

Abstract
Cholangiocarcinoma (CCA) mortality rates are increasing due to rising incidence and limited curative treatment(s) for patients with advanced disease. NOTCH pathway reactivation has been reported in biliary malignancies to conflicting degrees, hindering prioritization of key therapeutic targets within the network and identification of candidate responder patients for NOTCH-directed therapies. We analyzed genomic data from 341 CCA patients and identified NOTCH1 significantly increased in a subgroup characterized by distinct stromal infiltration. Network-wide imbalance of the NOTCH pathway was seen in CCA, including correlation of NOTCH1 with NOTCH3 and NOTCH ligands. Given the diversity of observed NOTCH receptor engagement, γ-secretase modulation was rationalized as a therapeutic option. Indeed, subcutaneous transplantation of sensitive and resistant CCA cell lines pre-treated with a γ-secretase inhibitor (GSi) cocktail demonstrated the antineoplastic effects of GSi in a subset of CCA and led to development of a 225-gene responder signature. This signature was validated in an independent cohort of 119 patients. Further, this signature was enriched in liver tumors initiated by hydrodynamic injections of activated-NOTCH as compared to the AKT-RAS-driven tumors. Candidate GSi-responder patients were characterized by distinct transcriptomes overlapping with previous hepatobiliary metastasis and stemness, unique stromal properties and dysfunctional intra-tumoral immune infiltration. Pan-cancer analysis identified 41.9% of cancer types to harbor prospective GSi-responder patients, which was adapted into a 20-gene GSi-sensitivity score matric capable of discriminating nanomolar versus micromolar sensitivity to a cell permeable GSi (Z-LLNle-CHO) across 60 diverse tumor lines (AUC=1). Conclusion: We have established a GSi-responder signature with evidence across several patient cohorts, as well as in vitro and in vivo models, to enable precision medicine application of NOTCH-directed therapy in CCA as well as prospectively across diverse malignancies.
Keywords: Cholangiocarcinoma, Bile duct, Notch, Gamma-secretase (GS) inhibitor, Theranostic response signature
INTRODUCTION
Cholangiocarcinoma (CCA) comprise a heterogeneous spectrum of adenocarcinoma arising within the biliary system, extending from intrahepatic ductules outwards towards the distal common bile. These aggressive tumors are characterized by intermediate mutational burden(1), extensive desmoplastic stroma(2), and atypically widespread cancer stem cell subpopulations within tumors(3). In contrast to general cancer trends, CCA mortality have progressively increased(4, 5), a phenomenon attributable at least in part to increasing incidence and failure to clearly advance curative or palliative clinical management. Currently, the only potential cures for CCA are surgical resection or liver transplantation, the former which is marred by a recurrence rate of 62% at 26 months post-operatively(6), and the latter which is still debated and rate-limited by donor availability and recipient eligibility. Despite these options, curative intervention is simply not possible for most patients due to presentation with locally advanced or metastatic disease. The intuitive solution to overcome such clinical stalemates would invoke enhanced diagnostics enabling earlier detection, a concept which is currently negated by the lack of non-invasive biomarkers and logistically due to limitations in population screening for this rare disease. Advancing chemotherapy and targeted therapies to elicit durable responses would enable chronic management of advanced disease and control the increasing mortality rate associated with biliary malignancies. Since the 1990s, gemcitabine has served as the standard palliative chemotherapy backbone which combined with cisplatin (GemCis) achieves median survival of 11.7 months post-diagnosis(7). No clear indications for second-line chemotherapy exist. Given the limited survival advantage of GemCis, it has been suggested that some patients should be enrolled in experimental trials in place of standard-of-care(8). Any experimental trials should require careful biomarker-driven stratification of prospective responders, given the potential of individual driver mutations to influence diverse pharmacologic responses(9). Presently, treatment of FGFR2 fusion-positive intrahepatic CCA (iCCA) patients with the FGFR2 inhibitor BGJ398 is the only current example of personalized translational success for biliary tumors, significantly extending progression-free survival(10). Inspired by a promise of targeted therapy, though constrained by the restricted number of recurrently dysregulated networks in CCA, it is now imperative to apply precision medicine strategies to revisit oncogenic networks that have traditionally been considered difficult to modulate and/or tolerate.
The NOTCH network ensures an intercellular communication initiated by in trans binding of 5 ligands (JAG1, JAG2, DLL1, DLL3, DLL4) to four corresponding transmembrane receptors (NOTCH1-4). Upon receptor-ligand engagement, NOTCH receptors undergo a series of extracellular (mediated by ADAM10 or ADAM17) and intracellular (mediated by γ-secretase (GS) complex) proteolytic processing events. This generates the NOTCH intracellular domain (NICD) fragment, which is rapidly shuttled to the nucleus to activate downstream targets, such as HES1 and HEY1 transcription factors. The NOTCH signaling pathway is closely associated with the biliary system, playing key roles in developmental biliary specification(11) and morphogenesis. Constitutive NOTCH activation in hepatocytes(12, 13) and hepatic progenitor cells(14) has been proven to induce iCCA in vivo. Animal models have identified transformative synergy of constitutive NOTCH activity in combination with specific genomic insults, including NICD/shP53/IDH1R132C in hydrodynamic models(15) and observation of aberrant post-transformation NOTCH activation in mutant PIK3CA/Yap mice(16) and following cholangiocyte-specific p53-loss in mice exposed to chemical insult(17). NOTCH receptors have been reported as upregulated to varying contradicting degrees(14, 18, 19), with NOTCH3 being the strongest patient-relevant receptor in CCA to date(18). Accordingly, the therapeutic potential of siRNA(20) and antibodies(21) directed against individual NOTCH receptors and ligands, as well as small molecule inhibitors of the GS complex(19) have been studied in CCA. However, translating NOTCH-directed therapies into the clinic has been far from successful with 40% clinical trials listed as terminated or withdrawn as of 2018. To make NOTCH-targeted therapy a viable clinical success, thorough guidelines are required for patient selection.
In this study, we performed transcriptomic characterization of the NOTCH receptors along extensive network analysis of the dominant signaling pathways in CCA patients. We rationalized GS as the most clinically feasible NOTCH component to target and confirmed its anti-tumor effects using in vivo and ex vivo CCA models. Finally, we derived a pan-(γ-secretase) inhibitor (GSi) responder signature capable of actively and prospectively predicting therapeutic response of various CCA models and diverse cancer types to GSi.
RESULTS
CLINICOPATHOLOGIC IMPLICATIONS OF NOTCH RECEPTOR EXPRESSION IN CHOLANGIOCARCINOMA
NOTCH receptor expression was assessed in resected tissue from 186 tumors and 131 paired surrounding livers (SL) across two independent cohorts of CCA patients: LEC2012(22) and LEC2018 that includes an additional 82 tumors and 71 SL tissues (Supplementary Table1). Comparison of clinicopathologic parameters showed that LEC2012 contained significantly higher numbers of patients with lymph node metastasis (P=0.000382) and perineural invasion (P=0.000839) indicating that LEC2012 comprises patients with more advanced disease, higher proportion of perihilar tumors (P<0.00001), and smaller tumor size (P=0.0013) (Supplementary Table2). Analysis of CCA samples in comparison with peri-tumoral SL tissues uncovered NOTCH1 (P<0.002, P<0.0001; Supplementary Figure1A) and NOTCH3 (P<0.0001, P<0.0001; Supplementary Figure1B) being significantly upregulated in in LEC2012 and LEC2018 cohorts, respectively. In contrast, neither NOTCH2 nor NOTCH4 were differentially expressed in either cohort (Supplementary Figure1C–D). Intra-patient expression of each NOTCH receptor was highly variable among patients (Supplementary Figure1E). Indeed, hierarchical clustering of inter-patient NOTCH receptor expression identified unique subgroups of CCA patients, namely a NOTCH1-overexpressing subgroup in LEC2012 (Figure1A) and two separate NOTCH1- and NOTCH3-overexpressing subgroups in LEC2018 (Figure1B). In univariate analysis, NOTCH1 expression was found to be associated with lymph node metastasis (P=0.0255), tumor necrosis (P=0.04506) and lower age at diagnosis (P=0.0241) (Supplementary Table3). Moreover, increased NOTCH3 was associated with poorly differentiated tumors (P=0.00105) in LEC2012, a finding in agreement with a previous immunohistochemical study in eCCA(19). Notably, no associations between NOTCH1 or NOTCH3 expression and tumor location (intrahepatic versus perihilar) were observed, though NOTCH3 trended towards association with intrahepatic disease (P=0.0639) in LEC2012. Kaplan-Meier analysis identified worse survival among NOTCH1high patients (defined as above median expression) in LEC2012 (P<0.03) (Figure1C), whereas no association between expression level and survival was noted for NOTCH3.
