

Abstract
Neuropeptides are important signaling molecules responsible for a wide range of functions within the nervous and neuroendocrine system. However, they are difficult to study due to numerous challenges, most notably their large degree of variability and low abundance in vivo. As a result, effective separation methods with sensitive detection capabilities are necessary for profiling neuropeptides in tissue samples, particularly those of simplified model organisms such as crustaceans. In order to address these challenges, this study utilized a capillary electrophoresis (CE)-matrix-assisted laser desorption/ionization (MALDI)-mass spectrometry imaging (MSI) platform, building upon our previous design for improved neuropeptidomic coverage. The capillary was coated with polyethylenimine (PEI) to reduce peptide adsorption and reverse the electroosmotic flow, and large volume sample stacking (LVSS) was used to load and pre-concentrate 1 μL of sample. The method demonstrated good reproducibility, with lower than 5% relative standard deviation for standards, and a limit of detection of approximately 100 pM for an allatostatin III peptide standard. The method was tested on brain and sinus gland (SG) tissue extracts and enabled detection of over 200 neuropeptides per run. When comparing the number detected in brain extracts in a direct spot, 60-second fractions, and 30-second fractions, the continuous trace collection afforded by the CE-MALDI-MSI platform yielded the largest number of detected neuropeptides. The method was compared to conventional LC-ESI-MS, and though the number of neuropeptides detected by LC-ESI-MS was slightly larger, the two methods were highly complementary, indicating the potential for the CE-MALDI-MSI method to uncover previously undetected neuropeptides in the crustacean nervous system. These results indicate the potential of CE-MALDI-MSI for routine use in neuropeptide research.
Keywords: Mass spectrometry, neuropeptides, capillary electrophoresis, crustacean, separations
Graphical Abstract
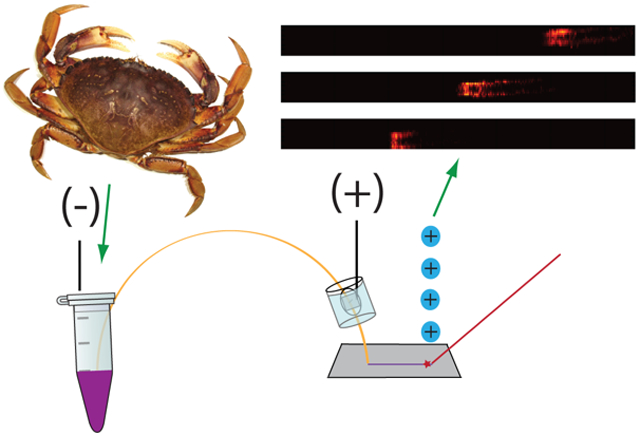
This work reports on a capillary electrophoresis (CE) separation method coupled to matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (MS) imaging for improved neuropeptide coverage in the model organism Cancer borealis.
Introduction:
Neuropeptides are one of the most diverse classes of signaling molecules. These short-chain amino acid sequences are responsible for facilitating cell-to-cell signaling, driving central pattern generators, and modulating the neuroendocrine system as long-range circulating hormones.1-4 However, studying neuropeptides remains challenging due to their low abundance in vivo, tendency toward rapid degradation, and signal suppression due to masking from other biomolecules present in their complex sample matrices.5 These challenges necessitate the development of rapid workflows, and having an effective separation method is critical to maximizing the number of detected neuropeptides. Furthermore, their large span of functions is a consequence of the extensive variability in their structures. As a result, finding a single technique able to reliably profile all neuropeptides is difficult.6 Model organisms are typically used in order to characterize the structure and function of neuropeptides. The crustacean model is commonly employed for neuropeptide research because crustaceans possess a simplified nervous system with many of their neuropeptides being either conserved in or homologous to those in mammals, such as RFamide and tachykinin families or the allatostatin/galanin family.7-9 However, even with this simplified model, coverage of the neuropeptidome remains incomplete, and it is expected that there are many more neuropeptides present that have not yet been detected and characterized.
