

Abstract
Background:
Traumatic brain injury (TBI) is an under-recognized public health threat. Even mild brain injuries can lead to long-term neurologic impairment. Microglia play a fundamental role in the development and progression of this ensuing neurologic impairment. Despite this, a microglia-specific injury signature has yet to be identified. We hypothesized that TBI would lead to long-term changes in the transcriptional profile of microglial pathways associated with the development of subsequent neurologic impairment.
Materials and Methods:
Male C57BL/6 mice underwent TBI via a controlled cortical impact and were followed longitudinally. FACSorted microglia from TBI mice were subjected to Quantiseq 3’-biased RNA-sequencing at 7, 30, and 90 days post-TBI. K-means clustering on 396 differentially expressed genes was performed and gene ontology enrichment analysis was used to determine corresponding enriched processes.
Results:
Differentially expressed genes in microglia exhibited 4 main patterns of expression over the course of TBI. In particular, we identified 4 gene clusters which corresponded to the host defense response, synaptic potentiation, lipid remodeling, and membrane polarization.
Conclusions:
Transcriptional profiling within individual populations of microglia after TBI remains a critical unmet research need within the field of traumatic brain injury. This focused study identified several physiologic processes within microglia that may be associated with development of long-term neurologic impairment after TBI. These data demonstrate the capability of longitudinal transcriptional profiling to uncover potential cell-specific targets for the treatment of TBI.
Keywords: TBI, Microglia, FACS, RNA-sequencing
Introduction
Traumatic brain injury is a growing and under recognized public health threat. The CDC estimates nearly 2 million people sustain a traumatic brain injury (TBI) each year in the United States, contributing to over 30% of all injury related deaths (1, 2). In fact, TBI related healthcare expenditures near 80 billion dollars annually with an average cost of 4 million dollars per person surviving a severe TBI (3-5). The impact of TBI is highlighted by both its high mortality rate and by the significant long-term complications suffered by its survivors with the progressive development of motor, cognitive, and behavioral disorders (6-10). Prior data from our laboratory has demonstrated persistent and progressive deficits in working memory, motor coordination, and skill acquisition in TBI mice even after resolution of the acute injury process (11). The immune response to TBI plays a fundamental role in the development and progression of this subsequent neurologic impairment and represents a complex interplay between the injured brain and the resident immune cells of the brain— microglia (12). Microglia are the first responders to brain injury and are capable of promoting inflammation as well as fostering wound repair (13). This generates an environment that can either propel the index injury or support repair and regeneration depending on the activation state of microglia and the resultant gene expression profile (14-16). The current manuscript is focused on developing this cell-type-specific understanding of the microglial response to injury. Here we highlight a focused study demonstrating the utility of longitudinal transcriptional profiling in assessing microglial gene expression over the course of TBI.
The first step towards any cell-specific transcriptional analysis relies on obtaining sufficient cells of interest with the highest purity. The historical standard for distinguishing between microglia and infiltrating macrophages is immunohistochemistry. Although immunohistochemistry is useful for cell characterization, it has a number of drawbacks limiting its use (17). Several investigative groups have focused on this problem including column free magnetic separation and CD11b immunomagnetic enrichment combined with the differential expression of CD45 with flow cytometry (17-19). However, CD45 expression has been reported to vary depending on the pathologic condition; thus, reliable separation of microglia from peripheral myeloid cells is impaired (20, 21). To overcome this issue, fluorescently marked myeloid cells, such as CX3CR1+/GFP/CCR2+/RFP, have been used. However, these mice are on mixed backgrounds, which could greatly affect the immune response. Furthermore, the presence of contaminated nonclassical monocytes could not be excluded (22, 23). Therefore, we developed head-shielded bone marrow chimeric mice with CD45.1 cells in the circulation and CD45.2 microglia in the brain allowing definitive and unambiguous differentiation between microglia and infiltrating bone-marrow derived myeloid cells (11). To the best of our knowledge, no cell-type-specific study has been conducted to specifically identify transcriptional changes in isolated populations of microglia over the course of TBI. We hypothesized that TBI would lead to long-term changes in the transcriptional profile of microglial pathways associated with the development of subsequent neurologic impairment. In the resultant focused data, we show that TBI-associated microglia do adopt longitudinal changes in their transcriptional profile. In particular, we found that differential genes exhibit 4 main patterns of expression. Most notable was an upregulation of genes associated with pathways linked to synaptic plasticity, a biologic process essential in learning and memory (24, 25). These focused data demonstrate the power of longitudinal transcriptional profiling in uncovering potential cell-specific targets for minimizing TBI-associated disabilities.
Material and Methods
Mice
All procedures were approved by the Northwestern University Institutional Animal Care and Use committee and all experiments were carried out in accordance with the ARRIVE guidelines on the reporting of in vivo experiments. Two mouse strains were used— C57BL/6 and B6.SJL-Ptprca Pepcb/BoyJ (CD45.1). All mice were male and purchased from the Jackson Laboratory and housed at a barrier facility at the Center for Comparative Medicine at Northwestern University (Chicago, IL, USA).
