7.1 kcal/mol) binding affinities. (G-I) Trimer-Y303, homology model constructed using CaiT trimer (PDB: 2WSX) as template. In G, DAT is shown in gray ribbons and CaiT in transparent gray surface. The EL3 loop (magenta) in DAT aligns with the H7 helix (yellow ribbon) of CaiT. Three Y303 residues (green wdW balls) cluster at the center. Inset in G displays that interfacial hDAT salt bridge R295-E307, and its counterpart in CaiT, D288-R299 (semi-transparent, vdW). A high affinity AIM-100 binding site (−7.0±0.5 kcal/mol) is observed at the interface of two subunits. AIM-100 binding energies are calculated using AutoDock.
As an alternative model, we constructed a third trimeric model for DAT by homology modeling, using as template the crystal structure resolved for CaiT. The latter, called trimer-Y303, is presented in Fig 1G–I. Like trimer-C306, EL3 loops assemble at the trimerization interface. The trimer is further stabilized by inter-protomer salt bridges R295-E307. The central cavity is more loosely packed compared to trimers-W238 and -C306 and open to the IC region. Notably, in CaiT, the EC-facing H7 helices (counterpart of EL3 in DAT; yellow ribbon in Fig 1G) span the trimeric interface, along with the salt-bridge R299-D288 (inset in Fig 1G). The results were further consolidated by comparison with BetP, also resolved in trimeric form (Ressl et al., 2009), which supported the robustness of the assembly exemplified by trimer-Y303 despite the weak associations between the protomers.
Overall, our computations suggest that two diametrically opposite packing of protomers, one placing EL2 loops at the trimerization interface (trimer-W238; the most probable conformer), and the other placing the EL3 loops at the interface (trimers-C306 and -Y303), are accessible to the protomers (Fig 1). Note that the latter is independently predicted by two different approaches: docking simulations (trimer-C306) and homology modeling (trimer-Y303) supported by two crystal structures resolved for paralogs. A systematic analysis using full-atomic MD simulations presented next established the high stability of trimer-W238 and -Y303, and relatively higher flexibility (and lower stability) of trimer-C306 in the PM.
3.2. Molecular dynamics simulations confirm the stability of the trimers and reveal key interfacial interactions.
MD simulations were performed to investigate the stability of the trimeric models in the neuronal membrane environment. We examined four systems in duplicate runs of 100–200 ns each: (1–2) two trimer-W238 models, one in the IF state (Figs 2B, D and E, and Supplementary Fig S2) and the other, in the OF state (Figs 2C and F, and S3); (3) OF trimer-C306 (Figs 1D–F and S4) and (4) OF trimer-Y303 (Figs 1G–H and S5). The time evolutions of the backbone atoms root-mean-square deviations (RMSDs) with respect to the initial conformations are presented in Fig. S6 for all cases, reproduced in duplicate or triplicate runs. Trimers-W238 and –Y303 were stabilized at 3.5±0.5Å irrespective of the starting state of the protomers or the simulation protocol; whereas trimer-C306 exhibited larger fluctuations (RMSD of 5.5±0.5Å) with a continuous drift in the RMSD over 100 ns, which suggested that the starting model generated by ClusPro had a lower stability compared to the other two trimers. Yet, the interfacial contacts were maintained in all three trimers; and no protomer dissociation nor noticeable unfolding was detected. While all trimers were originally formed by symmetrical packing of the protomers, they all underwent slight variations/shifts in their contacts at the trimerization interface, hence their description as pseudo-symmetric associations.
Fig. 2. Simulations of AIM-100 binding to DAT trimer-W238 highlight the involvement of EL2b, TM4 and TM9 in stabilizing the trimer and coordinating the drug.

(A) Typical set up for MD simulations. The trimer-W238 (protomers colored blue, red and orange) was embedded into neuronal lipids (licorice) containing POPE (green), POPC (lime), cholesterol (yellow), POPI (cyan), and PIP2 (purple) molecules. AIM-100 molecules (vDW, various colors) were initially distributed in both the EC and IC regions. Multiple binding events were observed. (B) AIM-100 (cyan, vdW) bound to the IC pore of IF trimer, stabilized by TM4 (solid) and TM9 (transparent); and (C) AIM-100 bound to the EC pore of the OF trimer, lined by TM9 (solid) and EL2. (D) Time evolution of contacts between IF trimer-W238 and AIM-100. Contacts are indicated by gray-shaded areas. (E) Interprotomer contacts between TM4 and TM9, observed during the simulations of the IF trimer (see also Fig S2). (F) Same as D for OF trimer shown in C (see also Fig S3). In D-F, contacts refer to atom-atom distances closer than 3.5 Å.
The histograms of pairwise amino acid (AA) contacts observed during the MD simulations are presented in Figs 2E and S2A (trimer-W238, IF), S3A (-W238, OF), S4A (-C306, OF) and S5B (-Y303, IF). The average numbers of interprotomer AA contacts recorded at 2 ns intervals during 100 ns trajectories were 330, 245, 300 and 118 for the respective trimers. Therefore, the IF trimer-W238 exhibited the largest number of contacts, mostly at TM4, TM9 and EL2b (Figs 2E and S2). The same secondary structural elements also lined the interface in OF-trimer-W238, with the addition of TM12 (Fig S3). In contrast, the trimer-Y303 had the smallest number of intersubunit contacts. Yet, despite its loose packing, intersubunit contacts between EL3 loops (L298SVDFYRLC306) involving the aromatic residues Y303 and F302 and the salt bridges R295-E307 (Fig 1G–H; Fig S5) were sufficiently strong to stably maintain the trimeric association, as also evidenced by the small RMSDs observed in Fig S6.
The intersubunit disulfide bridge C306-C306 has been proposed to stabilize the hDAT dimer (Hastrup et al., 2001), possibly complemented by the interfacial salt bridge R304-E307 (Cheng et al., 2017; Jayaraman et al., 2018).The simulations of the trimer-C306 showed close contacts between the three C306 (Fig. S4A–B) as well as the intermittent formation of the interprotomer salt bridges R304-E307 (Fig. S4D). These were complemented by TM2-TM11 contacts along the interfacial cavity; and the IC-exposed face of this OF trimer was stabilized by TM11-IL5 contacts involving W497 and K525 (Figs 1F and S4A–C).
3.3. All trimers harbor a hydrophobic cavity for binding AIM-100.
Docking simulations performed to explore the binding sites and affinities of AIM-100 with respect to the trimeric DAT models indicated a multiplicity of binding sites. The strongest binding was observed at the trimerization interface in all cases, with an estimated affinity of −7.0±1.0 kcal/mol. This supports earlier work (Sorkina et al., 2018), where AIM-100 was proposed to stabilize the oligomers via an allosteric mechanism upon binding the oligomerization interface. Yet, we also observed occasional binding to the vicinity of the substrate-binding (S1) and allosteric (S2) sites known for hSERT (Coleman et al., 2016) and other NSSs (Nyola et al., 2010). This would be consistent with the ability of AIM-100 to compete with the substrate binding on SERT (Wu et al., 2015).
Fig. 1C and F illustrate the hydrophobic pocket at the trimerization interface, where AIM-100 stably bound in trimers-W238 and -C306. The IF trimer-W238 exhibited a relatively larger pocket than the OF trimer, which, in turn, led to a higher AIM-100 occupancy. Binding onto the IF trimer-W238 was therefore entropically more favorable than that onto the OF trimer. This high affinity was robustly imparted by L450, L454 and L458, on the hydrophobic face of TM9 (Fig. 1C), as confirmed by MD simulations below. As to trimer-Y303, we identified a high affinity AIM-100 binding site at the interface of two subunits in the vicinity of the above mentioned R295-E307 salt bridge (Fig 1H). Double π-stacking interactions were observed between AIM-100 and W311 (Fig 1I), which apparently assisted in stabilizing AIM-100. These results motivated a closer examination of binding poses and affinities by full atomic simulations, presented next.
