

Abstract
Alkyne-Directed Chemo- and Enantioselectivity. Highly enantioselective catalytic reductive coupling of allyl acetate with acetylenic ketones occurs in a chemoselective manner in the presence of aliphatic or aromatic ketones. This method was used to construct C14-C23 of pladienolide D in half the steps previously required.
Keywords: Enantioselective, Iridium, Ketone Allylation, π-allyl, Transfer Hydrogenation
Graphical Abstract
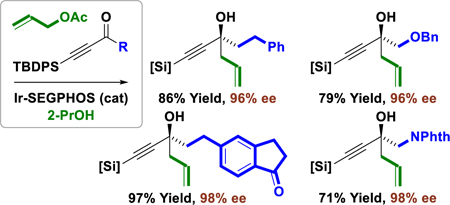
The catalytic enantioselective addition of C-nucleophiles to ketones represents a persistent challenge in chemical synthesis.[1] Differentiation of the acyl substituents by the chiral catalyst is often incomplete, leading to low C=O π-facial enantioselectivities. Additionally, for stabilized C-nucleophiles, ketone addition is frequently reversible, which erodes kinetic stereoselectivity. Consequently, work in this area is relegated to the use of non-stabilized C-nucleophiles in the form of stoichiometric organometallic reagents or related metalloids, which are often prepared via successive transmetallation.[2,3] In the specific context of enantioselective ketone allylation, reagents based on B, Si, and Sn have been described.[4] Metal-catalyzed carbonyl reductive couplings of π-unsaturated feedstocks potentially provides an alternative to the use of premetalated C-nucleophiles.[5] However, the requisite reductants utilized in these processes are typically metallic (Zn, Mn), pyrophoric (ZnEt2, BEt3), expensive or mass-intensive (R3SiH).[6] To address these limitations, we have developed metal-catalyzed carbonyl reductive couplings that utilize elemental hydrogen, 2-propanol or formate as terminal reductant, as well as related hydrogen auto-transfer processes wherein alcohols serve as hydrogen donor and carbonyl proelectrophile.[7] While this technology has enabled diverse catalytic enantioselective aldehyde allylations[7f] and propargylations,[7g] asymmetric transfer hydrogenative allylations of ketones are restricted to vicinal dicarbonyl compounds.[8]
It was posited that acetylenic ketones might participate in efficient alcohol-mediated carbonyl allylation due to electrophilic activation associated with the negative inductive effect of the alkyne.[9] Although isolated examples have been reported,[10] only one systematic study on the asymmetric allylation of acetylenic ketones appears in the literature, which exploits an allylzinc reagent bound by a stoichiometric quantities of a chiral modifier.[11] Given this lack of prior art and the utility of the resulting 1,5-enynes vis-á-vis diverse carbophilic metal-catalyzed transformations,[12] the 2-propanol-mediated transfer hydrogenative allylation of acetylenic ketones was explored. Here, we report that such processes not only occur with high levels of enantioselectivity, but may be conducted in a chemoselective manner in the presence of aliphatic and aromatic ketones (Figure 1).
Figure 1.
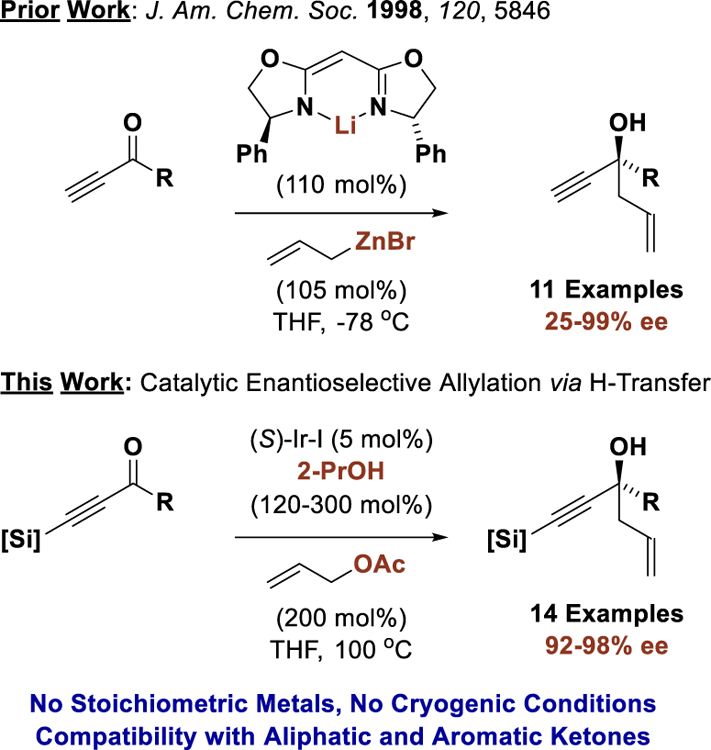
Asymmetric allylation of acetylenic ketones.
