

Abstract
Simultaneous enrichment of glyco- and phospho- peptides will benefit the studies of biological processes regulated by these post-translational modifications (PTMs). It will also reveal potential crosstalk between these two ubiquitous PTMs. Unlike custom designed multifunctional solid phase extraction (SPE) materials, operating strong-anion exchange (SAX) resin in electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) mode provides a readily available strategy to analytical labs for enrichment of these PTMs for subsequent mass spectrometry (MS)-based characterization. However, the choice of mobile phase has largely relied on empirical rules from hydrophilic interaction chromatography (HILIC) or ion-exchange chromatography (IEX) without further optimization and adjustments. In this study, ten mobile phase compositions of ERLIC were systematically compared; the impact of multiple factors including organic phase proportion, ion pairing reagent, pH and salt on the retention of glycosylated and phosphorylated peptides were evaluated. This study demonstrated good enrichment of glyco- and phospho- peptides from the nonmodified peptides in a complex tryptic digest. Moreover, the enriched glyco- and phospho- peptides elute in different fractions by orthogonal retention mechanisms of hydrophilic interaction and electrostatic interaction in ERLIC, maximizing the LC-MS identification of each PTM. The optimized mobile phase can be adapted to the ERLIC HPLC system, where the high resolution in separating multiple PTMs will benefit large-scale MS-based PTM profiling and in-depth characterization.
Graphical Abstract
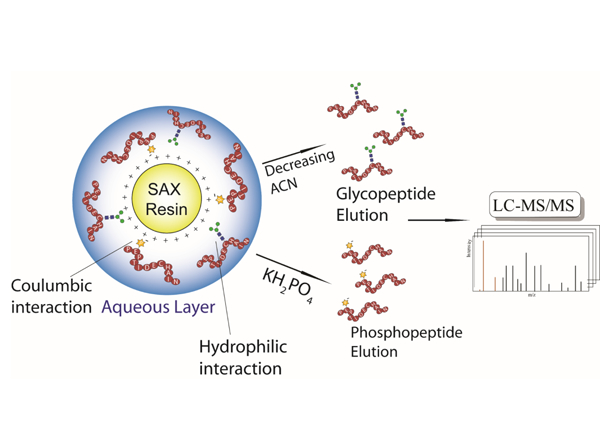
Introduction
Phosphorylation and glycosylation are two of the most ubiquitous and important protein post-translational modifications (PTMs) that can significantly alter protein tertiary structure, stability, turnover and protein-protein interactions [1]. It has been estimated that nearly one-third of human proteins are phosphorylated and more than one-half of mammalian proteins are glycosylated [2, 3]. They are highly associated with many biological processes, including signal transduction, enzyme activity regulation, protein folding and cell adhesion [4, 5], and related to various human diseases, including diabetes, Alzheimer’s disease, autoimmunity, congenital disorders of glycosylation, and cancer [6–14]. It also has been demonstrated that there is complex crosstalk between the two PTMs in biological regulation. [15][16]. In order to study the PTM related biological processes and further dissect the molecular mechanisms underlying the diseases involving these two modifications, the ability to simultaneously analyze both phosphorylation and glycosylation at the proteome level is essential.
Bottom-up proteomics (i. e., shotgun proteomics), which couples liquid chromatography (LC) and tandem mass spectrometry to analyze proteolytic peptides, has become a main-stream technology that enables rapid profiling of proteins in complex samples [17–19]. The technology has undergone rapid development in the past few decades, and its high-throughput allows the whole proteome from eukaryotic cells to be analyzed within just a few hours [20–22]. To better adjust the method for analyzing phosphate or glycan modified peptides, more sophisticated LC-MS experiments have been designed such as the ‘multistage activation (MSA)’ method [23], the electron-driven dissociation [24, 25], and the hybrid fragmentation strategies such as electron-transfer/higher-energy collision dissociation (EThcD) and activated ion electron-transfer dissociation (AI-ETD) that combine orthogonal types of activation on the modified precursor ions [26–33]. However, neither of the above strategies can avoid the increased duty cycle of MS/MS analysis, which will compromise the number of MS/MS events and reduce the large-scale profiling efficiency and overall coverage of modified peptides. Furthermore, MSn and electron-driven fragmentation capabilities are currently only available on a limited number of instruments. On the other hand, stepped-energy of higher-energy collision dissociation (stepped HCD or stepped NCE) approach, which combines the fragment ions from three different collision energies and records these fragments in a single MS/MS spectrum, was found to increase the diversity of the fragment ions without significantly increasing the MS analysis duty cycle or sacrificing the throughput, which benefits large scale PTM analysis. For instance, improved phospho site localization through increased sequence coverage on HEK 293T cell lysate was reported by applying a stepped HCD approach [34]. A recent application of stepped NCE to the human serum proteome also proved its capability to fully characterize intact glycopeptides on a large scale, while simultaneously yielding structural information of the peptides and the covalently linked glycans via high and low collision energies [35].
Despite the technical improvements to MS instruments, it is still challenging to analyze phosphorylated or glycosylated peptides by direct LC-MS without reducing sample complexity, due to the sub-stoichiometric level abundance of the two PTMs and poor ionization efficiency in positive ion mode MS derived from additional negative charges of these PTMs. Therefore, numerous enrichment techniques have been developed targeting phosphorylated or glycosylated peptides, including metal oxide affinity chromatography (MOAC) such as TiO2 and ZrO2 enrichment [36, 37], immobilized metal affinity chromatography (IMAC) [38–42], lectin affinity chromatography-based enrichment [43, 44], hydrazide chemistry based SPE method [45–48], boronic acid enrichment method [49–51] and hydrophilic interaction chromatography (HILIC) [52–55]. Most of the methods listed above were designed to target phosphorylation or glycosylation alone and each has its own limitations. MOAC and IMAC suffer from non-specific binding of aspartic acid and glutamic acid rich peptides. Lectin affinity chromatography exhibits biases towards specific glycan structures. Hydrazide chemistry-based approaches can enrich N-glycopeptides with high specificity and efficiency, but the release of enriched analytes relies on enzymatic digestion that makes intact glycopeptide analysis impossible. In recent years, novel MOAC and IMAC materials were synthesized with optimized morphology, ligand selection and density of functional groups to increase the selectivity and enrichment efficiency of both phosphorylated and glycosylated peptides [56][57] [58]. However, most of the applications of those novel enrichment materials were limited to MALDI-MS validation on mixtures of several phosphorylated and glycosylated peptide standards, while the in-depth analysis of the modified proteome was limited by the availability of the custom synthesized material to biological labs.
