

Abstract
The GE81112 tetrapeptides are a small family of unusual non-ribosomal peptide congeners with potent inhibitory activity against prokaryotic translation initiation. With the exception of the 3-hydroxy-L-pipecolic acid unit, little is known about the biosynthetic origins of the non-proteinogenic amino acid monomers of the natural product family. Here, we elucidate the biogenesis of the 4-hydroxy-L-citrulline unit and establish the role of an iron- and α-ketoglutarate-dependent enzyme (Fe/αKG) in the pathway. Homology modelling and sequence alignment analysis further facilitate the rational engineering of this enzyme to become a specific 4-arginine hydroxylase. We subsequently demonstrate the utility of this engineered enzyme in the synthesis of a dipeptide fragment of the antibiotic enduracidin. This work highlights the value of applying a bioinformatics-guided approach in the discovery of novel enzymes and engineering of new catalytic activity into existing ones.
Keywords: citrulline hydroxylase, non-heme dioxygenase, enzyme engineering, enduracidin
Graphical Abstract
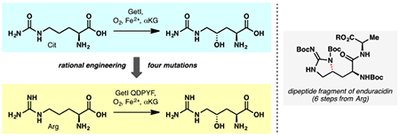
Citrulline hydroxylation:
A new citrulline 4-hydroxylase was identified from the biosynthesis of a non-ribosomal tetrapeptide, GE81112. Using bioinformatics-guided rational engineering, switching of the substrate specificity of the enzyme from citrulline to arginine was achieved in just four mutations. The utility of the engineered enzyme in chemical synthesis was demonstrated in the synthesis of a key dipeptide unit of enduracidin.
Identified in 2006 through a high throughput in vitro screening, the GE81112s (1) are a small family of three tetrapeptide congeners (A, B and B1) displaying prokaryotic-specific initiation inhibition (Figure 1A).1 Antibacterial profiling showed that each congener displays effective growth inhibition against several Gram-positive and Gram-negative pathogens.2 Recent studies indicated that GE81112 B stalls initiation in the unlocked 30S pre-initiation complex state and impedes its transition to the corresponding initiation complex.3 This process represents a unique mechanism of action relative to other antibiotics that target the ribosome.4 Thus, the GE81112 family represents an intriguing scaffold to further optimize as an antibacterial drug candidate.
Figure 1.

A. Structures of GE81112 A, B and B1 peptide antibiotics. B. The presence of two Fe/αKGs in the biosynthetic gene cluster of GE81112.
Structurally, the family consists of several highly unusual amino acid monomers, including 3-hydroxy-L-pipecolic acid, 4-hydroxy-L-citrulline, O-carbamoyl-α-amino-dihydroxyvaleric acid, 2-amino-L-histidine and β-hydroxy-2-chloro-L-histidine. Biosynthetic studies trace the production of these peptides to a non-ribosomal peptide synthetase (NRPS).5 In addition, the biosynthetic gene cluster of 1 also contains several genes encoding tailoring enzymes, including two iron- and α-ketoglutarate-dependent enzymes (Fe/αKGs), GetF and GetI (Figure 1B). GetF was recently characterized as an L-pipecolic acid hydroxylase responsible for the production of the 3-hydroxy-L-pipecolic acid monomer (2).6 Conversely, GetI was initially proposed to catalyze the β-hydroxylation of 2-chloro-L-histidine, either as the free or peptidyl carrier protein (PCP)-bound amino acid (3).5 Given the potential utility of GetI in the production of novel noncanonical amino acids, we became interested in its functional characterization and exploration of its biocatalytic utility.7
GetI is annotated as a member of clavaminate synthase-like protein (InterPro family IPR014503) in Uniprot. BLAST analysis revealed that GetI is 45% and 51% identical to VioC and OrfP, two arginine hydroxylases from the capreomycidine8 and streptolidine9 biosynthetic pathways, respectively. Furthermore, sequence alignment (Figure 2A and Figure S2) shows conservation of α-amino and carboxylate binding residues in GetI, VioC, and OrfP (Q124 and R322 in GetI, Q137 and R334 in VioC, Q123 and R321 in OrfP). Given this observation, GetI seems more likely to act on a free amino acid than on one that is bound to a PCP. At lower levels of sequence identity (28-40%), several hydroxylases that act on free-standing amino acids10 could also be located and no hydroxylases that act