Figure 1. NOTCH receptor expression and clinicopathologic implications in CCA.
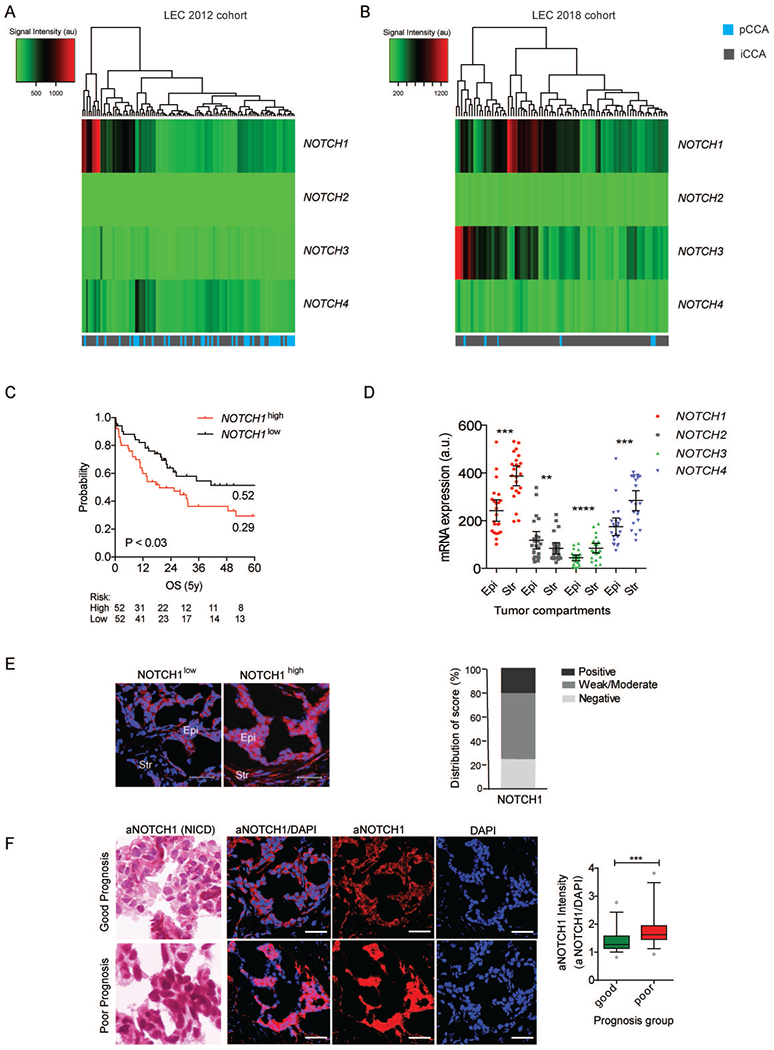
(A, B) Hierarchical clustering of the four NOTCH receptor genes in 104 CCA tissue samples in LEC2012 (A) and in 82 CCA tissue samples in LEC2018 (B). iCCA: intrahepatic CCA. pCCA: perihilar CCA. (C) Survival analysis of patients according to NOTCH hierarchical clustering. Patients were stratified by NOTCH1 median expression. Kaplan-Meier and Mantel-Cox statistics were used to determine levels of significance. (D) mRNA expression of four NOTCH receptors in matched tumor epithelia and tumor stroma from laser micro-dissected CCA patient tissue (n=23). Epi, Tumor epithelia; Str, Tumor stroma. (E) Immunofluorescent analysis of full-length NOTCH1 protein in CCA tumors (n=20, random from LEC2012). Representative images of high and low expression level tissues are shown. Epi, Tumor epithelia; Str, Tumor stroma. Scale bar: 50 µm. (F) Heamatoxylin staining and immunofluorescent analyses of activated NOTCH1 (aNOTCH1) protein in CCA tumors (LEC2012, n=24). Representative images of good and poor prognosis patient tissues are shown (n=12, each)(22). Scale bar: 50 µm. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Given our identification of a NOTCH1high CCA subgroup in both cohorts, and its association with adverse prognostic factors in more advanced disease (LEC2012), we next sought to confirm mRNA findings at the protein level. Normalization of levels from tumor epithelia and stroma through laser microdissection and transcriptome profiling of a subset of LEC2012 tumors (n=23 pairs, serial sections), higher NOTCH1 (P=0.0001), NOTCH3 (P<0.0001) and NOTCH4 (P=0.0002) expression was observed in the intra-tumor stromal compartment compared to matched tumor epithelia, whereas NOTCH2 was significantly higher in tumor epithelia (P=0.0043) (Figure1D). Western blot analysis of a subset of bulk tumor samples within LEC2012 indicated elevation of NOTCH1 protein level in 29.2% (7/24) patients, resembling the frequency of patients with increased NOTCH1 mRNA (Supplementary Figure 2). Similarly, immunofluorescent analysis of randomly selected tumors within LEC2012 confirmed 20% (4/20) of patients with strongly positive NOTCH1 particularly in epithelia (Epi) compared to stroma (Str), 55% (1½0) with moderate-to-weak NOTCH1 and 25% (5/20) with negative NOTCH1 staining (Figure 1E). Further, heamatoxylin and immunofluorescent staining of activated-NOTCH1 (aNOTCH1; NOTCH Intracellular Domain (NICD)) in patients from LEC2012 demonstrated significantly higher aNOTCH1 in poor prognosis patients (P=0.0006), as previously defined by our laboratory(22) (Figure1F).
Subsequent to our findings of NOTCH1 upregulation and activation in CCA patients, we sought to determine whether NOTCH1 upregulation could be attributed to aberrant genomic mechanisms. First, we analyzed matched whole exome- and RNA-seq data(1) and identified the highest (n=28) and lowest (n=25) NOTCH1 expression quartiles of patients. None of the NOTCH receptors were mutated among these samples. Furthermore, neither mutational signatures (Supplementary Figure3) nor recurrent mutational targets (Supplementary Figure4) substantially differed between the patient quartiles indicating NOTCH1 upregulation as largely independent of underlying mutational processes and programs. To investigate prospective epigenetic mechanisms for NOTCH1 upregulation in CCA, we analyzed DNA methylation from 118 independent patients with matched gene expression(23). Comparison of the highest (n=30) and lowest (n=27) NOTCH1 expression quartiles revealed no significant differences in methylation of either loci (cg07658207, cg16829154) located nearby the NOTCH1 transcriptional start site (Supplementary Figure5), thus negating DNA methylation-associated transcriptional activation. Lastly, given normalized NOTCH1 expression to be higher in tumor stroma than tumor epithelia in microdissected compartments (Figure1D), we asked whether biliary tumors with high NOTCH1 expression possessed higher stromal content. Based on our micro-dissected data, we established ‘reference’ transcriptomes for tumor epithelia and stroma and determined the relative proportion of these tumor compartments in 186 CCA patients using the CIBERSORT algorithm(24). NOTCH1 mRNA levels were found to be significantly positively correlated with tumor stroma content (LEC2012: Spearman r= 0.2325, P= 0.0175; LEC2018: Spearman r=0.3247, P = 0.0029) and negatively correlated with tumor epithelia content (LEC2012: Spearman r=−0.2325, P=0.0175; LEC2018: Spearman r=−0.3247, P=0.0029) in both cohorts (Supplementary Table4). The estimates of the stromal content significantly correlated with the stromal marker VIMENTIN (VIM) in both cohorts (LEC2012: Spearman r=0.5454, P<0.0001; LEC2018: Spearman r=0.5549, P<0.0001) supporting the robustness of this method (Supplementary Figure6). Immunohistochemical analysis of intra-tumoral immune infiltrates in an independent CCA tissue microarray (n=36) stratified based presence or absence of NOTCH1 staining identified increased numbers of FoxP3+ immune cells in NOTCH1-elevated tumors (P=0.0019) but no significant difference in immune cell NOTCH1-expression between tumor subgroups (Supplementary Figure7). Thus, high NOTCH1 expression in CCA appears to be mediated at least in part by or associated with enhanced desmoplastic stroma reaction and independent of infiltrative immune cell NOTCH1 expression.