Guided by our prior work on the transfer hydrogenative allylation of acetylenic aldehydes,[9] methyl ketone 1a (100 mol%, [Si] = iPr3Si) was exposed to allyl acetate 2a (200 mol%) and 2-propanol (200 mol%) in the presence of Cs2CO3 (50 mol%) and the commercially available π-allyliridium-C,O-benzoate modified by (S)-SEGPHOS, (S)-Ir-I (5 mol%), in anhydrous THF (Table 1, entry 1). The desired product of ketone allylation 3a was not observed. However, upon introduction of water (100 mol%), which may facilitate alkoxide exchange at the metal center and solubilize the carbonate base, the tertiary homoallylic alcohol 3a was obtained in 44% yield in highly enantiomerically enriched form (Table 1, entry 2). The mass balance in this experiment consisted primarily of unreacted 1a and the corresponding product of silyl cleavage. As acetylenic C-H moieties are not tolerated by the iridium catalyst, an effort was made to accelerate carbonyl addition with respect silyl cleavage through the introduction of a more inductive silyl groups. While use of the triphenylsilyl group, as in 1a ([Si] = Ph3Si), only exacerbated silyl cleavage to prevent formation of 3a (Table 1, entry 3), the corresponding tert-butyl-diphenylsilyl compound, 1a ([Si] = tBuPh2Si), better balanced inductive activation and stability toward cleavage, enabling formation of 3a in 48% yield (Table 1, entry 4). While adjusting the loading of water did not increase efficiency (Table 1, entries 5 and 6), decreased loadings of 2-propanol (120 mol%) and use of a slightly weaker base, K2CO3 (50 mol%) each led to modest improvements (Table 1, entries 7 and 8). Finally, introduction of 3-NO2-4-CN-BzOH (10 mol%), which presumably stabilizes the catalyst and attenuates silyl cleavage by buffering the medium, led to a much cleaner reaction, delivering 3a in 66% yield and 96% enantiomeric excess (Table 1, entry 9).
Table 1.
Selected optimization experiments in the enantioselective π-allyliridium-C,O-benzoate catalyzed allylation of acetylenic ketone 1a.a
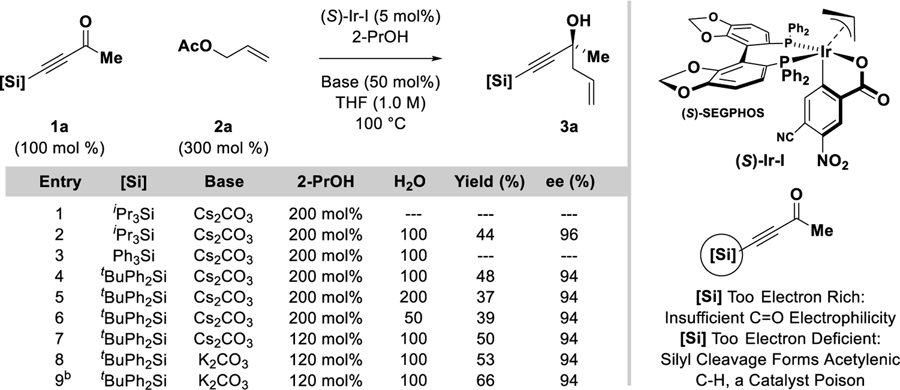 |
Yields are of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
3-NO2-4-CN-BzOH (10 mol%).