3.4. AIM-100 binding central pore is lined by TM4 and TM9/EL2b in the respective IF and OF states of trimer-W238.
We examined the migration and binding of AIM-100 molecules in multiple MD runs, initiated by placing 8–10 AIM-100 molecules approximately ~25 Å away from the putative binding sites, as illustrated in Fig 3A. Among the three models, trimer-W238 was found to readily attract AIM-100 molecules and bind one, or occasionally two, to the central pore; whereas the binding sites on trimers-C306 and -Y303 were inaccessible during 100 ns simulations.
Fig. 3. EL2b loop interactions drive the tight packing of the protomers, assisted by AIM-100 binding.

(A-C) Time evolution of protomer positions (top, viewed from EC region) and interfacial interactions (bottom) at t = 0, 10, and 180 ns to stabilize the trimer-W238. The surrounding lipids and other molecules included in simulations (see Fig 2A) are omitted for clarity. (A) Initially, protomers are separated by ~50 Å (center of mass distances) (B) AIM-100 (cyan, vDW), originally 25 Å away from the binding site, migrated to locate the interfacial region at the EC-exposed vestibule, stabilized by P235 and P236. (C) Inter-subunit salt bridges D231/D232-R237 consolidate the trimeric packing and the closer assembly of the protomers whose mass center distances are reduced by 5Å.
Fig 2 panels B and C display the most stable binding poses of AIM-100 to the respective IF and OF trimer-W238. The time evolution of the contacts made with AIM-100 molecules are presented in the respective panels D and F. In the IF state, AIM-100 molecules were mainly coordinated by V249, Y252-F253, and W256 on a hydrophobic face of TM4, and L450, V453, and F457 on TM9. Notably, W256 and F253 located at the pore entrance facing the IC region, may have served as attractors. AIM-100, once inserted into the hydrophobic cavity, remained stably bound for extended durations. Occasionally, two AIM-100 molecules simultaneously bound to the interfacial cavity, which further enhanced the stability of the trimer. As to the OF trimer-W238, the binding site is slightly shifted toward the EC entrance, coordinated by F457, L458, L461, F462 and T465 on TM9 and L233- Q239 on EL2B (Fig 2C and F).
As a further test, we examined whether AIM-100 was instrumental in bringing together (via EL2b associations) the three monomers when they were in close proximity but not assembled in a trimer. To this aim, we separated the monomers by moving them 5 Å away radially from their positions in trimer-W238 (OF) so that they would make no contacts, while keeping their orientations. Fig 3 displays three snapshots at t = 0, 10 ns and 180 ns, illustrating the gradual assembly of the monomers facilitated by AIM-100 binding and mediated by EL2b. We note that tight packing is ensured by intersubunit interactions involving P235 and P236, and further consolidated by the salt bridges D231/D232-R237. These results show that AIM-100 not only binds the trimerization pore but may also help assemble and stabilize the protomers in a trimer.
In the case of trimers-C306 and -Y303, AIM-100s occasionally formed aggregates, and bound to trimer surface or dimeric interfaces, especially near W497 on the loop IL5 and I595 at the C-terminal segment exposed to the cytoplasm but could not penetrate the central pore. The entrance near C306 (Fig 1E) remained occluded in OF trimer-C306. As to the trimer-Y303, the surrounding lipids quickly occupied the central opening, precluding AIM-100 binding. Note that lipid molecules were also found to occupy the inter-protomer interface of the trimeric NSS ortholog BetP (Koshy et al., 2013).
The inability of AIM-100 molecules to locate these binding sites might originate from the longer time required (beyond 100 ns simulations) for the exposure of the EC opening near C306, or for the displacement of lipid molecules that occupy the central cavity. Given the consistency between these two sets of results (trimers-C306 and -Y303) with regard to the interfacial elements (EL3) observed in both CaiT and BetP (Ressl et al., 2009; Schulze et al., 2010), we investigated possible significance of these pore-lining residues that stabilize the trimer and/or bind AIM-100 by site-directed mutagenesis experiments, presented in Subsection 3.7 below.
3.5. Differential affinity of DAT and SERT to bind AIM-100 can be traced to their sequence differences at TM9 and EL2B.
The above analysis showed that the hDAT trimer-W238 possesses the highest propensity to bind AIM-100, or even to assemble upon binding AIM-100. Residues lining the pore are contributed by TM9 in both OF and IF trimer, complemented by TM4 in the IF-state, and EL2B in the OF state (Fig 2). Of interest is to understand whether this behavior persists in SERT, and /or the origin of the experimentally observed differential behavior of DAT and SERT with regard to trimerization in the presence of AIM-100.
Normal mode analysis of SERT, DAT, and LeuT in OF and IF states using the ANM (Eyal et al., 2015) (Fig 4C) revealed three interesting features: (i) excessively high fluctuations at EL2a in SERT, compared to other NSS transporters with the LeuT fold, which might exert a destabilizing effect near the EC pore entrance; (ii) restricted mobility of EL2b in the IF state compared to the OF (Fig 4C, solid and dashed curves), which explains the inability of EL2b to adapt to AIM-100 binding in the IF trimer, while it plays an important role in the OF state (Fig 2); and (iii) the high correlations (r = 0.95 ± 0.04) between EL2b (e.g. W238) and TM9 (e.g. L450, L454, L461, F462 and T465) (Fig 4D), which suggest that LeuT fold provides a mechanical basis for pre-existing cooperative motions between EL2 and TM9 observed here to coordinate AIM-100 in hDAT trimer.
Fig. 4. Extracellular loop segment EL2b is distinguished by its propensity to make interfacial contacts specific to hDAT.

(A) The diagrams display (in vdW spheres) the EL2b residues that make intersubunit contacts in trimer models generated for DAT (left) and SERT (right), consistent with the trimer-W238 presented in Fig 1A–B. A protomer is shown for each transporter in tube representation. Hydrophobic, hydrophilic, acidic and basic residues are colored in white, green, red, and blue. (B) Multiple sequence alignment of human and fruit fly DATs (hDAT and dDAT), human SERT, NET, glycine transporter (Gly1) and GABA transporter (GAT1). (C). Comparison of residue fluctuation profiles for SERT, DAT and LeuT (labelled) in OF (solid) or IF (dashed) states, computed using the ANM. Curves are vertically shifted for clarity and represent the probability distributions of mean-square fluctuations of residues. Selected residues are indicated by arrows and dashed lines. EL2 exhibits transporter-specific fluctuations. (D) Cross-correlations between residue fluctuations averaged over the ensemble of transporters listed in Table S3. The map represents the signature dynamics of the shared fold, obtained by SignDy (Zhang et al., 2019). The region corresponding to the cross-correlations of EL2b with other residues is highlighted by the yellow rectangle.
However, the predisposition of EL2b and TM9 to undergo cooperative movements is not sufficient to tightly bind AIM-100 in SERT. Docking simulations showed that hSERT could assume the trimer-W238 geometry, but the propensity of EL2b to make interfacial contacts was weaker compared to hDAT. This can be attributed to the difference in their respective EL2b (D231DLGPPRW238 vs Q246DLGGISW253) and TM9 sequences (Fig 4A–B). hDAT residues D231 and R237 that form interprotomer salt bridges (see Fig 1A inset) are replaced by weaker Q246-S252 polar interactions in hSERT. Furthermore, EL2b residues P235 and P236 that tightly seal AIM-100 from the EC region in hDAT trimer (Fig 2C and F) are replaced by Gly and Ile in hSERT, which cannot play the same gating/lid closure role. Finally, TM9 residues F451, L454, L458 are not conserved in SERT.
Thus, overall, while the LeuT fold offers the adaptability to trimerize and potentially bind small molecules at the trimerization interface, as shown in hDAT, the specificity of amino acids is essential to modulate the differential behavior of different NSS family members. The amino acid variations between hDAT and hSERT at putative binding sites explain the ability of AIM-100 to bind and trimerize hDAT, but not hSERT. Consistently, the top AIM-100 binding site in SERT was not at the trimerization interface but overlapped with S1 or S2 site (not shown). Overall, these results are consistent with the ability of AIM-100 to compete with substrate binding onto SERT (Wu et al., 2015) and its inability to induce the oligomerization of SERT (Sorkina et al., 2018).