NETWORK-WIDE DYSREGULATION OF NOTCH PATHWAY IN CHOLANGIOCARCINOMA
Canonical NOTCH signaling may be deconstructed into groups of NOTCH ligands, NOTCH receptors and membrane proteolytic processing, alongside ubiquitin ligase modulators and nuclear interactors. Accordingly, we examined alterations in transcript levels of NOTCH pathway genes as defined by the Pathway Interaction Database. Among NOTCH pathway genes, 64.3% (36/56 targets) and 75% (42/57 targets) were aberrantly expressed in CCA compared with SL tissues (P<0.05) in both cohorts, confirming extensive network-wide imbalance in CCA (Figure 2A, Supplementary Table5). Further, 30 transcripts (24 upregulated, 6 downregulated) were commonly deregulated in CCA as compared with SL. Notably, 4 (JAG1, JAG2, DLL3, DLL4) out of 5 known NOTCH ligands were recurrently upregulated, indicating hyperstimulation of already elevated NOTCH receptor levels. Transcriptomics also confirmed upregulation of 4 (APH1A, APH1B, NCSTN, PSNEN) out of 5 known GS subunits, inferring increased NOTCH receptor cleavage and intracellular activity, as well as upregulation of the GS catalytic core, PSEN1, in LEC2018. These data collectively emphasize the fundamental importance of GS complex stabilization and activation. Notable transcriptional downregulation events included those of key transcriptional repressors (NCOR1, SPEN) and rate-limiting ubiquitin ligases (CUL1, ITCH). Given the prominence of NOTCH1-overexpressing subgroups in both patient cohorts, we next evaluated NOTCH network genes for which expression significantly correlated with NOTCH1 levels (Supplementary Table6). In total, 12 NOTCH pathway members were positively correlated (Figure2B) and 5 negatively correlated (Figure2C) with NOTCH1 levels across both cohorts. Remarkably, 4 (DLL1, DLL4, JAG1, JAG2) out of 5 NOTCH ligands were positively correlated with NOTCH1, inferring multiple putative ligand usage with the receptor. Moreover, NOTCH1 and NOTCH3 transcript levels were positively correlated, further suggesting a prospective co-operative interplay of these receptors during biliary transformation. Thus, monotherapies directed against individual receptors or ligands appear to be less likely to ensure durable responses in comparison to the agents targeting the fundamental NOTCH processes, such as membrane proteolytic cleavage mediated by the GS complex.
Figure 2. NOTCH network dysregulation in CCA.
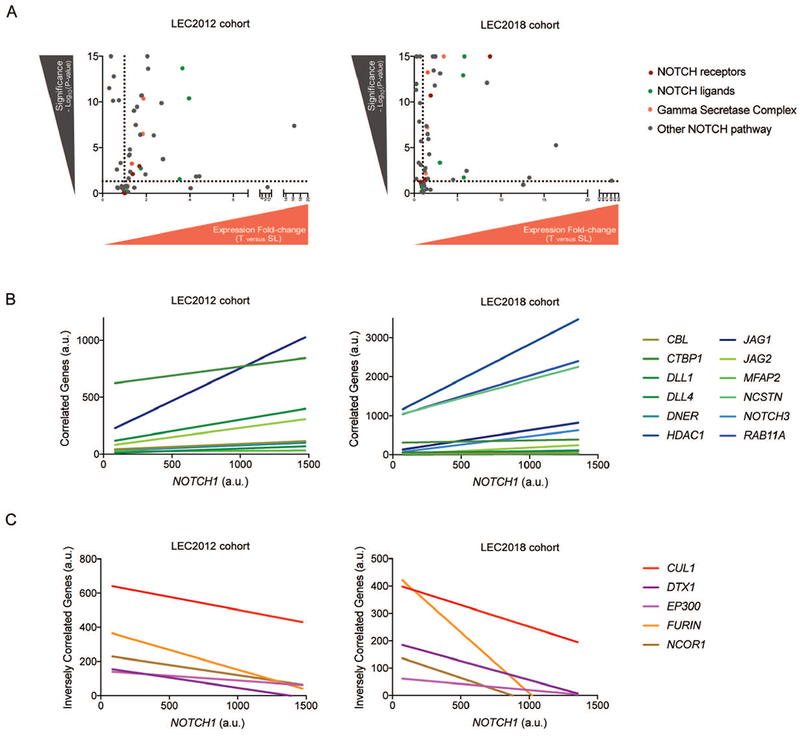
(A) NOTCH network genes differentially expressed between CCA and SL tissues in LEC2012 and LEC2018. (B) Significant positive correlation of NOTCH network genes with NOTCH1 mRNA levels common across both cohorts. (C) Significant negative correlation of NOTCH network genes with NOTCH1 mRNA levels common across LEC2012 and LEC2018.
CHOLANGIOCARCINOMA DISPLAYS HETEROGENEOUS THERAPEUTIC RESPONSE TO γ-SECRETASE INHIBITORS IN VITRO AND IN VIVO
Having rationalized targeting the GS complex to overcome the diversity of ligand and receptor usage observed in CCA patients, we evaluated the therapeutic potential of two small molecule γ-secretase inhibitors (GSi), R04929097 and YO-01027 (Dibenzazepine), in vitro and in vivo. In total, 13 different CCA cell line models were employed in order to recapitulate the inter-individual transcriptomic heterogeneity observed among patients and to identify the best-fit models of prospective chemosensitivity or chemoresistance to GSi treatment. Each cell line was treated with either drug over a dose range (0.1-=10μM) for 3 or 7 days to determine the gross sensitivity profiles across the models as judged by the cleaved NOTCH1 protein levels (Figure3A). Remarkably, dose response profiling (1nM-100μM) revealed only limited effects of treatment on viability (Supplementary Table7). Given our data on microdissected tissues, showing enhanced NOTCH receptor (particularly NOTCH1 and NOTCH3) expression in the stroma versus epithelia compartments, and the role of NOTCH signaling in the tumor microenvironment(25), we hypothesized that the anti-proliferative effects of GSi would be observed in vivo. From the 13 examined CCA lines, HuCCT-1 was selected as a model of GSi-sensitivity and WITT as a model of GSi-resistance. Both cell lines were pre-treated with a mixture of R04929097 and YO-01027 (1μM, 1:1 ratio) for 24hours and transplanted subcutaneously into right flanks of mice (Figure3B). Parental control cells were pre-treated with the vehicle (DMSO) and injected into the left flanks. Exposure to GSi significantly reduced tumor growth of the sensitive HuCCT-1 xenograft tumors compared with controls (P<0.001) (Figure3C). In contrast, the tumor growth initiated by GSi-resistant WITT cells was unaffected (Figure3D). Subsequent transcriptome profiling of control and GSi-sensitive tumors produced a 225-gene responder signature (P<1×10−4) (Figure4A–B, Supplementary Table8). In striking contrast, the same differential expression analysis of the GSi-resistant and control tumors identified only two significantly changed genes (HIST4H4, RPL13) (Figure4A–B). Gene set enrichment analysis (GSEA) confirmed that NOTCH was the most significantly affected pathway in GSi-sensitive tumors (P<0.001) (Figure4C). Among the most significantly enriched transcripts were key NOTCH receptors (NOTCH1, NOTCH3), NOTCH ligands (DLL1, JAG1) and GS subunits (APH1A, APH1B, NCSTN, PSEN1) (Figure4D).
Figure 3. Pharmacologic inhibition of γ-secretase in vitro and in vivo.