Ion exchange chromatography (IEX) such as strong cation exchange (SCX) or strong anion exchange (SAX) has been another widely used peptide and protein separation method in biological and analytical labs [59]. MS coupled with IEX separation has been successfully implemented in phosphorylated peptide analysis, due to the charge difference introduced by the phosphate group [60–62]. Electrostatic repulsion hydrophilic interaction chromatography (ERLIC) is a novel mode of separation utilizing IEX stationary phase with HILIC mobile phases (i.e. analytes are eluted by decreasing organic solvent gradient), which was first introduced by Alpert [63]. In ERLIC, hydrophilic interaction and electrostatic interaction between analytes and stationary phase are superimposed; and one can manipulate the charge state of analytes by operating at optimal pH to maximize the electrostatic attraction between analytes of interest and charged stationary phase, while the electrostatic repulsion will antagonize the retention of interfering molecules that cannot be separated by conventional HILIC with neutral stationary phase [63]. Anion exchange-ERLIC can selectively separate phosphorylated peptides from non-modified counterparts because of the increased hydrophilicity and additional negative charge introduced by a phosphate group at the operating pH [64]. Glycosylated peptide enrichment by its hydrophilicity is also compatible with ERLIC, and it has been reported that ERLIC outperforms HILIC by neutral stationary phases on isobaric labeled glycopeptide enrichment, which facilitated further PTM relative quantification [65, 66]. Furthermore, ERLIC by both SCX and WAX has been investigated for the simultaneous, selective isolation of both phosphopeptides and glycopeptides from tryptic digests [67–69]. Despite the plentiful application of ERLIC, the mobile phase constitution for simultaneous enrichment of multiple PTMs has not been thoroughly explored.
In this work, we examined the potential of coupling ERLIC SPE with RPLC-MS to achieve large-scale phosphorylation and N-glycosylation analysis. Different mobile phase compositions, including organic phase proportion, ion pairing reagent, salt and pH, were tested and evaluated to maximize the enrichment performance of both N-glycosylated and phosphorylated tryptic peptides from MM.1S cell lysate (a model cell line for multiple myeloma) [70].
Experimental Section
Materials.
Poly SAX LP™ bulk material (12μm, pore size 300Å) was obtained from PolyLC (Columbia, MD). 0.22 μm Millex-GV Hydrophilic Durapore (PVDF) Membrane filter units were purchased from Merck Millipore (Tullagreen, Ireland). Iodoacetamide (IAA), Roche protease inhibitor tablets and Roche PhosSTOP phosphatase inhibitor tablet were purchased from Sigma-Aldrich (St. Louis, MO). Phosphoric acid, ammonium acetate, tris base, urea, potassium phosphate monobasic, sodium chloride and calcium chloride were purchased from Fisher Scientific (Pittsburgh, PA). C18 SepPak cartridges were purchased from Waters (Milford, MA). C18 OMIX tips were purchased from Agilent (Santa Clara, CA). Dithiothreitol (DTT) and sequencing grade trypsin were purchased from Promega (Madison, WI). Protein and peptide BCA assays were purchased from Pierce (Rockford, IL).
Cell Culture.
MM.1S cells were cultured in RPMI-1640 medium (Corning) supplemented with 10% FBS, 1% sodium pyruvate, 1% penicillin/streptomycin and 10mM HEPES at 37 °C in humidified atmosphere with 5% CO2. When the MM.1S cells were confluent, they were harvested and then washed once with PBS to remove remaining media content. Resulting cell pellet(s) were stored under −80 °C until use.
Cell Lysis.
MM.1S cell pellets were homogenized in lysis buffer (8M urea, 50 mM Tris, pH=8, 5 mM CaCl2, 20 mM NaCl, 1 EDTA-free Roche protease inhibitor tablet and 1 Roche PhosSTOP phosphatase inhibitor tablet) with a probe sonicator for 3 pulses at 60W, 20 kHz for 15 s, each followed by a 30 s pause period for cooling at 4ºC. Crude lysates were then centrifuged at 14000 xg for 5 min, after which the supernatant was collected and protein concentrations were measured by Pierce BCA Protein Assay according to the manufacturer’s protocol.
Trypsin Digestion.
Lysate containing 2 mg protein was reduced in 5 mM DTT at room temperature for 1h, followed by alkylation in 15 mM IAA for 30 min in the dark. Alkylation was quenched by adding DTT to 5 mM. The resulting solution was then diluted with Tris buffer (pH=8) to 0.9 M urea and proteins were digested with trypsin at 1:50 enzyme to protein ratio at 37 ºC for 18 hours. Digestion was quenched by adding trifluoroacetic acid (TFA) to a final concentration of 0.3% and desalted with C18 SepPak cartridges. Digested peptide concentration was measured by Pierce Peptide BCA assay, peptides were aliquoted to 100 μg/tube and dried under vacuum before enrichment.
ERLIC Enrichment.
ERLIC enrichment of phospho- and N-glyco- peptides from MM.1S cell digest was performed in the custom packed spin-tips [71, 72]. As Figure 1B illustrates, 3mg of cotton wool was inserted into a 200 μL pipette tip. SAX LP bulk material was dispersed in 0.1% TFA as a 10 mg/ 200 μL slurry and activated for 15 min under vigorous vortexing. After activation, 60 μL slurry was added to the spin-tip. Solvent was removed by centrifugation at 1200 rpm for 2 min, leaving the SAX material packed above the cotton wool. The stationary phase was then conditioned by 150 μL ACN, 100 mM ammonium acetate, 1% TFA and the loading buffers, each repeated 3 times. 100 μg MM.1S cell digest was dissolved in 150 μL of the corresponding loading buffer and loaded onto the tips by centrifugation at 1200 rpm for 2 min, the flow-through was collected and loaded again to ensure complete retention. The tips were then washed with 150 μL loading buffers 6 times, after which four fractions were eluted with 300 μL 50/50 ACN/Water with 0.1% formic acid (FA), 0.1% FA in water, 0.1% TFA in water and 300 mM KH2PO4 (pH = 2) and were collected in four separate centrifuge tubes. Each fraction was filtered by a 0.22 μm Millex-GV Hydrophilic Durapore (PVDF) Membrane filter unit, the KH2PO4 fractions were desalted by C18 OMIX tips according to manufacturer’s protocol, after which all fractions were dried under vacuum before MS analysis.
Figure 1.

(A) Illustration of ERLIC enrichment and separation mechanism of phosphopeptides and glycopeptides; (B) Custom-made spin-tip composition; (C) Schematic workflow of spin-tip-based ERLIC SPE coupled with LC-MS/MS analysis on MM.1S cell lysate digest.