Mass spectrometry is useful for probing the neuropeptides in these biological samples due to its sensitivity, fast acquisition rate, and ability to discriminate between different neuropeptide isoforms with different masses.10-12 The development of robust reversed-phase liquid chromatography (LC)-mass spectrometry (MS) techniques made large-scale profiling of neuropeptides possible, vastly expanding neuropeptide identification and driving functional discovery of novel neuropeptides. Since that point, LC-MS has been the main-stream technology for large-scale neuropeptide studies, and most routine workflows employ some variation of this method for neuropeptidomics, with very successful results.13-17 However, alternative separation methods have been largely under-explored. Most notable of these is capillary electrophoresis (CE), which is a highly-promising method due to its rapid and highly-efficient separation mechanism. Furthermore, it separates molecules based on electrophoretic mobility, making it orthogonal to reversed-phase LC, which separates based on hydrophobicity. Many recent works have demonstrated the impressive capabilities of CE, but it still has yet to be incorporated as a routine method of analysis like LC.18-21
Most MS workflows utilizing either LC or CE incorporate electrospray ionization (ESI) to introduce analytes to the MS.22-26 The other most commonly-used ionization method, matrix-assisted laser desorption/ionization (MALDI), has not seen quite as many developments in recent years.27 As a solid-phase ionization method, MALDI cannot be easily coupled to online separation methods like ESI can. MALDI is a highly-suitable ionization method, though, because it is especially tolerant to salts and the formation of primarily singly-charged ions is useful for avoiding charge dilution, where the signal of a single peptide is spread out over several charge states. Furthermore, because MALDI uses a different ionization mechanism than ESI, the ionization efficiencies of molecules differ between the two sources, resulting in complementary detection of peptides.28 In order to couple MALDI with separation methods, offline fraction collection is typically performed.29-31 However, this limits the achievable separation resolution, which is particularly discouraging for CE, as one of the most notable benefits to CE is its sharp, highly-resolved peaks, which is compromised during fraction collection. A continuous collection and MS acquisition method is necessary to preserve the high resolution of CE and maximize separation of complex biological samples.
There have been several methods for achieving continuous analysis of CE samples with MALDI-MS in recent years. Many of these show highly promising results, including one online coupling with a rotating ball interface between the CE outlet and vacuum MALDI surface,32 and another implementing a vacuum deposition interface for continuous collection.33, 34 However, these technique requires sophisticated instrument modification, so cannot be easily incorporated into existing laboratory workflows. MS imaging (MSI) appears to be the most promising method for offline coupling of CE and MALDI, as it does not require additional instrumental modifications, and the only equipment required is a moving platform for continuous collection. The coupling of LC with MALDI-MSI has been demonstrated with a similar type of setup.35, 36 Previous studies in our group implementing CE-MALDI-MSI have shown excellent results with high separation resolution and detection sensitivity.37-39
Here, we improve upon our previous CE-MALDI-MSI method for the analysis of neuropeptides in crustacean tissue samples in order to increase the obtainable depth of neuropeptidomic coverage. This result was achieved by utilizing larger injection volumes and enhanced separation resolution with a positively-charged PEI capillary coating that reduces peptide adsorption and reverses electroosmotic flow (EOF). With this method, an increase in the number of detected neuropeptides in two tissue sample types was obtained with good reproducibility. When compared to LC-ESI-MS, the results yielded complementary identifications, showcasing the potential of the methods together to substantially improve neuropeptidomic profiling. These results indicate the potential of the CE-MALDI-MSI interface to address challenges associated with neuropeptide research.
Experimental Section:
Chemicals and Materials
Fused silica capillary with a 50 μm inner diameter and 360 μm outer diameter was purchased from Polymicro Technologies - Molex (Lisle, IL, USA). ACS-grade formic acid (FA), anhydrous methanol, and cellulose acetate were purchased from Sigma-Aldrich (St. Louis, MO, USA). Polyethylenimine (50/50 v/v in isopropanol) was purchased from Gelest, Inc. (Morrisville, PA, USA). All other chemicals and solvents were purchased from Fisher Scientific (Pittsburgh, PA, USA). Acidified methanol was prepared with 90/9/1 v/v/v water/methanol/acetic acid. For LC-ESI-MS instrumental analysis, Optima-grade solvents were used.
Tissue collection and sample preparation
C. borealis crabs were purchased from the Fresh Lobster Company, LLC (Gloucester, MA, USA) and housed in artificial seawater tanks with an alternating light/dark cycle of 12 hours. Animals were packed in ice for 30 minutes prior to dissection for tissue collection. The dissection took place in chilled physiological saline (440 mM NaCl, 11 mM KCl, 26 mM MgCl2, 13 mM CaCl2, 11 mM Trizma base, 5 mM maleic acid, adjusted to pH 7.45 with NaOH) as described previously.40 Brains and sinus glands (SG) were collected and stored in acidified methanol at −80° C until use. A total of 8 sinus glands and 10 brains were collected for the CE-MALDI-MSI experiments and comparison LC-ESI-MS experiment.