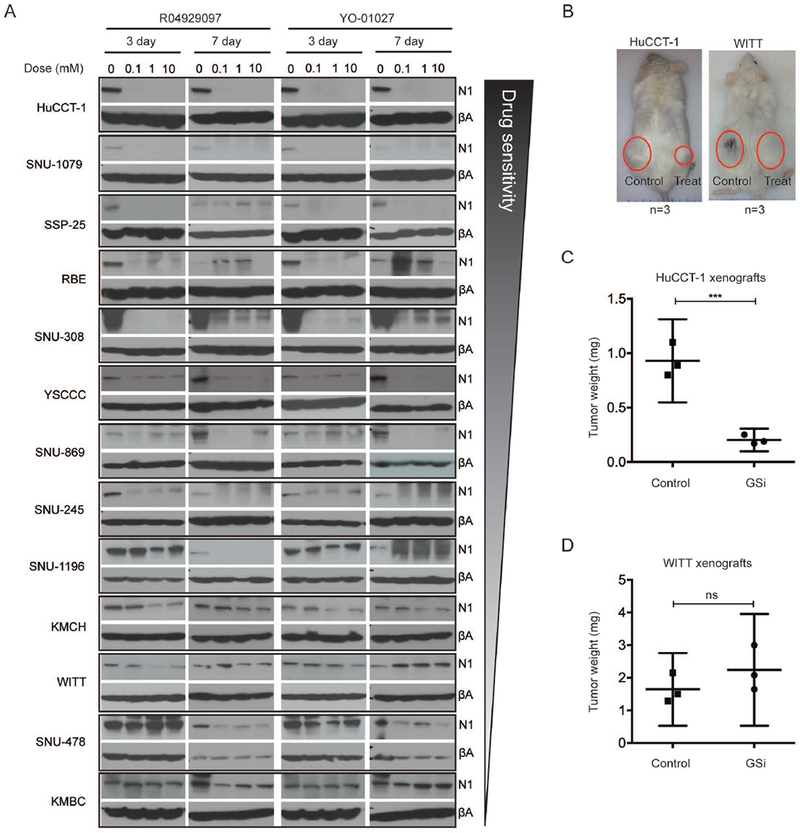
(A) NOTCH1 protein expression in 13 CCA cell lines treated with two different GSi (RO4929097 and YO-01027) for 3- and 7-days. GSi were tested over the dilution series (0.1-10μM). βA: β-actin. N1: NOTCH1. (B) Mice at time of sacrifice (maximum tumor size) after subcutaneous injection of with HuCCT-1 and WITT cell lines, each with and without prior treatment with GSi. (C, D) Effect of GSi pre-treatment on tumor growth initiated by HuCCT-1 (GSi sensitive) (C) and WITT (GSi resistant) cells (D). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Figure 4. Development and validation of a GSi-responder signature in vivo.
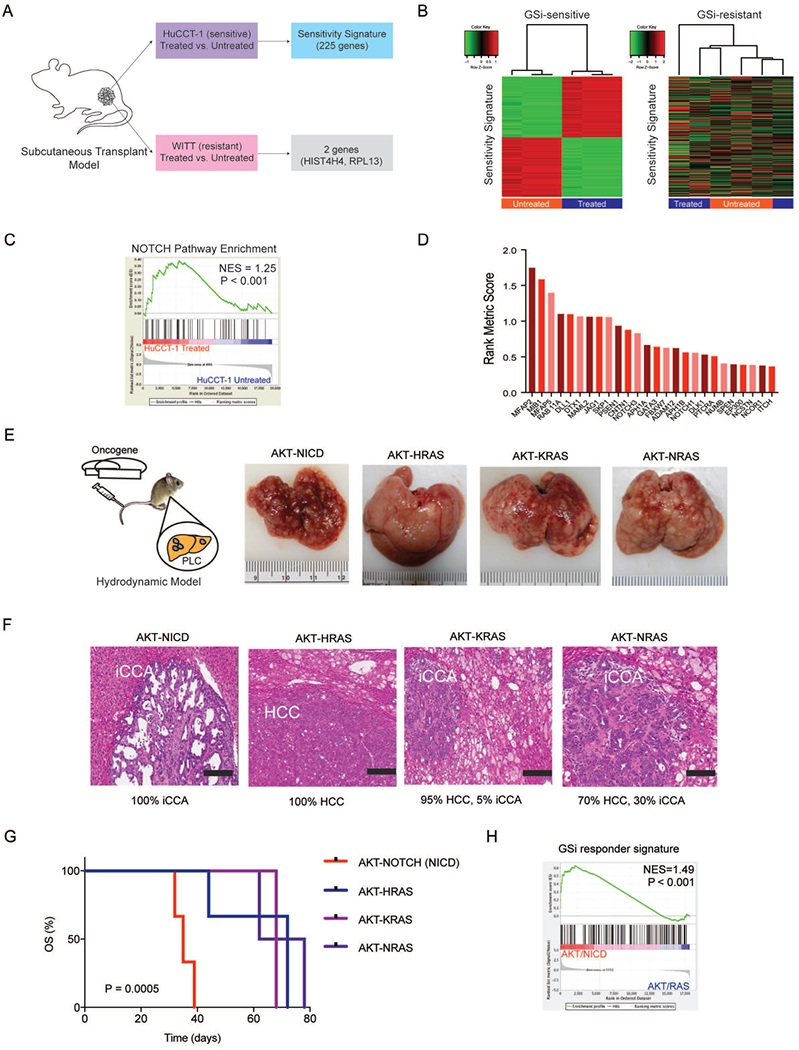
(A) Schematic overview of generating the GSi-sensitivity signature using xenograft models of GSi sensitive (HuCCT-1) and resistant (WITT) cell lines treated with GSi. (B) Hierarchical clustering using 225-gene responder signature in sensitive and resistant tumors. (C) Gene set enrichment analysis (GSEA) of NOTCH pathway in sensitive tumors with and without GSi treatment. NES: normalized enrichment score. (D) Rank metric scores of NOTCH pathway mRNA transcripts significantly altered in xenograft tumors initiated by GSi treatment. (E) Schematic overview of the hydrodynamic tail vein models (AKT/NICD, AKT/HRASG12R, AKT/KRASG12D, AKT/NRASG12V) used to generate primary liver tumors in vivo and gross liver morphology at the end of study. (F) Histopathological classification of primary liver tumors formed across each of the hydrodynamic mouse models. H&E staining (scale bar, 200 μm). (G) Survival analysis for each of the hydrodynamic mouse model groups. Kaplan-Meier and Mantel-Cox statistics were used to determine levels of significance. (H) GSEA of GSi-responder signature in NOTCH-driven tumors versus RAS-driven tumors. NES: normalized enrichment score.
GSi-RESPONDER SIGNATURE VALIDATION IN A NOTCH-DRIVEN MURINE MODEL OF CHOLANGIOCARCINOMA
Following identification of the 225-gene responder signature, we next aimed to investigate whether biliary tumors driven specifically by aberrant NOTCH signaling would respond to GSi. To accomplish this, we utilized hydrodynamic tail vein injection using Sleeping Beauty-mediated somatic integration of AKT-NICD as a model of NOTCH-driven CCA. For comparison, we employed several models of hepatocytic transformation by injections of AKT-HRASG12R, AKT-KRASG12D and AKT-NRASG12V(26) (Figure4E). All mice developed tumors but with profoundly different histopathology. AKT-NICD-driven transformation produced exclusively iCCA whereas AKT-HRAS generated 100% hepatocellular carcinoma (HCC), as verified by immunohistochemical staining for CK19 and HNF4α (Supplementary Figure8). In comparison, AKT-KRAS and AKT-NRAS caused development of both HCC and iCCA at different frequency (Figure 4F). Across mouse models, liver-to-body weight ratios significantly varied (P=0.0254) with lowest levels seen among the AKT-NICD mice (Supplementary Table9), mirroring findings in NotchIC::AlbCre mice(14). Overall survival was the worst in the AKT-NICD mice (P=0.0005) (Figure4G). Transcriptomics identified 178 mouse ortholog genes from our 225-gene GSi-responder signature (Supplementary Table10–11), which were significantly enriched in AKT-NICD tumors when compared to all other groups (P<0.001) (Figure4H).
IDENTIFICATION OF PROSPECTIVE GSi-RESPONDER CCA PATIENTS
After confirming the presence of the responder signature in GSi-naïve mice and linking it to NOTCH-driven CCA, we next investigate whether subgroups of GSi-naïve human CCA differ in GSi-responder signature expression, which in turn may render them sensitive to GSi. Transcriptomes of an independent cohort of 119 CCA patients(1) identified 20½25-responder genes. Non-negative matrix factorization (NMF) stratification of patients with the GSi-responder signature identified two groups with C1 containing 51.3% (61/119) and C2 containing 48.7% (58/119) patients (Figure5A). Both the GSi-responder signature (P=0.0232) and NOTCH pathway (P=0.0082) were significantly enriched in C2, inferring C2 as a prospective ‘responde’ subgroup of patients and C1 as the ‘non-responders’ (Figure5B). Notably, the prospective responder subgroup was characterized by significantly higher expression of all 4 NOTCH receptors (P<0.0001) (Supplementary Figure9A).
Figure 5. Identification of GSi-responder CCA patients ex vivo.