3.6. Trimerization shifts AIM-100 binding sites in favor of EL2b, TM4, EL3, IL5 and C-terminal residues.
As a further test in silico, we examined the ability of DAT monomers to bind AIM-100. To this aim, we performed MD simulations with a higher concentration of AIM-100 molecules (Fig S7). These simulations revealed several potential interaction sites, including Arg443 that may serve as an attractor at the IC opening facilitating the binding of AIM-100 to TM9 residues. Persistent interactions with H193 and T207 also suggest the role of the entire loop EL2 assisting the dominant role of the segment EL2b.
A summary of contact probabilities between AIM-100 molecules and all residues observed in all MD simulations is presented in Fig 5A–B. Panel A presents the results for the four trimeric models, and panel B for OF and IF monomers. Comparison of the two distributions helps us distinguish the recognition sites intrinsically favored by the monomeric structure itself (e.g. peaks at EL2 and EL4) presumably driven by the EC exposure of hydrophobic or aromatic residues (indicated by the light orange shades). Notably, trimerization exposes new sites, shifting the affinity toward EL2b portion of EL2, and toward EL3. We note the peaks at the IL5 loop (e.g. W497 and F498) and C-terminal segment (e.g. I595) exposed to the IC region, indicated by light green shades. Panels C and D display AIM-100 binding poses at these regions exposed to the cytoplasm observed in our MD simulations. Note the involvement of W497, which plays a central role in the trimer-C306 (Fig 1F).
Fig. 5. Histogram of contacts between AIM-100 molecules and DAT, and a high affinity binding sites near the C-terminus.

(A) The bars display the probability distribution of residues within 3.5 Å atom-atom distance from AIM-100, observed in the MD simulations of all four DAT trimers (over 2,000 frames). We note the preponderance of aromatic residues (highlighted in red). (B) Results from the MD simulations of IF (black) and OF (red) monomers in the presence of AIM-100s. Light orange and green shades in A and B highlight EC and IC-exposed regions distinguished by high-binding affinity. (C-D) The region between the C-terminus (pink) and TM10-IL5 (cyan) harbors a favorable AIM-100 binding site for (C) one AIM-100 (cyan sticks) or (D) two AIM-100s (cyan and purple sticks). This type of binding was consistently observed in all three OF trimer simulations, with I595 interacting intermittently with W497 or F498. Panels C and D display snapshots from the simulations of trimer-Y303 and -W238, both in OF state.
3.7. Site-directed mutagenesis experiments highlight the critical roles of EL2b and W497.
The above computations provided insights into alternative mechanisms of trimerization, driven by the association of the EL2b (trimers-W238), or EL3 (trimers-C306 and -Y303) loops and stabilized by TM4 and TM9 hydrophobic residues lining the trimerization pore, and/or consolidated (if not driven) by AIM-100 binding (see Fig 4). Computations also highlighted several residues implicated in those events. Here we test the role of selected residues listed in Table 1. The table lists the mutants tested in each case, including single-, double- and multiple-substitutions, and the corresponding secondary structural elements and specific role suggested by simulations (columns 1–3). The last three columns list the results from experiments assessing localization of DAT mutants transiently and/or stably expressed in PAE cells, and the effects of AIM-100 on their multimerization state and endocytosis.
Table 1.
Biological and biophysical data on mutants tested in the present study
| Mutant | Position in DAT | Role of mutation site inferred from simulations | Plasma membrane expression** | AIM-100 induced oligomerization***/endocytosis** | Phenotype |
|---|---|---|---|---|---|
| Wild-type (WT) DAT | N/A | N/A | + | +/+ | WT |
| C306A | EL3 | Stabilization of trimer-C306 (Fig 1D–E, S4) | + | +/+ | WT |
| W497A | TM10 | Coordination of AIM-100 (Fig 1F, 4) | − | N.D./N.D. | ER, IC aggregates |
| W497F | TM10 | Coordination of AIM-100 (Fig 1F, 4) | − | N.D./N.D. | ER |
| K525A | TM11 | Cation-π interactions with W497F (Fig 1F) | − | N.D./N.D. | ER, IC aggregates |
| D79A | TM1 | Critical (protonation) site for DA translocation (*) | (+) / − | N.D./N.D. | ER, IC aggregates |
| I595A | CT | IC-exposed high affinity site for AIM-100 binding (Fig 4) | − | N.D./N.D. | ER |
| I595V | C-terminus | See row above | + | N.D./+ | WT |
| W256A | TM4 | AIM-100 recognition/binding at IC end of trimeric interface, Figs 1B, 2B, D and 4A | + | N.D./+ | WT |
| W238A/W256A | TM4 | EC and IC exposed ends of TM4, AIM-100 binding | + | +/+ | WT |
| W238A | TM4 | Trimeric interface (close to EC vestibule) and AIM-100 binding (Fig1A–C, 2F, S2) | + | N.D./+ | WT |
| H223A | EL2 | + | +/+ | WT | |
| L450A/L454A/L458A | TM9 | Lining trimer hydrophobic pocket and/or coordinating AIM-100 (Figs 1C, 2B–F, S2, S3, 4) | + | +/+ | WT |
| L450A | TM9 | See row above | + | N.D./+ | WT |
| L454A | TM9 | See row above | + | N.D./+ | WT |
| L458A | TM9 | See row above | + | +/+ | WT |
| D231A/D232A/P235A/P236A/R237A | EL2b/TM4 | Stabilization of trimer, AIM-100 binding (Figs 1A, S1-S3, 3, 4A) | − | N.D./N.D. | ER, IC aggregates |
| D231A/D232A | EL2b | Intersubunit trimeric salt-bridges (Figs 1A, S1–S3, 3) | − | N.D./N.D. | ER, IC aggregates |
| P235A/P236A/R237A | EL2b/TM4 | Intersubunit association and AIM-100 binding (Figs 1A, S1, 2C and F, S3, 3, 4A) | − | N.D./N.D. | ER, IC aggregates |
“(+)/−”, partial phenotype – small pool of DAT is delivered to the plasma membrane but the bulk is retained in the ER (endoplasmic reticulum); N.D., not determined..
(Cheng and Bahar, 2015; Cheng et al., 2015; Cheng et al., 2017; Cheng et al., 2018; Gur et al., 2017; Kaya et al., 2018; Khelashvili et al., 2015; LeVine et al., 2018).
Imaging experiments were performed at least 3 times with each mutant;
Western blot analysis of each mutant was performed 2–3 times with similar results.
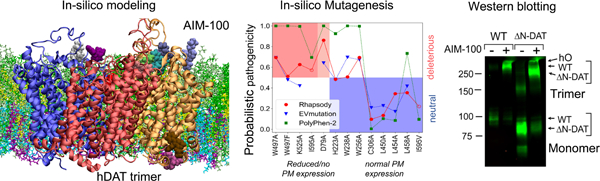
DAT mutants can be divided into two groups based on experimental observations: (1) mutants whose response to AIM-100 is similar to that of wt DAT (highlighted in gray in Table 1); and (2) mutants that are unable to exit the endoplasmic reticulum (ER) and be delivered to the PM (highlighted in orange). The first group, such as C306A and mutants with substitutions in TM4 or TM9 (e.g. W256A and W238A), predominantly localize in the PM, often concentrated in filopodia. AIM-100 treatment induced strong oligomerization and endocytosis in these mutants (Fig 6A–B; Table 1). These data suggest that the formation of C306 cross-links and the mediating role of TM4:TM9 interactions are either unlikely or redundant in stabilizing AIM-100-driven oligomerization.
Fig. 6. Examples of the effects of mutations listed in Table 1 on localization and oligomerization of YFP-HA-DAT.