NanoLC-MS/MS Analysis.
Peptides in each fraction were reconstituted in 0.1% FA, 3% ACN and subjected to reversed phase LC-MS/MS analysis with a Q-Exactive HF orbitrap mass spectrometer (Thermo Fisher Scientific, San Jose, CA) interfaced with a Dionex Ultimate 3000 UPLC system (Thermo Fisher Scientific, San Jose, CA). Peptides were loaded onto a 75 μm i.d. microcapillary column custom-packed with 15 cm of BEH C18 particles (1.7 μm, 130 Å, Waters). Peptides were separated with a 90 min gradient from 3% to 30% ACN with 0.1% FA, followed by 10 min to 75% ACN and then 10 min to 95% ACN. After that, the column was re-equilibrated with 3% ACN for 15 min to prepare for the next injection.
The mass spectrometer was operated in a top 15 data-dependent acquisition mode. Survey scans of peptide precursors from m/z 300 to 2000 were performed at a resolving power of 60K and an AGC target of 2×105 with a maximum injection time of 150 ms. The top 15 intense precursor ions were selected and subjected to the stepped HCD fragmentation at normalized collision energy of 22, 30, and 38% followed by tandem MS acquisition at a resolving power of 30K and an AGC target of 5×104, with a maximum injection time of 250 ms. Precursors were subjected to a dynamic exclusion of 15s with a 10 ppm mass tolerance.
Data Analysis
Raw files were processed with the Byonic search engine (Protein Metrics Inc, San Carlos, CA) embedded within Proteome Discoverer 2.1 (Thermo Fisher Scientific, San Jose, CA). Spectra were searched against the UniProt Homo sapiens proteome database (April 12, 2016; 16764 entries) with trypsin as the specific digestion enzyme and maximum two missed cleavages. The parent mass error tolerance was set to be 50 ppm and fragment mass tolerance was 0.02 Da. Fixed modifications are specified as carbamidomethylation on cysteine residues (+57.02146 Da). Dynamic modifications included oxidation of methionine residues (+15.99492 Da), phosphorylation on S, Y, and T, and N-glycosylation. It is reported that ERLIC can enrich for O- glycopeptides [73], but in this work, we only analyzed N-glycopeptides from each enrichment. Glycan modifications were specified as the common mammalian N-glycome (default N-glycome database in Byonic with removal of sodium mass, which contains overall 309 N-glycans), expanded with typical mannose-6-phosphate glycans including HexNAc (2)-Hex (4–9)-Phospho (1–2), HexNAc (3–4)-Hex (4–9)-Phospho (1–2), HexNAc (2) Hex (3–4) Phospho (1) and HexNAc (3) Hex (3–4) Phospho (1). Identifications were filtered at 1% false discovery rate (FDR).
Results and Discussion
Because multiple interactions such as hydrophilic interaction and electrostatic interaction are superimposed while operating ERLIC, selecting the best mobile phase is not straightforward. Here, we aim to examine the impact of organic phase proportion, ion pairing reagent, pH and salt content on the retention and elution of phospho- and N-glyco- peptides in ERLIC-RP-MS utilizing the new PolySAX LP material [64]. This work will benefit studies of the mechanism of ERLIC and broaden its application to simultaneous phosphorylation and N-glycosylation analysis on a large scale.
Loading buffer affects enrichment efficiency.
To evaluate the impact of mobile phase composition on the retention of modified peptides, 10 different loading buffers were compared according to Table 1. Since HILIC was named and defined in 1990 [74], its mechanism of retention has aroused the interest of many scientists, among whom the partitioning theory is the most supported. In this theory, a water-rich layer is immobilized on the stationary phase due to the high density of hydrophilic functional groups, then the retention of solutes was equilibrated by the partition between the bulk eluent and the water-rich layer, which is different from conventional normal phase chromatography where the retention is predominantly governed by surface adsorption [75]. According to the empirical observations, mobile phase with a higher content of aqueous phase serves as a stronger eluent. Most of the pioneering studies utilizing ERLIC SPE for N-glycopeptide enrichment used 95% ACN for loading and washing without thorough justification [65, 66]. However, we found this condition to be too retentive in that many non-modified peptides with minimal hydrophilicity were co-retained with the modified peptides of interest. In Figure 2, the loading buffers with high (95%) ACN (group c, d and e) generally retain more total peptides than the low (80%) ACN buffer conditions, where the difference in peptide retention is as high as 10 times greater with the 95% ACN combinations (except for combination f, with 1% FA added), but the LC-MS identified significantly fewer modified peptides in high organic phase loading conditions. The identification of phosphorylated or N-glycosylated peptides was largely suppressed by the numerous co-eluting and co-fragmenting non-modified tryptic peptides that ionized better or were more abundant. On the other hand, mobile phase with 80% ACN was adequate to retain the majority of the modified peptides while the non-modified peptides were removed during the washing step. By modulating the organic phase proportion in the mobile phase, the ERLIC enrichment specificity was greatly increased, benefiting the MS identification of peptides with phosphorylation and N-glycosylation.
Table 1.
ERLIC loading buffer composition
| Condition | Organic phase | Ion pairing regent | pH | Notes |
|---|---|---|---|---|
| a | 80% ACN | 1% TFA | 1 | |
| b | 80% ACN | 0.1% TFA | 2 | |
| c | 95% ACN | 1% TFA | 1 | |
| d | 95% ACN | 0.1% TFA | 2 | |
| e | 95% ACN | 0.1% FA | 2.7 | |
| f | 95% ACN | 1% FA | 2 | |
| g | 80% ACN | 1% FA | 2 | |
| h | 80% ACN | 0.1% FA | 2.7 | |
| i | 80% ACN | 1% TFA | 1 | Without ammonium acetate conditioning |
| j | 80% ACN | 1% TFA | 1 | Activated in 80% ACN 1% TFA |
Figure 2.

Identification results of peptide modifications in 100 μg of MM.1S cell lysate by ERLIC fractionation. ERLIC loading buffer conditions are provided in Table 1. (A) Total peptide identification; (B) Glycopeptide and phosphopeptide identification; (C) % specificity of ERLIC enrichment for each condition, calculated by dividing the number of modified peptides identified divided by the total number of peptides.