Tissues were extracted for neuropeptides with acidified methanol as the extraction solvent. Acidified methanol was added to each tissue in a glass homogenizer, and the tissue was manually homogenized, sonicated in a bath sonicator, and centrifuged for 15 minutes at 16.1 rcf. The supernatant was removed and the cycle repeated for two additional extractions. The supernatant was evaporated in a speedvac, reconstituted in 15 μL 0.1% FA, and desalted with C18 ziptips (Agilent Technologies, Santa Clara, CA, USA). The SG sample and one of the brain samples were divided into two separate aliquots. Samples were then evaporated again in a speedvac and reconstituted in water for CE-MALDI-MSI experiments. The second aliquot of SG and brain extract were reconstituted in 0.1% FA for LC-ESI-MS runs.
All standards used in the CE-MALDI-MSI experiments were diluted from stock solutions (1 mg/mL) to the appropriate concentration with water.
CE experiments
For the CE-MALDI interface, a fracture was made 10 cm from the outlet of a 110 cm capillary with a glass cutter, as described elsewhere.41 Briefly, the glass cutter was used to scratch the coating off the capillary. The capillary was then gently bent to fracture the silica without breaking all the way through the capillary. The fractured part was affixed to a piece of plastic with glue, and a 120 mg/mL cellulose acetate solution was dripped over the fracture to create an ion-permeable membrane. The area was allowed to dry for 30 minutes prior to use. A Hewlett Packard 3D CE (Palo Alto, CA, USA) was used for all CE runs. The capillary was conditioned before the first used by flushing with each of the following for 4 minutes: methanol, water, 0.1 M NaOH, water, 0.1 M HCl, water, methanol. To coat the capillary with polyethylenimine (PEI) for a positively-charged surface, the capillary was flushed with 1 M NaOH for 30 minutes, followed by water for 10 minutes, and a 10% PEI solution (diluted with anhydrous methanol) for 2 hours. The coating was allowed to dry for 1 hour, and then the capillary was flushed with water for 25 minutes, followed by background electrolyte (BGE) solution for 20 minutes.42 The coating was replenished before each day’s CE runs. As this coating generates a positive charge on the inner wall of the capillary, the electroosmotic flow was reversed such that the bulk solution migrated from the negatively-charged cathode to the positively-charged anode.
Samples were injected using pressure injection such that the total volume injected was approximately 1 μL, 12% of the total capillary volume. The inlet of the capillary was then moved to a vial of BGE solution and −20 kV was applied for 30 minutes for CZE separation. The BGE solution used was a 50 mM acetic acid buffer solution at pH 4.5, prepared by diluting glacial acetic acid with water and titrating the solution with 0.1 M sodium hydroxide to the desired pH. The outlet of the capillary was placed in a reservoir filled with BGE solution such that the fractured part was submerged in solution and the outlet tip was outside of the reservoir (Figure 1A). The capillary was flushed with BGE for 10 minutes between subsequent analyses. For fraction collection, the outlet tip of the capillary was placed on a spot on a MALDI target plate for either 30 seconds or 60 seconds and then moved to the adjacent spot. Each fraction was mixed on the spot with 0.2 μL DHB matrix (150 mg/mL 2,5-dihydroxybenzoic acid in 50/50/0.1 water/methanol/FA). For continuous trace collection, the capillary tip was placed on a plain, stainless steel MALDI target plate attached to a syringe pump with a platform (Figure 1B). During the separation, the syringe pump was set to move at 4.2 mm/min, resulting in continuous collection of liquid emerging from the capillary tip. The plate was then coated with 12 layers of DHB matrix (40 mg/mL in 50/50/0.1 water/methanol/FA) using an automated TM sprayer set to 80° C with 30 s drying time between passes. The entire workflow is shown in Figure 1C.
Figure 1.

Depiction of the CE-MALDI-MSI set up, including a photograph of the interface of the capillary outlet with the MALDI plate (A), a close-up photograph of the capillary outlet and MALDI plate during trace collection (B), and a workflow showing the experimental steps, including CE separation (with arrows indicating direction of migration for electroosmotic flow (EOF), peptides, and net migration), matrix application, MALDI-MS analysis, generation of images, and database matching (C).