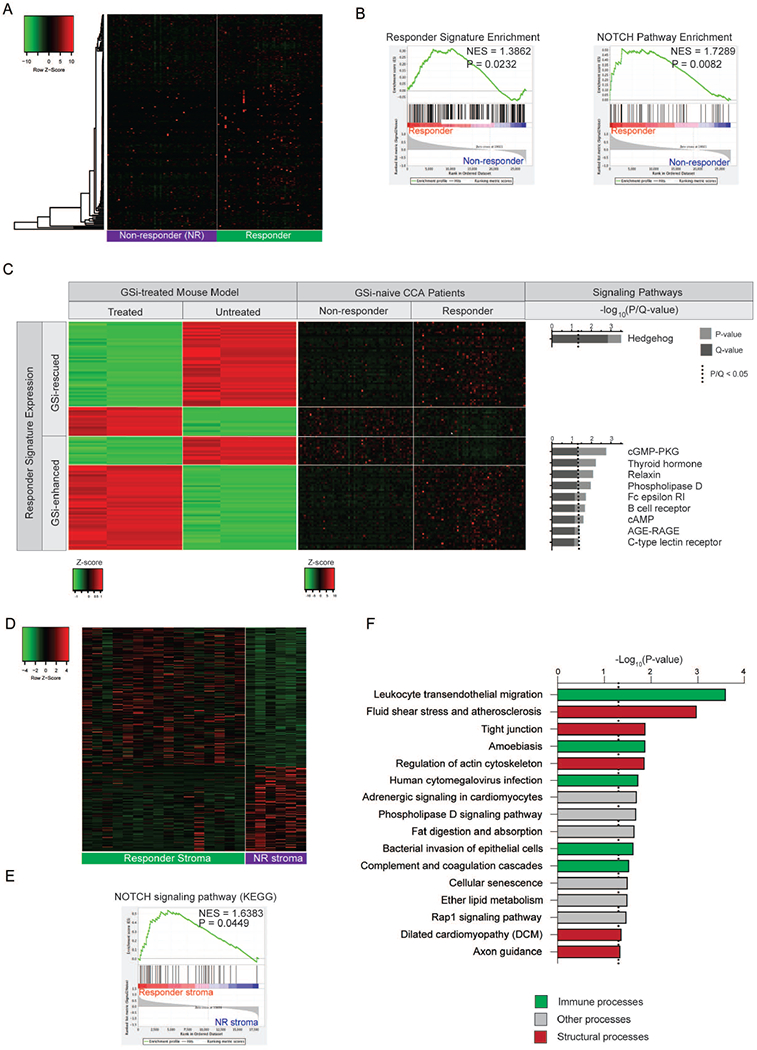
(A) Heatmap of mouse-derived GSi-responder signature expression in predicted responder and non-responder patient subgroups. NMF-based clustering using the responder signature was utilized to stratify patients. (B) Gene set enrichment analysis (GSEA) of mouse-derived GSi-responder signature and NOTCH pathway in predicted responder versus non-responder patients. NES, normalized enrichment score. (C) Heatmaps illustrating expression of 124-gene responder signature (classified as GSi-enhanced or GSi-rescued) genes mutually differentially expressed in patients (responders versus non-responders) and rescued subcutaneous mouse models (treated versus untreated) with corresponding over-represented signaling pathways. (D) Heatmap of 331-gene GSi-responder stromal signature in 22 microdissected CCA stroma samples. (E) Gene set enrichment analysis (GSEA) of NOTCH signaling pathway (KEGG) in GSi-responder versus non-responder stroma. NES: normalized enrichment score. (F) KEGG pathway over-representation analysis of the 331-gene GSi-responder stromal signature.
To evaluate the robustness of this classification, we identified the three most GSi-sensitive (HuCCT-1, SNU-1079, SSP-25) and three most GSi-resistant (KMBC, SNU-478, WITT) cell lines (Figure3A), and established reference sensitivity and resistance based on the transcriptomes, respectively. Cellular deconvolution using the corresponding reference transcriptomes confirmed that predicted responder patients had a significantly higher proportion of GSi sensitive-like CCA cells (P=0.0011) whereas the non-responder patients had a significantly higher proportion of GSi resistant-like CCA cells (P=0.0011) within tumors (Supplementary Figure9B). Since gross transcriptome signatures supported differential GSi responsiveness between the proposed subgroups, we next determined whether an innate sensitivity signature could be derived based on the treatment naïve cells. Inter-group class comparison identified 861 genes (Supplementary Table12) differentially expressed (P<0.05) between GSi-sensitive and -resistant cell lines, in which the predicted responder patients were significantly enriched (NES=1.7735, P<0.001) (Supplementary Figure9C). Thus, these analyses support that tumors from predicted GSi-responder patients closely mirror the CCA cell models of GSi-sensitivity.
Next, we virtually dissected the individual components of the xenograft-derived GSi-responder signature. From 124 genes differentially expressed (P<0.05) in both mouse and human, we defined genes based on their innate expression in the predicted responder patients and expression directionality after GSi treatment in mice (Figure5C). One subset (50%, 62/124) of genes displayed significantly different expression in responder versus non-responder patients and became progressively more dissimilar in GSi-treated versus control mice. The ‘GSi-enhanced’ genes, 25.8% (16/62) were significantly lower in GSi-naïve responder patients and became further attenuated in GSi-treated xenograft tumors, whereas the opposite trend was observed for the remaining 74.2% (46/62) of genes. These genes include an overrepresentation of immune (B cell receptor, C-type lectin receptor, Fc epsilon RI) and signaling (cGMP-PKG, AGE-RAGE) processes. In contrast, the ‘GSi-rescu’ subset of signature genes displayed significantly different expression in responder versus non-responder patients albeit with the inverse direction in GSi-treated tumors. Importantly, the core set of ‘GSi-rescu’ genes underpins a substantive component of the therapeutic response observed in mice. Remarkably, these genes are exclusively enriched in the Hedgehog (Hh) pathway (P=0.0003), previously reported as activated following prolonged GSi treatment in resistant glioblastomas through direct Hes1-binding and activation of Hh activator Gli1(27). Here, we observed 3 key Hh components (BOC, BCL2, KIF7) that were expressed at high levels in predicted responder patients and became downregulated in mice following GSi treatment. As BOC and BCL2 are positively associated with Hh signaling, their downregulation in CCA models indicates that active therapeutic response may be dependent on Hh silencing following GSi exposure. Accordingly, chemosensitization strategies for GS-directed therapies could include Hh inhibitors (vismodegib), which was evaluated and deemed tolerable in a phaseI/II trial for advanced and metastatic sarcoma ( NCT01154452).
Lastly, we evaluated the potential functional characteristics of candidate GSi-responder stroma given the positivity of NOTCH expression at mRNA and protein level within the desmoplasia. In LEC2012, 22 patients had matched bulk tumor tissue and microdissected tumor stroma. Using our original 225-gene GSi-responder signature, we stratified patients (k-means) into responders (n=17) and non-responders (n=5) as well as subsequently compared the corresponding stromal compartment between predicted GSi-sensitive tumors. As such, we unravelled a 331-gene stromal signature that discriminated GSi-responder from non-responder stroma (Figure5D, Supplementary Table13). Importantly, tumor stroma from responder patients were significantly enriched in NOTCH signalling when compared with non-responders (P=0.04; Figure5E), confirming that the stroma of GSi-responder tumors are engaging in active NOTCH-driven cross-talk. Further analysis of the 331-gene stromal signature indicated significant differences in various structural and immune signalling pathways between stromal subtypes (Figure5F), though we observed no differences in immune infiltrates by cellular deconvolution (Supplementary Table14). Given we observed no differences in intratumoral stromal content between responder and non-responder bulk tumors (Supplementary Figure10), these data indicate that GSi-responder stroma exhibit unique pathobiological profiles that may contribute to GSi-sensitivity.
PATHOBIOLOGICAL CHARACTERIZATION OF PROSPECTIVE GSi-RESPONDER CCA PATIENTS
To characterize biological differences between GSi response-based subgroups, we performed GSEA (Supplementary Table15). NOTCH was the most significantly enriched signaling pathway in predicted responders, highlighting that GSi-responder status in CCA is mediated through NOTCH, as distinct from modulation of other GS targets (Figure6A). The prospective responders were significantly enriched in many other core-signaling pathways (ErbB, Hedgehog, JAK-STAT, MAPK, MTOR, VEGF, WNT), immune (adipocytokine, B-cell receptor, FC epsilon RI, T-cell receptor, Toll-like receptor), and hormonal (GNRH, insulin, neurotrophin) processes. In contrast, limited pathways were enriched in non-responders.
Figure 6. Characterization of GSi-responder subgroup of CCA patients.