Ion pairing has been reported as an effective strategy to increase the specificity in HILIC based enrichment of glycosylated peptides, and ion pairing HILIC has been successfully applied to mammalian and plant glycoproteomics in various forms including SPE and HPLC [52, 76–78]. Wimley-White water/octanol free energy scale of amino acids has shown theoretical evidence that peptides with charged moieties have lower hydrophobicity than their uncharged forms [77, 79], which is consistent with the experimental results in ion-pairing normal phase LC that the hydrophobicity of charged non-glycopeptides increase and the separation between glycopeptides and non-glycopeptides were achieved when efficient ion pairing reagents were added [77]. Ion pairing reagents such as monovalent salt ions including NaCl, LiCl and KCl or acids including acetic acid, formic acid and TFA will “neutralize” the charges of peptides by forming ion-pairs with the positively charged groups such as the N-termini, lysine and arginine residues, and the negatively charged groups including C-termini, aspartate and glutamate residues. With the non-modified peptides’ charges neutralized, their hydrophilicity is largely reduced, and the glycopeptide enrichment specificity is improved because the hydrophilic retention of analytes will predominantly depend on the existing glycans whose hydrophilicity is moderately affected by ion pairing reagents [52, 77, 78]. Moreover, a recent study systematically evaluated the impact of different salts on the retention of HILIC and ERLIC, where they found well-hydrated counterions of charged analytes usually increase the hydrophilic retention while poorly-hydrated counterions decrease it [80]. The similar concept of counterion pairing was adapted to ERLIC here, with the choice of the poorly-hydrated TFA and weakly-hydrated FA as the ion pairing reagents being compared. On the other hand, the two acid modifiers resulted in different pH of the mobile phase and shifted the protonation-dissociation equilibrium of phosphate and other functional groups with acidic protons in the analytes, which would potentially influence the electrostatic interaction-based retention. As the most hydrophilic group in peptides except for basic residues, charged phosphate groups can increase the hydrophilicity of tryptic peptides [74], However, the phosphate group alone does not suffice to permit the separation of phosphopeptides from nonphosphopeptides as a set in HILIC. In ERLIC, the concurrent electrostatic interaction plays a significant role in phosphopeptide retention. Phosphate has a pKa1 of 2.15, which is lower than aspartic and glutamic residues and the C-terminus. With an appropriate pH in between them, the majority of peptides with acidic residues can be uncharged while the charged phosphopeptides can be strongly attracted by the positively charged SAX stationary phase. Thus the enrichment selectivity of phosphopeptides and highly hydrophilic glycopeptides over non-modified peptides can be enhanced with the selection of pH at 2–3. As Figure 2B indicates, TFA serves as the best ion pairing reagent for selective N-glycosylated peptide retention, where use of loading buffer combinations of a, b, i and j, containing either 1% or 0.1% TFA resulted in more than 300 N-glycopeptide identifications. For phosphorylated peptides, the retention depends on hydrophilic interaction and electrostatic interaction synergistically. At high organic phase loading conditions, most of the phosphorylated peptides can be retained and identified regardless of the TFA content, because the strong hydrophilic interaction plays a predominate role. At low organic phase and stronger hydrophilic eluent loading conditions as in groups a, b and g-j, the electrostatic interaction became a more significant component of the mechanism of phosphopeptide retention. At lower TFA or FA concentration, pH was higher and phosphate groups were more highly deprotonated and better attracted by the SAX resin. For example, in group b, 165 phosphopeptides were identified with high confidence whereas there were only 54 phospphopeptides identified in group a. Group h with even higher pH of 3 of 0.1% FA also showed great phosphopeptide retention where 171 phosphopeptides were identified in the total four fractions.
Conditioning is an essential preparation step before operating SAX resins. A highly concentrated salt buffer such as triethylammonium acetate is usually used to flush SAX resin for various reasons [65, 66]. Some of the main reasons include: (1) It can wash off the byproducts that came from SAX resin synthesis before the first time usage; (2) Well-hydrated counter ions can partition into the immobilized aqueous layer on the stationary phase and pulls the charged solutes into the aqueous layer thus increase the HILIC retention, while poorly hydrated counter ions have the opposite effect [80]. To prove the concept, we incorporated a control group i without flushing with salt in the experimental procedure before sample loading. Surprisingly, we found both N-glycosylated and phosphorylated peptide identifications from group i actually had a moderate increase over group a, which could arise from the more well-hydrated counterions that PolySAX LP has from manufacturer actually outperform acetate. It is worth mentioning that a large amount of 1% FA as the mobile phase modifier in groups f and g resulted in largely reduced retention of both modified and non-modified peptides comparing to groups e and h. We hypothesize that high concentration of FA partitioned in the immobilized aqueous layer can compete the electrostatic interaction between charged analytes and stationary phase to a large extent, while the retention of hydrophilic peptides is reduced by “salt out” effect [80]. In contrast, these effects are not observed with 1% TFA groups a, c and j, which is because TFA tends to partition mostly into the organic mobile phase of ERLIC. Figure 2C shows the proportion of identified N-glycosylated or phosphorylated peptides within the total identified peptides for each combination. Group b is the optimal combination among the ten tested, with enrichment specificity of 24.8 % and 11.0 % for N-glycosylated and phosphorylated peptides, respectively.
Eluting profile reveals the key retention mechanism.
To further separate the modified peptides and simplify the MS based analysis, four crude elution fractions were collected with mobile phase conditions as indicated in Table 2. The peptide spectral matches (PSMs) of each modification-containing peptide are counted and the distributions of the PSMs among the eluting fractions are shown in Figure 3. Figure 3A summarizes total PSM distribution in four fractions. Figures 3B shows that the majority of N-glycosylated peptides elute in the first two fractions with the decreasing gradient of organic phase, indicating the retention mechanism for N-glycopeptides is predominately by hydrophilic interaction. On the other hand, in Figure 3C, phosphopeptides aggregate in the fractions 3 and 4, where the pH is lowered and the competitive salt is added to reduce the column interaction with the SAX resin. The results show that ERLIC can separate peptides with the two types of PTMs, which improves MS analysis and minimizes interference between peptides with the two modifications. Sialic acid containing N-glycopeptides have additional acidic groups of which the pKa is 2.6. According to Figure 3D, the elution profiles of sialylated N-glycopeptides are consistent with those of the bulk N-glycopeptides from each condition in Figure 3B. The results indicate the predominant mechanism for ERLIC to retain sialic acid containing N-glycopeptides at pH 1~3 is through hydrophilic interaction. We hypothesize that if pH were further increased to >3, the electrostatic interaction would have more contribution.
Table 2.