MALDI-MSI analysis
Samples were run on a MALDI LTQ-Orbitrap XL (Thermo Scientific, Bremen, Germany) in positive mode with an m/z range of 500 to 2000 and resolution of 60,000. The MALDI source was equipped with a 337.1 nm, 60 Hz nitrogen laser, set to 20 μJ laser energy. For spot analysis, 50 scans were acquired for each spot. For imaging, MS spectra were acquired at a raster size of 100 μm, with the exception of the images in Figure 2C, which were acquired at a raster size of 200 μm. MS imaging run times took approximately 6 hours to complete. LC-ESI-MS data was acquired on an Orbitrap Elite (Thermo Scientific, Bremen, Germany). Details of the parameters can be found in the Supporting Information.
Figure 2.

Electropherogram and MS images of CE traces of standard peptides, demonstrating the separation of a mixture of 5 standard peptides (A) with the corresponding MS images (B), the consistency of migration times of two standards run on different days (C), and the detection of a standard run at different concentrations (D). In each MS image, green circles indicate the location of the maximum peak intensity for the m/z value.
Data Analysis
Spot data was processed by accurate mass-matching peaks to our lab’s in-house crustacean neuropeptide database within ± 5 ppm mass error. Images were generated for the CE traces in ImageQuest (Thermo Scientific, Bremen, Germany). The image files were exported as centroid imzml files, which were uploaded into MSiReader.43 MS heat map images were generated for each m/z in the neuropeptide database. All images were normalized to the total ion current (TIC), with a mass error set to ± 5 ppm, and 5th order linear smoothing applied. Electropherograms were constructed by exporting the intensity matrix in MSiReader to Excel for each detected neuropeptide m/z. For each x-axis coordinate, the y-value intensities were summed to make a 2-dimensional electropherogram, with different colors representing different m/z values. LC-ESI-MS data was processed in MaxQuant44 for feature detection, and then accurate mass-matched to the neuropeptide database within ± 5 ppm mass error.
Results and Discussion
Optimization of separation conditions
CE separation of neuropeptides was employed with continuous collection of liquid coming out of the capillary for subsequent MALDI-MSI in order to maximize separation resolution. Figure 1A shows the overall setup of the CE, capillary outlet, and MALDI target plate. Figure 1B shows a close-up of the CE outlet allowing for the interface with MALDI. The overall workflow of the method is depicted in Figure 1C. The purpose of this study was to improve upon our previous designs for the CE-MALDI interface in order to gain a more in-depth profiling of neuropeptides. The key aspects to achieve such improvements include the incorporation of a positively-charged coating and by increasing the injection volume with LVSS.
The capillaries used were bare fused silica, which carries a negative charge. The peptides traveling through the capillary are predominantly positively-charged, and so electrostatic interactions cause them to adhere to the capillary. This phenomenon results in peak broadening, which causes poorer separation resolution and detection sensitivity. To overcome this problem, we coated the capillary with PEI, creating a positive charge on the inner wall of the capillary. The positive charge repels the positive charge of the peptides, resulting in sharper, more resolved peaks. In addition to preventing inner-wall adsorption, the PEI coating improves separation by reversing the EOF in the capillary. Due to the reversed charge of the surface, the EOF migrates from the negatively-charged electrode (cathode) to the positively-charged electrode (anode), while the peptides still flow anode to cathode. As a result, the separation is lengthened when performed under reverse polarity, allowing the peptides longer distance to separate within the capillary, thus improving resolution. Figure S1 (See Supporting Information) shows a comparison of electropherograms of a neuropeptide extract with and without the PEI coating, demonstrating the improvement in peak shape and resolution.
In order to increase the amount of peptides that could be loaded onto the capillary, we employed the LVSS injection and pre-concentration method. This method works with low-concentration samples dissolved in water, as the low concentration causes the sample plug to have a higher field strength than the surrounding BGE due to its lower conductivity. Instead of dissolving the sample in 0.1% formic acid, as is done for LC analysis, the sample was dissolved in pure water. As the neuropeptides in crustacean tissue sample are present in low abundance, the method is well-suited for this analysis. After the sample plug is loaded on the capillary and a voltage applied, the more highly-concentrated BGE travels quickly through the capillary. As current needs to be uniform at all points in a closed circuit, ions in the dilute sample plug move at a faster rate until the ions reach the interface of the sample plug and BGE, thus concentrating the sample plug until it is at the same concentration as the BGE. At that point, the peptides in the sample plug separate based on their individual electrophoretic mobilities.45-47 Because the EOF and peptide migration were in opposite directions, the peptides moved toward the entrance of the capillary until they reached the BGE solution. At that point, their net migration was toward the capillary outlet as they were carried by the EOF. In this setup, the BGE was 50 mM pH 4.5 acetic acid/ammonium acetate buffer. This pH was selected because it reduced the EOF enough such that the peptides could separate within a 30-minute separation window. The LVSS method enabled an injection of 1 μL of sample, making the method more comparable to nanoLC methods while still providing excellent separation and high-efficiency peak shapes. The MS images in Figure 2A show the separation of a mixture of 5 standard peptides (20 μM concentration) in a single CE trace. An improvement in peak area of approximately 2-fold was observed for standard peptides compared to a conventional 20 nL injection at this concentration. It is likely that this improvement would be even larger at lower concentrations, as the standards were not detected at lower concentrations with the 20 nL injection volume.