(A) wt DAT (WT), L450/454/458A (LLL), C306A or W238/256A mutants were stably expressed as clonal pools in PAE cells (approximately 10–20% of cells are expressing DAT or its mutants). The cells were incubated with DMSO (vehicle; “-” AIM-100) or 20 μM AIM-100 for 2 hours at 37°C and lysed. Western blotting of cell lysates was performed using GFP antibody. hO, higher oligomers. All lanes are from the same blot. *Non-specific bands (detected due to low levels of expression of DAT and its mutants). The results are representative of 2–3 experiments with each mutant. (B-C) wt DAT (WT) and mutants H223A, D231/232A, or D231A/D232A/P235A/P236A/R237A (DDPPR) were transiently expressed in PAE cells. The cells were fixed untreated (C) or fixed after incubation with DMSO (vehicle) or 20 μM AIM-100 at 37 °C for 2 hours (B). Individual confocal sections from 3D images of YFP fluorescence are shown. Images are representative of at least 3 experiments with each mutant. Scale bars, 10 μm. (D) wt DAT (WT), and YFP-ΔN-DAT mutant expressing cells were incubated with DMSO (vehicle) or 20 μM AIM-100 for 2 hours at 37 °C and lysed. Western blotting of cell lysates was performed using GFP antibody. The results are representative of 2 experiments.
The second group of mutations, W497A, K525A and mutations of EL2b residues D231-R237, including P235A-P236A-R237A, resulted in a dramatic retention of the mutants in the ER, manifested by an extensive IC tubular network, and virtually a lack of detectable DAT at the cell surface (Fig 6C and Table 1). Several ER-retained DAT mutants were additionally accumulated in IC aggregates in a population of transiently transfected cells (e.g. the DDPPR mutant in Fig 6C). Typically, retention in the ER is the result of misfolding of a TM protein and/or inability to assemble as a multimer. In the case of DAT, it has been proposed that DAT oligomerization is necessary for efficient exit from the ER (Sorkina et al., 2003; Torres et al., 2003). Therefore, if a mutation results in ER retention, it is likely that the mutant is not properly folded, and it also indirectly indicates its oligomerization is inhibited. Thus, the inability of W497A and K525A (Fig 1F), and D231A-R237A mutants (Figs 1A, 2, 3C and F, S2–S4, 4 and 5A) to exit ER can be viewed as indirect evidence in support of the role of these residues in the stabilization of the respective trimers-C306 and -W238 upon AIM-100 binding.
Of note, if a DAT mutant is unfolded and stuck in the ER, the expression level is low due to its aggregation and degradation in ER. This precludes generation of stable mutant-expressing cells, and therefore, biochemical measurements of AIM-100 induced oligomerization. Therefore, we can only use single-cell assay to analyze the effects of AIM-100 on ER-retained DAT mutants. Indeed, when a mutant was efficiently delivered to the cell surface, its AIM-100 induced oligomerization and endocytosis were found to be comparable to the effects of AIM-100 on wt DAT (see Fig 6A–C, and Table 1).
Finally, we used YFP-ΔN-HA-DAT mutant lacking N-terminal residues 1–65, that is incapable of substrate uptake because of the destabilized OF state, to demonstrate that the N-terminal residues supporting the OF state (R60 and W63) are not necessary for AIM-100-induced oligomerization (Fig 6D). Moreover, because of the smaller apparent molecular weight of YFP-ΔN-HA-DAT (~80–83 kDa) compared to wt YFP-HA-DAT, the apparent molecular weight of the corresponding minimal oligomer recovered from AIM-100 treated cells was better defined (exactly as the 250 kDa marker) (Fig 6D). This result further confirmed that AIM-100-induced SDS-resistant DAT oligomer is a trimer.
Among the mutants tested, we note the sensitivity of the phenotypic behavior to the type of substitution, I595A versus I595V. This residue exposed to the cytoplasm, presumably affected by a hydrophobic mismatch, shows a deleterious effect if mutated to alanine; whereas substitution by valine retains wild-type-like properties. Our in silico saturation mutagenesis tool, Rhapsody v2, captured this subtle behavior, and provided a description of the complete spectrum of behavior as a function of all possible substitutions, as presented next.
3.8. In silico saturation mutagenesis analysis corroborates the critical sites.
We used our novel approach developed for estimating the pathogenicity of SAVs (Ponzoni and Bahar, 2018) to scan all possible types of substitutions at the structurally resolved parts of DAT. Fig. 7A presents the results from this so-called in silico saturation mutagenesis experiment. The ordinate lists all possible amino acid substitutions and the abscissa shows the sequence position. The entries represent the color-coded probability of being ‘pathogenic’ predicted by Rhapsody v2, with higher values (red) indicating increased probabilities of deleterious effects on function and vice versa. Wild-type residue are shown in white.
Fig. 7. In silico saturation mutagenesis results and comparison with experimental data.

(A) Heatmap displaying the pathogenicity probabilities for all possible amino acid substitutions (y-axis) at every residue (x-axis) of DAT, on a scale from 0 (blue, neutral) to 1 (red, deleterious). The red curve in the bottom plot shows the average pathogenicity over all 19 substitutions in each column, computed with Rhapsody v2 (solid line) when possible, or v1 (dotted line, mainly chain termini) in the absence of Pfam data. The pink shade shows the range of values at each column. The blue curve represents an analogous profile computed using EVmutation. Residues 170–212 in EL2 have been reconstructed and their secondary structure is not defined. (B) The PM expression of 13 SAVs (Table 1) has been measured in vitro. On the abscissa, these variants are sorted based on the change in their expression levels (left, red and light-red shades: no or partial expression; right, blue shade: normal/wild-type expression). On the y-axis, the functional impact as predicted by Rhapsody v2 (red circles), EVmutation (blue inverted triangles) and PolyPhen-2 (green squares) is shown. The EVmutation epistatic score is normalized such that the optimal cutoff between predicted neutral and deleterious effects matches that for Rhapsody v2 and PolyPhen-2 (y = 0.5). Correct predictions lie within the two shaded areas, red (true positives) and blue (true negatives). (C) DAT monomer color-coded by the average pathogenicity shown in A (red/blue: high/low pathogenicity probability). The 13 mutations sites are labelled and shown in licorice.
The red curve in the lower part of Fig. 7A displays the average of each column and the light shade shows its overall variation range, thus providing an estimate on the significance and specificity of the particular residue. It is interesting to see how the predicted pathogenicity roughly matches the secondary structure, with the TM helices more likely to be impacted by mutations. A closer look at the average pathogenicity curve reveals even the 3–4 residues periodicity consistent with the α-helix turns, with the residues at the exposed face exhibiting lower sensitivity to mutations (see Fig. S8 for magnified version of Fig. 7A).
The blue curve in Fig. 7A (bottom panel) shows the average profile predicted by EVmutation (Hopf et al., 2017), a method that estimates the fitness of a specific SAV based on co-evolutionary patterns. The similarity between the two profiles is remarkable, considering the differences in the underlying algorithms and features (Rhapsody takes account of structure and dynamics, in addition to sequence features). Moreover, a relatively high correlation is still observed when completely excluding Pfam features from the Rhapsody classifier (see Fig. S9).
Fig 7B compares the results from experiments and computations for the 13 SAVs listed in Table 1. Panel C displays these residues, color-coded by their average behavior in Fig 7A. Based on experimental data, the variants were grouped into three subsets, corresponding to those displaying a total lack of PM expression accompanied by retention at the ER or aggregation at the cytoplasm (red shade in Fig 7C), a strongly reduced expression level again manifested by ER retention or cytoplasmic aggregation (light red shade), and a normal/wild-type expression level and endocytic behavior (blue shade). The hypothesis is that lower expression levels might be attributed to (partial) misfolding or reduced stability, which prevents the transport from the ER to the PM. Alternatively, the impairment of oligomerization otherwise induced upon AIM-100 binding may lead to ER retention phenotype. In other words, the former group (red and light red) refers to pathogenic/deleterious mutations that impair the correct folding and/or multimerization at the ER, and the latter, to neutral mutations. With these inputs, we compare the experimental observations and computational predictions from Rhapsody v2, EVmutation and PolyPhen-2.