ERLIC eluting buffer composition
| Fraction | Organic phase | Ion pairing regent | pH | Salt |
|---|---|---|---|---|
| 1 | 50% ACN | 0.1% FA | 3 | |
| 2 | - | 0.1% FA | 3 | |
| 3 | - | 0.1% TFA | 2 | |
| 4 | - | - | 2 | 300mM KH2PO4 |
Figure 3.

PSM distribution of modifications among fractions. ERLIC loading buffer conditions are given in Table 1. Eluting buffer conditions are given in Table 2. (A) total PSMs; (B) glycopeptide PSMs; (C) phosphopeptide PSMs; (D) Sialic acid containing glycopeptide PSMs.
Furthermore, we manually checked some PSMs of N-glyco-, phospho-, and sialylated N-glycopeptides from the Byonic search engine output. Figure 4 shows three representative spectra, where the HCD with stepped collisional energy of 22%, 30% and 38% resulted in sufficient peptide backbone fragments with clear modification site information and glycan fragments cleaved at the glycosidic linkage. The results support stepped HCD as being a highly efficient and simple MS fragmentation method for simultaneous profiling of multiple PTMs without tedious optimization when coupled with ERLIC fractionation and separation.
Figure 4.

Representative stepped HCD MS/MS spectra with site-specific identifications of glycopeptides, phosphopeptides and those with sialic acid containing glycopeptides. (A) MS/MS of N-glycopeptide TN[+876.32]STFVQALVEHVKEECDR (AA 214–232) from human prosaposin, spectra of +4 charged precursor at m/z 785.36; (B) MS/MS of phosphopeptide LQQGAGLESPQGQPEPGAAS[+79.97]PQR (AA 72–94) from human coiled-coil domain containing protein 86, spectra of +3 charged precursor at m/z 795.04; (C) MS/MS of sialylated glycopeptide AN[+2204.77]C[+57.02]SVYESC[+57.02]VDC[+57.02]VLAR (AA 495–510) from human semaphorin-4A, spectra of +4 charged precursor at m/z 1027.65; (I) m/z 150 to 410 of (C) with oxonium ions and NeuAc reporter ions resolved; (II) 7-fold zoom-in of (C) at m/z 410 to 2000 with most of the peptide backbone fragment ions being detected and resolved.
Conclusions
In this study, we demonstrated the great potential of ERLIC for simultaneous enrichment of phosphorylated and N-glycosylated peptides. Ten different loading buffer conditions were examined, thoroughly considering the factors of organic phase proportion, ion pairing reagents, pH and salt. While a high concentration of organic phase (95% ACN) has commonly been used in previous studies, it is outperformed here by 80% ACN in terms of hydrophilic interaction-based enrichment’s selectivity and specificity for modified versus non-modified peptides. The use of a less well-hydrated ion pairing reagent benefits N-glycosylation enrichment by reducing non-specific retention. With weak hydrophilic eluent, phosphopeptides’ retention is independent of ion-pairing reagents; with intermediate hydrophilic eluent, the pH of mobile phase has a stronger impact on electrostatic interaction-based retention of phosphorylated peptide, where pH between 2–3 ensures a decent extent of phosphate group dissociation and phosphopeptide retention, while the majority of acidic peptides remains to be uncharged, which enhances the enrichment selectivity of phosphopeptides by ERLIC. With an inappropriate selection of counterions before or during the enrichment, the retention of modified peptides of interest can be largely hampered by competitive binding to charged resins and “salting out” effect. In the future, it is worth exploring the types of salt modifier that benefit the selective retention of N-glyco- and phospho- peptides in ERLIC.
The PSMs in the elution profiles of phosphorylated and N-glycosylated peptides showed a decent level of separation, where the majority of N-glycopeptides were eluted by disrupting hydrophilic interaction with decreasing organic phase gradient and most phosphopeptides were eluted by disrupting coulombic interaction with increased protonation and competitive salt content. The elution by multiple steps in the ERLIC SPE should be applicable to an HPLC system. We anticipate our optimized ERLIC SPE method will produce great resolution when adapted to HPLC and enable simultaneous enrichment and separation of N-glycosylated and phosphorylated peptides in complex mixtures, with improved coverage.
Acknowledgments
We would like to acknowledge Dr. Andrew Alpert from PolyLC Inc. for helpful discussions. This research was supported in part by the National Institutes of Health grants U01CA231081, R01 DK071801, R21 AG055377, and RF1 AG052324 (to LL). The Orbitrap instruments were purchased through the support of an NIH shared instrument grant (NIH-NCRR S10RR029531 to LL) and Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin-Madison. LL acknowledges a Vilas Distinguished Achievement Professorship and a Charles Melbourne Johnson Distinguished Chair Professorship with funding provided by the Wisconsin Alumni Research Foundation and University of Wisconsin-Madison School of Pharmacy.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References
- 1.Ke M, Shen H, Wang L, Luo S, Lin L, Yang J, Tian R: Identification, Quantification, and Site Localization of Protein Posttranslational Modifications via Mass Spectrometry-Based Proteomics. In: Mirzaei H and Carrasco M (eds.) Modern Proteomics -- Sample Preparation, Analysis and Practical Applications pp. 345–382. Springer International Publishing, Cham; (2016) [DOI] [PubMed] [Google Scholar]
- 2.Cohen P: The origins of protein phosphorylation. Nat. Cell Biol 4, E127–E130 (2002) [DOI] [PubMed] [Google Scholar]
- 3.Apweiler R, Hermjakob H, Sharon N: On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta - Gen. Subj 1473, 4–8 (1999) [DOI] [PubMed] [Google Scholar]