Shielded Bone Marrow Chimeras
Bone marrow chimeric mice were generated as previously described by our laboratory (11). In brief, bone marrow was aseptically harvested from the tibias and femurs of eight-week-old B6.CD45.1 donor mice (n=18). Erythrocytes were lysed and the cells were counted using a Countess automated cell counter. Eight-week-old B6.CD45.2 mice (n=9) received a single 1000-cGy γ-irradiation dose using a Cs-137-based Gammacell 40 irradiator. The mice heads were shielded with a lead bar so as to deliver the irradiation to the body only. Six hours after shielded irradiation, busulfan (30 mg/kg) was administered to completely ablate the bone marrow of the recipient mice. Donor bone marrow (CD45.1) was transplanted twelve hours after busulfan ablation. Shielded bone marrow chimeras were maintained on antibiotics trimethoprim/sulfamethoxazole (40 mg/5 mg, respectively). Eight weeks after irradiation, 95% of the circulating monocytes were of donor origin (Fig. 1).
Figure 1:
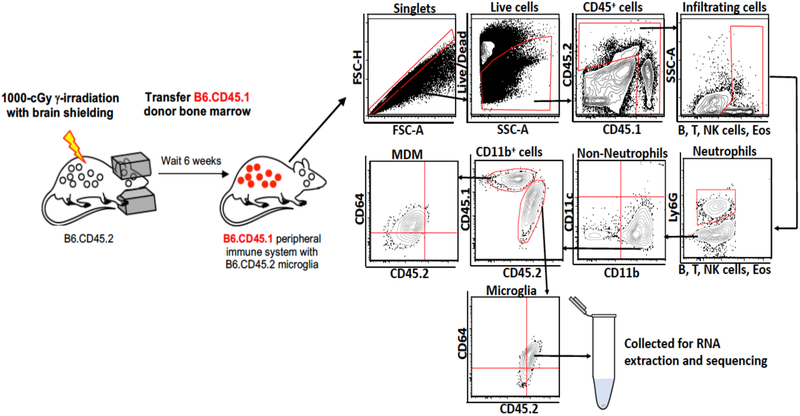
Microglia from head-shielded bone marrow chimeric mice are distinct from infiltrating macrophages after TBI. Brains isolated from chimeric mice post TBI were analyzed by flow cytometry. CD45.1neg ‘B, T, NK cells and Eosinophils’ acted as the gating control for CD45.2hi cells, showing that resident microglia (CD45lo) can be unambiguously differentiated from infiltrating monocyte-derived macrophages (CD45.1+). Arrows indicate the directionality of gating.
Controlled cortical impact
Controlled cortical impact was induced as previously described by our laboratory (11, 26). Sixteen-week-old chimeric mice (n = 9) were used for all RNA-seq experiments. In brief, mice were anesthetized with 100 mg/kg Ketamine and 10 mg/kg Xylazine via intraperitoneal injection. A 1cm scalp incision was performed and a 5mm craniectomy was performed 2 mm left of the sagittal suture and 2 mm rostral to the coronal suture. The dura was left intact. Mice were then placed in a stereotaxic operating frame and the impactor (Leica Biosystems Inc., Buffalo Grove, IL) was maneuvered into position. A controlled cortical impact was then applied with a 3mm impacting tip at a velocity of 2.5m/s and an impacting depth of 2mm with the dwell time set at 0.1s. Immediately following injury all animals had their scalps sealed with VetBond (3M). All animals received post injury analgesia with Buprenorphine SR via subcutaneous injection and were allowed to recover in separate cages over a warming pad. Mice were euthanized via carbon dioxide inhalation at 7, 30, and 90 days post TBI and brains were harvested.
Tissue preparation and fluorescence activated cell sorting
Immediately following euthanasia, mice were perfused with ice-cold PBS via transcardial puncture. Brains were harvested, weighed, morcellated and placed into digestion buffer (2.5 mg/mL Liberase TL (Roche, Basel, Switzerland), and 1 mg/mL of DNase I in HBSS). The digested brains were then placed on a MACS dissociator (Miltenyi Biotec) using the brain dissociation protocol per the manufacturer instructions. Following dissociation they were incubated in a shaker at 200 rpm for 30 minutes at 37°C. Following incubation, the brains were replaced on the MACS dissociator using the brain dissociation protocol per the manufacturer instructions. The tissue slurry was then strained through a 40 μm nylon mesh strainer and washed with wash buffer (1% BSA in HBSS). Microglia and infiltrating leukocytes were separated using a 30/70 percoll gradient (Percoll Plus, GE Healthcare). Microglia and infiltrating leukocytes were then collected from the interphase of the gradient and washed with HBSS. The cells were counted with a Countess automated cell counter (Invitrogen) and dead cells were identified with trypan blue. Live/dead Aqua (Invitrogen) viability dye, Fc-Block (BD Bioscience), and fluorochrome-conjugated antibodies were applied (Table I). Data were acquired, and microglia sorted, on a BD FACSAria cell sorter (BD Biosciences, San Jose, CA). “Fluorescence minus one” controls were used to establish gates. Sorted microglia were lysed and RNA extracted with a PicoPure RNA isolation kit (Arcturus Bioscience). Flow cytometry data was analyzed with Flowjo software (TreeStar, Ashland, OR).