The red circles in Fig. 7B represent the predictions from Rhapsody v2, mapped onto the interval [0, 1]. Values greater than 0.5 indicate deleterious effects, with higher values corresponding to stronger effects. Except for W256A, the predictions are in good agreement with experiments, i.e. all pathogenic mutations lie in the red shaded region, and all neutral mutations lie in the blue-shaded region, with three borderline assignments (W497F, H223A, W238A). We note the high confidence in attributing damaging effect to mutation D79A, which exhibited a strongly reduced PM expression. This variant occurs in a site buried deep within the transporter (Fig. 7C), on the kink of TM1. The critical role of this helix, and, in particular, this site in DAT and all members of the LeuT fold family is well established (Cheng and Bahar, 2014; Cheng and Bahar, 2015; Penmatsa et al., 2013, 2015; Ponzoni et al., 2018; Wang et al., 2015; Yamashita et al., 2005). Of note, all TM1 residues share similarly exhibit high sensitivity to mutations, as can be seen in A, consistent with the central role of TM1 for transport activity. We also note the ability of the classifier to distinguish between I595A (dysfunctional) and I595V (functional), in accord with experimental data.
Figure 7B also shows the predictions from EVmutation (blue inverted triangles) and PolyPhen-2 (green squares). We selected these two methods for their relatively high accuracy and their distinctive and complementary approaches. PolyPhen-2 is a broadly used tool that integrates sequence conservation patterns with local structural features into a probabilistic naïve Bayes classifier, and EVmutation rigorously takes account of coevolution patterns provided that extended sequence data or MSAs are available in Pfam. In the current application, EVmutation outputs partially overlap with Rhapsody v2, except for two cases that actually disagree with experiments, one false negative (K525A) and one false positive (W238A); PolyPhen-2 displays here a lower sensitivity/recall, in that it incorrectly assigns a high pathogenicity to H223A, W238A, W256A and L458A that exhibited wt-like behavior in in vitro testing.
4. Discussion
The functional, physiological and pharmacological roles of DAT oligomerization still remain elusive due to the lack of structural and sub-cellular location information. Earlier studies suggested oligomerization as a determinant for efficient ER export of DAT, surface expression and endocytosis (Jayaraman et al., 2018; Sitte et al., 2004; Sorkina et al., 2003; Torres et al., 2003). Oligomerization-induced endocytosis may serve as part of the regulatory and quality-control mechanisms that remove excess DAT from the cell surface under conditions of low levels of extracellular DA and may clear DAT oligomers from the cell surface if they are functionally impaired, e.g. in IF state (Sorkina et al., 2018; Zhen and Reith, 2018). The present study attempts to take advantage of AIM-100 effects on DAT oligomerization (Sorkina et al., 2018) to advance our understanding of oligomerization mechanisms. Determining the oligomerization state of a membrane protein is a very difficult task, especially if it is induced by a compound in intact cells but not in solution. Indeed, additional high-resolution studies are necessary to unequivocally demonstrate that trimers are the minimal oligomerization state of DAT induced by AIM-100 in intact cells. In the following sections, we will discuss the limitations and strategy of our methodologies, the new insights into the potential oligomerization geometry of hDAT and AIM-100 binding properties, and the interpretation of computational and experimental results.
4.1. Methodology limitations and challenges.
Full atomic MD simulations are usually considered as the ground-truth in molecular computations as they take account of atomic structure with detailed force field, but they are known to have limitations concerning sampling inaccuracies, computational time and memory costs that restrict simulations to relatively short time scales (e.g. up to hundreds of nanoseconds in membrane proteins, when multiple runs under different states are performed), and approximations in the force fields and parameters (Corradi et al., 2019; Marrink et al., 2019). The current runs of 100 – 200 ns are too short to visualize complete ligand binding processes, and accompanying protein-lipid, and protein-protein reconfigurations, especially in multi-lipid bilayers where relaxation times could be far exceeding the simulation duration (Corradi et al., 2019; Marrink et al., 2019). A continuous RMSD drift in the 100 ns runs of trimer-C306 (Fig S6A) suggested that that particular ClusPro model might not be as stable as the other trimeric models, and should be cautiously interpreted. Yet, many runs carried in duplicates or triplicates (see Table S2) consistently maintained the integrity of the trimeric structures and permitted us to examine the diffusion of AIM-100 molecules, and even the assembly of protomers in close proximity (Figs 2–3).
Atomic simulations were complemented by ANM normal mode analyses (Fig 4) and in silico saturation mutagenesis experiments (Fig 7) to consolidate the results from simulations and interpret experimental observations. While ANM-based approaches lack atomic details, they provide robust analytical solutions for the collective motions uniquely defined by the inter-residue network connectivity and reveal coupled movements beyond the reach of atomic simulations. Furthermore, we repeated the in silico saturation mutagenesis computations with other established tools to corroborate the results. Apart from one discrepancy (W256A) (Fig 7), experimental (qualitative) and computational (quantitative) results obtained by Rhapsody (Ponzoni and Bahar, 2018) were generally consistent.
Whereas the trimeric models constructed here are broadly symmetric, we cannot exclude the possibility of asymmetric trimer constructs, as observed by the self-assembly of SERT (Periole et al., 2018) and DAT (Jayaraman et al., 2018) dimeric constructs in coarse-grained MD simulations. The pseudo-symmetric arrangements resolved for ion channels, receptors or transporters (Jayaraman et al., 2018) may not necessarily hold for DAT trimers or higher oligomers.
We also note that ClusPro favors close packing of subunits such that the trimeric interface would not allow for the insertion of lipids during short simulations. Such lipid-insertion events were observed only in the trimer-Y303 constructed by homology modeling, and they prevented the binding of AIM-100 molecules to the trimeric interface during MD simulations. In contrast, AIM-100 could bind to trimer-W238 (IF or OF), as the trimeric cavity remained accessible in the absence of interstitial lipid molecules. Out of seven runs performed for trimer-W238, five confirmed the accessibility of the trimeric interface, either from the EC or IC region, but no transport was observed (Table S2). The simulations were not long enough to observe unbinding or translocation events. The estimated AIM-100 binding affinities (~7.1±0.9 kcal/mol) are broadly consistent with the μM range inferred from cocaine-competition experiments (Sorkina et al., 2018).
Accurate assessment of discriminative interfacial sites susceptible to modulation without complete eradication of functionality is a challenge. We should exclude conserved sites that may result in misfolding or retention of DAT in the ER; or those directly interfering with substrate/ion binding (e.g. D79A). However, because oligomerization is necessary for the ER exit of DAT (Sorkina et al., 2003; Torres et al., 2003), disruption of folding and ER exit by mutations affecting oligomerization may be unavoidable. It would be ideal to perform binding studies of hDAT ligands to purified ER membranes in order to confirm the correct folding of hDATs in the ER. Unfortunately, experiments with purified ER membrane are technically impossible because ER-retained DAT mutants are expressed only in a small population of transfected cells and at low levels, likely due to ER stress, and we are limited to single-cell experiments.
4.2. DAT can adapt to different trimerization geometries by virtue of LeuT fold adaptability.
Consistent with the diversity of DAT dimeric models recently shown by the computations of Stockner, Sitte and coworkers (Jayaraman et al., 2018) and the adaptability of LeuT fold to different oligomerization states (Ponzoni et al., 2018), the co-existence or transient stabilization of DAT oligomers with different packing geometries seems plausible. The present study suggests that two alternative packing geometries, one placing the EL2 loops at the trimeric interface (trimer-W238), and the other placing the EL3 loop (trimers-C306 and -Y303), may be selectively stabilized upon AIM-100 binding. Notably, trimer-Y303 also conforms to the trimerization geometry of the structural homologs CaiT and BetP.
Notably, a number of residues making trimeric contacts here have been also observed to make interfacial contacts in DAT dimers (Cheng et al., 2017; Jayaraman et al., 2018). Examples are a series of hydrophobic residues along TM9 in trimer-W238 (Fig S3) or the salt-bridges between EL3 residues in trimer-C306 (Fig S4). Overall, these findings reveal the conformational malleability and adaptability of the oligomers and the avidity of selected residues to make intersubunit contacts, either dimeric or trimeric.