on (PCP)-bound amino acid could be identified in the BLAST analysis. Interestingly, neither 2-chloro-L-histidine nor L-histidine provided any desired hydroxylation product when subjected to reaction with GetI, αKG, O2, and Fe2+ at various different pHs (Figure 2B). A control experiment showed that the S-N-acetylcysteamine (SNAc) derivative of L-histidine is not accepted as a substrate by GetI either. These results led us to suspect that GetI might be involved in the biogenesis of a different monomer. At this stage, we realized that the origins of the 4-hydroxy-L-citrulline and O-carbamoyl-α-amino-dihydroxyvaleric acid were unaccounted for in the original biosynthetic proposal of GE81112. Given its high sequence identity to arginine hydroxylases, it seemed likely that GetI would act on structurally-related δ-carbamoyl amino acids. Indeed, treatment of L-citrulline (Cit) with Fe2+, O2, and αKG in the presence of GetI led to the formation of a hydroxylated product as judged by LC/MS. Subsequent 1H NMR analysis confirmed the C4 selectivity of the hydroxylation reaction, suggesting that GetI is responsible for the production of the 4-hydroxy-L-citrulline monomer prior to its loading to the NRPS assembly line. A similar outcome could also be observed when α-amino-δ-carbamoylhydroxyvaleric acid was employed as substrate. Prior to this work, an Fe/αKG from polyoxin biosynthesis, PolL, was previously characterized as a α-amino-δ-carbamoylhydroxyvaleric acid 4-hydroxylase.11 However, this enzyme is classified as a member of the PF10014 family, shares only minimal sequence identity with GetI and affords product with an opposite stereochemical configuration at C4 to GetI. Finally, several other polar and charged amino acids were also tested for reaction with GetI (See Table S1 in the Supporting Information), but among those tested, only L-arginine (Arg) yielded low levels of hydroxylation activity.
Figure 2.

A. Sequence alignment of VioC, GetI and OrfP, highlighting key residues that are involved in iron and amino acid substrate binding. B. LCMS profile of hydroxylation of select amino acids with GetI, suggesting the role of GetI in the biogenesis of the 4-hydroxy-L-citrulline monomer. C. Homology model of GetI using a solved crystal structure of OrfP as the template (PDB ID: 4M26).
To rationalize this outcome, we constructed a homology model of GetI using a solved crystal structure of OrfP as the template and performed virtual docking of Cit into the predicted active site of the enzyme (Figure 2C). Our model suggested that several hydrogen bonding and ionic interactions are potentially in play for Cit recognition. We propose that the α-amino and carboxylate groups of Cit form salt bridges with E157 and R322 in the active site. With respect to side-chain engagement, T153 and T258 are predicted to act as hydrogen bond donors to the δ-carbamoyl group and D256 is predicted to serve as a hydrogen bond acceptor to the ε-nitrogen of Cit. This binding mode stands in stark contrast to what was previously observed in Arg binding to VioC and OrfP, whereby aspartate residues (D268 and D270 in VioC, and D255 in OrfP, respectively) are involved in salt bridge formation with the guanidine side-chain. This observation also raises the possibility of a ‘specificity determinant’ loop in the active site of clavaminate synthase-like amino acid hydroxylases that serves to govern their substrate and/or reaction pathway specificity. A similar phenomenon has been observed previously in NRPS adenylation domains, whereby critical active site residues serve as specificity determinant to govern substrate recognition in the active site.12 This observation has led to the development of a predictive model for substrate specificity and forms the basis for existing NRPS predictor tools.13