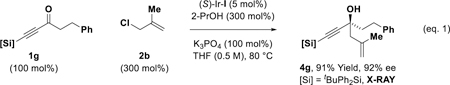
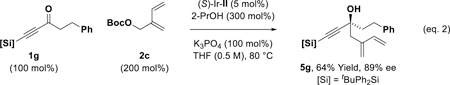
With these optimal conditions in hand, the enantioselective allylation of diverse acetylenic ketones was explored (Table 2). While simple alkyl-substituted acetylenic ketones 1a and 1b provide moderate yields of the corresponding tertiary homoallylic alcohols 3a and 3b, acetylenic ketones 1c-1g, which incorporate substituents that exert a negative σ-inductive effect, deliver the tertiary homoallylic alcohols 3c-3g, respectively, excellent yields. For α-alkoxy ketones 1c and 1d, which exert a relatively strong negative σ-inductive effect, the less inductive and more stable iPr3Si moiety can be used instead of the tBuPh2Si moiety. Remarkably, ketone 1h, which incorporate acetylenic and vinylic acyl substituents, is an effective partner of carbonyl allylation, forming the tertiary propargylic-allylic-homoallylic alcohol 3h in good yield. Tolerance of N-heterocycles and acetals are illustrated by the formation of 3i and 3j, respectively. Perhaps most significant, however, are the reactions of ketones 1k-1n. The acetylic ketone moieties of 1k-1n undergo completely chemoselective allylation in the presence of aliphatic or aromatic ketone moieties to form 3k-3n as single constitutional isomers. For all adducts 3a-3n, uniformly high levels of enantioselectivity are observed. In lower yielding transformations, unreacted ketone along with trace quantities of tert-butyldiphenylsilanol (<5 %) were observed. The absolute stereochemistry of tertiary alcohols 3a-3n was assigned in analogy to that determined for adducts 3g, which was determined by single crystal X-ray diffraction analysis. Aryl substituted acetylenic ketones did not participate in allylation. Additionally, formation of compounds 3a-3n from the propargyl alcohols corresponding to ketones 1k-1n via hydrogen auto-transfer was inefficient, as the negative inductive effect of the alkyne impedes dehydrogenation. Finally, acetylenic ketones are competent partners in related enantioselective transfer hydrogenative allylations. For example, exposure of acetylenic ketone 1g to methallyl chloride 2b and 2-propanol (300 mol%) in the presence of (S)-Ir-I (5 mol%) provides the product of methallylation 4g in 91% yield and 92% ee (eq. 1).[13] Additionally, the reductive coupling of acetylenic ketone 1g with isoprenyl tert-butoxy carbonate 2c mediated by 2-propanol (300 mol%) and catalyzed by the corresponding (S)-DM-SEGPHOS-modified catalyst (S)-Ir-II (5 mol%) provides the product of isoprenylation 5g in 64% yield and 89% ee (eq. 1).[14]
Table 2.