Table I.
List of antibody conjugated fluorochromes used to differentiate microglia from infiltrating leukocytes.
| Antibody | Fluorochrome | Company and Clone |
|---|---|---|
| CD45.1 | FITC | A-20 / BD Bioscience |
| CD45.2 | BV421 | 104 / Biolegend |
| CD64 | APC | X54-5/7.1/ BD Bioscience |
| CD11b | APC- Cy7 | M1/70 / BD Biosciences |
| CD11c | PE-Cy7 | HL3 / BD Bioscience |
| Ly6G | Alexa Flour 700 | 1A8 / BD Bioscience |
| MHC II | Percp- Cy 5.5 | M5/114.15.2 / Biolegend |
| Siglec H | PE | 551 / Biolegend |
| B220 | PECF594 | RA3-6B2 / BD Bioscience |
| Siglec F | PECF594 | E50-2440 / BD Bioscience |
| CD4 | PECF594 | RM4-5/ BD Bioscience |
| CD8 | PECF594 | 53-6.7 / BD Bioscience |
| NK1.1 | PECF594 | PK136/ BD Bioscience |
| Viability | e-bioscience Fixable Viability Dye eFluor 506 | Invitrogen |
RNA sequencing
RNA from the FACSorted microglia was isolated with Picopure RNA isolation kits. RNA quality and quantity was confirmed using a High Sensitivity RNA ScreenTape System. Sample quality control, processing and library preparation (Quantseq 3’-biased protocol) were performed by the Division of Rheumatology and Pulmonary and Critical Care Sequencing Facility. Libraries were multiplexed and sequenced on an Illumina NextSeq 500 instrument to an average depth of 20×106 reads per sample.
RNA sequencing analysis
RNA-seq processing was performed using an established computational pipeline described previously (27). Briefly, the BCL files generated from sequencing were demultiplexed (bcl2fastq), trimmed (bbduk: http://jgi.doe.gov/data-and-tools/bb-tools/), aligned to mm10 (STAR), mapped to genes (HTSeq), and normalized to CPM (28, 29). We performed transcriptional analyses on n=3 for each time point. We defined expressed genes as those with an average expression at any time point greater than 7 cpm (or log2(CPM+1 ≥ 3). We defined differentially expressed genes by ANOVA (p<0.05) across time points. We used GENE-E (https://software.broadinstitute.org/GENE-E/) with default settings to perform K-means clustering (k=4). Functional annotations of Gene Ontology processes were determined by GOrilla(30). Normalized fold-change and p-values associated with pairwise differential genes between any 2 time points were determined using DEseq2 (31). Volcano plots were visualized using the ggplot2 package from R Studio software. To investigate relationships between the genes in our dataset as well as to visualize potential gene networks, filtered genes were input into the ‘build a network’ tool in the Metacore software package (Clarivate Analytics. Philadelphia, PA). Pathway maps and network associations were generated.
Data Availability
RNA sequencing data is available through the NCBI Sequence Read Archive (SRA accession number: SRP160379). The remaining data that support the findings in this manuscript are available from the corresponding author upon request.
Results
Global patterns of gene expression from isolated populations of microglia over the time course of TBI
FACSorted microglia were subjected to unbiased transcriptional profiling (RNA-seq) on 7, 30, and 90 days after TBI. We defined 396 genes that change expression over the course of injury (see methods). We clustered these differentially expressed genes into 4 main patterns of expression (Fig. 2). Clusters 1 and 3 represent genes involved in the host response to injury and lipid remodeling. Representative genes found in this pathway include Lcn2, Bst2, Apoe, and Lpl and are progressively downregulated over the course of injury. Cluster 2 represents genes involved in synaptic plasticity such as Ptpn5, Sqstm1, and Shank3 and are progressively upregulated over the course of injury. Cluster 4 represents genes involved in the regulation of membrane polarization such as Grin2a, Crtc1, and Hcn1 with gene expression that is upregulated at 30 days post-injury and then downregulated by 90 days post injury. These data provide new insights into the biology of microglial activation over the course of TBI.