- 4.Moremen KW, Tiemeyer M, Nairn AV: Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol. Cell Biol 13, 448–462 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee RT, Lauc G, Lee YC: Glycoproteomics: protein modifications for versatile functions. EMBO Rep 6, 1018–1022 (2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ihara Y, Nukina N, Miura R, Ogawara M: Phosphorylated Tau Protein Is Integrated into Paired Helical Filaments in Alzheimer’s Disease. J. Biochem 99, 1807–1810 (1986) [DOI] [PubMed] [Google Scholar]
- 7.Maverakis E, Kim K, Shimoda M, Gershwin ME, Patel F, Wilken R, Raychaudhuri S, Ruhaak LR, Lebrilla CB: Glycans in the immune system and The Altered Glycan Theory of Autoimmunity: a critical review. J. Autoimmun 57, 1–13 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hakomori S: Glycosylation defining cancer malignancy: new wine in an old bottle. Proc. Natl. Acad. Sci. USA 99, 10231–10233 (2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schedin-Weiss S, Winblad B, Tjernberg LO: The role of protein glycosylation in Alzheimer disease. FEBS J 281, 46–62 (2014) [DOI] [PubMed] [Google Scholar]
- 10.Lassen PS, Thygesen C, Larsen MR, Kempf SJ: Understanding Alzheimer’s disease by global quantification of protein phosphorylation and sialylated N-linked glycosylation profiles: A chance for new biomarkers in neuroproteomics? J. Proteomics 161, 11–25 (2017) [DOI] [PubMed] [Google Scholar]
- 11.Karlsson HKR, Zierath JR, Kane S, Krook A, Lienhard GE, Wallberg-Henriksson H: Insulin-Stimulated Phosphorylation of the Akt Substrate AS160 Is Impaired in Skeletal Muscle of Type 2 Diabetic Subjects. Diabetes 54, 1692–1697 (2005) [DOI] [PubMed] [Google Scholar]
- 12.Boucher J, Kleinridders A, Kahn CR: Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol 6, a009191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freeze HH, Eklund EA, Ng BG, Patterson MC: Neurology of inherited glycosylation disorders. Lancet Neurol 11, 453–466 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanninen K, Goldsteins G, Auriola S, Alafuzoff I, Koistinaho J: Glycosylation changes in Alzheimer’s disease as revealed by a proteomic approach. Neurosci. Lett 367, 235–240 (2004) [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Li D, Wu G, Bazer FW: Functional roles of fructose: Crosstalk between O-linked glycosylation and phosphorylation of Akt-TSC2-mtor cell signaling cascade in ovine trophectoderm cells. Biol. Reprod 95 (2016); 102:1–17 [DOI] [PubMed] [Google Scholar]
- 16.Hang Q, Isaji T, Hou S, Im S, Fukuda T, Gu J: Integrinα5 suppresses the phosphorylation of epidermal growth factor receptor and its cellular signaling of cell proliferation via N-glycosylation. J. Biol. Chem 290, 29345–29360 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aebersold R, Mann M: Mass spectrometry-based proteomics. Nature 422, 198–207 (2003) [DOI] [PubMed] [Google Scholar]
- 18.Domon B, Aebersold R: Mass spectrometry and protein analysis. Science 312, 212–217 (2006) [DOI] [PubMed] [Google Scholar]
- 19.Yates JR, Ruse CI, Nakorchevsky A: Proteomics by Mass Spectrometry: Approaches, Advances, and Applications. Annu. Rev. Biomed. Eng 11, 49–79 (2009) [DOI] [PubMed] [Google Scholar]
- 20.Bekker-Jensen DB, Kelstrup CD, Batth TS, Larsen SC, Haldrup C, Bramsen JB, Sørensen KD, Høyer S, Ørntoft TF, Andersen CL, Nielsen ML, Olsen JV: An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Syst 4, 587–599.e584 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riley NM, Hebert AS, Coon JJ: Proteomics Moves into the Fast Lane. Cell Syst 2, 142–143 (2016) [DOI] [PubMed] [Google Scholar]
- 22.Hebert AS, Richards AL, Bailey DJ, Ulbrich A, Coughlin EE, Westphall MS, Coon JJ: The One Hour Yeast Proteome. Mol. Cell. Proteomics 13, 339–347 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schroeder MJ, Shabanowitz J, Schwartz JC, Hunt DF, Coon JJ: A neutral loss activation method for improved phosphopeptide sequence analysis by quadrupole ion trap mass spectrometry. Anal. Chem 76, 3590–3598 (2004) [DOI] [PubMed] [Google Scholar]
- 24.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF: Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U.S.A 101, 9528–9533 (2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zubarev RA: Electron-capture dissociation tandem mass spectrometry. Curr. Opin. Biotechnol 15, 12–16 (2004) [DOI] [PubMed] [Google Scholar]
- 26.Reiding KR, Bondt A, Franc V, Heck AJR: The benefits of hybrid fragmentation methods for glycoproteomics. Trend. Anal. Chem 108, 260–268 (2018) [Google Scholar]
- 27.Riley NM, Hebert AS, Dürnberger G, Stanek F, Mechtler K, Westphall MS, Coon JJ: Phosphoproteomics with Activated Ion Electron Transfer Dissociation. Anal. Chem 89, 6367–6376 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glover MS, Yu Q, Chen Z, Shi X, Kent KC, Li L: Characterization of intact sialylated glycopeptides and phosphorylated glycopeptides from IMAC enriched samples by EThcD fragmentation: Toward combining phosphoproteomics and glycoproteomics. Int. J. Mass Spectrom 427, 35–42 (2018) [Google Scholar]
- 29.Yang Y, Liu F, Franc V, Halim LA, Schellekens H, Heck AJR: Hybrid mass spectrometry approaches in glycoprotein analysis and their usage in scoring biosimilarity. Nat. Commun 7 (2016); 13397: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu Q, Shi X, Feng Y, Kent KC, Li L: Improving data quality and preserving HCD-generated reporter ions with EThcD for isobaric tag-based quantitative proteomics and proteome-wide PTM studies. Anal. Chim. Acta 968, 40–49 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mommen GPM, Frese CK, Meiring HD, van Gaans-van den Brink J, de Jong APJM, van Els CACM, Heck AJR: Expanding the detectable HLA peptide repertoire using electron-transfer/higher-energy collision dissociation (EThcD). Proc. Natl. Acad. Sci. U.S.A 111, 4507–4512 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu Q, Wang B, Chen Z, Urabe G, Glover MS, Shi X, Guo L-W, Kent KC, Li L: Electron-Transfer/Higher-Energy Collision Dissociation (EThcD)-Enabled Intact Glycopeptide/Glycoproteome Characterization. J. Am. Soc. Mass Spectrom 28, 1751–1764 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu Q, Canales A, Glover MS, Das R, Shi X, Liu Y, Keller MP, Attie AD, Li L: Targeted Mass Spectrometry Approach Enabled Discovery of O-Glycosylated Insulin and Related Signaling Peptides in Mouse and Human Pancreatic Islets. Anal. Chem 89, 9184–9191 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diedrich JK, Pinto AFM, Yates JR III: Energy dependence of HCD on peptide fragmentation: Stepped collisional energy finds the sweet spot. J. Am. Soc. Mass Spectrom 24, 1690–1699 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang H, Yang C, Sun T: Characterization of glycopeptides using a stepped higher-energy C-trap dissociation approach on a hybrid quadrupole orbitrap. Rapid Commun. Mass Sp 32, 1353–1362 (2018) [DOI] [PubMed] [Google Scholar]