In order to demonstrate the reliability and reproducibility of the separation, three runs of the same standard peptides were performed on different days with different capillaries. Figure 2B shows heat map MS images of two standard peptides from each of the three runs. As can be seen, the migration time of each peptide is approximately the same on different days. The relative standard deviation, based on the migration time of the peak maximum, was 0.67% for FMRFamide and 2.31% for allatostatin III. While the peak migrations shifted a small amount from day to day, likely due to slight variations in capillary length, they remained generally consistent, and the reproducibility was found suitable for separating peptide mixtures.
With the increased loading capacity of the CE method, it was expected that the detection of low-abundance neuropeptides would be possible. In order to evaluate this expectation, the limit of detection was approximated using a standard peptide, allatostatin III. Subsequent runs were analyzed with varying concentrations. Solutions of 1 pM, 10 pM, 100pM, 1 nM, and 1 μM were analyzed. The standard was able to be easily detected at levels down to 100 pM, but at 10 pM and 1 pM, no signal was observed. Figure 2C shows the MS images of the standard at the three concentrations of the standard peptide. The migration times changes slightly for lower concentrations of peptide due to the increased amount of stacking that occurs within the capillary. From these data, it was determined that the approximate limit of detection is 100 pM, though the actual concentration will likely vary based on ionization efficiencies of peptides and complexity of samples.
Application of the CE-MALDI-MSI method to profiling neuropeptides in crustacean tissue extracts
The CE-MALDI-MSI method was evaluated on two tissue extracts, brain and sinus gland. Approximately 1.72 μg of SG extract was loaded onto the capillary. For the brain extract, approximately 0.44 μg was loaded onto the capillary. For each sample, three replicate injections were run in order to obtain three technical replicates for each. Two additional biological replicates were performed with the brain extract on different days using different brain tissue samples. All sample runs resulted in the detection of over 200 neuropeptides, a substantial improvement to the 67 neuropeptides detected with the previous MALDI-MSI method, with relatively low standard deviations. The results are summarized in Figure 3, which shows the number of neuropeptides detected in each sample, with error bars indicating the standard deviation between the three technical replicates. The number of detected neuropeptides in each sample had low relative standard deviations (3.9%, 12.4%, and 1.5% for the brain samples and 6.8% for the SG sample), indicating good technical reproducibility. The relative standard deviation of peaks areas between samples were also relatively low, 27.6%, 22.4%, and 31.8% for the brain samples and 32.3% for the SG sample. Figure 4 shows electropherograms from representative runs of both the brain and tissue extract, with different colors indicating different neuropeptides detected. The electropherograms were constructed by exporting the intensity of each m/z value at each pixel. The intensities were summed across the Y axis for each m/z value to generate a plot of peak intensity as a function of the X axis. As can be seen, the separations led to highly-resolved peptide signals in the MS imaging trace. The narrowness of the peak shapes is likely due to the large degree of stacking that took place with neuropeptides in low abundance. Furthermore, the migration times of the neuropeptides appear to be evenly distributed throughout the entire separation time for both tissue samples, indicating that the optimized parameters are suitable for separating the neuropeptides within a relatively short separation window (30 minutes).
Figure 3.

Number of detected neuropeptides in samples of neuropeptide tissue extract, with the total representing the average of three replicate injections of the same sample and error bars indicating the standard deviation.
Figure 4.

Electropherograms constructed from MS images of detected neuropeptides in representative brain (top) and sinus gland (bottom) CE-MALDI-MSI runs, with each color indicating a different neuropeptide.