Figure 2:
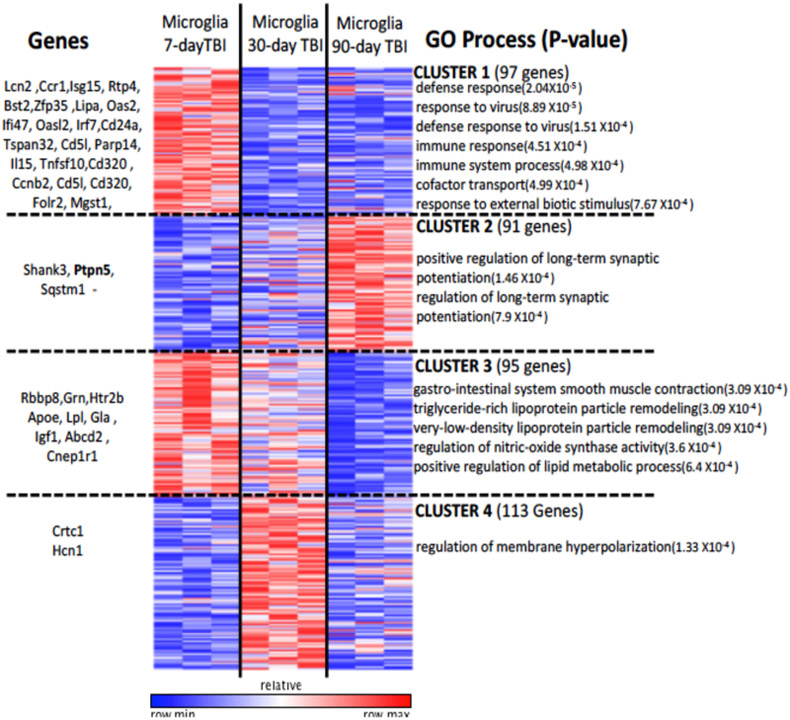
Microglia from TBI mice show distinct time-dependent transcriptional profiles. K-means clustering (k=4): Four clusters representing 396 genes are shown with distinct time-specific expression patterns. Example genes from each cluster (left) and significant GO processes (right) are shown.
Pairwise comparison of microglia gene expression between time points post-TBI
To further evaluate gene expression over time in TBI-associated microglia, we investigated differentially expressed genes between microglia across time points (Fig. 3). We found 187 genes between days 7 and 30 that are positively differentiated and are likely involved in channel transport activity, as well as defense response. Several genes including genes involved in immune response and tissue repair such as Tnfsf10, Bst2, Igf1, and Ccr1 were upregulated at the earlier time points and then progressively downregulated over the course of injury. However, several genes implicated in the development of long-term neurodegenerative diseases such as Ptpn5 (STEP), Shank3, and Sqstm1 were upregulated by 90 days compared to 7 days post TBI. To determine whether gene expression trends at 30 days post-TBI were predictive of gene expression at 90 days post-TBI, we compared the fold changes between 7 vs. 30 days post-TBI with those between 7 vs. 90 days post-TBI (Fig. 4). We find that these gene expression changes were significantly correlated (R=0.537, p=2.2 × 10−16). This indicates that genes which were upregulated at 90 days post-TBI had already started to increase as early as 30 days post-injury. Conversely, those genes that were downregulated at 30 days post-TBI remained downregulated or continued to fall over time.
Figure 3:
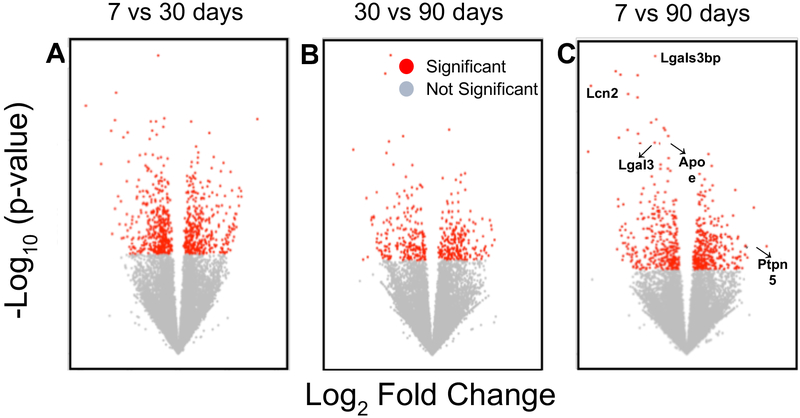
Differentially expressed genes over the time course of injury. Volcano plots of differentially expressed genes in microglia (n=9) between 7D and 30D post-TBI (A), 30D and 90D post-TBI (B), and 7D and 90D post-TBI (C). Examples of genes with a p-value <0.05 (as calculated by DEseq model) are shown in red.
Figure 4:
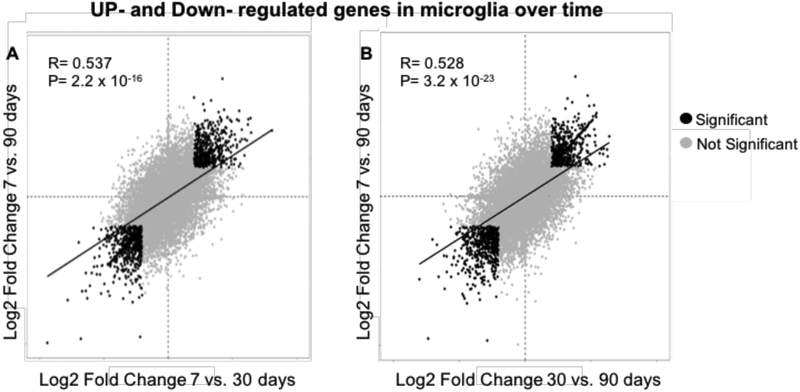
Correlational analysis of change in gene expression microglia over time after TBI.
Scatter plot to examine the relationship between the genes that are up-regulated or down-regulated in the microglia of mice at A) 7 vs 30 days post-TBI compared to 7 vs 90 days post-TBI and at B) 30 vs 90 days post-TBI compared to 7 vs 90 days. A log2 fold change of 1 is equal to a 2-fold change between time point mean expression (indicated in black dots) (n=9).