Comparative analysis of the structural dynamics of DAT and other transporters that share the LeuT fold (Ponzoni et al., 2018; Zhang et al., 2019) reveal the collective motions intrinsically accessible to the monomers, which are exploited for stabilizing trimers. For example, a highly cooperative mode of motion predicted for the LeuT fold is the reconfiguration of EL3 to stabilize the BetP and CaiT trimers. Notably, the loop gains structure (H7 helix in BetP) upon trimerization, consistent with the concept of folding upon binding. A similar role for EL3 emerges here in trimer-Y303. The insertion of lipid molecules into the IC-exposed opening of the trimeric cavity made it difficult to observe the diffusion and binding of AIM-100 molecules from the IC environment in silico. Yet, the interfacial R295-E307 (counterpart of D288-R299 in CaiT) salt bridges prevented the dissociation of subunits regardless of AIM-100 binding. Mutations that disrupt these interfacial salt bridges may help assess the stability and/or functionality of the hDAT trimer-Y303.
A diversity of oligomerization mechanisms have been reported (Hashimoto and Panchenko, 2010; Marsh and Teichmann, 2015), including domain swapping, ligand-induced oligomerization, posttranslational modifications (e.g, phosphorylation or disulfide bonds formation), and specific insertions/deletions which favor oligomeric or monomeric states. Relatively small variations in interfacial loop regions may have a profound effect on enabling/disabling complex formation or multimerization; and frequently polar and charged residues, and glycine and proline serve as enablers (Hashimoto and Panchenko, 2010). In line with such findings (Hashimoto and Panchenko, 2010), our simulations (Fig. 4) suggest that prolines (e.g. P235 and P236) and charged residues (e.g. D231/D232 and R237) in an EC loop (EL2b) play an important role in coordinating AIM-100 and/or triggering oligomerization; binding of AIM-100 to the trimeric interfacial cavity may indeed selectively stabilize the trimer-W238. P235-P236 further emerge here as critical residues underlying the specificity of AIM-100-induced trimerization to DAT, but not hSERT where they are not conserved.
Collectively, we suggest that the LeuT fold endows an adaptability to form multimers and bind ligands, but the stability of the multimers/complexes depends on the specific sequences. Sequence variations can alter oligomerization propensities, evidenced by EL2b, TM9 or EL3 residues that differ between hDAT and hSERT which may form potential interfacial salt bridges and intersubunit contacts.
4.3. AIM-100 binding propensities implicate multiple roles.
AIM-100 is an amphipathic membrane-penetrating molecule (Sorkina et al., 2018), with neutral-charge assigned in the ZINC database (Irwin and Shoichet, 2005). Not surprisingly, AIM-100 is in silico predicted to bind the interfacial hydrophobic cavities (Fig. 1). In addition to serving as a potential oligomerization agent (Sorkina et al., 2018), AIM-100 is well-known to target ATP-binding sites (Wu et al., 2015) and one would expect either zwitter-ion or cationic form to be a dominant form, especially in polar/charged microenvironment. In MD simulations, we observed that AIM-100 would preferentially bind aromatic residues and be coordinated by acidic and/or basic residues (Fig. 5). Notably, an intermittent formation of cation-π interactions between AIM-100 and K525 (Fig. 1F) or R588 (Fig. 5D) facilitated its binding. In the future, it might be useful to fine-tune the protonation state of AIM-100 to explore the modulation of its multiple roles.
Our previous study (Sorkina et al., 2018) demonstrated that AIM-100 induced DAT oligomerization is strongly inhibited by cocaine, which stabilizes the OF state. Together with the ability of AIM-100 to cause oligomerization of the W63A mutant and ΔN-DAT (Fig 6D) mutant (where the OF state is disrupted), we suggested that the IF state might have a relatively higher propensity of multimerization (Sorkina et al., 2018). In accord, we observed in silico that AIM-100 bound to multiple interfacial sites with high affinity estimation (comparable to its binding to S1 site); and AIM-100 bound to the IF trimer-W238 was distinguished by a high docking occupancy (or favorable entropy).
Besides the interfacial pockets, multiple AIM-100 binding sites were observed in hDAT protomers. AIM-100 binding to the S1 or S2 site might not be associated with oligomerization since AIM-100-triggered oligomerization also occurred in W63A DAT mutant (binding at S1/S2 is blocked) (Sorkina et al., 2018) and ΔN-DAT (Fig 6D). Notably AIM-100 exhibited a high affinity to bind the C-terminus (Fig. 5) near the F587REK590 motif (Fig. 5D), exposed to the IC region. The FREK motif serves as kinase binding motif (Kristensen et al., 2011; Pramod et al., 2013) and was suggested to form a potential hDAT dimer interface (Jayaraman et al., 2018). It would be of interest to investigate further the role of hDAT C-terminus in mediating AIM-100 triggered oligomerization.
4.4. W497 and K525 emerge as critical sites in imparting stability and mediating AIM-100 binding to trimer-C306.
An early experimental study by Javitch and coworkers (Hastrup et al., 2001) found that DAT dimers formed inter-protomeric cross-links between C306 residues belonging to two protomers, in agreement with our in silico modeling of DAT dimer (Cheng et al., 2017). Interestingly, our trimer-C306 model also allows for such potential inter-protomeric disulfide bridge formation in the OF state (Fig 1A), and not in the IF state. On the other hand, our in silico analysis pointed to inter-subunit cation-π interactions between W497 and K525 (Fig 1F), which might stabilize the trimer-C306. C306A mutant did not generate any phenotypic effects, but W497A and W497F, and K525A did. The significance of these residues was also supported by the independent in silico saturation mutagenesis analysis (Fig 7). These findings suggest that disulfide bond formation among C306 residues may not be required for forming/stabilizing DAT trimers; whereas the critical role of W497 and K525 may be attributed to their high affinity to bind AIM-100, which, in turn, stabilizes the trimer. Mutants W497A, W497F and K525A were indeed observed by experiments to impair the migration from the ER to the PM, or to form aggregates in the cytoplasm, in line with their possible role in stabilization assisted by AIM-100 binding.
5. Conclusion
Modulation of DAT multimerization by small molecule binding emerges as a new possible therapeutic strategy, supported by the direct effect on DAT internalization. The current study suggests that D231-R237 and W497 are important for proper oligomerization or folding of DAT. Notably, two substitutions for I595 exhibited completely opposite phenotypes: I595V was functional, like the wt DAT, and I595A was not. The fact that such subtle differences could be correctly captured by in silico saturation mutagenesis experiments (Fig 7) suggests that the latter could provide guidance for selecting mutation sites and specific substitutions for probing the response of the investigated protein. Such studies may become increasingly important for identifying sites susceptible to inducing cooperative responses (e.g. trimerization) upon small molecule binding.
Supplementary Material
Highlights.
Combination of modeling and experiments clarify DAT trimerization mechanisms
DAT can adapt to different trimerization geometries by virtue of the adaptability of the LeuT fold
Extracellular loops EL2 or EL3 facilitate the assembly of DATs into trimers
W497 and K525 emerge as critical sites in imparting structural stability and/or binding AIM-100.
In silico saturation mutagenesis data provide new hypotheses for future assays
Acknowledgments
Funding
This work was supported by the National Institutes of Health awards P41GM103712 and P30DA035778 (IB), and R01DA014204 (AS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
All authors declare no conflict of interest.