Enantioselective π-allyliridium-C,O-benzoate catalyzed allylation of acetylenic ketones 1a-1n via 2-propanol-mediated reductive coupling with allyl acetate 2a.a
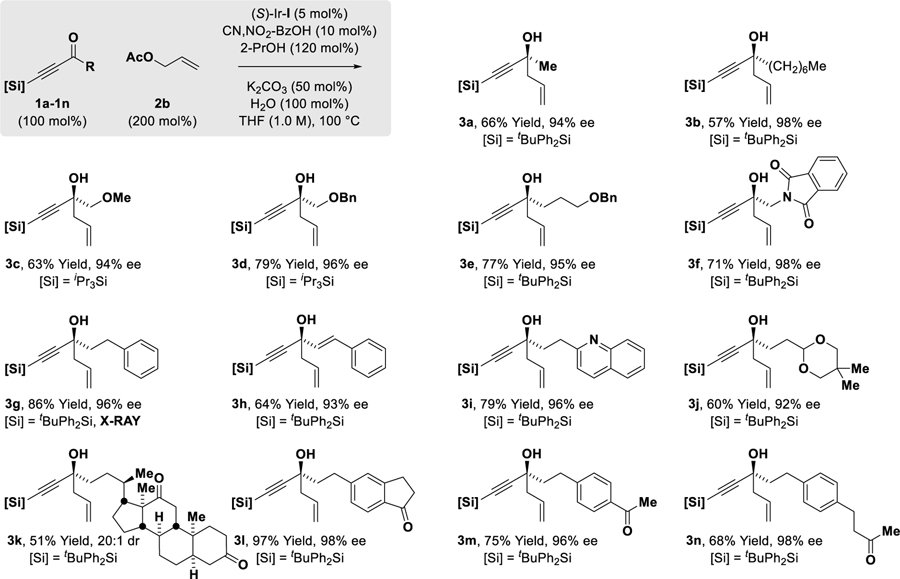 |
Yields are of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
The utility of the present ketone allylation method to natural product total synthesis is illustrated by the rapid conversion of adduct ent-3a to the C14-C23 side chain of pladienolide D (Scheme 1). The pladienolides were isolated in 2004 from the culture broth of Mer-11107, an engineered strain of Streptomyces platensis,[15] and were found to inhibit the proliferation of multiple drug resistant human cancer cells with low nanomolar IC50 values.[16] These compounds operate through a novel mechanism of action, involving binding to the splicing factor 3b (SF3b) subunit of the spliceosome.[17] Clinical evaluation of pladienolide analogue E7107 was undertaken but discontinued due to vision loss.[18] More recently, however, clinical trials were initiated with another pladienolide analogue, H3B-8800, which was granted orphan drug status by the FDA in August 2017 for treatment of myelogenous leukemia and chronic myelomonocytic leukemia.[19] Cross-metathesis of ent-3a with compound 6 (conveniently prepared via butadiene-mediated crotylation of propanal),[20] followed by Shi epoxidation[21] of the resulting olefin, delivers acetylenic epoxide 7 with high levels of stereocontrol. To corroborate the stereochemical assignment of acetylenic epoxide 7, it was subjected to silyl deprotection and Lindlar reduction to furnish the previously reported tertiary allylic alcohol 8,[21] which embodies C14-C23 of pladienolide D. Whereas the prior synthesis of tertiary allylic alcohol 8 required 16 steps (LLS), the present synthesis of 8 is completed in only 8 steps (LLS).
Scheme 1.
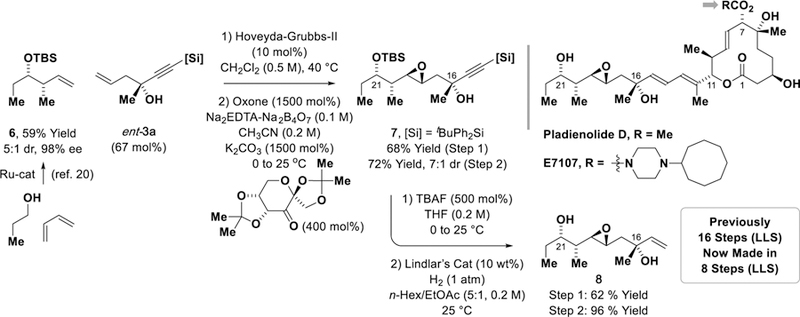
Enantioselective synthesis of C14-C23 side chain of pladienolide D.
In summary, we report the first catalytic enantioselective allylations of acetylenic ketones. Specifically, using the commercially available π-allyliridium-C,O-benzoate modified by (S)-SEGPHOS, (S)-Ir-I, as catalyst, allyl acetate 2a engages acetylenic ketones 1a-1n in enantioselective 2-propanol-mediated reductive coupling to form tertiary propargyl alcohols 3a-3n. Additionally, owing the highly chemoselective nature of this process, we demonstrate that acetylenic ketones undergo allylation in the presence of aliphatic or aromatic ketone functional groups to form single constitutional isomers. Using this protocol, a concise enantioselective synthesis of C14-C23 side chain of pladienolide D is achieved. More broadly, these studies contribute to a growing body of alcohol-mediated carbonyl additions that collectively signify a departure from the use of premetalated reagents C-C bond formation.[5d]
Supplementary Material
Acknowledgments
The Welch Foundation (F-0038) and the NIH (RO1-GM069445) are acknowledged for partial support of this research. Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) is acknowledged for postdoctoral fellowship support (G.B. 2017/00734-5).