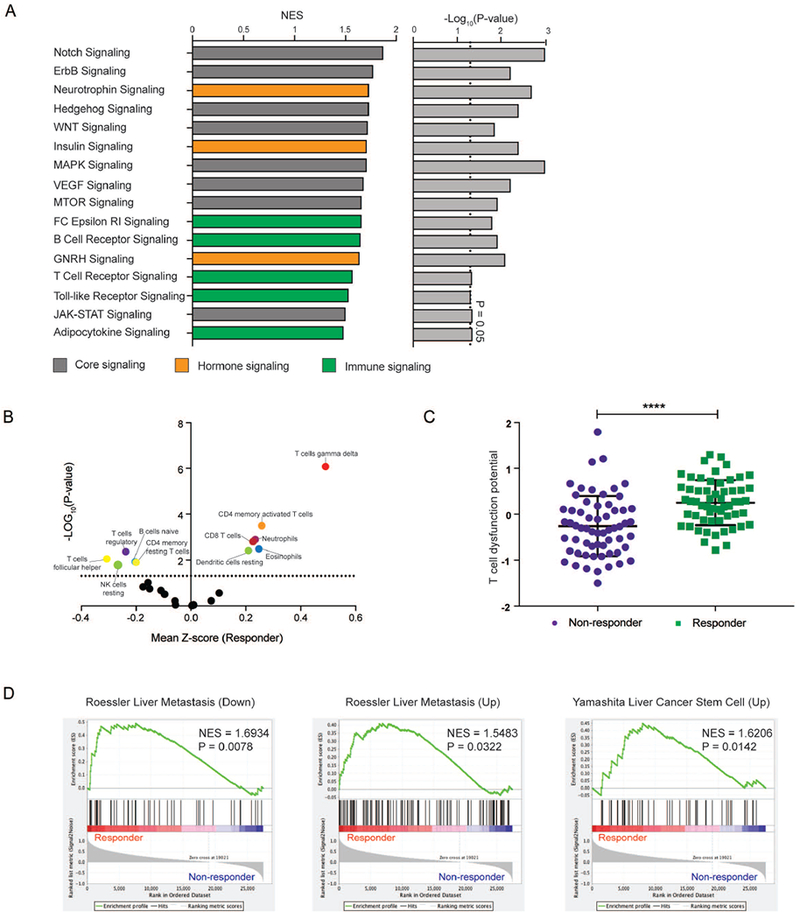
(A) Normalized enrichment scores (NES) and confidence level for significantly enriched KEGG pathways in predicted GSi-responder versus non-responder patients, as determined by GSEA. (B) Differential enrichment and depletion of immune subpopulations in predicted GSi-responders compared to non-responders. (C) Comparison of T cell dysfunction scores between predicted GSi-responder and non-responder tumors, as determined by TIDE algorithm. (D) Enrichment analysis of previously reported prognostic liver cancer signatures in predicted GSi-responder versus non-responder CCA patients. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Given the difference in immunological pathways between CCA patient subgroups and a reported link between GSi treatment and immune networks in breast cancer(28), we questioned whether these differences were caused by distinct intra-tumoral immune infiltrates. Remarkably, cellular deconvolution predicted GSi-responders appeared relatively immunogenic, with significant enrichment of γδT-cells (P=8.42×10−7), CD4 memory activated T-cells (P=3.21×10−4), eosinophils (P=3.31×10−3), neutrophils (P=1.27×10−3), CD8 T-cells (P=1.57×10−3) and resting dendritic cells (P=3.9×10−3) (Figure6B). In contrast, responder tumors were depleted in follicular helper T-cells (P=8.93×10−3), resting NK cells (P=1.63×10−2), regulatory T-cells (4.31×10−3), naïve B-cells (P=1.19×10−2) and memory resting CD4 T-cells (P=1.27×10−2). However, as presence of immune infiltrates does not indicate immune-functionality, we applied the TIDE algorithm(29) to CCA patient transcriptomes which unveiled that predicted GSi-responders have significantly greater T-cell dysfunction score than non-responders (P<0.0001, Figure6C). This implicates candidate GSi-responder tumors as highly immune-active but T-cell dysfunctional.
Next, we investigated whether the GSi-responder subgroups were associated with previously published hepatobiliary gene-signatures (Supplementary Table16). Among 11 signatures tested, 3 were found to be significantly enriched in our prospective responders: ‘ROESSLER_LIVER_CANCER_METASTASIS_DN’ (P=0.0078), ‘YAMASHITA_LIVER_CANCER_STEM_CELL_UP’ (P=0.01417), ‘ROESSLER_LIVER_CANCER_METASTASIS_UP’ (P=0.0322) (Figure6D). Thus, our GSi-responder signature appears to be reflective of the entire tumor-compartment rather than individual cellular components such that responders possess unique signaling activity, stromal biology, intra-tumoral infiltrates and pathobiological tumor properties which may influence GSi chemosensitivity.
PROSPECTIVE THERANOSTIC IMPLICATIONS OF GSi-RESPONDER SIGNATURE PAN-CANCER
To assess the potential of our signature to personalize GSi-therapy pan-cancer, we analyzed 9409 tumor transcriptomes (31 cancer types) generated by TCGA (Supplementary Table17). Similar to our analysis of GSi-naïve CCAs (Figure5A), NMF clustering on the responder signature was used to stratify patients into subgroups and subsequently tested for signature-enrichment. In total, 41.9% (13/31) of cancers (including TCGA_CHOL, n=36) were found to include a responder signature-enriched patient subgroup (P<0.05) with predicted tumor GSi-sensitivity (Figure7A). On average, responders comprised 46.2% patients ranging from 32.6% (cervical, endocervical cancers) up to 59.6% (uterine carcinosarcoma). Indeed, prospective pan-cancer GSi-responder patients were found recurrently enriched in immune pathways (consistent with CCA), in which responders were shown to be enriched in tumor immune-infiltration (Figure7B). In CCA (TCGA_CHOL, n=36), NOTCH1 (P=0.0422) and NOTCH3 (P=0.0457) were elevated in predicted responder patients (Supplementary Figure11A). In HCC (TCGA_LIHC, n=371), all 4 NOTCH receptors (P<0.0001) displayed increased expression in predicted responders (Supplementary Figure11B). Given the pleiotropic role of GS-signaling, we assessed the expression status of other known GS interactors (APP, CD44, CDH1, CDH2, EFNB2, ERBB4) along with NOTCH receptors. Ranking of tumors based on elevated NOTCH1 expression, a characteristic of predicted GSi-responders, revealed an inconsistent pattern of expression of other known GS-interacting genes both in CCA (Figure7C) and HCC (Figure7D), supporting NOTCH-directed GSi-activity in hepatobiliary cancers.
Figure 7. Prospective theranostic applications of GSi-responder signature in diverse cancer types.
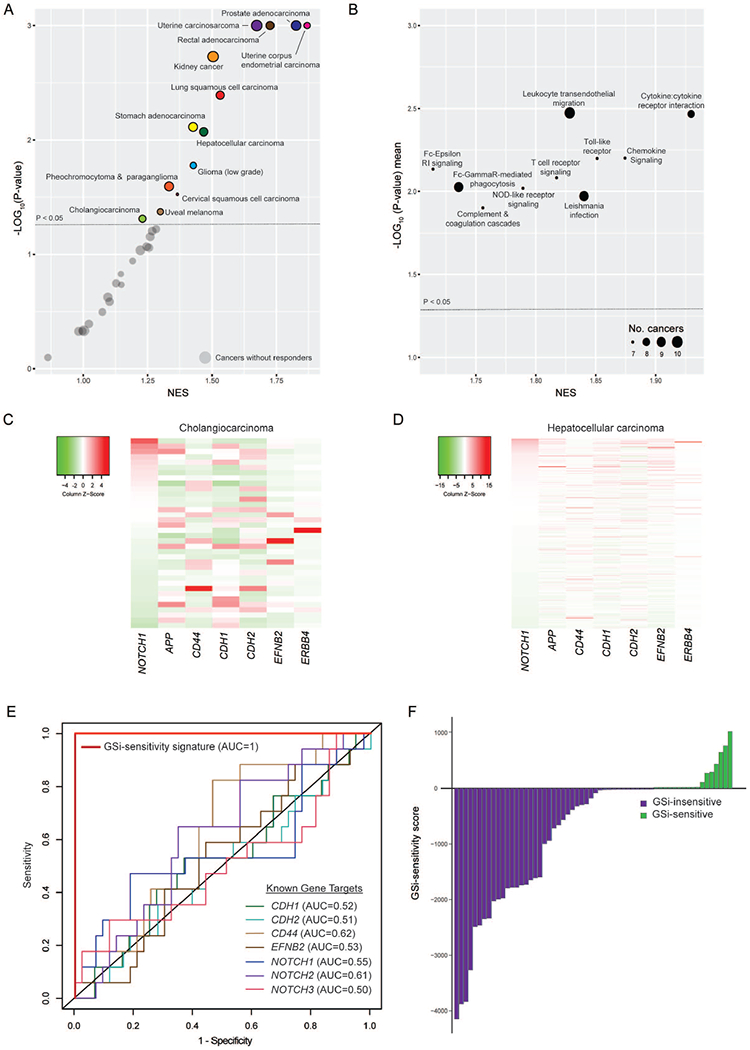
(A) Enrichment and significance of GSi-responder signature in patient subgroups across 31 cancer types. Circle area indicates relative proportion of intra-cancer responders. (B) Enrichment and significance of recurrently significant immune processes (KEGG) in prospective responder versus non-responder tumors across GSi-responder cancer types. NES and significance are presented as mean values across cancers with significant responder-enrichment. Circle area indicates number of cancer types with significant responder-enrichment. (C) Heatmap of NOTCH1 and other known GS-interactors in TCGA CCA_CHOL. (D) Heatmap of NOTCH1 and other known GS-interactors in TCGA_LIHC. (E) ROC curve highlighting the predictive performance of an optimized 20-gene sub-signature to discriminate nanomolar from micromolar GSi-sensitivity across 60 diverse cancer cell lines. Matched ROC curves are also shown for expression of key GS targets. (F) Prediction of GSi-sensitivity in 60 diverse cancer cell lines using the GSi-sensitivity score.