Trem2-APOE gene expression is not upregulated in microglia over the course of traumatic brain injury.
There has been considerable interest in the Trem2-APOE pathway in the generation of a neurodegenerative microglial phenotype in both Alzheimer’s Disease (AD) and multiple sclerosis (MS). In fact, recent data has identified the Trem2-APOE pathway as a pivotal regulator of microglial phenotype in both of these disease processes (32). Additionally, gene co-expression networks have shown Trem2 to be a major hub in APOE expressing mice after TBI and TREM2 deficiency has also been shown to alter peripheral macrophage responses after TBI (33, 34). Therefore, we aimed to determine if the Trem2-APOE pathway was a major regulator of microglial phenotype in our model TBI. However, unlike prior studies, our data demonstrates no significant change in TREM2 expression as well as a progressive decrease in APOE expression over the course of TBI (Fig. 5B). These seemingly contradictory results emphasize the need for microglia-specific transcriptional studies in the setting TBI.
Figure 5.
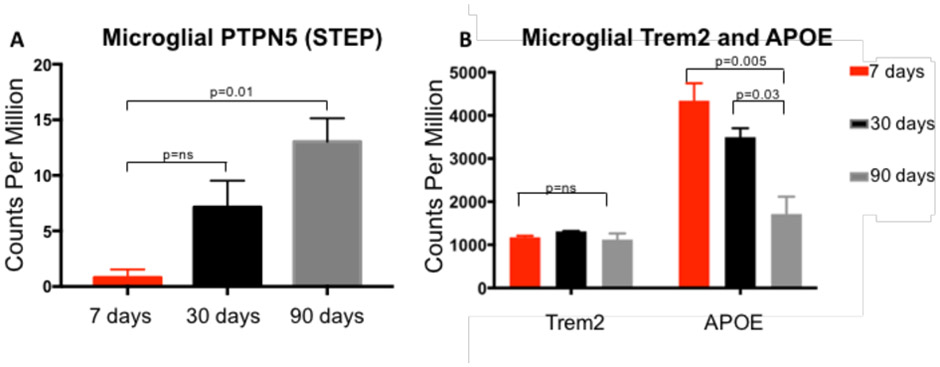
Normalized expression of Microglial Ptpn5, Trem2 and APOE over the course of TBI.
Ptpn5 expression from sequenced FACSorted microglia progressively increased from 7 days post-TBI to 90 days post-TBI; p=0.01, Trem2 did not significantly change over the course of brain injury while APOE expression progressively decreased over time following TBI p=0.005. Ordinary one way ANOVA with Bonferroni’s multiple comparison test (n=9).
Microglial STEP expression is progressively upregulated over the course of brain injury.
While our data failed to demonstrate a common pathway with Alzheimer’s disease and multiple sclerosis through Trem2-APOE, a closer examination of Cluster 2 (Fig. 2) revealed a number of upregulated genes associated with long-term synaptic potentiation, including PTPN5 also known as STEP (STriatial-Enriched protein tyrosine Phosphatase), Shank3 (SH3 and multiple ankyrin repeat domains 3), and Sqstm1 (Sequestosome-1). Each of the proteins are key regulators of cognitive impairment in Alzheimer’s disease, schizophrenia and autism, respectively. Our data show that expression of each of these proteins is progressively increased in microglia over time in our model of TBI (Fig. 5C). These data suggest that STEP, Shank3, Sqstm1, and other genes identified in our sequencing analysis may share a common molecular pathway connecting TBI with other known neurodegenerative disorders.
Pathway analysis suggests down-regulation of NMDA receptor expression.
Genes that were differentially expressed across all three time points were input into the Metacore database to investigate potential relationships between these genes and biological pathways. The top ten downregulated pathways were related to the complement mediated immune response, suggesting an increase in negative regulation of complement activation after TBI (Fig. 6A). The top ten upregulated pathways were related to cell remodeling and neurophysiologic processes (Fig. 6B). Notably, two of the genes identified in this pathway were Ptpn5 which codes for the STEP protein, and Grin2a which codes for the Nr2a subunit of the NMDA receptor. Both of these genes are critical determinates of synaptic plasticity via the regulation of NMDA receptor expression. We observed that Ptpn5 increased sharply from 7-30 days, with an even higher increase from 30-90 days post-TBI. Grin2a however was slightly decreased from 7-30 days with an even sharper decrease from 30-90 days post-TBI (Fig. 6C).
Figure 6.

Pathway map analysis of differentially expressed microglial genes after TBI. Expressed genes were input into the Metacore software and pathway map analyses was performed. The red highlighted lines show inhibition or negative effect, the green lines show activation or positive effect, the grey lines show an unspecified relationship between the two linked elements.
All highlighted interactions have been implicated in brain and neurodegenerative diseases as curated by Metacore analytics.