References
- [1].a) For selected reviews on catalytic enantioselective ketone addition, see:Betancort JM, Garcia C, Walsh PJ, Synlett 2004, 749–760;; b) Ramon DJ, Yus M, Angew. Chem, 2004, 116, 286–289; Angew. Chem., Int. Ed. 2004, 43, 284–287; [Google Scholar]; c) Garcia C, Martin VS, Curr. Org. Chem 2006, 10, 1849–1889; [Google Scholar]; d) Riant O, Hannedouche J, Org. Biomol. Chem 2007, 5, 873–888; [DOI] [PubMed] [Google Scholar]; e) Cozzi PG, Hilgraf R, Zimmermann N, Eur. J. Org. Chem 2007, 5969–5994;; f) Hatano M, Ishihara K, Synthesis 2008, 1647–1675;; g) Shibasaki M, Kanai M, Chem. Rev 2008, 108, 2853–2873; [DOI] [PubMed] [Google Scholar]; h) Adachi S, Harada T, Eur. J. Org. Chem 2009, 3661–3671; [Google Scholar]; i) Wisniewska HM, Jarvo ER, J. Org. Chem 2013, 78, 11629–11636; [DOI] [PubMed] [Google Scholar]; j) Rong J, Pellegrini T, Harutyunyan SR, Chem. Eur. J 2016, 22, 3558–3570; [DOI] [PubMed] [Google Scholar]; k) Thaima T, Zamani F, Hyland CJT, Pyne SG, Synthesis 2017, 49, 1461–1480; [Google Scholar]; l) Liu Y-L, Lin X-T, Adv. Synth. Catal 2019, 361, 876–918. [Google Scholar]
- [2].A previously reported method for catalytic enantioselective carbonyl vinylation requires stoichiometric quantities of zirconium and zinc reagents. Cryogenic conditions are required and the zinc reagent is pyrophoric: Wipf P, Ribe S, J. Org. Chem 1998, 63, 6454–6455. [Google Scholar]
- [3].a) The preparation of Brown’s reagent for enantioselective carbonyl crotylation requires use of three premetallated reagents (n-BuLi, KOC4H7, Ipc2BOMe) under cryogenic conditions: Brown HC, Bhat KS, J. Am. Chem. Soc 1986, 108, 293–294; [DOI] [PubMed] [Google Scholar]; b) Brown HC, Bhat KS, J. Am. Chem. Soc 1986, 108, 5919–5923. [DOI] [PubMed] [Google Scholar]
- [4].a) For selected examples of catalytic enantioselective ketone allylation using premetallated reagents, see: Allylboration: Wada R; Oisaki K; Kanai M; Shibasaki M J. Am. Chem. Soc 2004, 126, 8910–8911; [DOI] [PubMed] [Google Scholar]; b) Lou S; Moquist PN; Schaus SE J. Am. Chem. Soc 2006, 128, 12660–12661; [DOI] [PubMed] [Google Scholar]; c) Allylsilation: >Wadamoto M; Yamamoto H J. Am. Chem. Soc 2005, 127, 14556–14557; [DOI] [PubMed] [Google Scholar]; d) Allylstannation: Casolari S; D’Addario D; Tagliavini E Org. Lett 1999, 1, 1061–1063; [Google Scholar]; e) Kim JG; Waltz KM; Kwiatkowski D; Walsh PJ J. Am. Chem. Soc 2004, 126, 12580–12585. [DOI] [PubMed] [Google Scholar]
- [5].a) For general reviews on metal-catalyzed carbonyl reductive coupling, see: Krische MJ, Jang H-Y, Metal-Catalyzed Reductive Cyclization (C=C, C≡C, C=O Bonds), Comprehensive Organometallic Chemistry III, Vol. 10 (Eds.: Mingos M, Crabtree R), Elsevier, Amsterdam, 2006, pp 493–536; [Google Scholar]; b) Metal-Catalyzed Reductive C–C Bond Formation, Topics in Current Chemistry, Vol. 279 (Ed.: Krische MJ) Springer, Berlin, Heidelberg, 2007; [Google Scholar]; c) Moragas T, Correa A, Martin R, Chem. Eur. J 2014, 20, 8242–8258; [DOI] [PubMed] [Google Scholar]; d) Nguyen KD, Park BY, Luong T, Sato H, Garza VJ, Krische MJ, Science 2016, 354, aah5133; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Holmes M, Schwartz LA, Krische