Next, we used the Genomics-of-Drug-Sensitivity-in-Cancer Database to identify 60 solid cancer cell lines with available transcriptomes and drug screening data with Z-LLNle-CHO, a cell-permeable GSi (Supplementary Figure12A, Supplementary Table18). Identical to our patient analyses, NMF-based stratification using the responder signature (147/225-genes identified) segregated cell lines into two clusters (35% in cluster C1, 65% in cluster C2). Inter-group comparison of drug responses revealed C2 to have significantly lower AUC (P=0.0132) and corresponding IC50 values (P=0.0467), predicting C2 as GSi-sensitive (Supplementary Figure12B). We refined the theranostic signature applying Akaike Information Criterion (AIC) backwards elimination to generate an optimal sub-signature to predict whether cell lines displayed nanomolar or micromolar GSi-sensitivity in vitro. Accordingly, 20 core-signature genes individually displayed modest predictive capacity (AUC=0.4514-=0.7209; Supplementary Table19) but combined discriminated GSi-response with AUC=1 (Figure7E). In contrast, NOTCH1 (AUC=0.55, 95%CI:0.37-=0.74), NOTCH2 (AUC=0.61, 95%CI:0.46-=0.76) and NOTCH3 (AUC=0.50, 95%CI:0.32-=0.68) expression in addition to other GS-interactors poorly discriminate GSi-sensitivity. Crucially, we adapted the 20-gene signature into a linear model generating a GSi-sensitivity score (Figure7F). These data support the probability of our GSi-responder signature to identify prospective responder patients across diverse cancers, laying the foundation for GSi-based precision medicine.
DISCUSSION
In chemotherapeutic management of advanced cancer patients, it is imperative to clearly demarcate the extent of acceptable toxicity versus clinical response. In contrast to most other cancers, CCA is unique because there are no viable treatment options for more than 70% patients with advanced and metastatic disease. In this study, we identified a subgroup of advanced CCA patients with adverse prognostic features which is distinguished by extensive NOTCH network dysregulation. Given the modest survival advantage offered by current GemCis regimens(7), CCA patients may represent an ideal demographic for drug repositioning trials, such as adaptation of GSi. Successful implementation of GSi in other cancer types has been limited due to adverse toxicities, in particular affecting the gastrointestinal tract, shifting support to more targeted therapies, such as monoclonal antibodies directed against individual NOTCH receptors or ligands. However, here we provide data which indicate significant correlation between NOTCH1 and NOTCH3 expression as well as multiple ligands in patient tumors, thus undermining prospective therapeutic potential of individual targets. In comparison, GSi can block activation of all four NOTCH receptors preventing compensatory effects and diminishing potential for developing resistance mechanisms. Still, GSi usage may be confounded by the greater diversity of GS-targets which are reportedly in excess of 100 proteins(30). Indeed, our data consistently indicated that the NOTCH pathway is the most significantly affected pathway following GSi treatment and that predicted GSi-sensitive samples are significantly enriched in NOTCH pathway activity across patients, CCA cell lines and mouse models. Of significance, the GSi-responder signature could significantly predict classification in diverse solid malignancies, although the expression status of alternative GS targets should also be considered. Overall, given the poor classification power of NOTCH receptor expression to predict sensitivity to GSi, it is clear that GSi response is defined by the signature rather than the NOTCH receptor alone.
Prospective clinical applications of GSi for CCA should be directed towards the patients with advanced disease which comprise the majority of cases at diagnosis. One contextualized application of GSi could involve cancer stem cell (CSC) depletion, thereby acting as a cellular differentiation therapy. Here, we observed a significant association between NOTCH3 and tumor differentiation, and demonstrate an enrichment of a liver CSC signature previously identified by Yamashita and colleagues(31) in predicted GSi-responder patients. CCA has been reported to display 10- to 20-fold greater numbers of cells expressing cancer stemness markers compared to other malignancies(3), suggesting enhanced sensitivity to compounds with differentiating effects. Another potential application of GSi treatment based on our study is related to the capacity to inhibit metastasis, thus defining GSi as a ‘migrastatić drug. Increasing evidence supports a role for NOTCH signaling in metastasis, exemplified by the capacity of NOTCH1 to trigger pre-metastatic niche formation(32). In CCA, we observed elevated NOTCH1 to be associated with lymph node metastasis in advanced disease patients as well as enrichment of metastasis signatures in predicted GSi-responders. GSi could also be administered as a synergistic chemo-sensitizing agent along with a more standard chemotherapy. Preliminary evidence in vitro has indicated that gemcitabine monotherapy may result in elevated numbers of CCA cells with CSC markers, an effect abolished by GSi combination therapy(19). Opposingly, our data indicated that GSi-responder tumors are not likely to be valid immune checkpoint blockade (ICB) candidates due to predicted infiltrative T cell dysfunction, though immune reinvigoration through epigenomic agents(33) or in epigenetically-compromised patients, such as ARID1A mutants(34), could potentially be feasible.
While the differentiating, migrastatic, and chemo-sensitizing prospects of GSi are highly appealing for CCA, the dose and scheduling of such treatment appear to be a challenge, which is reflected in the historically poor performance of GSi in clinical trials. However, it is inappropriate to dismiss this class of compounds on the basis of the highly heterogeneous, under-powered and non-biomarker-guided clinical trials to date. Acute side effects, in particular gastrointestinal toxicities and diarrhea, plagued early trials due to poor dosing and administration schedules. Rather, sub-acute intermittent dosing, as well as co-administration of glucocorticoids, have been found to substantially minimize toxicities, as reported from multiple phase I studies(35, 36). Further, among mixed solid malignancies treated with RO4929097 (one of the GSi used in this study), the differentiation effects on tumor tissues were found to persist after end treatment. This suggests that a temporal window of drug exposure may only be required to exert long-lasting anti-tumor effects(36). Thus, biomarker-guided patient selection, optimized dosing schedules and careful management of adverse events together hold great potential to revisit and reinvigorate GSi as viable agents for advanced CCA.
In conclusion, reassessment of the NOTCH pathway classically considered as difficult to modulate and tolerate now appears as a feasible and much needed therapeutic avenue for CCA patients. Given the status of CCA as a rare orphan disease, and encouraged by the results from our pan-cancer analyses, stratification of diverse cancer patients using our GSi-responder signature may facilitate a basket trial approach to accelerate the critical assessment of NOTCH-directed GS modulation potentially in a combination therapy with other agents.
MATERIALS & METHODS
CCA PATIENT COHORTS & TRANSCRIPTOME PROFILING
In total, whole transcriptome data from 341 resected CCA tissues across 4 cohorts were analyzed in this study. Discovery cohort 1 (LEC2012) comprised of our previously published dataset of 104 tumors and 60 surrounding normal (SN) samples subjected to whole transcriptome profiling using humanRef-8v2 BeadChips (Illumina)(22). Discovery cohort 2 (LEC2018), included additionally 82 surgically resected CCAs (Supplementary TableS1–2), profiled along with 71 SN with Human HT-12 v3 Expression BeadChips (Illumina), as per manufacturer’s instructions. Validation cohorts comprising RNA-seq data of 119 CCA samples(1) and 36 (TCGA_CHOL network)(37) were employed to evaluate our GSi-responder signature developed from subcutaneous in vivo models. For further information, see Supplementary Materials & Methods.