- 36.Palmisano G, Lendal SE, Engholm-Keller K, Leth-Larsen R, Parker BL, Larsen MR: Selective enrichment of sialic acid–containing glycopeptides using titanium dioxide chromatography with analysis by HILIC and mass spectrometry. Nat. Protoc 5, 1974–1982 (2010) [DOI] [PubMed] [Google Scholar]
- 37.Kweon HK, Håkansson K: Selective Zirconium Dioxide-Based Enrichment of Phosphorylated Peptides for Mass Spectrometric Analysis. Anal. Chem 78, 1743–1749 (2006) [DOI] [PubMed] [Google Scholar]
- 38.Feng S, Ye M, Zhou H, Jiang X, Jiang X, Zou H, Gong B: Immobilized Zirconium Ion Affinity Chromatography for Specific Enrichment of Phosphopeptides in Phosphoproteome Analysis. Mol. Cell. Proteomics 6, 1656–1665 (2007) [DOI] [PubMed] [Google Scholar]
- 39.Zhou H, Ye M, Dong J, Corradini E, Cristobal A, Heck AJR, Zou H, Mohammed S: Robust phosphoproteome enrichment using monodisperse microsphere–based immobilized titanium (IV) ion affinity chromatography. Nat. Protoc 8, 461–480 (2013) [DOI] [PubMed] [Google Scholar]
- 40.Hong Y, Yao Y, Zhao H, Sheng Q, Ye M, Yu C, Lan M: Dendritic Mesoporous Silica Nanoparticles with Abundant Ti4+ for Phosphopeptide Enrichment from Cancer Cells with 96% Specificity. Anal. Chem 90, 7617–7625 (2018) [DOI] [PubMed] [Google Scholar]
- 41.Iliuk AB, Martin VA, Alicie BM, Geahlen RL, Tao WA: In-depth analyses of kinase-dependent tyrosine phosphoproteomes based on metal ion functionalized soluble nanopolymers. Mol. Cell. Proteomics 9, 2162–2172 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tao WA, Wollscheid B, O’Brien R, Eng JK, Li X. j., Bodenmiller B, Watts JD, Hood L, Aebersold R: Quantitative phosphoproteome analysis using a dendrimer conjugation chemistry and tandem mass spectrometry. Nat. Methods 2, 591–598 (2005) [DOI] [PubMed] [Google Scholar]
- 43.Zhu F, Trinidad JC, Clemmer DE: Glycopeptide Site Heterogeneity and Structural Diversity Determined by Combined Lectin Affinity Chromatography/IMS/CID/MS Techniques. J. Am. Soc. Mass Spectrom 26, 1092–1102 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaji H, Saito H, Yamauchi Y, Shinkawa T, Taoka M, Hirabayashi J, Kasai K.-i., Takahashi N, Isobe T: Lectin affinity capture, isotope-coded tagging and mass spectrometry to identify N-linked glycoproteins. Nat. Biotechnol 21, 667–672 (2003) [DOI] [PubMed] [Google Scholar]
- 45.Chen R, Jiang X, Sun D, Han G, Wang F, Ye M, Wang L, Zou H: Glycoproteomics Analysis of Human Liver Tissue by Combination of Multiple Enzyme Digestion and Hydrazide Chemistry. J. Proteome Res 8, 651–661 (2009) [DOI] [PubMed] [Google Scholar]
- 46.Zhang H, Li X.-j., Martin DB, Aebersold R: Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol 21, 660–666 (2003) [DOI] [PubMed] [Google Scholar]
- 47.Tian Y, Zhou Y, Elliott S, Aebersold R, Zhang H: Solid-phase extraction of N-linked glycopeptides. Nat. Protoc 2, 334–339 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou Y, Aebersold R, Zhang H: Isolation of N-Linked Glycopeptides from Plasma. Anal. Chem 79, 5826–5837 (2007) [DOI] [PubMed] [Google Scholar]
- 49.Chen W, Smeekens JM, Wu R: A universal chemical enrichment method for mapping the yeast N-glycoproteome by MS. Mol. Cell. Proteomics 13, 1563–1572 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiao H, Chen W, Smeekens JM, Wu R: An enrichment method based on synergistic and reversible covalent interactions for large-scale analysis of glycoproteins. Nat. Commun 9 (2018); 1692: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao H, Wu R: Simultaneous Quantitation of Glycoprotein Degradation and Synthesis Rates by Integrating Isotope Labeling, Chemical Enrichment, and Multiplexed Proteomics. Anal. Chem 89, 10361–10367 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mysling S, Palmisano G, Højrup P, Thaysen-Andersen M: Utilizing Ion-Pairing Hydrophilic Interaction Chromatography Solid Phase Extraction for Efficient Glycopeptide Enrichment in Glycoproteomics. Anal. Chem 82, 5598–5609 (2010) [DOI] [PubMed] [Google Scholar]
- 53.Hägglund P, Bunkenborg J, Elortza F, Jensen ON, Roepstorff P: A New Strategy for Identification of N-Glycosylated Proteins and Unambiguous Assignment of Their Glycosylation Sites Using HILIC Enrichment and Partial Deglycosylation. J. Proteome Res 3, 556–566 (2004) [DOI] [PubMed] [Google Scholar]
- 54.Calvano CD, Zambonin CG, Jensen ON: Assessment of lectin and HILIC based enrichment protocols for characterization of serum glycoproteins by mass spectrometry. J. Proteomics 71, 304–317 (2008) [DOI] [PubMed] [Google Scholar]
- 55.Yu L, Li X, Guo Z, Zhang X, Liang X: Hydrophilic Interaction Chromatography Based Enrichment of Glycopeptides by Using Click Maltose: A Matrix with High Selectivity and Glycosylation Heterogeneity Coverage. Chem. – A Eur. J 15, 12618–12626 (2009) [DOI] [PubMed] [Google Scholar]
- 56.Xu D, Yan G, Gao M, Deng C, Zhang X: Highly selective SiO2–NH2@TiO2 hollow microspheres for simultaneous enrichment of phosphopeptides and glycopeptides. Anal. Bioanal. Chem 409, 1607–1614 (2017) [DOI] [PubMed] [Google Scholar]