Under the hypothesis that the substrate specificity of clavaminate synthase-like amino acid hydroxylases is governed by their specificity determinant loops, we asked if the substrate specificity of GetI could be altered via a simple loop-grafting procedure. Given its low levels of hydroxylation activity on Arg, we targeted the conversion of GetI to an Arg-specific hydroxylase. To date, no dedicated Arg C4-hydroxylase has been identified: VioC hydroxylates exclusively at C3 and OrfP provides predominantly dihydroxylation at C3 and C4. A biocatalyst that can catalyze a selective C4 oxygenation of Arg could find useful application in the chemoenzymatic synthesis of L-enduracididine or L-allo-enduracididine,14 a key motif in enduracidin,15 teixobactin16 and mannopeptimycin17 antibiotics. Additionally 4-hydroxyarginine can also be found in several peptide antibiotics, such as argimicins A and B,18 and K-582.19 To this end, we performed sequential site-directed mutagenesis to incrementally incorporate the guanidine binding sequence of VioC or OrfP into GetI (Figure 3). Towards a VioC-like enzyme, mutation T258D (‘DAD’) led to a 2-fold reduction in total turnover number (TTN) for Cit hydroxylation and a marginal increase in TTN for Arg hydroxylation. Interestingly, introduction of A257G mutation into this variant (‘DGD’) resulted in complete abolition of Cit hydroxylation activity and 1.4-fold increase in TTN for Arg hydroxylation. Finally, the triple mutant A257G/T258D/H259F (‘DGDF’) was found to catalyze the C4 hydroxylation of Arg with 87 TTN. However, this reaction was also accompanied by appreciable dihydroxylation (ca. ~ 4:1 monohydroxylation at C4: dihydroxylation at C3 and C4 by 1H NMR). Given the sub-optimal site-selectivity of this engineered enzyme, we next investigated the conversion of GetI to an OrfP-like enzyme. The double mutant A257P/T258Y (‘DPY’) was found to be completely unreactive towards Arg and Cit. However, introduction of L143Q mutation into this variant (‘QDPY’) rescued hydroxylation activity towards Arg (TTN = 42). One additional mutation, H259F, afforded variant ‘QDPYF’, which is able to catalyze selective C4-hydroxylation of Arg with 94 TTN without any observable activity on Cit. Steady-state kinetic analyses revealed an apparent KM of 4.8 ± 1.5 mM and kcat of 21 ± 2.3 min−1 for hydroxylation of Arg with GetI QDPYF. In contrast, wild-type GetI shows a slightly lower apparent KM for Cit (2.1 ± 0.41 mM) and a much higher kcat (69 ± 5.3 min−1). The loop-grafting approach therefore results in reduced substrate affinity but is also accompanied by an even larger decrease in turnover efficiency. This observation suggests the presence of non-obvious secondary interactions that contribute in accelerating the various elementary steps in the catalytic cycle. We anticipate that further directed evolution of GetI QDPYF could result in the identification of an enzyme variant with improved catalytic efficiency.
Figure 3.

Rational engineering of GetI to convert its substrate specificity from L-citrulline to L-arginine. Conditions for total turnover number (TTN) measurements: free amino acid (20 mM, 1 equiv), αKG (60 mM, 3 equiv), sodium ascorbate (10 mM, 0.5 equiv), FeSO4 (1 mM, 0.05 equiv), GetI variant (0.1–0.2 mol%), kPi buffer (pH = 8.0, 50 mM, 3.0 mL total volume), 12 h at 20°C. TTNs were determined by 1H NMR analysis of the reaction conversion.
Notwithstanding the modest TTN, GetI QDPYF could catalyze C4 hydroxylation of Arg to full conversion when the reaction was conducted in unclarified cell lysate (pre-lysis OD600 = 30). Encouraged by this observation, we next pursued a chemoenzymatic synthesis of a dipeptide fragment of enduracidin starting from 4-hydroxyarginine (8). We initially focused on the global Boc protection20 of the α-amino and guanidine side chain of 8. However, attempts to effect this transformation were plagued by low conversion and yield, as well as formation of NBoc regioisomer on the side chain. This mixture proved to be problematic to carry forward for subsequent manipulations. As a workaround, we elected to first selectively protect the α-amino group as the corresponding Boc derivative (Scheme 1A). Treatment19 of 9 with N2H4 afforded clean removal of the guanidine side chain to provide N2-Boc-4-OH-L-ornithine (10). In contrast, these conditions proved unreactive for removing the ureido group of Cit. While resulting in increased step count, this sequence resulted in superior material throughput and higher overall yield to 10 relative to the initial global Boc protection approach. Furthermore, the sequence allowed the most direct access to 4-OH-L-ornithine to date21 and could also be adapted to the formation of alkylated guanidine side chain by simply switching the coupling partner (e.g., to 13).