References
- Adkins EM, Samuvel DJ, Fog JU, Eriksen J, Jayanthi LD, Vaegter CB, Ramamoorthy S, Gether U, 2007. Membrane mobility and microdomain association of the dopamine transporter studied with fluorescence correlation spectroscopy and fluorescence recovery after photobleaching. Biochemistry 46, 10484–10497. [DOI] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR, 2010. A method and server for predicting damaging missense mutations. Nat Methods 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amara SG, Sonders MS, 1998. Neurotransmitter transporters as molecular targets for addictive drugs. Drug Alcohol Depend 51, 87–96. [DOI] [PubMed] [Google Scholar]
- Anderluh A, Hofmaier T, Klotzsch E, Kudlacek O, Stockner T, Sitte HH, Schütz GJ, 2017. Direct PIP2 binding mediates stable oligomer formation of the serotonin transporter. Nat. Commun 8, 14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderluh A, Klotzsch E, Reismann AW, Brameshuber M, Kudlacek O, Newman AH, Sitte HH, Schutz GJ, 2014. Single molecule analysis reveals coexistence of stable serotonin transporter monomers and oligomers in the live cell plasma membrane. J. Biol. Chem 289, 4387–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar I, Lezon TR, Yang LW, Eyal E, 2010. Global dynamics of proteins: bridging between structure and function. Annu Rev Biophys 39, 23–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakan A, Dutta A, Mao W, Liu Y, Chennubhotla C, Lezon TR, Bahar I, 2014. Evol and ProDy for bridging protein sequence evolution and structural dynamics. Bioinformatics 30, 2681–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baucum AJ II, Rau KS, Riddle EL, Hanson GR, Fleckenstein AE, 2004. Methamphetamine increases dopamine transporter higher molecular weight complex formation via a dopamine- and hyperthermia-associated mechanism. J Neurosci 24, 3436–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SP, Farrell K, Conant D, Kempner ES, Paul SM, 1994. Radiation inactivation studies of the dopamine reuptake transporter protein. Mol. Pharmacol 46, 726–731. [PubMed] [Google Scholar]
- Caltagarone J, Ma SQ, Sorkin A, 2015. Dopamine transporter is enriched in filopodia and induces filopodia formation. Mol. Cell. Neurosci 68, 120–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng MH, Bahar I, 2014. Complete Mapping of Substrate Translocation Highlights the Role of LeuT N-terminal Segment in Regulating Transport Cycle. PLoS Comput. Biol 10, e1003879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng MH, Bahar I, 2015. Molecular mechanism of dopamine transport by human dopamine transporter. Structure 23, 2171–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng MH, Block E, Hu F, Cobanoglu MC, Sorkin A, Bahar I, 2015. Insights into the modulation of dopamine transporter function by amphetamine, orphenadrine and cocaine binding. Front. Neurol 6, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng MH, Garcia-Olivares J, Wasserman S, DiPietro J, Bahar I, 2017. Allosteric modulation of human dopamine transporter activity under conditions promoting its dimerization. J. Biol. Chem 292, 12471–12482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng MH, Kaya C, Bahar I, 2018. Quantitative assessment of the energetics of dopamine translocation by human dopamine transporter. J Phys Chem B 122, 5336–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JA, Green EM, Gouaux E, 2016. X-ray structures and mechanism of the human serotonin transporter. Nature 532, 334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeau SR, Gatchell DW, Vajda S, Camacho CJ, 2004. ClusPro: a fully automated algorithm for protein–protein docking. Nucleic Acids Res 32, W96–W99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradi V, Sejdiu BI, Mesa-Galloso H, Abdizadeh H, Noskov SY, Marrink SJ, Tieleman DP, 2019. Emerging diversity in lipid–protein interactions. Chem Rev 119, 5775–5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das AK, Kudlacek O, Baumgart F, Jaentsch K, Stockner T, Sitte HH, Schütz GJ, 2019. Dopamine transporter forms stable dimers in the live cell plasma membrane in a phosphatidylinositol-4,5-bisphosphate independent manner. J Biol Chem 294, 5632–5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn SD, Wahl LM, Gloor GB, 2008. Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction. Bioinformatics 24, 333–340. [DOI] [PubMed] [Google Scholar]
- El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, Sonnhammer ELL, Hirsh L, Paladin L, Piovesan D, Tosatto SCE, Finn RD, 2019. The Pfam protein families database in 2019. Nucleic Acids Res 47, D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyal E, Lum G, Bahar I, 2015. The anisotropic network model web server at 2015 (ANM 2.0). Bioinformatics 31, 1487–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiser A, Sali A, 2003. ModLoop: automated modeling of loops in protein structures. Bioinformatics 19, 2500–2501. [DOI] [PubMed] [Google Scholar]
- Forrest LR, Rudnick G, 2009. The rocking bundle: a mechanism for ion-coupled solute flux by symmetrical transporters. Physiology 24, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest LR, Zhang Y-W, Jacobs MT, Gesmonde J, Xie L, Honig BH, Rudnick G, 2008. Mechanism for alternating access in neurotransmitter transporters. Proc. Natl. Acad. Sci. U.S.A 105, 10338–10343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gether U, Andersen PH, Larsson OM, Schousboe A, 2006. Neurotransmitter transporters: molecular function of important drug targets. Trends Pharmacol. Sci 27, 375–383. [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG, 1996. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 379, 606–612. [DOI] [PubMed] [Google Scholar]
- Gloor GB, Martin LC, Wahl LM, Dunn SD, 2005. Mutual information in protein multiple sequence alignments reveals two classes of coevolving positions. Biochemistry 44, 7156–7165. [DOI] [PubMed] [Google Scholar]
- Gur M, Cheng MH, Zomot E, Bahar I, 2017. Effect of Dimerization on the Dynamics of Neurotransmitter:Sodium Symporters. J. Phys. Chem. B 121, 3657–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haliloglu T, Bahar I, 2015. Adaptability of protein structures to enable functional interactions and evolutionary implications. Curr. Opin. Struct. Biol 35, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Panchenko AR, 2010. Mechanisms of protein oligomerization, the critical role of insertions and deletions in maintaining different oligomeric states. PNAS 107, 20352–20357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastrup H, Karlin A, Javitch JA, 2001. Symmetrical dimer of the human dopamine transporter revealed by cross-linking Cys-306 at the extracellular end of the sixth transmembrane segment. Proc. Natl. Acad. Sci. U. S. A 98, 10055–10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf TA, Ingraham JB, Poelwijk FJ, Scharfe CP, Springer M, Sander C, Marks DS, 2017. Mutation effects predicted from sequence co-variation. Nat Biotechnol 35, 128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K, 1996. VMD: Visual molecular dynamics. J. Mol. Graph 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Ingólfsson HI, Carpenter TS, Bhatia H, Bremer P-T, Marrink SJ, Lightstone FC, 2017. Computational lipidomics of the neuronal plasma membrane. Biophys J 113, 2271–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin JJ, Shoichet BK, 2005. ZINC– a free database of commercially available compounds for virtual screening. Journal of chemical information and modeling 45, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iversen L, 2006. Neurotransmitter transporters and their impact on the development of psychopharmacology. Br. J. Pharmacol 147, S82–S88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardetzky O, 1966. Simple allosteric model for membrane pumps. Nature 211, 969–970. [DOI] [PubMed] [Google Scholar]
- Jayaraman K, Morley AN, Szollosi D, Wassenaar TA, Sitte HH, Stockner T, 2018. Dopamine transporter oligomerization involves the scaffold domain, but spares the bundle domain. PLoS Comput Biol 14, e1006229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanner BI, Zomot E, 2008. Sodium-coupled neurotransmitter transporters. Chem. Rev 108, 1654–1668. [DOI] [PubMed] [Google Scholar]
- Kaya C, Cheng MH, Block ER, Bartol TM, Sejnowski TJ, Sorkin A, Faeder JR, Bahar I, 2018. Heterogeneities in Axonal Structure and Transporter Distribution Lower Dopamine Reuptake Efficiency. eNeuro 5, ENEURO.0298–0217.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelashvili G, Stanley N, Sahai MA, Medina J, LeVine MV, Shi L, De Fabritiis G, Weinstein H, 2015. Spontaneous inward opening of the dopamine transporter is triggered by PIP2-regulated dynamics of the N-terminus. ACS Chem. Neurosci 6, 1825–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilic F, Rudnick G, 2000. Oligomerization of serotonin transporter and its functional consequences. Proc. Natl. Acad. Sci. U. S. A 97, 3106–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshy C, Schweikhard ES, Gärtner RM, Perez C, Yildiz Ö, Ziegler C, 2013. Structural evidence for functional lipid interactions in the betaine transporter BetP. EMBO J 32, 3096–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, Vajda S, 2017. The ClusPro web server for protein-protein docking. Nat. Protoc 12, 255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy H, Gouaux E, 2012. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 481, 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen AS, Andersen J, Jørgensen TN, Sørensen L, Eriksen J, Loland CJ, Strømgaard K, Gether U, 2011. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol. Rev 63, 585–640. [DOI] [PubMed] [Google Scholar]