Discussion
Microglia are the resident innate immune cells of the CNS. They are ontologically distinct from peripheral bone marrow-derived monocytes and macrophages, arising from the yolk sac as opposed to the developing liver in the embryo (35). In fact, microglia rely on a distinctive set of transcription factors during development resulting in a lineage of tissue macrophages (microglia) derived from the yolk sac that are genetically distinct from bone marrow-derived macrophages (23, 36-39). Additionally, microglia are self-renewing suggesting that monocyte-derived macrophages do not contribute to the maintenance of the mature microglia pool (13, 40). Distinct developmental origin and renewal mechanisms may suggest that microglia possess discrete functions in pathological processes (35). Despite this, the cellular mechanisms by which microglia promote or attenuate the progression of injury are largely unknown (41). Our pilot experiments presented here are the first we know of to use unbiased transcriptional profiling of isolated populations of microglia to define the genes/pathways/signatures involved in the generation of TBI-associated microglia over the course of injury.
To adequately capture the heterogeneity and complexity of microglia at different stages of injury, a comprehensive, genome-wide sampling of individual cell types is required (23, 42). A cell-specific delineation of innate immune function based on transcriptional profiling in TBI has yet to be undertaken. Therefore, we combined our ability to discriminate and sort microglia from infiltrating monocytes and monocyte-derived macrophages with unbiased transcriptional profiling (RNA-seq) on FACSorted microglia.
Our analysis identified 4 sequentially upregulated or downregulated gene clusters involved in various biological processes such as the host defense response, synaptic potentiation, lipid remodeling, and membrane polarization each providing new insights into the biology of microglial activation over the course of TBI (Fig. 2). While there has recently been considerable recent interest in the Trem2-APOE pathway in the generation of a neurodegenerative microglial phenotype in Alzheimer’s Disease and multiple sclerosis, our data demonstrates no significant change in Trem2 expression as well as a progressive decrease in APOE expression over the course of TBI (Fig. 5B) (32). However, an examination of cluster II revealed a number of genes associated with long-term synaptic potentiation including Ptpn5 (STEP), Shank3, and Sqstm1. STEP is a brain-specific phosphatase that is highly expressed within the striatum, cortex, hippocampus, and amygdala (43, 44). STEP is critical in the long-term depression, or weakening, of synaptic efficacy between neurons—a process fundamental to learning, memory, and cognition (24, 25). Elevated STEP is associated with the pathophysiology of Alzheimer’s disease (AD), schizophrenia, and ischemic brain injury in both human cortex and mouse models. In fact, genetic knockout of STEP reverses many of the cognitive and behavioral deficits in AD models (45, 46). To the best of our knowledge, STEP expression has never been studied within the context of TBI, nor has STEP been identified as a key protein within microglia. Our RNA-seq analysis demonstrates a 13-fold increase in microglial expression of STEP over the course of injury (Fig. 5A). Previous studies have shown that STEP affects neuronal communication by opposing synaptic strengthening. High levels of STEP are believed to disrupt synaptic function and to contribute to learning deficits in neurodegenerative disease (47, 48). When STEP activity is elevated, several substrates are inactivated resulting in the internalization of NMDA glutamate receptors (49),(50). This disrupts synaptic function and contributes to cognitive deficits (25, 51). In other words, STEP activation modulates learning and memory by removing glutamate receptors from synaptic membranes. Additionally, we found that expression of Grin2a, which codes for the Nr2a subunit of the NMDA receptor, is significantly down-regulated by 90 days post-TBI (Fig. 6A). This further suggests a role for microglia in the disruption of synaptic function after TBI. This focused dataset is remarkable in demonstrating the power of longitudinal transcriptional profiling in identifying the genes/pathways/signatures involved in the generation of TBI-associated microglia as well as potential links with other neurogenerative diseases previously regarded as distinct.
Conclusions
Our data suggest that TBI-associated microglia adopt longitudinal transcriptional changes in the host defense response, synaptic potentiation, lipid remodeling, and membrane polarization. The contribution of altered microglial gene expression to the pathogenesis of TBI has not been previously investigated. These focused data suggest that TBI-associated microglia may play a previously unknown role in the weakening of synaptic efficacy between neurons after brain injury. As a result, learning, memory, and cognitive performance may all be affected leading to long-term neurocognitive impairments after TBI. Due to the preliminary nature and high cost of this study, the gene expression of age-matched sham and naive mice were not performed. This is a critical limitation of the current study precluding any assessment of causation. However, these early data are still valuable to research community at large as they provide strong rationale for larger, well controlled, studies of isolated populations of microglia after TBI. Moving forward, it will be essential for future experiments to include side-by-side comparisons with age-matched sham and naive control mice to account for baseline transcriptional changes as well as changes associated with aging. Additional time points will also be necessary to ensure that transcriptional changes are captured throughout the entire course of TBI—from acute injury process to chronic neurologic disease. Furthermore, it will be important to measure synaptic efficacy directly as well as study larger cohorts of brain-injured mice during both the acute and chronic phases of TBI. It has also been suggested that bulk analysis of microglia in certain disease processes may encompass a heterogeneous population of microglia with subpopulations demonstrating unique expression profiles (52). This may require a single-cell RNA-seq approach to account for inherent microglial heterogeneity at the site of injury. This approach could allow for the identification of novel microglial subpopulations within and surrounding the site of injury. Regardless of the techniques used, once the molecular mechanisms underlying the transcriptional changes in microglia after injury are further delineated, targeting the microglial response after TBI may soon represent a target for future therapeutic intervention.