Scheme 1.

A. Chemoenzymatic synthesis of lactones 12 and 14 featuring regioselective C4 hydroxylation of L-arginine with GetI QDPYF. B. Conversion of lactone 12 to the L-End15-D-Ala16 dipeptide fragment of enduracidin and related dipeptide 19 via AlMe3-mediated lactone opening. C. Utility of AlMe3-mediated lactone opening in the conversion of 14 to dipeptide 20.
In our previous work,7 we have found that 4 and 5-hydroxyacids could be activated for subsequent peptide coupling via intramolecular lactonization, followed by treatment with the appropriate amine nucleophile. In the same vein, the free 4-hydroxyacids from 10 were lactonized by treatment with EDC. Lactone opening of 12 with H-Ala-OtBu (15) could be carried out in the presence of AlMe3 to afford alcohol 16 in 63% yield (Scheme 1B).22 Finally, use of Mitsunobu conditions23 (DIAD, PPh3) on 16 effected an intramolecular displacement of the secondary alcohol by the pendant guanidine to complete the synthesis of the L-enduracididine15-D-alanine16 dipeptide fragment of enduracidin (17). Our chemoenzymatic route compares favorably to previous synthetic approaches14 to L-enduracididine (See Table S2 for comparison), which typically result in poor stereocontrol at C4. Highly diastereoselective and high-yielding syntheses24 of L-allo-enduracididine from N-Boc-trans-4-hydroxy-L-proline have recently been developed. However, these approaches require at least eight steps and their adaptation to the synthesis of L-enduracididine would necessitate the use of the more expensive N-Boc-cis-4-hydroxy-L-proline diastereomer ($15/g) as the starting material.
To test the versatility of our methodology, 12 was submitted to the same AlMe3-assisted coupling conditions with H-L-Orn(Z)-OtBu (18) as the reaction partner. Smooth formation of dipeptide 19 could be realized in 71% isolated yield. The reaction was highly chemoselective as no side products arising from attack by δ-amino group of 18 could be observed. Lactone 14 was also found as an able coupling partner in this reaction, affording dipeptide 20 in 48% yield when 18 was used as the nucleophile (Scheme 1C). Thus, we believe that this methodology could be adapted to the preparation of enduracidin analogues and related 4-hydroxyarginine-containing peptides.
Selective remote oxidation of amino acids still remains an unmet challenge in the field of C-H functionalization. Though the hydroxylation activity of the Fe/αKGs has been known as early as 1966, many Fe/αKG amino acid hydroxylases25 have remained uncharacterized to date. By relying on bioinformatics analysis and chemical intuition, we have performed the first functional characterization of GetI and revised its functional annotation from chlorohistidine hydroxylase to citrulline hydroxylase. Further sequence similarity analysis allowed us to predict GetI’s substrate recognition ensemble (specificity determinant) and facilitated its rational engineering to become a specific 4-arginine hydroxylase with just four mutations. The utility of this engineered enzyme is highlighted in the concise chemoenzymatic synthesis of several novel dipeptides related to enduracidin. This work expands the catalytic repertoire of the IPR014503 family and lays the groundwork for rational discovery of novel enzymatic reactions within this family through further phylogenetic and sequence similarity network analysis.26
Experimental Section
See Supporting Information for Experimental Details.
Supplementary Material
Acknowledgements
Financial support for this work was generously provided by The Scripps Research Institute and the National Institutes of Health (grant GM128895). We thank Dr. Hajeung Park for assistance in homology modelling and in silico docking. We acknowledge the Shen lab and the Roush lab for generous access to their instrumentation.