MJ, Chem. Rev 2018, 118, 6026–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Recently, a (MeO)2MeSiH-mediated allene-ketone reductive coupling was reported (ref. c). In prior work from our laboratory, gaseous allene, 1-methylallene, and 1,1-dimethylallene were utilized as allyl-pronucleophiles (ref. a), and catalytic enantioselective tert-prenylations based on 1,1-dimethylallene-alcohol were developed (ref. b). These hydrogen auto-transfer processes do not require an exogenous reductant: Bower JF, Skucas E, Patman RL, Krische MJ, J. Am. Chem. Soc 2007, 129, 15134–15135; [DOI] [PubMed] [Google Scholar]; b) Han SB, Kim IS, Han H, Krische M,J, J. Am. Chem. Soc 2009, 131, 6916–6917; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu RY, Zhou Y, Yang Y, Buchwald SL, J. Am. Chem. Soc 2019, 141, 2251–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) For selected reviews on metal-catalyzed carbonyl reductive coupling via hydrogenation, transfer hydrogenation and hydrogen auto-transfer see: Ngai M-Y, Kong J-R, Krische MJ, J. Org. Chem 2007, 72, 1063–1072; [DOI] [PubMed] [Google Scholar]; b) Iida H, Krische MJ, Top. Curr. Chem 2007, 279, 77–104; [Google Scholar]; c) Bower JF, Krische MJ, Top. Organomet. Chem 2011, 43, 107–138; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hassan A, Krische MJ, Org. Proc. Res. Devel 2011, 15, 1236–1242; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Ketcham JM, Shin I, Montgomery TP, Krische MJ, Angew. Chem 2014, 126, 9294–9302; Angew. Chem. Int. Ed. 2014, 53, 9142–9150; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Kim SW, Zhang W, Krische MJ, Acc. Chem. Res 2017, 50, 2371; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Ambler BR, Woo SK, Krische MJ ChemCatChem 2019, 11, 324–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Itoh J, Han SB, Krische MJ, Angew. Chem 2009, 121, 6431–6434; Angew. Chem. Int. Ed. 2009, 48, 6313–6316. [Google Scholar]
- [9].For prior studies on the catalytic enantioselective transfer hydrogenative allylation of acetylenic aldehydes, see: Brito GA, Della-Felice F, Luo G, Burns AS, Pilli R, Rychnovsky SD, Krische MJ, Org. Lett 2018, 20, 4144–4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) For isolated examples of catalytic enantioselective allylation of acetylenic ketones, see: Jadhav PK, Bhat KS, Perumal PT, Brown HC, J. Org. Chem 1986, 51, 432–439. [Google Scholar]; b) Alam R, Raducan M, Eriksson L, Szabó KJ, Org. Lett 2013, 15, 2546–2549. [DOI] [PubMed] [Google Scholar]; c) Robbins DW, Lee K, Silverio DL, Volkov A, Torker S, Hoveyda AH, Angew. Chem 2016, 128, 9762–9766. Angew. Chem. Int. Ed. 2016, 55, 9610–9614. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) van der Mei FW, Qin C, Morrison RJ, Hoveyda AH, J. Am. Chem. Soc 2017, 139, 9053–9065 and reference 4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nakamura M, Hirai A, Sogi M, Nakamura E, J. Am. Chem. Soc 1998, 120, 5846–5847. [Google Scholar]