Acknowledgments
Flow cytometry cell sorting was performed in the Northwestern University Lurie Cancer Center Flow Cytometry Core Facility. RNA sequencing was performed in the Northwestern University Division of Rheumatology and Pulmonary and Critical Care sequencing facility.
Funding Sources
This study was supported by NIH grant GM117341 and The American College of Surgeons C. James Carrico Research Fellowship to S.J.S .
Footnotes
Author Disclosures: The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Faul M Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002-2006. Atlanta (GA): Centers for Disease Control and Prevention, National Center for Injury Prevention and Control, 2010. [Google Scholar]
- 2.Roozenbeek B, Maas AI, Menon DK Changing patterns in the epidemiology of traumatic brain injury. Nat Rev Neurol 2013:9:231–236. [DOI] [PubMed] [Google Scholar]
- 3.Corso P, Finkelstein E, Miller T, Fiebelkorn I, Zaloshnja E Incidence and lifetime costs of injuries in the United States. Injury prevention : journal of the International Society for Child and Adolescent Injury Prevention 2006:12:212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pearson WS, Sugerman DE, McGuire LC, Coronado VG Emergency department visits for traumatic brain injury in older adults in the United States: 2006-08. The western journal of emergency medicine 2012:13:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whitlock JA Jr., Hamilton BB Functional outcome after rehabilitation for severe traumatic brain injury. Archives of physical medicine and rehabilitation 1995:76:1103–1112. [DOI] [PubMed] [Google Scholar]
- 6.Schwarzbold M, Diaz A, Martins ET, Rufino A, Amante LN, et al. Psychiatric disorders and traumatic brain injury. Neuropsychiatric disease and treatment 2008:4:797–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whelan-Goodinson R, Ponsford J, Johnston L, Grant F Psychiatric disorders following traumatic brain injury: their nature and frequency. The Journal of head trauma rehabilitation 2009:24:324–332. [DOI] [PubMed] [Google Scholar]
- 8.Peskind ER, Brody D, Cernak I, McKee A, Ruff RL Military- and sports-related mild traumatic brain injury: clinical presentation, management, and long-term consequences. The Journal of clinical psychiatry 2013:74:180–188; quiz 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin LA, Neighbors HW, Griffith DM The experience of symptoms of depression in men vs women: analysis of the National Comorbidity Survey Replication. JAMA psychiatry 2013:70:1100–1106. [DOI] [PubMed] [Google Scholar]
- 10.Makinde HM, Just TB, Cuda CM, Perlman H, Schwulst SJ The Role of Microglia in the Etiology and Evolution of Chronic Traumatic Encephalopathy. Shock 2017:48:276–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Makinde HM, Cuda CM, Just TB, Perlman HR, Schwulst SJ Nonclassical Monocytes Mediate Secondary Injury, Neurocognitive Outcome, and Neutrophil Infiltration after Traumatic Brain Injury. J Immunol 2017:199:3583–3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hernandez-Ontiveros DG, Tajiri N, Acosta S, Giunta B, Tan J, et al. Microglia activation as a biomarker for traumatic brain injury. Front Neurol 2013:4:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aguzzi A, Barres BA, Bennett ML Microglia: scapegoat, saboteur, or something else? Science 2013:339:156–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perry VH, Holmes C Microglial priming in neurodegenerative disease. Nat Rev Neurol 2014:10:217–224. [DOI] [PubMed] [Google Scholar]
- 15.Lozano D, Gonzales-Portillo GS, Acosta S, de la Pena I, Tajiri N, et al. Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr Dis Treat 2015:11:97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kokaia Z, Martino G, Schwartz M, Lindvall O Cross-talk between neural stem cells and immune cells: the key to better brain repair? Nat Neurosci 2012:15:1078–1087. [DOI] [PubMed] [Google Scholar]
- 17.Nikodemova M, Watters JJ Efficient isolation of live microglia with preserved phenotypes from adult mouse brain. Journal of neuroinflammation 2012:9:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bedi SS, Smith P, Hetz RA, Xue H, Cox CS Immunomagnetic enrichment and flow cytometric characterization of mouse microglia. Journal of neuroscience methods 2013:219:176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gordon R, Hogan CE, Neal ML, Anantharam V, Kanthasamy AG, et al. A simple magnetic separation method for high-yield isolation of pure primary microglia. Journal of neuroscience methods 2011:194:287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turtzo LC, Lescher J, Janes L, Dean DD, Budde MD, et al. Macrophagic and microglial responses after focal traumatic brain injury in the female rat. Journal of neuroinflammation 2014:11:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trahanas DM, Cuda CM, Perlman H, Schwulst SJ Differential Activation of Infiltrating Monocyte-Derived Cells After Mild and Severe Traumatic Brain Injury. Shock 2015:43:255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noristani HN, Gerber YN, Sabourin JC, Le Corre M, Lonjon N, et al. RNA-Seq Analysis of Microglia Reveals Time-Dependent Activation of Specific Genetic Programs following Spinal Cord Injury. Front Mol Neurosci 2017:10:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016:353:aad8670. [DOI] [PubMed] [Google Scholar]