References
- [1].(a) Brandi L, Fabbretti A, La Teana A, Abbondi M, Losi D, Donadio S, Gualerzi CO, Proc. Natl. Acad. Sci. USA 2006, 103, 39–44; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brandi L, Lazzarini A, Cavaletti L, Abbondi M, Corti E, Ciciliato I, Gastaldo L, Marazzi A, Feroggio M, Fabbretti A, Maio A, Colombo L, Donadio S, Marinelli F, Losi D, Gualerzi CO, Selva E, Biochemistry 2006, 45, 3692–3702. [DOI] [PubMed] [Google Scholar]
- [2].Maio A, Brandi L, Donadio S, Gualerzi CO, Antibiotics 2016, 5, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Fabbretti A, Schedlbauer A, Brandi L, Kaminishi T, Guiliodori AM, Garofalo R, Ochoa-Lizarralde B, Takemoto C, Yokoyama S, Connell SR, Gualerzi CO, Fucini P, Proc. Natl. Acad. Sci. USA 2016, 113, E2286–E2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Arenz S, Wilson DN, Cold Spring Harb. Perspect. Med 2016, 6, a025361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Binz TM, Maffioli SI, Sosio M, Donadio S, Müller R, J. Biol. Chem 2010, 285, 32710–32719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mattay J, Hüttel W, ChemBioChem 2017, 18, 1523–1528. [DOI] [PubMed] [Google Scholar]
- [7].For recent examples of biocatalytic hydroxylation for novel amino acid synthesis:; (a) Zwick CR III, Renata H, J. Am. Chem. Soc 2018, 140, 1165–1169; [DOI] [PubMed] [Google Scholar]; (b) Amatuni A, Renata H, Org. Biomol. Chem 2019, 17, 1736–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].(a) Ju J, Ozanick SG, Shen B, Thomas MG, ChemBioChem 2004, 5, 1281–1285; [DOI] [PubMed] [Google Scholar]; (b) Yin X, Zabriskie TM, ChemBioChem 2004, 5, 1274–1277; [DOI] [PubMed] [Google Scholar]; (c) Dunham NP, Chang W-C, Mitchell AJ, Martinie RJ, Zhang B, Bergman JA, Rajakovich LJ, Wang B, Silakov A, Krebs C, Boal AK, Bollinger JM Jr., J. Am. Chem. Soc 2018, 140, 7116–7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chang C-Y, Lyu S-Y, Liu Y-C, Hsu N-S, Wu C-C, Tang C-F, Lin K-H, Ho J-Y, Wu C-J, Tsai M-D, Li T-L, Angew. Chem. Int. Ed 2014, 53, 1943–1948. [DOI] [PubMed] [Google Scholar]
- [10].For characterization of other amino acid hydroxylases from clavaminate synthase-like protein family:; (a) Haltli B, Tan Y, Magarvey NA, Wagenaar M, Yin X, Greenstein M, Hucul JA, Zabriskie TM, Chem. Biol 2005, 12, 1163–1168; [DOI] [PubMed] [Google Scholar]; (b) Strieker M, Kopp F, Mahlert C, Essen L-O, Marahiel MA, ACS Chem. Biol 2007, 2, 187–196; [DOI] [PubMed] [Google Scholar]; (c) Baud D, Saaidi PL, Monfleur A, Harari M, Cuccaro J, Fossey A, Besnard M, Debard A, Mariage A, Pellouin V, Petit J-L, Salanoubat M, Weissenbach J, de Berardinis V, Zaparucha A, ChemCatChem 2014, 6, 3012–3017. [Google Scholar]
- [11].Qi J, Wan D, Ma H, Liu Y, Gong R, Qu X, Sun Y, Deng Z, Chen W, Cell Chem. Biol 2016, 23, 935–944. [DOI] [PubMed] [Google Scholar]
- [12].Challis GL, Ravel J, Townsend CA, Chem. Biol 2000, 7, 211–224. [DOI] [PubMed] [Google Scholar]