Comparison of different collection methods
While the CE-MALDI interface has been explored by numerous groups in a variety of contexts in the past, the coupling with MSI is a relatively recent technique. The improved number of identifications in tissue extract is largely attributed to the improved separation resolution. Other methods often couple CE with MALDI using fraction collection. By continuously collecting liquid from the capillary onto the plate and performing MS imaging, the resolution is expanded drastically (with the limit being determined instead by the raster size of the instrument). In our method, the raster size was set to 100 μm, which corresponds to a time resolution of approximately 0.01 s. In order to determine if this enhanced resolution is beneficial, a comparison was made between different time resolutions, including a direct spot (no resolution), 60 s fraction collections, 30 s fraction collections, and the continuous trace. Brain tissue extract was used for all collections. The resulting number of detected neuropeptides with each method is shown in Figure 5. As is expected, the number of neuropeptides increases with increasing separation resolution. This observation was confirmed by performing a one-way ANOVA test (p-value = 1.0×10−4), followed by a Dunnett post-hoc test in which the continuous trace was compared to the three other methods. Each comparison was determined to be statistically significant below a threshold of 0.05. Figure 6 shows the MS spectra acquired from 10 of the 60 s fractions collected of the brain sample showing the difference in peptides detected within each fraction of the CE separation.
Figure 5.

Bar graph indicating the number of neuropeptides detected in brain tissue extract samples when analyzing a direct spot, 60 s CE fractions, 30 s CE fractions, and a continuous CE trace using MSI. Error bars represent the standard deviation, and n corresponds to the number of replicates performed. An ANOVA test was performed (p-value 1.0×10−4) followed by a Dunnett’s post-hoc test comparing each method to the continuous trace. Asterisks indicate statistical significance at α < 0.05.
Figure 6.

Mass spectra from CE separation of brain tissue extract collected as 60 second fractions and analyzed with MALDI-MSI. Shown are fractions 14 – 24, showing the difference in peptides identified in each fraction (A), as well as a mass spectrum of brain tissue extract without CE separation (B).
Comparison of CE-MALDI-MSI to LC-ESI-MS
Online LC coupled to ESI-MS is the gold standard for peptide separations and is routinely performed with excellent results. In order to determine how the CE-MALDI-MSI method compared, brain and SG samples were run on an Orbitrap Elite MS coupled to a Waters nanoAcquity LC system. This instrument was chosen for comparison because it has the same geometry as the MALDI LTQ-Orbitrap XL instrument, with all aspects of the instrument being approximately the same except for the ionization source. A standard 2-hour LC gradient was used with a self-packed 14 cm C18 column. While the results showed a slightly greater number of neuropeptides detected in the LC-ESI-MS samples, it is interesting to note that the results were highly complementary, with less than a third of the neuropeptides being detected with both methods. Figure 7 shows the overlap of neuropeptides detected with the two methods. These results indicate that neither method is necessarily superior to the other, but combining the two may substantially improve neuropeptidomic coverage. Furthermore, applying the CE-MALDI-MSI method toward additional neuropeptide samples may result in the detection of neuropeptides that have not been previously detected.
Figure 7.
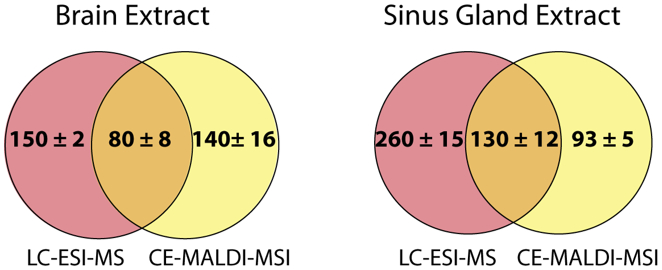
Venn diagrams indicating the overlap of neuropeptides detected in neuropeptide extracts from brain tissue (left) and sinus gland tissue (right) using LC-ESI-MS and CE-MALDI-MSI. Values indicate the number detected ± standard deviation (n = 3 technical replicates).
The neuropeptides detected in LC and CE were compared in order to identify any trends in characteristics causing certain neuropeptides to be identified with one method over the other. The mass, m/z, family, and retention/migration times were compared. The range and average masses and m/z values were approximately the same for all samples with both methods, lying near the median value. The neuropeptide families were also compared, as neuropeptides are grouped into families based on shared sequence motifs. For all samples, similar numbers of neuropeptides were detected belonging to the different common neuropeptide families. The average LC retention times and CE migration times were close to the median values, indicating that there was no bias related to the time of separation. Therefore, it is assumed that the differences in detected neuropeptides between the two methods is due to the orthogonality of the separation mechanisms resulting in different cohorts of peptides to be better separated and more easily detected.