- [12].a) For selected metal-catalyzed reactions of 1,5-enynes, see: Wang S, Zhang L, J. Am. Chem. Soc 2006, 128, 14274–14275. [DOI] [PubMed] [Google Scholar]; b) Debleds O, Campagne J-M, J. Am. Chem. Soc 2008, 130, 1562–1563. [DOI] [PubMed] [Google Scholar]; c) Nelsen DL, Gagne MR, Organometallics 2009, 28, 950–952. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Li J, Liu X, Lee D, Org. Lett 2012, 14, 410–413. [DOI] [PubMed] [Google Scholar]; e) Chen Z-Z, Liu S, Hao W-J, Xu G, Wu S, Miao J-N, Jiang B, Wang S-L, Tu S-J, Li G, Chem. Sci 2015, 6, 6654–6658. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Chen G-Q, Fang W, Wei Y, Tang X-Y, Shi M, Chem. Sci 2016, 7, 4318–4328. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Bohan PT, Toste FD, J. Am. Chem. Soc 2017, 139, 11016–11019. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Lu X-L, Lyu M-Y, Peng X-S, Wong HNC, Angew. Chem 2018, 130, 11535–11538. Angew. Chem. Int. Ed. 2018, 57, 11365–11368. [Google Scholar]
- [13].Hassan A, Townsend IA, Krische MJ, Chem. Comm 2011, 10028–10030. [DOI] [PMC free article] [PubMed]
- [14].Xiang M, Luo G, Wang Y, Krische MJ, Chem. Comm 2019, 55, 981–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) For isolation of pladienolides A-G, see: Sakai T, Sameshima T, Matsufuji M, Kawamura N, Dobashi K, Mizui Y, J. Antibiot 2004, 57, 173–179; [DOI] [PubMed] [Google Scholar]; b) Sakai T, Asai N, Okuda A, Kawamura N, Mizui Y, J. Antibiot 2004, 57, 180–187; [DOI] [PubMed] [Google Scholar]; c) Asai N, Kotake Y, Niijima J, Fukuda Y, Uehara T, Sakai T, J. Antibiot 2007, 60, 364–369. [DOI] [PubMed] [Google Scholar]
- [16].Mizui Y; Sakai T; Iwata M; Uenaka T; Okamoto K; Shimizu H; Yamori T; Yoshimatsu K; Asada M J. Antibiot 2004, 57, 188–196. [DOI] [PubMed] [Google Scholar]
- [17].For a recent investigation into the binding mode of pladienolide and related splicing modulators, see: Teng T, Tsai JHC, Puyang X, Seiler M, Peng S, Prajapati S, Aird D, Buonamici S, Caleb B, Chan B, Corson L, Feala J, Fekkes P, Gerard B, Karr C, Korpal M, Liu X, Lowe JT, Mizui Y, Palacino J, Park E, Smith PG, Subramanian V, Wu ZJ, Zhou J, Yu L, Chicas A, Warmuth M, Larson N, Zhou P, Nature Comm 2017, 8, 15522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].For a review of pladienolide and related natural products that inhibit splicing factor 3B subunit 1, see: Pham D, Koide K, Nat. Prod. Rep 2016, 33, 637–647. [DOI] [PubMed] [Google Scholar]
- [19].a) ClinicalTrials.gov Identifier: ; [Google Scholar]; b) Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, Agrawal AA, Caleb B, Csibi A, Sean E, Fekkes P, Karr C, Klimek V, Lai G, Lee L, Kumar P, Lee SC-W, Liu X, Mackenzie C, Meeske C, Mizui Y, Padron E, Park E, Pazolli E, Peng S, Prajapati S, Taylor J, Teng T, Wang J, Warmuth M, Yao H, Yu L, Zhu P, Abdel-Wahab O, Smith PG, Buonamici S, Nature Med 2018, 24, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) McInturff EL, Yamaguchi E, Krische MJ, J. Am. Chem. Soc 2012, 134, 20628; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gao X, Woo SK, Krische MJ, J. Am. Chem. Soc 2013, 135, 4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].For a related Shi epoxidation, see: Kanada RM, Itoh D, Nagai M, Niijima J, Asai N, Mizui Y, Abe S, Kotake Y, Angew. Chem 2007, 119, 4428–4433; Angew. Chem. Int. Ed. 2007, 46, 4350–4355. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.