- [13].(a) Bachmann BO, Ravel J, Methods Enzymol. 2009, 458, 181–217; [DOI] [PubMed] [Google Scholar]; (b) Knudsen M, Søndergaard D, Tofting-Olesen C, Hansen FT, Brodersen DE, Pedersen CN, Bioinformatics 2016, 32, 325–329; [DOI] [PubMed] [Google Scholar]; (c) Röttig M, Medema MH, Blin K, Weber T, Rausch C, Kohlbacher O, Nucleic Acids Res. 2011, 39, W362–W367; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Minowa Y, Araki M, Kanehisa M, J. Mol. Biol 2007, 368, 1500–1517. [DOI] [PubMed] [Google Scholar]
- [14].Atkinson DJ, Naysmith BJ, Furkert DP, Brimble MA, Beilstein J. Org. Chem 2016, 12, 2325–2342 and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hori M, Iwasaki H, Horii S, Yoshida I, Hongo T, Chem. Pharm. Bull 1973, 21, 1175–1183. [Google Scholar]
- [16].Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, Mueller A, Schäberle TF, Hughes DE, Epstein S, Jones M, Lazarides L, Steadman VA, Cohen DR, Felix CR, Fetterman KA, Millett WP, Nitti AG, Zullo AM, Chen C, Lewis K, Nature 2015, 517, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].He HY, Williamson RT, Shen B, Graziani EL, Yang HY, Sakya SM, Petersen PJ, Carter GT, J. Am. Chem. Soc 2002, 124, 9729–9736. [DOI] [PubMed] [Google Scholar]
- [18].Yamaguchi T, Kobayashi Y, Adachi K, Imamura N, J. Antibiot 2003, 56, 655–657. [DOI] [PubMed] [Google Scholar]
- [19].Kawauchi H, Tohno M, Tsuchiya Y, Hayashida M, Adachi Y, Mukai T, Hayashi I, Kimura S, Kondo S, Int. J. Peptide Protein Res 1983, 21, 546–554. [DOI] [PubMed] [Google Scholar]
- [20].(a) Wu Y, Matsueda GR, Bernatowicz M, Synth. Commun 1993, 23, 3055–3060. [Google Scholar]; (b) Cho J, Coats SJ, Schinazi RF, Tet. Lett 2015, 56, 3587–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].For examples of previous synthetic approaches to 4-OH-L-ornithine:; (a) Waldmann H, He Y-P, Tan H, Arve L, Arndt H-D, Chem. Commun 2008, 5562–5564; [DOI] [PubMed] [Google Scholar]; (b) Aouadi K, Msaddek M, Praly J-P, Tetrahedron 2012, 68, 1762–1768; [Google Scholar]; (c) Rudolph J, Hannig F, Theis H, Wischnat R, Org. Lett 2001, 3, 3153–3155. [DOI] [PubMed] [Google Scholar]
- [22].Martin SF, Dwyer MP, Lynch CL, Tet. Lett 1998, 39, 1517–1520. [Google Scholar]
- [23].Hirose T, Sunazuka T, Tsuchiya S, Tanaka T, Kojime Y, Mori R, Iwatsuki M, Omura S, Chem. Eur. J 2008, 14, 8220–8238. [DOI] [PubMed] [Google Scholar]
- [24].(a) Craig W, Chen J, Richardson D, Thorpe R, Yuan Y, Org. Lett 2015, 17, 4620–4623; [DOI] [PubMed] [Google Scholar]; (b) Gao B, Chen S, Hou YN, Zhao YJ, Ye T, Xu Z, Org. Biomol. Chem 2019, 17, 1141–1153. [DOI] [PubMed] [Google Scholar]
- [25].(a) Hibi M, Ogawa J, Appl. Microbiol. Biotechnol 2014, 98, 3869–3876; [DOI] [PubMed] [Google Scholar]; (b) Islam MS, leissing TM, Chowdhury R, Hopkinson RJ, Schofield CJ, Annu. Rev. Biochem 2018, 87, 585–620. [DOI] [PubMed] [Google Scholar]
- [26].(a) Atkinson HJ, Morris JH, Ferrin TE, Babbitt PC, PLoS One 2009, 4, e4345; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gerlt JA, Bouvier JT, Davidson DB, Imker HJ, Sadkhin B, Slater DR, Whalen KL, Biochim. Biophys. Acta 2015, 1854, 1019–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.