- 24.Bliss TV, Collingridge GL A synaptic model of memory: long-term potentiation in the hippocampus. Nature 1993:361:31–39. [DOI] [PubMed] [Google Scholar]
- 25.Silva AJ Molecular and cellular cognitive studies of the role of synaptic plasticity in memory. J Neurobiol 2003:54:224–237. [DOI] [PubMed] [Google Scholar]
- 26.Makinde HM, Just TB, Cuda CM, Bertolino N, Procissi D, et al. Monocyte depletion attenuates the development of posttraumatic hydrocephalus and preserves white matter integrity after traumatic brain injury. PLoS One 2018:13:e0202722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mike EV, Makinde HM, Gulinello M, Vanarsa K, Herlitz L, et al. Lipocalin-2 is a pathogenic determinant and biomarker of neuropsychiatric lupus. J Autoimmun 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anders S, Pyl PT, Huber W HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015:31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013:29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 2009:10:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anders S, Huber W Differential expression analysis for sequence count data. Genome Biol 2010:11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017:47:566–581 e569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castranio EL, Mounier A, Wolfe CM, Nam KN, Fitz NF, et al. Gene co-expression networks identify Trem2 and Tyrobp as major hubs in human APOE expressing mice following traumatic brain injury. Neurobiol Dis 2017:105:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saber M, Kokiko-Cochran O, Puntambekar SS, Lathia JD, Lamb BT Triggering Receptor Expressed on Myeloid Cells 2 Deficiency Alters Acute Macrophage Distribution and Improves Recovery after Traumatic Brain Injury. J Neurotrauma 2017:34:423–435. [DOI] [PubMed] [Google Scholar]
- 35.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010:330:841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012:336:86–90. [DOI] [PubMed] [Google Scholar]
- 37.Gomez Perdiguero E, Schulz C, Geissmann F Development and homeostasis of "resident" myeloid cells: the case of the microglia. Glia 2013:61:112–120. [DOI] [PubMed] [Google Scholar]
- 38.Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nature neuroscience 2014:17:131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014:159:1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nature neuroscience 2007:10:1538–1543. [DOI] [PubMed] [Google Scholar]
- 41.Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nature neuroscience 2011:14:1142–1149. [DOI] [PubMed] [Google Scholar]
- 42.Zeisel A, Munoz-Manchado AB, Codeluppi S, Lonnerberg P, La Manno G, et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015:347:1138–1142. [DOI] [PubMed] [Google Scholar]
- 43.Boulanger LM, Lombroso PJ, Raghunathan A, During MJ, Wahle P, et al. Cellular and molecular characterization of a brain-enriched protein tyrosine phosphatase. J Neurosci 1995:15:1532–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lombroso PJ, Naegele JR, Sharma E, Lerner M A protein tyrosine phosphatase expressed within dopaminoceptive neurons of the basal ganglia and related structures. The Journal of neuroscience : the official journal of the Society for Neuroscience 1993:13:3064–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goebel-Goody SM, Wilson-Wallis ED, Royston S, Tagliatela SM, Naegele JR, et al. Genetic manipulation of STEP reverses behavioral abnormalities in a fragile X syndrome mouse model. Genes Brain Behav 2012:11:586–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Kurup P, Xu J, Carty N, Fernandez SM, et al. Genetic reduction of striatal-enriched tyrosine phosphatase (STEP) reverses cognitive and cellular deficits in an Alzheimer's disease mouse model. Proc Natl Acad Sci U S A 2010:107:19014–19019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurup P, Zhang Y, Xu J, Venkitaramani DV, Haroutunian V, et al. Abeta-mediated NMDA receptor endocytosis in Alzheimer's disease involves ubiquitination of the tyrosine phosphatase STEP61. J Neurosci 2010:30:5948–5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poddar R, Deb I, Mukherjee S, Paul S NR2B-NMDA receptor mediated modulation of the tyrosine phosphatase STEP regulates glutamate induced neuronal cell death. Journal of neurochemistry 2010:115:1350–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tian M, Xu J, Lei G, Lombroso PJ, Jackson MF, et al. STEP activation by Galphaq coupled GPCRs opposes Src regulation of NMDA receptors containing the GluN2A subunit. Sci Rep 2016:6:36684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Braithwaite SP, Adkisson M, Leung J, Nava A, Masterson B, et al. Regulation of NMDA receptor trafficking and function by striatal-enriched tyrosine phosphatase (STEP). The European journal of neuroscience 2006:23:2847–2856. [DOI] [PubMed] [Google Scholar]
- 51.Chin J, Palop JJ, Puolivali J, Massaro C, Bien-Ly N, et al. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer's disease. The Journal of neuroscience : the official journal of the Society for Neuroscience 2005:25:9694–9703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer's Disease. Cell 2017:169:1276–1290 e1217. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
RNA sequencing data is available through the NCBI Sequence Read Archive (SRA accession number: SRP160379). The remaining data that support the findings in this manuscript are available from the corresponding author upon request.