- 57.Zou X, Jie J, Yang B: Single-Step Enrichment of N-Glycopeptides and Phosphopeptides with Novel Multifunctional Ti4+-Immobilized Dendritic Polyglycerol Coated Chitosan Nanomaterials. Anal. Chem 89, 7520–7526 (2017) [DOI] [PubMed] [Google Scholar]
- 58.Hong Y, Zhao H, Pu C, Zhan Q, Sheng Q, Lan M: Hydrophilic Phytic Acid-Coated Magnetic Graphene for Titanium(IV) Immobilization as a Novel Hydrophilic Interaction Liquid Chromatography-Immobilized Metal Affinity Chromatography Platform for Glyco- and Phosphopeptide Enrichment with Controllable Selectivity. Anal Chem 90, 11008–11015 (2018) [DOI] [PubMed] [Google Scholar]
- 59.Di Palma S, Hennrich ML, Heck AJR, Mohammed S: Recent advances in peptide separation by multidimensional liquid chromatography for proteome analysis. J. Proteomics 75, 3791–3813 (2012) [DOI] [PubMed] [Google Scholar]
- 60.Taouatas N, Altelaar AFM, Drugan MM, Helbig AO, Mohammed S, Heck AJR: Strong Cation Exchange-based Fractionation of Lys-N-generated Peptides Facilitates the Targeted Analysis of Post-translational Modifications. Mol. Cell. Proteomics 8, 190–200 (2009) [DOI] [PubMed] [Google Scholar]
- 61.Hennrich ML, Groenewold V, Kops GJPL, Heck AJR, Mohammed S: Improving Depth in Phosphoproteomics by Using a Strong Cation Exchange-Weak Anion Exchange-Reversed Phase Multidimensional Separation Approach. Anal. Chem 83, 7137–7143 (2011) [DOI] [PubMed] [Google Scholar]
- 62.Mohammed S, Heck AJR: Strong cation exchange (SCX) based analytical methods for the targeted analysis of protein post-translational modifications. Curr. Opin. Biotechnol 22, 9–16 (2011) [DOI] [PubMed] [Google Scholar]
- 63.Alpert AJ: Electrostatic repulsion hydrophilic interaction chromatography for isocratic separation of charged solutes and selective isolation of phosphopeptides. Anal. Chem 80, 62–76 (2008) [DOI] [PubMed] [Google Scholar]
- 64.Alpert AJ, Hudecz O, Mechtler K: Anion-exchange chromatography of phosphopeptides: weak anion exchange versus strong anion exchange and anion-exchange chromatography versus electrostatic repulsion-hydrophilic interaction chromatography. Anal. Chem 87, 4704–4711 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Totten SM, Feasley CL, Bermudez A, Pitteri SJ: Parallel Comparison of N-Linked Glycopeptide Enrichment Techniques Reveals Extensive Glycoproteomic Analysis of Plasma Enabled by SAX-ERLIC. J. Proteome Res 16, 1249–1260 (2017) [DOI] [PubMed] [Google Scholar]
- 66.Yang W, Shah P, Hu Y, Toghi Eshghi S, Sun S, Liu Y, Zhang H: Comparison of Enrichment Methods for Intact N- and O-Linked Glycopeptides Using Strong Anion Exchange and Hydrophilic Interaction Liquid Chromatography. Anal. Chem 89, 11193–11197 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tran TH, Hwang I-J, Park J-M, Kim J-B, Lee H-K: An Application of Electrostatic Repulsion Hydrophilic Interaction Chromatography in Phospho- and Glycoproteome Profiling of Epicardial Adipose Tissue in Obesity Mouse. Mass Spectro. Lett 3, 39–42 (2012) [Google Scholar]
- 68.Zhang H, Guo T, Li X, Datta A, Park JE, Yang J, Lim SK, Tam JP, Sze SK: Simultaneous Characterization of Glyco- and Phosphoproteomes of Mouse Brain Membrane Proteome with Electrostatic Repulsion Hydrophilic Interaction Chromatography. Mol. Cell. Proteomics 9, 635–647 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hao P, Guo T, Sze SK: Simultaneous Analysis of Proteome, Phospho- and Glycoproteome of Rat Kidney Tissue with Electrostatic Repulsion Hydrophilic Interaction Chromatography. PLoS One 6, e16844 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ge F, Tao S, Bi L, Zhang Z, Zhang XE: Proteomics: addressing the challenges of multiple myeloma. Acta Bioch. Bioph. Sin 43, 89–95 (2011) [DOI] [PubMed] [Google Scholar]
- 71.Reiding KR, Blank D, Kuijper DM, Deelder AM, Wuhrer M: High-Throughput Profiling of Protein N-Glycosylation by MALDI-TOF-MS Employing Linkage-Specific Sialic Acid Esterification. Anal. Chem 86, 5784–5793 (2014) [DOI] [PubMed] [Google Scholar]
- 72.Selman MHJ, Hemayatkar M, Deelder AM, Wuhrer M: Cotton HILIC SPE Microtips for Microscale Purification and Enrichment of Glycans and Glycopeptides. Anal. Chem 83, 2492–2499 (2011) [DOI] [PubMed] [Google Scholar]
- 73.Darula Z, Sherman J, Medzihradszky KF: How to Dig Deeper? Improved Enrichment Methods for Mucin Core-1 Type Glycopeptides. Mol. Cell. Proteomics 11, 1–10 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Alpert AJ: Hydrophilic-interaction chromatography for the separation of peptides, nucleic acids and other polar compounds. J. Chromatogr. A 499, 177–196 (1990) [DOI] [PubMed] [Google Scholar]
- 75.Hemström P, Irgum K: Hydrophilic interaction chromatography. J. Sep. Sci 29, 1784–1821 (2006) [DOI] [PubMed] [Google Scholar]
- 76.Thannhauser TW, Shen M, Sherwood R, Howe K, Fish T, Yang Y, Chen W, Zhang S: A workflow for large-scale empirical identification of cell wall N-linked glycoproteins of tomato (Solanum lycopersicum) fruit by tandem mass spectrometry. Electrophoresis 34, 2417–2431 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ding W, Hill JJ, Kelly J: Selective Enrichment of Glycopeptides from Glycoprotein Digests Using Ion-Pairing Normal-Phase Liquid Chromatography. Anal. Chem 79, 8891–8899 (2007) [DOI] [PubMed] [Google Scholar]
- 78.Ding W, Nothaft H, Szymanski CM, Kelly J: Identification and Quantification of Glycoproteins Using Ion-Pairing Normal-phase Liquid Chromatography and Mass Spectrometry. Mol. Cell. Proteomics 8, 2170–2185 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wimley WC, Gawrisch K, Creamer TP, White SH: Direct measurement of salt-bridge solvation energies using a peptide model system: implications for protein stability. PNAS 93, 2985 (1996) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alpert AJ: Effect of salts on retention in hydrophilic interaction chromatography. J. Chromatogr. A 1538, 45–53 (2018) [DOI] [PubMed] [Google Scholar]