Conclusion
The purpose of this study was to address challenges in neuropeptide detection related to the large degree of diversity and low abundances in vivo. To overcome these challenges, an MS platform was established that is orthogonal and complementary to conventional LC-ESI-MS. The developed CE-MALDI-MSI method further improved on our previous platform by utilizing larger injection volumes and enhanced separation resolution with a PEI capillary coating. The resulting method showed effective separation of both standards and tissue extracts with excellent reproducibility. Furthermore, the results yielded complementary identifications to that of LC-ESI-MS, indicating that combining the platforms can lead to substantial improvements in neuropeptidomic coverage and potentially novel, previously-undiscovered neuropeptides. While this method used accurate mass-matching to identify neuropeptides, it is expected that the incorporation of MS/MS acquisitions will result in more confident identifications, as well as the identification of novel neuropeptides with the assistance of de novo sequencing. This method demonstrates the potential of CE-MALDI-MSI as a routine method for neuropeptide profiling in a variety of samples.
Supplementary Material
Acknowledgements
This work was supported by a National Science Foundation (CHE-1710140) and National Institutes of Health through grants R01DK071801 and R01NS029436. The Orbitrap instruments were purchased through the support of an NIH shared instrument grant (NIH-NCRR S10RR029531). KD acknowledges a predoctoral fellowship supported by the National Institutes of Health, under Ruth L. Kirschstein National Research Service Award T32 HL 007936 from the National Heart Lung and Blood Institute to the University of Wisconsin-Madison Cardiovascular Research Center and the National Institutes of Health-General Medical Sciences F31 National Research Service Award (1F31GM126870-01A1) for funding. LL acknowledges a Vilas Distinguished Achievement Professorship and Charles Melbourne Johnson Professorship with funding provided by the Wisconsin Alumni Research Foundation and University of Wisconsin-Madison School of Pharmacy.
Abbreviations:
- BGE
Background electrolyte
- CE
capillary electrophoresis
- DHB
2,5-dihydroxybenzoic acid
- EOF
electroosmotic flow
- ESI
electrospray ionization
- FA
formic acid
- LVSS
large volume sample stacking
- LC
liquid chromatography
- MS
mass spectrometry
- MSI
mass spectrometry imaging
- MALDI
matrix-assisted laser desorption/ionization
- PEI
polyethylenimine
- SG
sinus gland
Footnotes
Electronic supplementary information (ESI) is available for the online version of this article through publisher’s web site.
Details of LC-ESI-MS method used for comparison, electropherograms of tissue extracted separated with uncoated and PEI-coated capillaries.
References
- 1.Blitz DM, Marder E and Nusbaum MP, Nature Reviews Neuroscience, 2017, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van den Pol AN, Neuron, 2012, 76, 98–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hook V and Bandeira N, J Am Soc Mass Spectrom, 2015, 26, 1970–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beal MF, M. G. H. a. H. M. S. Department of Neurology, Boston MA, M. G. H. Neurology Research 4, Fruit St, Boston MA: 02114, [Google Scholar]; Martin JB and M. G. H. a. H. M. S. Department of Neurology, Boston MA, Annals of Neurology, 2016, 20, 547–565. [Google Scholar]
- 5.Fricker LD, Lim JY, Pan H and Che FY, Mass Spectrometry Reviews, 2006, 25, 327–344. [DOI] [PubMed] [Google Scholar]
- 6.DeLaney K, Buchberger AR, Atkinson L, Grunder S, Mousley A and Li L, J Exp Biol, 2018, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birgül N, Weise C, Kreienkamp HJ and Richter D, EMBO J, 1999, 18, 5892–5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elphick MR and Mirabeau O, Front Endocrinol (Lausanne), 2014, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steinhoff MS, von Mentzer B, Geppetti P, Pothoulakis C and Bunnett NW, Physiol Rev, 2014, 94, 265–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmerberg CM and Li LJ, Protein and Peptide Letters, 2013, 20, 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buchberger A, Yu Q and Li L, Annu Rev Anal Chem (Palo Alto Calif), 2015, 8, 485–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee JE, Genomics Inform, 2016, 14, 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Secher A, Kelstrup C, Conde-Frieboes K, Pyke C, Raun K, Wulff B and Olson J, Nature Communications, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnsen E, Leknes S, Wilson SR and Lundanes E, Scientific Reports, 2015, 5, 9308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dasgupta S, Castro LM, Tashima AK and Fricker L, Methods Mol Biol, 2018, 1719, 161–174. [DOI] [PubMed] [Google Scholar]