STATISTICAL TESTS
Statistical analyses were performed using GraphPad Prism 7, unless otherwise stated. Gaussian distribution was tested using D’Agostino-Pearson omnibus normality test, with appropriate T tests subsequently applied. Degrees of statistical significance are indicated using standardized asterisk nomenclature (*P<0.05; **P<0.01; ***P<0.001; ****P<0.0001). Univariate analysis of binarized clinicopathologic parameters and NOTCH receptor expression were performed using glm function in R(v3.5.0). ROC curves and AUC values were computed using ‘pROC’, ‘ROCR’ and ‘ModelGood’ R packages.
Supplementary Material
ACKNOWLEDMENTS
JBA thanks the animal facility at the Center for Cancer Research, NCI. The results published here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Funding: The laboratory of JBA is supported by competitive funding from the Danish Medical Research Council, Danish Cancer Society, Novo Nordisk Foundation, and A.P. Møller Foundation. CJO is funded by a Marie Sklodowska-Curie postdoctoral fellowship. CN is supported by the Danish Medical Research Council individual postdoctoral fellowship.
Footnotes
Disclosure: The authors have nothing to disclose
Financial & competing interest’s disclosure: The authors have no relevant affiliation or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in this manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
REFERENCES
- 1.Nakamura H, Arai Y, Totoki Y, Shirota T, Elzawahry A, Kato M, Hama N, et al. Genomic spectra of biliary tract cancer. Nat Genet 2015;47:1003–1010. [DOI] [PubMed] [Google Scholar]
- 2.Hogdall D, Lewinska M, Andersen JB. Desmoplastic Tumor Microenvironment and Immunotherapy in Cholangiocarcinoma. Trends Cancer 2018;4:239–255. [DOI] [PubMed] [Google Scholar]
- 3.Cardinale V, Renzi A, Carpino G, Torrice A, Bragazzi MC, Giuliante F, DeRose AM, et al. Profiles of cancer stem cell subpopulations in cholangiocarcinomas. Am J Pathol 2015;185:1724–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yao KJ, Jabbour S, Parekh N, Lin Y, Moss RA. Increasing mortality in the United States from cholangiocarcinoma: an analysis of the National Center for Health Statistics Database. BMC Gastroenterol 2016;16:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Llovet JM, Villanueva A, Lachenmayer A, Finn RS. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol 2015;12:408–424. [DOI] [PubMed] [Google Scholar]
- 6.Endo I, Gonen M, Yopp AC, Dalal KM, Zhou Q, Klimstra D, D’Angelica M, et al. Intrahepatic cholangiocarcinoma: rising frequency, improved survival, and determinants of outcome after resection. Ann Surg 2008;248:84–96. [DOI] [PubMed] [Google Scholar]
- 7.Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, Madhusudan S, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 2010;362:1273–1281. [DOI] [PubMed] [Google Scholar]
- 8.Rizvi S, Khan SA, Hallemeier CL, Kelley RK, Gores GJ. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol 2018;15:95–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nepal C, O’Rourke CJ, Oliveira D, Taranta A, Shema S, Gautam P, Calderaro J, et al. Genomic perturbations reveal distinct regulatory networks in intrahepatic cholangiocarcinoma. Hepatology 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Javle M, Lowery M, Shroff RT, Weiss KH, Springfeld C, Borad MJ, Ramanathan RK, et al. Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced Cholangiocarcinoma. J Clin Oncol 2018;36:276–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zong Y, Panikkar A, Xu J, Antoniou A, Raynaud P, Lemaigre F, Stanger BZ. Notch signaling controls liver development by regulating biliary differentiation. Development 2009;136:1727–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sekiya S, Suzuki A. Intrahepatic cholangiocarcinoma can arise from Notch-mediated conversion of hepatocytes. J Clin Invest 2012;122:3914–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan B, Malato Y, Calvisi DF, Naqvi S, Razumilava N, Ribback S, Gores GJ, et al. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest 2012;122:2911–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zender S, Nickeleit I, Wuestefeld T, Sorensen I, Dauch D, Bozko P, El-Khatib M, et al. A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell 2013;23:784–795. [DOI] [PubMed] [Google Scholar]
- 15.Ding N, Che L, Li XL, Liu Y, Jiang LJ, Fan B, Tao JY, et al. Oncogenic potential of IDH1R132C mutant in cholangiocarcinoma development in mice. World J Gastroenterol 2016;22:2071–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, Tao J, Cigliano A, Sini M, Calderaro J, Azoulay D, Wang C, et al. Co-activation of PIK3CA and Yap promotes development of hepatocellular and cholangiocellular tumors in mouse and human liver. Oncotarget 2015;6:10102–10115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guest RV, Boulter L, Kendall TJ, Minnis-Lyons SE, Walker R, Wigmore SJ, Sansom OJ, et al. Cell lineage tracing reveals a biliary origin of intrahepatic cholangiocarcinoma. Cancer Res 2014;74:1005–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guest RV, Boulter L, Dwyer BJ, Kendall TJ, Man TY, Minnis-Lyons SE, Lu WY, et al. Notch3 drives development and progression of cholangiocarcinoma. Proc Natl Acad Sci U S A 2016;113:12250–12255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aoki S, Mizuma M, Takahashi Y, Haji Y, Okada R, Abe T, Karasawa H, et al. Aberrant activation of Notch signaling in extrahepatic cholangiocarcinoma: clinicopathological features and therapeutic potential for cancer stem cell-like properties. BMC Cancer 2016;16:854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Dong M, Xu Z, Song X, Zhang S, Qiao Y, Che L, et al. Notch2 controls hepatocyte-derived cholangiocarcinoma formation in mice. Oncogene 2018;37:3229–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huntzicker EG, Hotzel K, Choy L, Che L, Ross J, Pau G, Sharma N, et al. Differential effects of targeting Notch receptors in a mouse model of liver cancer. Hepatology 2015;61:942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andersen JB, Spee B, Blechacz BR, Avital I, Komuta M, Barbour A, Conner EA, et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 2012;142:1021–1031 e1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jusakul A, Cutcutache I, Yong CH, Lim JQ, Huang MN, Padmanabhan N, Nellore V, et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov 2017;7:1116–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meurette O, Mehlen P. Notch Signaling in the Tumor Microenvironment. Cancer Cell 2018. [DOI] [PubMed] [Google Scholar]
- 26.Matter MS, Marquardt JU, Andersen JB, Quintavalle C, Korokhov N, Stauffer JK, Kaji K, et al. Oncogenic driver genes and the inflammatory microenvironment dictate liver tumor phenotype. Hepatology 2016;63:1888–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schreck KC, Taylor P, Marchionni L, Gopalakrishnan V, Bar EE, Gaiano N, Eberhart CG. The Notch target Hes1 directly modulates Gli1 expression and Hedgehog signaling: a potential mechanism of therapeutic resistance. Clin Cancer Res 2010;16:6060–6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watters JW, Cheng C, Majumder PK, Wang R, Yalavarthi S, Meeske C, Kong L, et al. De novo discovery of a gamma-secretase inhibitor response signature using a novel in vivo breast tumor model. Cancer Res 2009;69:8949–8957. [DOI] [PubMed] [Google Scholar]
- 29.Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 2018;24:1550–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haapasalo A, Kovacs DM. The many substrates of presenilin/gamma-secretase. J Alzheimers Dis 2011;25:3–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY, Jia H, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology 2009;136:1012–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wieland E, Rodriguez-Vita J, Liebler SS, Mogler C, Moll I, Herberich SE, Espinet E, et al. Endothelial Notch1 Activity Facilitates Metastasis. Cancer Cell 2017;31:355–367. [DOI] [PubMed] [Google Scholar]
- 33.Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, Carter R, et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017;170:142–157 e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, Nagel ZD, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med 2018;24:556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sahebjam S, Bedard PL, Castonguay V, Chen Z, Reedijk M, Liu G, Cohen B, et al. A phase I study of the combination of ro4929097 and cediranib in patients with advanced solid tumours (PJC-004/NCI 8503). Br J Cancer 2013;109:943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tolcher AW, Messersmith WA, Mikulski SM, Papadopoulos KP, Kwak EL, Gibbon DG, Patnaik A, et al. Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J Clin Oncol 2012;30:2348–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Farshidfar F, Zheng S, Gingras MC, Newton Y, Shih J, Robertson AG, Hinoue T, et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep 2017;18:2780–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.