- Kuhn HW, 1955. The Hungarian method for the assignment problem. Naval research logistics quarterly 2, 83–97. [Google Scholar]
- LeVine MV, Cuendet MA, Razavi AM, Khelashvili G, Weinstein H, 2018. Thermodynamic coupling function analysis of allosteric mechanisms in the human dopamine transporter. Biophys. J 114, 10–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Cheng SY, Chen N, Reith ME, 2010. Interrelation of dopamine transporter oligomerization and surface presence as studied with mutant transporter proteins and amphetamine. J Neurochem 114, 873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrink SJ, Corradi V, Souza PC, Ingólfsson HI, Tieleman DP, Sansom MS, 2019. Computational modeling of realistic cell membranes. Chem Rev 119, 6184–6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh JA, Teichmann SA, 2015. Structure, dynamics, assembly, and evolution of protein complexes. Annu. Rev. Biochem 84, 551–575. [DOI] [PubMed] [Google Scholar]
- Nyola A, Karpowich NK, Zhen J, Marden J, Reith ME, Wang D-N, 2010. Substrate and drug binding sites in LeuT. Curr. Opin. Struct. Biol 20, 415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penmatsa A, Wang KH, Gouaux E, 2013. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503, 85–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penmatsa A, Wang KH, Gouaux E, 2015. X-ray structures of Drosophila dopamine transporter in complex with nisoxetine and reboxetine. Nat Struct Mol Biol 22, 506–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez C, Khafizov K, Forrest LR, Krämer R, Ziegler C, 2011. The role of trimerization in the osmoregulated betaine transporter BetP. EMBO Rep 12, 804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periole X, Zeppelin T, Schiøtt B, 2018. Dimer interface of the human serotonin transporter and effect of the membrane composition. Sci. Rep 8, 5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K, 2005. Scalable Molecular Dynamics with NAMD. J. Comput. Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponzoni L, Bahar I, 2018. Structural dynamics is a determinant of the functional significance of missense variants. Proc. Natl. Acad. Sci. U.S.A 115, 4164–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponzoni L, Zhang S, Cheng MH, Bahar I, 2018. Shared dynamics of LeuT superfamily members and allosteric differentiation by structural irregularities and multimerization. Phil. Trans. R. Soc. B 373, 20170177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramod AB, Foster J, Carvelli L, Henry LK, 2013. SLC6 transporters: structure, function, regulation, disease association and therapeutics. Mol. Aspects Med 34, 197–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahbek-Clemmensen T, Lycas MD, Erlendsson S, Eriksen J, Apuschkin M, Vilhardt F, Jørgensen TN, Hansen FH, Gether U, 2017. Super-resolution microscopy reveals functional organization of dopamine transporters into cholesterol and neuronal activity-dependent nanodomains. Nat. Commun 8, 740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressl S, van Scheltinga ACT, Vonrhein C, Ott V, Ziegler C, 2009. Molecular basis of transport and regulation in the Na+/betaine symporter BetP. Nature 458, 47–52. [DOI] [PubMed] [Google Scholar]
- Roberts E, Eargle J, Wright D, Luthey-Schulten Z, 2006. MultiSeq: unifying sequence and structure data for evolutionary analysis. BMC Bioinformatics 7, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RB, Barton GJ, 1992. Multiple protein sequence alignment from tertiary structure comparison: assignment of global and residue confidence levels. Proteins: Structure, Function, and Bioinformatics 14, 309–323. [DOI] [PubMed] [Google Scholar]
- Schmid JA, Scholze P, Kudlacek O, Freissmuth M, Singer EA, Sitte HH, 2001. Oligomerization of the human serotonin transporter and of the rat GABA transporter 1 visualized by fluorescence resonance energy transfer microscopy in living cells. J. Biol. Chem 276, 3805–3810. [DOI] [PubMed] [Google Scholar]
- Scholze P, Nørregaard L, Singer EA, Freissmuth M, Gether U, Sitte HH, 2002. The Role of Zinc Ions in Reverse Transport Mediated by Monoamine Transporters. J. Biol. Chem 277, 21505–21513. [DOI] [PubMed] [Google Scholar]
- Schulze S, Köster S, Geldmacher U, van Scheltinga A. C. Terwisscha, Kühlbrandt W, 2010. Structural basis of Na+-independent and cooperative substrate/product antiport in CaiT. Nature 467, 233. [DOI] [PubMed] [Google Scholar]
- Seidel S, Singer EA, Just H, Farhan H, Scholze P, Kudlacek O, Holy M, Koppatz K, Krivanek P, Freissmuth M, Sitte HH, 2005. Amphetamines take two to tango: an oligomer-based counter-transport model of neurotransmitter transport explores the amphetamine action. Mol. Pharmacol 67, 140–151. [DOI] [PubMed] [Google Scholar]
- Siciliano CA, Saha K, Calipari ES, Fordahl SC, Chen R, Khoshbouei H, Jones SR, 2018. Amphetamine Reverses Escalated Cocaine Intake via Restoration of Dopamine Transporter Conformation. J Neurosci 38, 484–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitte HH, Farhan H, Javitch JA, 2004. Sodium-dependent neurotransmitter transporters: oligomerization as a determinant of transporter function and trafficking. Molecular interventions 4, 38. [DOI] [PubMed] [Google Scholar]
- Sitte HH, Freissmuth M, 2010. The reverse operation of Na(+)/Cl(−)-coupled neurotransmitter transporters--why amphetamines take two to tango. J Neurochem 112, 340–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitte HH, Freissmuth M, 2015. Amphetamines, new psychoactive drugs and the monoamine transporter cycle. Trends Pharmacol Sci 36, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkina T, Caltagarone J, Sorkin A, 2013. Flotillins regulate membrane mobility of the dopamine transporter but are not required for its protein kinase C dependent endocytosis. Traffic 14, 709–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkina T, Doolen S, Galperin E, Zahniser NR, Sorkin A, 2003. Oligomerization of dopamine transporters visualized in living cells by fluorescence resonance energy transfer microscopy. J. Biol. Chem 278, 28274–28283. [DOI] [PubMed] [Google Scholar]
- Sorkina T, Ma S, Larsen MB, Watkins SC, Sorkin A, 2018. Small molecule induced oligomerization, clustering and clathrin-independent endocytosis of the dopamine transporter. eLife 7, e32293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkina T, Miranda M, Dionne KR, Hoover BR, Zahniser NR, Sorkin A, 2006. RNA interference screen reveals an essential role of Nedd4–2 in dopamine transporter ubiquitination and endocytosis. J Neurosci 26, 8195–8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, 2011. How Addictive Drugs Disrupt Presynaptic Dopamine Neurotransmission. Neuron 69, 628–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres GE, Carneiro A, Seamans K, Fiorentini C, Sweeney A, Yao WD, Caron MG, 2003. Oligomerization and trafficking of the human dopamine transporter. Mutational analysis identifies critical domains important for the functional expression of the transporter. J. Biol. Chem 278, 2731–2739. [DOI] [PubMed] [Google Scholar]
- Trott O, Olson AJ, 2010. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KH, Penmatsa A, Gouaux E, 2015. Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 521, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Bellve KD, Fogarty KE, Melikian HE, 2015. Ack1 is a dopamine transporter endocytic brake that rescues a trafficking-dysregulated ADHD coding variant. Proceedings of the National Academy of Sciences 112, 15480–15485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E, 2005. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature 437, 215–223. [DOI] [PubMed] [Google Scholar]
- Youden WJ, 1950. Index for rating diagnostic tests. Cancer 3, 32–35. [DOI] [PubMed] [Google Scholar]
- Zhang S, Li H, Krieger J, Bahar I, 2019. Shared signature dynamics tempered by local fluctuations enables fold adaptability and specificity. Mol Biol Evol, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen J, Antonio T, Cheng SY, Ali S, Jones KT, Reith ME, 2015. Dopamine transporter oligomerization: impact of combining protomers with differential cocaine analog binding affinities. J Neurochem 133, 167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen J, Reith ME, 2018. Functional properties of dopamine transporter oligomers after copper linking. Journal of neurochemistry 144, 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.