- 16.Skold K, Svensson M, Kaplan A, Bjorkesten L, Astrom J and Andren PE, Proteomics, 2002, 2, 447–454. [DOI] [PubMed] [Google Scholar]
- 17.Finoulst I, Pinkse M, Van Dongen W and Verhaert P, J Biomed Biotechnol, 2011, 2011, 245291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu G, Sun L, Yan X and Dovichi NJ, Anal Chem, 2013, 85, 2569–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun L, Hebert AS, Yan X, Zhao Y, Westphall MS, Rush M, Zhu G, Champion M, Coon J and Dovichi N, Angewandte Chemie-International Edition, 2014, 53, 13931–13933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ludwig KR, Sun L, Zhu G, Dovichi NJ and Hummon AB, Anal Chem, 2015, 87, 9532–9537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lubeckyj RA, McCool EN, Shen X, Kou Q, Liu X and Sun L, Anal Chem, 2017, 89, 12059–12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang P, Pan JZ and Fang Q, Talanta, 2018, 180, 376–382. [DOI] [PubMed] [Google Scholar]
- 23.Causon TJ, Maringer L, Buchberger W and Klampfl CW, J Chromatogr A, 2014, 1343, 182–187. [DOI] [PubMed] [Google Scholar]
- 24.Tseng MC, Chen YR and Her GR, Electrophoresis, 2004, 25, 2084–2089. [DOI] [PubMed] [Google Scholar]
- 25.Liu CC, Zhang J and Dovichi NJ, Rapid Commun Mass Spectrom, 2005, 19, 187–192. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Fonslow BR, Wong CC, Nakorchevsky A and Yates JR 3rd, Anal Chem, 2012, 84, 8505–8513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong X, Zhang Z, Jiang S and Li L, Electrophoresis, 2014, 35, 1214–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nadler WM, Waidelich D, Kerner A, Hanke S, Berg R, Trumpp A and Rösli C, 2017. [DOI] [PubMed] [Google Scholar]
- 29.Zuberovic A, Wetterhall M, Hanrieder J and Bergquist J, Electrophoresis, 2009, 30, 1836–1843. [DOI] [PubMed] [Google Scholar]
- 30.Pourhaghighi MR, Busnel JM and Girault HH, Electrophoresis, 2011, 32, 1795–1803. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, DeLaney K, Hui L, Wang J, Sturm RM and Li L, J Am Soc Mass Spectrom, 2018, 29, 948–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Musyimi HK, Narcisse DA, Zhang X, Stryjewski W, Soper SA and Murray KK, Anal Chem, 2004, 76, 5968–5973. [DOI] [PubMed] [Google Scholar]
- 33.Preisler J, Hu P, Rejtar T and Karger BL, Anal Chem, 2000, 72, 4785–4795. [DOI] [PubMed] [Google Scholar]
- 34.Rejtar T, Hu P, Juhasz P, Campbell JM, Vestal ML, Preisler J and Karger BL, J Proteome Res, 2002, 1, 171–179. [DOI] [PubMed] [Google Scholar]
- 35.Smith DF, Aizikov K, Duursma MC, Giskes F, Spaanderman DJ, McDonnell LA, O’Connor PB and Heeren RM, J Am Soc Mass Spectrom, 2011, 22, 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weidner SM and Falkenhagen J, Anal Chem, 2011, 83, 9153–9158. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Z, Ye H, Wang J, Hui L and Li L, Anal Chem, 2012, 84, 7684–7691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang JH, Ye H, Zhang ZC, Xiang F, Girdaukas G and Li LJ, Analytical Chemistry, 2011, 83, 3462–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang S, Liang Z, Hao L and Li L, ELECTROPHORESIS, 2016, 37, 1031–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kutz KK, Schmidt JJ and Li LJ, Analytical Chemistry, 2004, 76, 5630–5640. [DOI] [PubMed] [Google Scholar]
- 41.Whang CW and Chen IC, Anal Chem, 1992, 64, 2461–2464. [DOI] [PubMed] [Google Scholar]
- 42.Spanila M, Pazourek J and Havel J, J Sep Sci, 2006, 29, 2234–2240. [DOI] [PubMed] [Google Scholar]
- 43.Robichaud G, Garrard KP, Barry JA and Muddiman DC, Journal of the American Society for Mass Spectrometry, 2013, 24, 718–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cox J and Mann M, Nature Biotechnology, 2008, 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- 45.He Y and Lee HK, Anal Chem, 1999, 71, 995–1001. [DOI] [PubMed] [Google Scholar]
- 46.Burgi DS and Chien RL, Analytical Chemistry, 1991, 63, 2042–2047. [Google Scholar]
- 47.Chien RL and Burgi DS, Analytical Chemistry, 1992, 64, 1046–1050. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.